 Justin G. Boyer1,2
Justin G. Boyer1,2 Rashmi Kothary
Rashmi Kothary- 1Ottawa Hospital Research Institute, Regenerative Medicine Program, Ottawa ON, Canada
- 2Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, ON, Canada
- 3Department of Medicine, University of Ottawa, Ottawa, ON, Canada
Spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), and spinal-bulbar muscular atrophy (SBMA) are devastating diseases characterized by the degeneration of motor neurons. Although the molecular causes underlying these diseases differ, recent findings have highlighted the contribution of intrinsic skeletal muscle defects in motor neuron diseases. The use of cell culture and animal models has led to the important finding that muscle defects occur prior to and independently of motor neuron degeneration in motor neuron diseases. In SMA for instance, the muscle specific requirements of the SMA disease-causing gene have been demonstrated by a series of genetic rescue experiments in SMA models. Conditional ALS mouse models expressing a muscle specific mutant SOD1 gene develop atrophy and muscle degeneration in the absence of motor neuron pathology. Treating SBMA mice by over-expressing IGF-1 in a skeletal muscle-specific manner attenuates disease severity and improves motor neuron pathology. In the present review, we provide an in depth description of muscle intrinsic defects, and discuss how they impact muscle function in these diseases. Furthermore, we discuss muscle-specific therapeutic strategies used to treat animal models of SMA, ALS, and SBMA. The study of intrinsic skeletal muscle defects is crucial for the understanding of the pathophysiology of these diseases and will open new therapeutic options for the treatment of motor neuron diseases.
Introduction
Everything from physical exercise to daily chores and even breathing depends on force generated by skeletal muscles. For a skeletal muscle to produce a contraction, a signal in the form of an action potential is required. The motor neuron is responsible for providing this required signal. The site where the motor neuron joins the muscle is called the neuromuscular junction and together, the motor neuron and the muscle are referred to as the motor unit.
Defects in the motor unit can seriously impact muscle contraction generation and lead to severely disabling diseases. Motor neuron diseases consist of a group of conditions characterized by motor neuron loss and atrophy of the associated musculature. Spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), and spinal-bulbar muscular atrophy (SBMA) are examples of such motor neuron diseases. Although these diseases have quite different etiologies, SMA, ALS, or SBMA are all typified by progressive paralysis resulting in severe disability, and are commonly fatal.
Although the importance of motor neuron pathology is well-established in these diseases, recent work has revealed an involvement of other cell types, including myocytes, in the pathogenic process (Bricceno et al., 2012a; Hamilton and Gillingwater, 2013). Muscle weakness and atrophy are often tightly associated with motor neuron pathology. Importantly, however, use of both cell culture and conditional mouse models has revealed defects in skeletal muscle that occur in the absence of defective motor neurons. Such studies highlight a potential contribution of skeletal muscle defects to the symptoms of SMA, ALS, and SBMA patients. These findings have major implications for the development of therapeutics for these diseases. In the current review, we discuss the latest findings regarding intrinsic muscle defects as well as muscle-specific therapeutic strategies to treat SMA, ALS, and SBMA.
Spinal Muscular Atrophy
Disease Characteristics
SMA represents the leading genetic cause of infant death, affecting 1 in 10,000 live births per year (Pearn, 1978; Prior et al., 2010). Clinically, it is typified by progressive muscle weakness and loss of alpha motor neurons from the spinal cord. SMA is caused by mutations or deletions in the SMN1 gene (Lefebvre et al., 1995). Due to a second, partially functionally copy, named SMN2, SMA is a disease of low SMN levels, rather than no SMN (Brzustowicz et al., 1993; Rochette et al., 2001). The clinical severity of SMA is categorized into 4 main types, which vary in their time of onset and expected prognosis (reviewed in Boyer et al., 2010). Furthermore, SMN2 serves as a disease modifier since the copy number of the SMN2 gene in SMA patients modulates disease severity.
