
- Department of Psychiatry, Brudnick Neuropsychiatric Research Institute, University of Massachusetts Medical School, Worcester, MA, USA
Ion channels modulate ion flux across cell membranes, activate signal transduction pathways, and influence cellular transport—vital biological functions that are inexorably linked to cellular processes that go awry during carcinogenesis. Indeed, deregulation of ion channel function has been implicated in cancer-related phenomena such as unrestrained cell proliferation and apoptotic evasion. As the prototype for ligand-gated ion channels, nicotinic acetylcholine receptors (nAChRs) have been extensively studied in the context of neuronal cells but accumulating evidence also indicate a role for nAChRs in carcinogenesis. Recently, variants in the nAChR genes CHRNA3, CHRNA5, and CHRNB4 have been implicated in nicotine dependence and lung cancer susceptibility. Here, we silenced the expression of these three genes to investigate their function in lung cancer. We show that these genes are necessary for the viability of small cell lung carcinomas (SCLC), the most aggressive type of lung cancer. Furthermore, we show that nicotine promotes SCLC cell viability whereas an α3β4-selective antagonist, α-conotoxin AuIB, inhibits it. Our findings posit a mechanism whereby signaling via α3/α5/β4-containing nAChRs promotes lung carcinogenesis.
Introduction
Lung cancer remains the leading cause of cancer-related deaths worldwide (WHO, 2011). Despite considerable research efforts to elucidate the molecular underpinnings of the disease, the 5-year survival rate for lung cancer has not changed appreciably over the past three decades and persists at a dismal 15%. The two major types of lung cancer are non-small cell lung carcinoma (NSCLC) and SCLC. The former consists of a heterogeneous group of tumors that account for 80% of lung cancer cases while the latter is less common (15–20% of cases) but is particularly aggressive (Rom et al., 2000; Sandler, 2003). SCLC is characterized by rapid growth and early dissemination resulting in an extremely poor prognosis for which no effective treatments are currently available (Rudin et al., 2008).
Cigarette smoking is the major risk factor associated with lung cancer. This is not surprising given that tobacco contains ~250 damaging chemicals and ~50 carcinogens (Hecht, 1999). In the United States alone, over 45 million adults continue to smoke while globally, 10 million cigarettes are sold every minute, making tobacco use the leading cause of preventable deaths (WHO, 2011). Prevention efforts are hampered, however, by the strong reinforcing effects of nicotine, the primary psychoactive component in tobacco.
Nicotine's effects are mediated by nAChRs that are expressed in the reward circuitry and other areas of the brain (Albuquerque et al., 2009). nAChRs are also activated by the endogenous ligand acetylcholine (ACh), hence their name. Additionally, nAChRs are activated by the nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), the most potent carcinogen in tobacco (Schuller, 2007). Receptor activation allows the flow of sodium, potassium and calcium ions down their electrochemical gradients.
nAChRs are composed of transmembrane subunits that share a common evolutionary origin (Le Novere et al., 2002). In mammalian systems, these subunits are encoded by eleven genes located across different chromosomes (Table 1). The genes encoding the human α3, α5, and β4 subunits are found in a gene cluster in chromosome 15q24 and are thought to be both independently and coordinately regulated (Boulter et al., 1990; Scofield et al., 2008). The α3 subunit is usually co-expressed with the β4 subunit while α5 serves as an auxiliary subunit, whose incorporation modifies the calcium permeability of the receptor and its affinity to and desensitization by agonists (Ramirez-Latorre et al., 1996; Yu and Role, 1998). α3β4-containing nAChRs exhibit lower affinity for nicotine and are less desensitized by it, suggesting that this receptor subtype may mediate nicotine's rewarding effects after high affinity nAChR subtypes have been desensitized (Paradiso and Steinbach, 2003; Rose, 2007). Moreover, α3β4α5 nAChRs are thought to play a role in nicotine withdrawal and consistently, are highly expressed in brain regions associated with nicotine withdrawal, such as the medial habenula and the interpeduncular nucleus (Damaj et al., 2003; Salas et al., 2009).
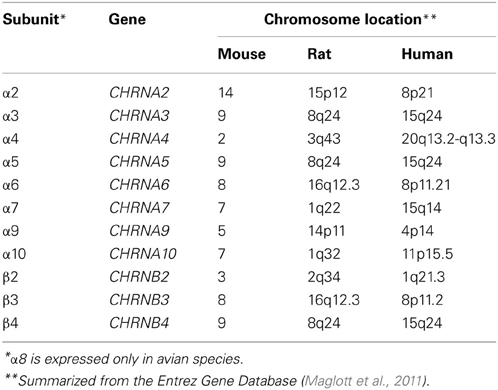
Table 1. Chromosomal locations of genes encoding nAChR subunits.