The SMN protein is ubiquitously expressed and localizes to the nucleus and cytoplasm (Liu and Dreyfuss, 1996). In the nucleus, SMN is present in gemini of coiled bodies (gems) which are structures associated with Cajal bodies (Liu and Dreyfuss, 1996; Coovert et al., 1997; Lefebvre et al., 1997). Here, SMN forms a complex with Gemin 2–8 as well as with Sm proteins to regulate small nuclear ribonucleoprotein (snRNP) biogenesis (Liu and Dreyfuss, 1996; Meister et al., 2000; Pellizzoni et al., 2002; Ogawa et al., 2007). SnRNPs are essential for pre-mRNA splicing and a decrease in SMN produces a reduction in snRNP assembly in SMA patients (Will and Luhrmann, 2001; Wan et al., 2005). In additional to SMN's established role in snRNP biogenesis, it has also been suggested to have a role in transcription and mRNA transport (Pellizzoni et al., 2001; Meister et al., 2002; Paushkin et al., 2002; Rossoll et al., 2003; Kariya et al., 2008).
Since the identification of SMN as the disease-causing gene in SMA, a number of mouse models have been generated to gain insight into the pathogenic process. Smn knockout mice (Smn−/−) are embryonic lethal while heterozygous (Smn+/−) mice can be used as a model of very mild SMA (Schrank et al., 1997). Mice do not harbor the SMN2 gene as it is unique to humans, but by introducing low copies of the human SMN2 gene into the Smn−/−background, researchers were able generate a mouse model (Smn−/−;SMN2) mimicking the severe SMA phenotype (Monani et al., 2000). Although the Smn−/−;SMN2 model is genetically representative of human patients, these mutant mice are very small in size and have a short life span making them difficult models to work with, especially when assessing therapeutics. The recent creation of milder SMA model mice has led to the discovery that cell types other than motor neurons are also affected in SMA (Hammond et al., 2010; Michaud et al., 2010; Bowerman et al., 2012a,b; Osborne et al., 2012; Cobb et al., 2013). These mice will also allow for the identification of new mechanisms of disease and will facilitate testing of therapeutic approaches.
Muscle Intrinsic Defects
As motor neurons and skeletal muscle are both functionally and structurally connected, it is difficult to study skeletal muscle defects independently from diseased motor neurons. The use of cell culture and conditional mouse models has, however, allowed researchers to study the role of Smn in skeletal muscle. Shafey et al. (2005) generated a series of hypomorphic cell lines in which Smn expression was depleted to varying levels in C2C12 myoblasts. Upon reducing Smn levels, defects such as decreased proliferation, aberrant myoblast fusion and malformed myotubes became apparent. These findings demonstrate that low Smn levels can result in intrinsic muscle defects and suggest a role for Smn in myoblast fusion. In vivo, the importance of Smn has been highlighted by conditional knockout mice in which the muscle-specific knockout of Smn was achieved using a floxed Smn allele with Cre recombinase controlled by the human skeletal actin (HSA) muscle-specific promoter (Cifuentes-Diaz et al., 2001). These mice develop a phenotype reminiscent of that observed in mouse models of muscular dystrophy. Skeletal muscle fibers from these mice have a disorganized sarcolemma and the mice have a reduced lifespan without displaying any overt neuropathology (Cifuentes-Diaz et al., 2001; Nicole et al., 2003). Although the results from conditional mouse studies have demonstrated a need for Smn in skeletal muscle, advances in animal genetic manipulation now allows for conditional deletion of Smn with residual SMN2 expression. Therefore, we propose that skeletal muscle conditional SMA model mice be revisited using genetics reminiscent of the human disease.
Studies demonstrating muscle intrinsic defects have also been performed using human primary myoblasts from SMA patients. Upon differentiation of human myoblasts, myotubes from SMA patients displayed a reduction in fusion (Arnold et al., 2004), a result reminiscent of what was observed in the C2C12 Smn-knockdown cells. Furthermore, skeletal muscle defects such as vacuolization and compromised sarcomere structures were reported in human SMA muscle cells co-cultured with rat spinal cord explants (Braun et al., 1995). Although these primary cell culture studies were fairly concise, they nonetheless provide valuable insight into intrinsic muscle abnormalities. Collectively, the cell culture and conditional mouse studies were the first to show muscle intrinsic defects in SMA, and establish and validate a strong rationale to study muscle defects in the context of SMA.