Multiple genome-wide association studies (GWAS) have implicated the CHRNB4/A3/A5 locus in nicotine dependence and lung cancer (Amos et al., 2008; Hung et al., 2008; Thorgeirsson et al., 2008). A particularly interesting variant in this locus is the non-synonymous single nucleotide polymorphism (SNP) that lies in the fifth exon of CHRNA5 (rs16969968). This variant encodes a change from an aspartic acid to an asparagine residue at amino acid position 398 (D398N). The asparagine risk allele is associated with decreased maximal response to agonists, indicating altered receptor function (Bierut et al., 2008; George et al., 2012). Additionally, the genotype in this locus appears to correlate with mRNA levels suggesting that rs16969968 may influence CHRNA5 expression as well (Falvella et al., 2009; Wang et al., 2009). In α3β4α5 nAChRs, the 398 residue also lies close to a β4 residue that is necessary for β 4′s ability to increase nicotine-evoked currents, which subsequently leads to nicotine aversion (Frahm et al., 2011). Notably, this increase in current is maximally competed by the D398N variant, resulting in reversal of nicotine aversion. Altogether, these results support the functional relevance of the rs16969968 variant.
The association of nAChR variants with both nicotine dependence and lung cancer susceptibility prompts two hypotheses regarding the role of nAChRs in lung cancer. One hypothesis is that nicotine mainly influences nAChRs in the brain, such that increased levels of nicotine dependence consequently lead to greater exposure to tobacco carcinogens and to lung cancer development (Le Marchand et al., 2008). Consistently, α3, α5, and β4 nAChR subunits are predominantly expressed in select neural circuits that control nicotine intake in rodent nicotine dependence models (Fowler et al., 2011; Frahm et al., 2011). An alternative hypothesis is that the association between nAChR variants and lung cancer is direct, in that altered nAChR function, as encoded by risk alleles, promote carcinogenic processes in the lungs and airway tissues (Schuller, 2009). The following sections discuss evidence in the literature as well as primary data that support a direct role for nAChRs in lung cancer.
Expression and Function of nAChRs in Lung Cancer
The first hint that nAChRs play a direct role in lung cancer comes from several studies demonstrating nAChR expression in several types of cancers (Table 2). In lung cancer, we have detected the expression of several nAChR subunit genes, in particular CHRNA3, CHRNA5, and CHRNB4 (Improgo et al., 2010). Differences in nAChR gene expression between smokers and non-smokers have also been reported (Lam et al., 2007).
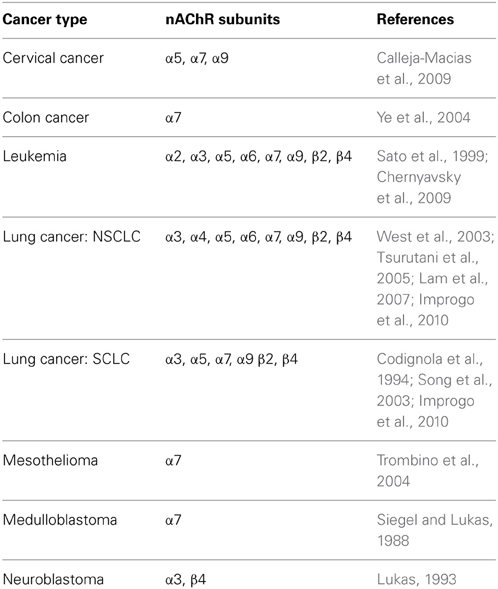
Table 2. Types of cancer cells expressing nAChR subunits.
Another line of evidence stems from studies showing that nAChR ligands promote several hallmarks of cancer (Hanahan and Weinberg, 2000; Schuller, 2009). Nicotine induces cell proliferation in lung cancer cells via protein kinase C (Schuller, 1989; Codignola et al., 1994) and Akt (West et al., 2003; Tsurutani et al., 2005) activation. Nicotine's carcinogenic metabolites have also been shown to promote cell proliferation in lung cancer cells via serotonin-induced stimulation of the Raf-1/MAPK/c-myc pathway (Schuller and Orloff, 1998; Jull et al., 2001) and the Akt pathway (West et al., 2003; Tsurutani et al., 2005).
In addition, nicotine has been shown to inhibit apoptosis by phosphorylation of Bcl-2 family members (Jin et al., 2004a). Apoptotic evasion potentially contributes to nicotine-induced chemoresistance (Maneckjee and Minna, 1990, 1994). Similarly, NNK inhibits apoptosis by Bcl-2 phosphorylation (Jin et al., 2004b). Moreover, both nicotine and NNK promote cell survival via the NF-kB pathway (Tsurutani et al., 2005).
Intriguingly, the complete cholinergic system is expressed and functional in lung cells, where ACh acts both as an autocrine and paracrine growth factor (Song et al., 2003; Proskocil et al., 2004). Once activated, this growth loop may provide endogenous mitogenic signaling without further nicotine activation. Such a mechanism may explain residual risk for lung cancer even after smoking cessation.
ACh is also thought to act as a pro-angiogenic signal via autocrine and paracrine signaling in endothelial cells (Heeschen et al., 2002). Nicotine promotes angiogenesis in a PI3-kinase and MAPK-dependent manner (Heeschen et al., 2001). Moreover, nicotine and its metabolite cotinine have been shown to up-regulate the expression of the pro-angiogenic factor VEGF (Conklin et al., 2002).
Many of these cancer-promoting processes are abrogated by pan-nAChR or subtype-selective antagonists. Nicotine-induced cell proliferation via fibronectin up-regulation can be abolished by α-BTx (Zheng et al., 2007). Both α-BTx and the non-selective nAChR inhibitor, mecamylamine, also hinder angiogenic growth in vitro (Heeschen et al., 2002). Additionally, nicotine-induced Akt activation and airway cell transformation can be inhibited by the α4β2-selective antagonist, DHβE (West et al., 2003). These observations collectively indicate that many of the above cancer-related processes are mediated by nAChRs.
CHRNA3/A5/B4 in SCLC
Given the GWAS association between the CHRNB4/A3/A5 locus and lung cancer risk, we pursued the hypothesis that nAChRs containing the α3, α5, and β4 subunits play a direct role in the development of lung cancer. We focused on SCLC as we had previously observed high expression of CHRNA5 and upregulation of CHRNA3 and CHRNB4 in SCLC (Improgo et al., 2010). We investigated the role of CHRNA3, CHRNA5, and CHRNB4 in SCLC by silencing the expression of these three genes in the SCLC cell line, DMS-53. Three distinct siRNAs against each gene were used (Figure 1A). We assessed the effect of knockdown using a bioluminescence-based cell viability assay as previously described (Improgo et al., 2011). All the siRNAs that yielded > 90% knockdown levels produced corresponding decreases in SCLC cell viability (n = 5 each, Figure 1B), suggesting that expression of CHRNA3, CHRNA5, and CHRNB4 is vital for SCLC cell viability, at least in the context of the DMS-53 cell line.
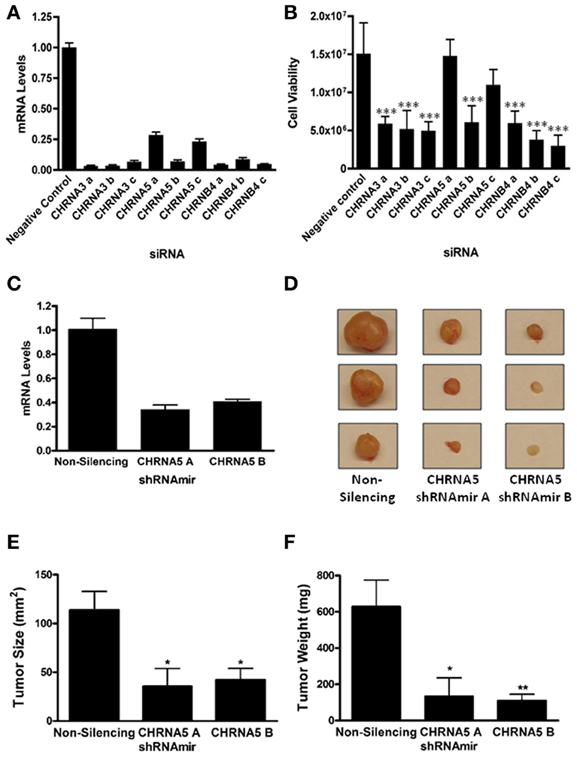
Figure 1. CHRNA3, CHRNA5, and CHRNB4 depletion decreases SCLC cell growth. DMS-53 cells were treated with three distinct CHRNA3, CHRNA5, and CHRNB4 siRNAs or a negative control siRNA (Applied Biosystems). (A) Quantitative RT-PCR was performed to determine mRNA levels upon knockdown. (B) A bioluminescence-based cell viability assay was performed to determine the effect of siRNA treatment on SCLC cell viability (***p < 0.001, ANOVA, Tukey post-hoc test). (C) CHRNA5 mRNA levels in DMS-53 cells stably expressing a non-silencing shRNAmir or two CHRNA5 shRNAmirs (Open Biosystems). shRNAmirs were transduced into DMS-53 cells using a lentiviral delivery system for stable expression (Open Biosystems Trans-Lentiral Packaging System). (D) DMS-53 cells stably expressing a non-silencing shRNAmir or two CHRNA5 shRNAmirs were subcutaneously injected into the hind flanks of athymic nude mice. Tumors were harvested after 2 months (representative images shown). Tumor size (E) and weight (F) were significantly lower in samples treated with CHRNA5 shRNAmirs vs the non-silencing control (*p < 0.05; **p < 0.01, ANOVA, Tukey post-hoc test).