Abnormal Skeletal Muscle Development in SMA Mice
A growing body of evidence suggests that Smn-depletion leads to aberrant skeletal muscle development in SMA model mice. In the Smn−/−;SMN2;Δ7 mouse model, Lee et al. (2011) showed that the cross-sectional area of myofibers did not increase from P5 to P13 suggesting impaired muscle growth. Similarly, the tibialis anterior muscle from phenotype stage Smn−/−;SMN2 mice show similar cross-sectional area as that in pre-symptomatic mice (Dachs et al., 2011). A study on patient samples confirms that SMA muscles demonstrate impaired growth and maturation characterized by smaller and disorganized myotubes (Martinez-Hernandez et al., 2009). Collectively, these studies point to defects in muscle growth, however, the molecular basis for this is still unclear. One possibility for the reduced myofiber size in muscles from SMA model mice is that it is due to a defect in post-natal muscle development. Delayed muscle development is supported by the increased expression of immature myosin heavy chain (MHC) isoform in SMA model mice. Sustained expression of the embryonic and perinatal MHC isoforms has been detected at the transcript and protein level in phenotype stage Smn−/−;SMN2;Δ7 mice (Kong et al., 2009; Lee et al., 2011). Furthermore, reduced expression of fast MHC isoforms has been reported in skeletal muscles of SMA model mice (Biondi et al., 2008; Lee et al., 2011). The transition between developmental MHC isoforms and fast MHC isoforms occurs later in development compared to slow MHC isoforms (Schiaffino and Reggiani, 2011), therefore delayed muscle development could account for the decreased expression of type 2B and 2× fibers in SMA muscles. The aberrant MHC expression profile was also associated with a delay in postsynaptic endplate development. The expression of the immature acetylcholine receptor persists in severe SMA model mice and is accompanied by impaired postsynaptic maturation at the morphological level (Kariya et al., 2008; Kong et al., 2009; Dachs et al., 2011; Bowerman et al., 2012a). A recent study assessed the functional capacities of skeletal muscle and demonstrated muscle weakness was an early feature of pathology in multiple mouse models of SMA (Boyer et al., 2013). Moreover, the severe muscle weakness was associated with delayed expression of mature isoforms of muscle function proteins such as ryanodine receptors and sodium channels. For the moment, direct evidence demonstrating that muscle weakness in SMA model mice is due to the delayed myogenesis and aberrant expression of muscle function proteins is lacking. Nonetheless, together these results suggest a role for Smn in skeletal muscle development. Further, it suggests that low levels of Smn lead to impaired muscle maturation, and thereby causing muscle weakness.
Hayhurst et al. (2012) have provided evidence suggesting that Smn-depletion leads to accelerated muscle differentiation in vivo. Satellite cells, identified by the paired-box transcription factor Pax7, were increased in number and expressed myogenic markers earlier in Smn−/−;SMN2 mice compared to controls. Despite the accelerated expression of the myogenic regulatory factors in Smn−/−;SMN2 satellite cells, these cells failed to grow upon differentiation in culture (Hayhurst et al., 2012).
How low levels of Smn lead to specific developmental defects in SMA model mice is unclear at the moment. However, several possible scenarios can be envisaged. First, Smn has specific function(s) during development through its association with one or more binding partners or second that low levels of Smn disrupts a major cellular process such as splicing which could impact on the expression of multiple genes important for muscle development and function. Thirdly, as muscle development is orchestrated by activity, defects at the levels of the motor neuron may negatively impact upon the expression of factors controlling post-natal muscle maturation.
Targeting Pathways of Muscle Atrophy in SMA
Although several attempts have been made to promote muscle growth in SMA model mice, few studies have begun addressing the underlying mechanisms leading to muscle atrophy in the disease. During skeletal muscle atrophy caused by denervation, myogenin is up-regulated and directly stimulates the expression of muscle-specific ubiquitin E3 ligases, namely atrogin 1 and muscle RING-finger protein 1 (MuRF1), collectively referred to as atrogenes (Moresi et al., 2010). Substrates for atrogin 1 include MyoD, and eIF3f, which is an activator of protein synthesis, while sarcomeric proteins such as myosins and troponins are substrates of MuRF1 (Bonaldo and Sandri, 2013). Deletion of either atrogin 1 or MuRF1 leads to complete protection from denervation-induced muscle atrophy (Bodine et al., 2001).