The two siRNAs that yielded the least knockdown (CHRNA5 a and c) did not significantly affect cell viability, suggesting that certain CHRNA5 depletion thresholds may need to be reached to obtain an observable phenotype. To therefore obtain more robust silencing, we utilized shRNAmirs, hairpins that are designed with flanking miRNA sequences that can harness the cell's endogenous RNAi machinery and promote more efficient knockdowns. In addition, we introduced these shRNAmirs to DMS-53 cells via lentiviral delivery to allow stable expression. For a more physiological approach, we used a tumor xenograft model for in vivo propagation of tumors. Cells were implanted into immunocompromised mice and tumor growth was monitored. CHRNA5 shRNAmir A treatment (n = 5) caused a 59% decrease in CHRNA5 levels while CHRNA5 shRNAmir B treatment (n = 9) caused a 66% decrease in mRNA levels (Figure 1C). Quite strikingly, tumor size and tumor weight (Figures 1D–F) were significantly lower in cells treated with the CHRNA5 shRNAmirs vs. the non-silencing shRNAmir control (n = 9). These results further support the in vitro data described above.
We next tested the effect of nAChR ligands on SCLC cell viability. Using the same bioluminescence assay as above, we observed that nicotine treatment increased SCLC cell viability (Figure 2A), consistent with aforementioned reports. To perform the converse experiment, we utilized the α3β4-selective ligand, α-conotoxin AuIB. α-conotoxins are derived from the venom of cone snails, a valuable source for disulfide-bonded peptides that target nAChRs in a highly subtype-selective manner (Azam and McIntosh, 2009). α-conotoxin AuIB, in particular, was isolated from the snail-eating cone Conus aurilicus and blocks α3β4 nAChRs with > 100-fold higher potency compared to other nAChR subtypes (Luo et al., 1998). Treatment with α-conotoxin AuIB led to decreased viability of DMS-53 cells (Figure 2A), indicating that functional α3β4 nAChRs are present in SCLC cells and are important for the maintenance of SCLC cell viability. In agreement with our genetic approach, this pharmacological approach suggests that activation and blockade of α3α5β4 nAChRs modulates SCLC cell viability.
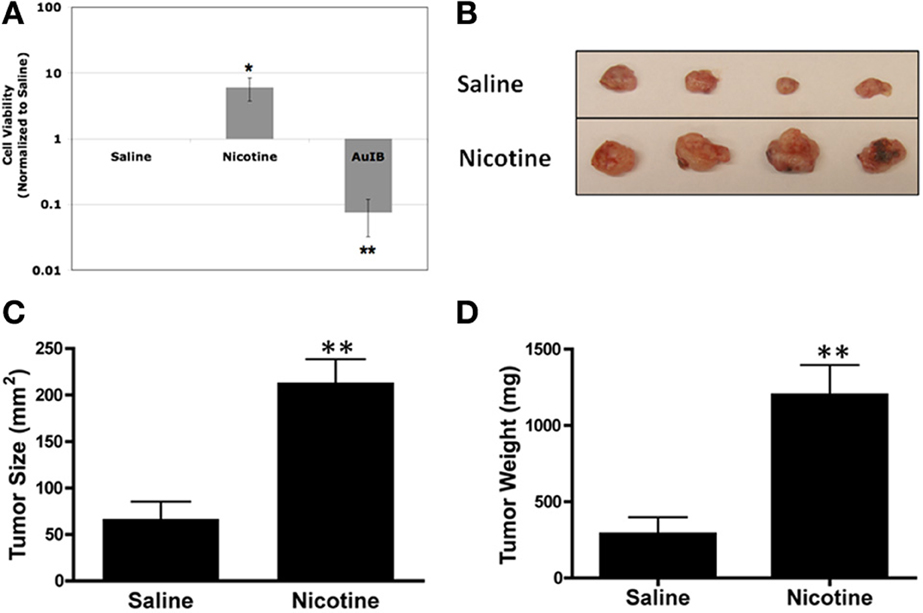
Figure 2. Pharmacological activation or inhibition of nAChRs modulates SCLC growth. (A) DMS-53 cells were treated daily for 1 week with 1 μM nicotine or 2 μM α-conotoxin AuIB. Cell viability assays show that nicotine increases while AuIB decreases SCLC cell viability (values normalized to saline control; *p < 0.05, **p < 0.01). (B) DMS-53 cells were injected subcutaneously into the hind flanks of athymic nude mice. Mice were then implanted with osmotic minipumps that delivered either saline or 24 mg/kg of nicotine daily. Tumors were harvested after 1 month (representative images shown). (C,D). Chronic nicotine exposure increased both tumor size and weight (**p < 0.01, Student's t-test).
We then tested the effect of chronic nicotine treatment on tumor growth in vivo. We used osmotic minipumps to deliver nicotine as these devices allow continued dosing of drugs while eliminating repeated injections (Salas et al., 2004). Using the same xenograft tumor model as above, we found that chronic nicotine treatment increased tumor size and weight in vivo compared to saline controls (Figures 2B–D). This is in line with previous findings showing that nicotine promotes tumor growth in various in vivo models (Davis et al., 2009; Al-Wadei et al., 2012).
Our results show that CHRNA3, CHRNA5, and CHRNB4 expression is critical for SCLC cell viability. These findings lend mechanistic support to the correlative link between the CHRNB4/A3/A5 locus and lung cancer susceptibility. That α3β4α5 nAChRs play a direct role in lung cancer, in addition to their role in the brain, points to the pleiotropic function of these genes. Along with published reports, our work suggests a mechanism by which cholinergic signaling via α3β4α5 nAChRs promotes SCLC growth. Though this may raise questions regarding the use of nicotine-based smoking cessation approaches, it also indicates the potential of nAChR antagonists for SCLC therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We express our sincere appreciation to Dr. J. Michael McIntosh for his generous gift of α-conotoxin AuIB.
Abbreviations
NSCLC, non-small cell lung carcinoma; SCLC, small cell lung carcinoma; ACh, acetylcholine; nAChRs, nicotinic acetylcholine receptors; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; GWAS, genome-wide association studies.
References
Albuquerque, E. X., Pereira, E. F., Alkondon, M., and Rogers, S. W. (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120. doi: 10.1152/physrev.00015.2008
Al-Wadei, H. A., Al-Wadei, M. H., Ullah, M. F., and Schuller, H. M. (2012). Gamma-amino butyric acid inhibits the nicotine-imposed stimulatory challenge in xenograft models of non-small cell lung carcinoma. Curr. Cancer Drug Targets 12, 97–106. doi: 10.2174/156800912799095171
Amos, C. I., Wu, X., Broderick, P., Gorlov, I. P., Gu, J., Eisen, T., et al. (2008). Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 40, 616–622. doi: 10.1038/ng.109
Azam, L., and McIntosh, J. M. (2009). Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 30, 771–783. doi: 10.1038/aps.2009.47
Bierut, L. J., Stitzel, J. A., Wang, J. C., Hinrichs, A. L., Grucza, R. A., Xuei, X., et al. (2008). Variants in nicotinic receptors and risk for nicotine dependence. Am. J. Psychiatry 165, 1163–1171. doi: 10.1176/appi.ajp.2008.07111711
Boulter, J., O'Shea-Greenfield, A., Duvoisin, R. M., Connolly, J. G., Wada, E., Jensen, A., et al. (1990). Alpha 3, alpha 5, and beta 4: three members of the rat neuronal nicotinic acetylcholine receptor-related gene family form a gene cluster. J. Biol. Chem. 265, 4472–4482.