Recently, a study by Bricceno et al. (2012b) demonstrated that following phenotype onset, a robust increase in atrogene expression was observed in SMA model mice (Table 1). Increased myogenin expression correlated with the up-regulation of the atrogenes, suggesting that the atrophy was mediated by muscle denervation. The expression of myogenin and atrogenes was also increased in skeletal muscle samples from human patients. Administration of the histone deacetylase inhibitor trichostatin A (TSA) prior to muscle atrophy onset attenuated atrogene expression in a mouse model of SMA (Bricceno et al., 2012b). TSA treatment decreased atrogene expression in denervated mice in which atrophy is mediated by myogenin but had no effect in a starved model of atrophy. Thus, this result suggests that TSA directly impacts on the myogenin dependent atrogene pathway. Direct evidence supporting denervation of the muscles used in the study by Bricceno et al. (2012b) was not presented. Therefore, it is not fully understood whether the up-regulation of myogenin in SMA model mice is attributable to impaired myogenesis, muscle denervation or both. Currently, understanding mechanisms of atrophy in SMA is an understudied area of research. For instance, whether the autophagic pathway or calpain-mediated muscle breakdown contribute to muscle atrophy in SMA is not known but could be of therapeutic importance. Further dissection of the molecular pathways responsible for the muscle atrophy, and testing therapeutics to treat atrophy in SMA is definitely warranted.
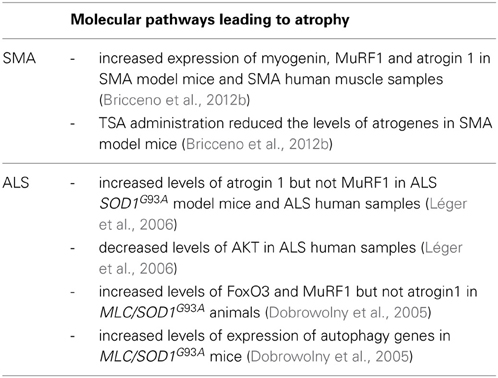
Table 1. Summary of pathways leading to muscle atrophy in motor neuron diseases.
A Role for SMN in Muscle
By isolating single myofibrils from myofibers, Walker et al. (2008) were able to identify Smn as a sarcomeric protein in striated muscle from mice. Specifically, Smn localizes to the Z-disc where it interacts with the actin crosslinking protein α-actinin (Rajendra et al., 2007; Walker et al., 2008; Shafey et al., 2010). Interestingly, other members of the SMN complex such as Gemins 2, 3, 4, and 6 are also present at Z-discs. However, other proteins essential for snRNP assembly are absent, suggesting that Smn plays a novel role at this adhesion site.
Currently, it is unclear whether there are any overt cytoskeletal structural defects in muscles from SMA model mice. Since mature muscles require less Smn expression than developing muscle (La Bella et al., 1998), the residual full-length protein produced by SMN2 may be sufficient to fulfill its role at the Z-disc. However, given the localization of Smn in skeletal muscle, the complete absence of full-length Smn protein in skeletal muscle may lead to cytoskeletal defects reminiscent of what was observed in Smn muscle-specific depleted mice. Given the domains present in the Smn protein, it is unlikely that Smn plays a structural role in mature myofibers. However, a role for Smn in mechanosensing may be possible.
Increased Cell Death in SMA Muscle
To gain insight into intrinsic molecular changes in skeletal muscle prior to motor neuron degeneration, Mutsaers et al. (2011) performed a proteomic screen using the rostral band of the levator auris longus muscle in pre-symptomatic severe SMA model mice. Several proteins involved in cell death pathways were aberrantly expressed, some of which were validated in skeletal muscle from human patients. Furthermore, the authors highlighted changes in protein expression that were not identified in muscle samples from denervated mice, suggesting these changes are unlikely to be due to defective muscle innervation. In a separate study, increased apoptotic cell death was observed in muscles from phenotype stage Smn−/−;SMN2 mice but was not detected at the pre-symptomatic P0-P1 time point. To demonstrate that the presence of apoptotic cell death was a muscle intrinsic phenomenon, Dachs et al. (2011) performed sciatic nerve denervation in neonatal mice. Forty-eight hours post-denervation, no increase in apoptotic cells was observed in the experimentally denervated muscles compared to sham controls. Therefore, the increased cell death correlates with disease progression in Smn−/−;SMN2 mice and is not attributed to acute muscle denervation per se. That said, an investigation of cell death in Smn-depleted C2C12 cells and cultured satellite cells isolated from Smn−/−;SMN2 mice revealed normal proportion of apoptotic cells (Shafey et al., 2005; Hayhurst et al., 2012). Furthermore, the number of apoptotic cells in type 1 human SMA muscle samples was comparable to controls (Martinez-Hernandez et al., 2009). Therefore, it would appear that increased cell death in skeletal muscle might be limited to mouse models of SMA. How the absence of Smn leads to increased apoptosis and whether apoptosis in skeletal muscle is primary or secondary to the SMA pathogenesis is unclear and requires further study.