Calleja-Macias, I. E., Kalantari, M., and Bernard, H. U. (2009). Cholinergic signaling through nicotinic acetylcholine receptors stimulates the proliferation of cervical cancer cells: an explanation for the molecular role of tobacco smoking in cervical carcinogenesis? Int. J. Cancer 124, 1090–1096. doi: 10.1002/ijc.24053
Chernyavsky, A. I., Arredondo, J., Galitovskiy, V., Qian, J., and Grando, S. A. (2009). Structure and function of the nicotinic arm of acetylcholine regulatory axis in human leukemic T cells. Int. J. Immunopathol. Pharmacol. 22, 461–472.
Codignola, A., Tarroni, P., Cattaneo, M. G., Vicentini, L. M., Clementi, F., and Sher, E. (1994). Serotonin release and cell proliferation are under the control of alpha-bungarotoxin-sensitive nicotinic receptors in small-cell lung carcinoma cell lines. FEBS Lett. 342, 286–290. doi: 10.1016/0014-5793(94)80518-0
Conklin, B. S., Zhao, W., Zhong, D. S., and Chen, C. (2002). Nicotine and cotinine up-regulate vascular endothelial growth factor expression in endothelial cells. Am. J. Pathol. 160, 413–418. doi: 10.1016/S0002-9440(10)64859-6
Damaj, M. I., Kao, W., and Martin, B. R. (2003). Characterization of spontaneous and precipitated nicotine withdrawal in the mouse. J. Pharmacol. Exp. Ther. 307, 526–534. doi: 10.1124/jpet.103.054908
Davis, R., Rizwani, W., Banerjee, S., Kovacs, M., Haura, E., Coppola, D., et al. (2009). Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 4:e7524. doi: 10.1371/journal.pone.0007524
Falvella, F. S., Galvan, A., Frullanti, E., Spinola, M., Calabro, E., Carbone, A., et al. (2009). Transcription deregulation at the 15q25 locus in association with lung adenocarcinoma risk. Clin. Cancer Res. 15, 1837–1842. doi: 10.1158/1078-0432.CCR-08-2107
Fowler, C. D., Lu, Q., Johnson, P. M., Marks, M. J., and Kenny, P. J. (2011). Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature 471, 597–601. doi: 10.1038/nature09797
Frahm, S., Slimak, M. A., Ferrarese, L., Santos-Torres, J., Antolin-Fontes, B., Auer, S., et al. (2011). Aversion to nicotine is regulated by the balanced activity of beta4 and alpha5 nicotinic receptor subunits in the medial habenula. Neuron 70, 522–535. doi: 10.1016/j.neuron.2011.04.013
George, A. A., Lucero, L. M., Damaj, M. I., Lukas, R. J., Chen, X., and Whiteaker, P. (2012). Function of human alpha3beta4alpha5 nicotinic acetylcholine receptors is reduced by the alpha5(D398N) variant. J. Biol. Chem. 287, 25151–25162. doi: 10.1074/jbc.M112.379339
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57–70. doi: 10.1016/S0092-8674(00)81683-9
Hecht, S. S. (1999). Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 91, 1194–1210. doi: 10.1093/jnci/91.14.1194
Heeschen, C., Jang, J. J., Weis, M., Pathak, A., Kaji, S., Hu, R. S., et al. (2001). Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 7, 833–839. doi: 10.1038/89961
Heeschen, C., Weis, M., Aicher, A., Dimmeler, S., and Cooke, J. P. (2002). A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Invest. 110, 527–536. doi: 10.1172/JCI200214676
Hung, R. J., McKay, J. D., Gaborieau, V., Boffetta, P., Hashibe, M., Zaridze, D., et al. (2008). A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637. doi: 10.1038/nature06885
Improgo, M. R., Johnson, C. W., Tapper, A. R., and Gardner, P. D. (2011). Bioluminescence-based high-throughput screen identifies pharmacological agents that target neurotransmitter signaling in small cell lung carcinoma. PLoS ONE 6:e24132. doi: 10.1371/journal.pone.0024132
Improgo, M. R., Schlichting, N. A., Cortes, R. Y., Zhao-Shea, R., Tapper, A. R., and Gardner, P. D. (2010). ASCL1 regulates the expression of the CHRNA5/A3/B4 lung cancer susceptibility locus. Mol. Cancer Res. 8, 194–203. doi: 10.1158/1541-7786.MCR-09-0185
Jin, Z., Gao, F., Flagg, T., and Deng, X. (2004a). Nicotine induces multi-site phosphorylation of Bad in association with suppression of apoptosis. J. Biol. Chem. 279, 23837–23844. doi: 10.1074/jbc.M402566200
Jin, Z., Gao, F., Flagg, T., and Deng, X. (2004b). Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 279, 40209–40219. doi: 10.1074/jbc.M404056200
Jull, B. A., Plummer, H. K. 3rd., and Schuller, H. M. (2001). Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 127, 707–717.
Lam, D. C., Girard, L., Ramirez, R., Chau, W. S., Suen, W. S., Sheridan, S., et al. (2007). Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 67, 4638–4647. doi: 10.1158/0008-5472.CAN-06-4628
Le Marchand, L., Derby, K. S., Murphy, S. E., Hecht, S. S., Hatsukami, D., Carmella, S. G., et al. (2008). Smokers with the CHRNA lung cancer-associated variants are exposed to higher levels of nicotine equivalents and a carcinogenic tobacco-specific nitrosamine. Cancer Res. 68, 9137–9140. doi: 10.1158/0008-5472.CAN-08-2271
Le Novere, N., Corringer, P. J., and Changeux, J. P. (2002). The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 53, 447–456. doi: 10.1002/neu.10153
Lukas, R. J. (1993). Expression of ganglia-type nicotinic acetylcholine receptors and nicotinic ligand binding sites by cells of the IMR-32 human neuroblastoma clonal line. J. Pharmacol. Exp. Ther. 265, 294–302.
Luo, S., Kulak, J. M., Cartier, G. E., Jacobsen, R. B., Yoshikami, D., Olivera, B. M., et al. (1998). alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 18, 8571–8579.
Maglott, D., Ostell, J., Pruitt, K. D., and Tatusova, T. (2011). Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 39, D52–D57. doi: 10.1093/nar/gkq1237
Maneckjee, R., and Minna, J. D. (1990). Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc. Natl. Acad. Sci. U.S.A. 87, 3294–3298. doi: 10.1073/pnas.87.9.3294
Maneckjee, R., and Minna, J. D. (1994). Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ. 5, 1033–1040.
Paradiso, K. G., and Steinbach, J. H. (2003). Nicotine is highly effective at producing desensitization of rat alpha4beta2 neuronal nicotinic receptors. J. Physiol. 553, 857–871. doi: 10.1113/jphysiol.2003.053447
Proskocil, B. J., Sekhon, H. S., Jia, Y., Savchenko, V., Blakely, R. D., Lindstrom, J., et al. (2004). Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 145, 2498–2506. doi: 10.1210/en.2003-1728
Ramirez-Latorre, J., Yu, C. R., Qu, X., Perin, F., Karlin, A., and Role, L. (1996). Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature 380, 347–351. doi: 10.1038/380347a0
Rom, W. N., Hay, J. G., Lee, T. C., Jiang, Y., and Tchou-Wong, K. M. (2000). Molecular and genetic aspects of lung cancer. Am. J. Respir. Crit. Care Med. 161, 1355–1367. doi: 10.1164/ajrccm.161.4.9908012
Rose, J. E. (2007). Multiple brain pathways and receptors underlying tobacco addiction. Biochem. Pharmacol. 74, 1263–1270. doi: 10.1016/j.bcp.2007.07.039
Rudin, C. M., Hann, C. L., Peacock, C. D., and Watkins, D. N. (2008). Novel systemic therapies for small cell lung cancer. J. Natl. Compr. Canc. Netw. 6, 315–322.