The Therapeutic Requirements of SMN in Skeletal Muscle
Several therapeutic approaches currently being developed for the treatment of SMA involve increasing SMN levels. In an attempt to demonstrate the therapeutic requirements of SMN in neuronal and muscle tissue, a series of genetic rescues in mouse models were performed. In an initial study, the expression of Smn was driven by either the muscle-specific promoter HSA or by the neuron-specific prion protein promoter (PrP) (Gavrilina et al., 2008). Crossing HSA and PrP rescue mice onto the Smn−/−;SMN2 background allowed for a direct comparison between both the neuronal and muscle approaches on survival. The longest surviving PrP rescued line lived an average of 210 days while the best surviving HSA rescued line lived an average of 160 days. Thus, these results demonstrate that rescue in either neurons or muscle can offer significant improvements in survival. However, the results from this study are challenging to interpret given that both the longest surviving muscle and neuronal rescue lines demonstrated leaky Smn protein expression in the spinal cord and in muscle, respectively. Moreover, the use of the HSA promoter to express Smn in skeletal muscle may not be early enough in development, and may therefore not be representative of when Smn is needed in skeletal muscle. In addition, the use of the PrP promoter makes it difficult to conclude that Smn is required precisely in the spinal cord since this promoter targets multiple cell types in the nervous system.
More recently, a similar study was performed using conditional SMA model mice expressing tissue-specific Cre drivers, including the motor neuron-specific choline acetyltransferase promoter (ChATcre), and the muscle-specific myogenic determination 1 (MyoDcre) and myogenic factor 5 (Myf5cre). MyoD and Myf5 are myogenic regulatory factors expressed very early during myogenesis and it was therefore anticipated that they would yield more significant improvements in the survival of SMA mice (Martinez et al., 2012). Interestingly, restoring Smn expression in motor neurons of ChATcre mice led to a very modest increase in survival (from 15 to 23 days) compared to the survival reported using the PrP promoter. The ChATcre extended survival 2 days more than the muscle-specific promoters MyoDcre and Myf5cre. ChATcre conditional mice showed increased SMN expression in motor neurons and improved neuromuscular junction function and morphology. Despite these improvements, only the MyoDcre and the Myf5cre lines produced a robust increase in the cross-sectional area of myofibers. It should be noted that Smn expression from the Myf5cre driver was detectable in the neuronal tissue making it somewhat difficult to interpret the results from this particular conditional mouse model (Martinez et al., 2012). Nonetheless, the results from MyoDcre and Myf5cre demonstrate the importance of restoring Smn expression in skeletal muscle and suggest that a combinatorial expression of Smn in both myofibers and motor neurons may provide a more impressive extension of life in mouse models of SMA. These data, suggest that restoration of Smn expression in skeletal muscle should not be overlooked when developing therapeutic strategies.
Inducing Muscle Growth Pathways in SMA Model Mice
In an effort to reverse the muscle atrophy phenotype in mouse models of SMA, researchers have attempted to stimulate muscle growth pathways by various means. Myostatin is a potent negative regulator of muscle mass and the modulation of the myostatin pathway leads to dramatic muscle growth (Lee and McPherron, 2001). It was therefore hypothesized that modulation of the myostatin pathway may lead to phenotypic improvements in mouse models of SMA. Interestingly, myostatin and follistatin, a myostatin antagonist, were both found to be increased in mouse models of SMA suggesting a possible compensatory attempt by the animal to minimize muscle size loss (Sumner et al., 2009). Genetic overexpression of myostatin in SMA model mice proved to have a modest effect on muscle weight but offered no improvements in motor function and survival (Rindt et al., 2012). In a similar study, the genetic overexpression of follistatin in SMA model mice did not increase myofiber size or improve lifespan (Sumner et al., 2009). However, administration of recombinant follistatin in the same SMA mouse models used as in the genetic overexpression studies had a positive effect on the disease phenotype and lifespan (Rose et al., 2009). Specifically, delivery of the recombinant follistatin increased muscle mass, body weight, improved motor function and also increased motor neuron numbers and size in SMA model mice. Collectively, these changes led to an increase in median survival time. The differences in results between both follistatin studies are unclear but are likely attributed to the different methods used to overexpress follistatin that is, genetic overexpression versus administration of recombinant protein.
Insulin-like growth factor 1 (IGF-1) is a robust positive regulator of muscle mass and the overexpression of IGF-1 in mice causes a dramatic increase in musculature (Musaro et al., 2001). Interestingly, circulating IGF-1 levels are reduced in multiple mouse models of SMA (Hua et al., 2011; Murdocca et al., 2012). Delivery of IGF-1 to the central nervous system of type III SMA model mice using an adeno-associated virus vector proved beneficial for motor neuron health but had little effect on myofiber integrity and motor function (Tsai et al., 2012). The systemic administration of IPLEX (recombinant human IGF-1 complexed with recombinant human IGF-1 binding protein 3) by intraperitoneal injection in Smn−/−;SMN2;Δ7 animals led to increased myofiber size, reduced motor neuron cell loss but did not impact on life span and body weight of SMA model mice (Murdocca et al., 2012). Finally, combining SMN trans-splicing with an IGF-1 vector increased life span and body mass in a severe mouse model of SMA (Shababi et al., 2011). It is difficult to partial out whether the improvements in myofiber size were due to the IGF-1 protein acting on myofibers directly or rather, increased muscle size was a secondary consequence from the healthier motor neurons. The genetic muscle overexpression of IGF-1 onto the same SMA background used in the follistatin studies led to increased muscle mass as well as an increase in the median lifespan (Bosch-Marce et al., 2011). Overexpression of muscle IGF-1 had little effect on markers of muscle maturity. Therefore, IGF-1 cannot restore proper muscle development in SMA model mice. IGF-1 may prove to be of greater benefit to diseases in which myofibers have formed and matured properly and are subsequently affected by intrinsic and extrinsic pathology. In fact, promoting muscle growth using IGF-1 overexpression before the muscle has fully matured may be detrimental to the tissue. Furthermore, it should be noted that the SMA model mice were crossed to IGF-1 overexpressing heterozygotes. Therefore, the benefits of IGF-1 may have been more obvious had the mice been bred to homozygosity.
Perhaps targeting molecules more downstream where myostatin and IGF-1 pathways converge might yield more robust results. For example, Akt is downstream of both myostatin and IGF-1, and overexpression of Akt in mice has led to increased muscle size (Lai et al., 2004; Schiaffino and Mammucari, 2011). In addition to promoting muscle growth, Akt also plays an important role in inhibiting mechanisms of atrophy including inhibiting atrogenes and the autophagic pathway (Schiaffino and Mammucari, 2011).
Amyotrophic Lateral Sclerosis (ALS)
Disease Characteristics
ALS is a progressive adult-onset fatal disease characterized by the selective degeneration of upper motor neurons in the cerebral cortex and lower motor neurons of the brain stem and spinal cord (Rowland and Shneider, 2001). Features of the disease include muscle weakness and atrophy, spasticity and paralysis. It has an incidence of two per 100,000 people per year in the United States (Rowland and Shneider, 2001).
The term “amyotrophic” refers to the muscle atrophy, weakness and fasciculation that signify disease of lower motor neurons, while “lateral sclerosis” refers to the hardening of lateral columns, where gliosis follows the degeneration of corticospinal tracts (Rowland and Shneider, 2001). The mean duration of survival for ALS patients is 3–5 years following disease onset, with denervation of the respiratory muscles and diaphragm generally representing the fatal event of the disease. Despite intense research efforts, limited therapeutic options remain in attenuating disease progression; nevertheless advances are being made in palliative therapy (Carter et al., 2012).