Salas, R., Cook, K. D., Bassetto, L., and De Biasi, M. (2004). The α3 and β4 nicotinic receptor subunits are necessary for nicotine-induced seizures and hypolocomotion in mice. Neuropharmacology 47, 401–407. doi: 10.1016/j.neuropharm.2004.05.002
Salas, R., Sturm, R., Boulter, J., and De Biasi, M. (2009). Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J. Neurosci. 29, 3014–3018. doi: 10.1523/JNEUROSCI.4934-08.2009
Sandler, A. B. (2003). Chemotherapy for small cell lung cancer. Semin. Oncol. 30, 9–25. doi: 10.1053/sonc.2003.50012
Sato, K. Z., Fujii, T., Watanabe, Y., Yamada, S., Ando, T., Kazuko, F., et al. (1999). Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci. Lett. 266, 17–20. doi: 10.1016/S0304-3940(99)00259-1
Schuller, H. M. (1989). Cell type specific, receptor-mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem. Pharmacol. 38, 3439–3442. doi: 10.1016/0006-2952(89)90112-3
Schuller, H. M. (2007). Nitrosamines as nicotinic receptor ligands. Life Sci. 80, 2274–2280. doi: 10.1016/j.lfs.2007.03.006
Schuller, H. M. (2009). Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 9, 195–205. doi: 10.1038/nrc2590
Schuller, H. M., and Orloff, M. (1998). Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem. Pharmacol. 55, 1377–1384. doi: 10.1016/S0006-2952(97)00651-5
Scofield, M. D., Bruschweiler-Li, L., Mou, Z., and Gardner, P. D. (2008). Transcription factor assembly on the nicotinic receptor beta4 subunit gene promoter. Neuroreport 19, 687–690. doi: 10.1097/WNR.0b013e3282fbcef7
Siegel, H. N., and Lukas, R. J. (1988). Nicotinic agonists regulate alpha-bungarotoxin binding sites of TE671 human medulloblastoma cells. J. Neurochem. 50, 1272–1278. doi: 10.1111/j.1471-4159.1988.tb10604.x
Song, P., Sekhon, H. S., Jia, Y., Keller, J. A., Blusztajn, J. K., Mark, G. P., et al. (2003). Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res. 63, 214–221.
Thorgeirsson, T. E., Geller, F., Sulem, P., Rafnar, T., Wiste, A., Magnusson, K. P., et al. (2008). A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452, 638–642. doi: 10.1038/nature06846
Trombino, S., Cesario, A., Margaritora, S., Granone, P., Motta, G., Falugi, C., et al. (2004). Alpha7-nicotinic acetylcholine receptors affect growth regulation of human mesothelioma cells: role of mitogen-activated protein kinase pathway. Cancer Res. 64, 135–145. doi: 10.1158/0008-5472.CAN-03-1672
Tsurutani, J., Castillo, S. S., Brognard, J., Granville, C. A., Zhang, C., Gills, J. J., et al. (2005). Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis 26, 1182–1195. doi: 10.1093/carcin/bgi072
Wang, J. C., Cruchaga, C., Saccone, N. L., Bertelsen, S., Liu, P., Budde, J. P., et al. (2009). Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum. Mol. Genet. 18, 3125–3135. doi: 10.1093/hmg/ddp231
West, K. A., Brognard, J., Clark, A. S., Linnoila, I. R., Yang, X., Swain, S. M., et al. (2003). Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Invest. 111, 81–90. doi: 10.1172/JCI200316147
Ye, Y. N., Liu, E. S., Shin, V. Y., Wu, W. K., and Cho, C. H. (2004). The modulating role of nuclear factor-kappaB in the action of alpha7-nicotinic acetylcholine receptor and cross-talk between 5-lipoxygenase and cyclooxygenase-2 in colon cancer growth induced by 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. J. Pharmacol. Exp. Ther. 311, 123–130. doi: 10.1124/jpet.104.068031
Yu, C. R., and Role, L. W. (1998). Functional contribution of the alpha5 subunit to neuronal nicotinic channels expressed by chick sympathetic ganglion neurones. J. Physiol. 509(Pt 3), 667–681. doi: 10.1111/j.1469-7793.1998.667bm.x
Keywords: nicotinic acetylcholine receptor, ligand-gated ion channel, lung cancer, small cell lung carcinoma, CHRNA5
Citation: Improgo MR, Soll LG, Tapper AR and Gardner PD (2013) Nicotinic acetylcholine receptors mediate lung cancer growth. Front. Physiol. 4:251. doi: 10.3389/fphys.2013.00251
Received: 15 May 2013; Accepted: 26 August 2013;
Published online: 17 September 2013.
Edited by:
Andrea Becchetti, University of Milano-Bicocca, ItalyReviewed by:
Michael P. Blanton, Texas Tech University Health Sciences Center, USAEliot Spindel, Oregon Health and Science University, USA
Scott W. Rogers, University of Utah School of Medicine, USA
Copyright © 2013 Improgo, Soll, Tapper and Gardner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul D. Gardner, Department of Psychiatry, Brudnick Neuropsychiatric Research Institute, University of Massachusetts Medical School, 303 Belmont Street, Worcester, MA 01604, USA e-mail: paul.gardner@umassmed.edu
†Present address: Ma. Reina Improgo, Dana-Farber Cancer Institute, Harvard Medical School, Boston, USA