A vast majority of ALS cases are sporadic and have no known genetic component, except for missense mutations in the TAR-DNA binding protein (Kabashi et al., 2008). Inherited forms of ALS (fALS) have an autosomal dominant or recessive pattern of inheritance and constitute ~10% or less of the remaining ALS cases (Rowland and Shneider, 2001). In 1993, a breakthrough in ALS research was made with the discovery of missense mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene of a subset of fALS cases (Rosen et al., 1993). It is estimated that 15–20% of fALS patients harbor missense mutations in the SOD1 gene (equating to 2% of all ALS cases).
SOD1 is a ubiquitously expressed cytosolic metalloprotease that non-covalently binds Cu and Zn. The enzyme functions by detoxifying and maintaining intracellular superoxide anions (O2−) by catalyzing the dismutation of O2− to molecular oxygen and hydrogen peroxide. As such, mutations in SOD1 impart oxidative stress, which has become a key mechanism underlying disease pathogenesis (Barber et al., 2006). That said, to date, a number of different pathogenic mechanisms are believed to trigger ALS pathogenesis (Ilieva et al., 2009).
Most of our current knowledge of ALS pathogenic mechanisms comes from transgenic mice expressing various forms of mutant SOD1. Studies with these mice have highlighted multiple targets of damage in disease including mitochondria, proteasomes, and secretory pathways. Furthermore, while expression of mutant SOD1 within motor neurons is a primary determinant of disease onset and of an early phase of disease progression, the expression of mutant SOD1 also affects structural, physiological, and metabolic parameters in other cell types (e.g., glia and skeletal muscle) (see reviews Boillee et al., 2006; Ilieva et al., 2009). ALS is now considered a multisystemic disease, wherein a variety of cell types act synergistically to exacerbate disease pathogenesis (Ilieva et al., 2009).
The progressive paralysis in ALS is the result of degeneration and demise of motor neurons (Rowland and Shneider, 2001). Data from multiple studies suggest that toxicity is non-cell-autonomous, meaning toxicity to motor neurons derives from damage developed within cell types beyond the motor neuron (see reviews Fuchs et al., 1994; Boillee et al., 2006; Ilieva et al., 2009). Studies supporting this notion showed that ubiquitous transgenic overexpression of SOD1 mutations causing fALS leads to an ALS phenotype in mice (Wong et al., 1995), however, restricted expression of mutant SOD1 in neurons alone is not sufficient to cause this phenotype. It is perhaps important to note, however, that levels of mutant SOD1 selectively expressed in motor neurons may have been too low to initiate disease (Pramatarova et al., 2001; Lino et al., 2002). More recent studies suggest that neuron-specific expression of human mutant SOD1 in mice triggers motor neuron degeneration (Jaarsma et al., 2008; Wang et al., 2008). These discrepant studies indicate that mutant SOD1-induced motor neuron degeneration is at least partly cell autonomous, and non-neuronal mutant SOD1 expression is also likely required for disease manifestation.
Muscle Defects in ALS
Numerous studies support the notion that multiple tissues outside the CNS, including skeletal muscle (Wiedemann et al., 1998; Krasnianski et al., 2005; Dupuis et al., 2006), fibroblasts (Aguirre et al., 1998; McEachern et al., 2000), and lymphocytes (Cova et al., 2006) are affected in human ALS. In both sporadic and fALS, functional aberrations and skeletal muscle pathology are present including neurogenic-induced muscle pathology and mitochondria dysfunction (Vielhaber et al., 1999; Krasnianski et al., 2005; Echaniz-Laguna et al., 2006; Corti et al., 2009). Similarly, transgenic mice expressing mutant SOD1 recapitulate functional and metabolic deficits in skeletal muscle as seen in human ALS patients (Derave et al., 2003; Dupuis et al., 2004; Mahoney et al., 2006).
Selective expression of mutant SOD1 in mouse skeletal muscle using the myosin light chain (MLC) promoter (MCL/SOD1G93A) induced ALS-like muscle pathologies, including progressive muscle atrophy, reduced muscle strength, impaired contractility, and mitochondrial dysfunction (Dobrowolny et al., 2008). Interestingly, exclusive expression of mutant SOD1 in skeletal muscle did not trigger the degeneration of motor neurons. This finding contrasts with a similar study from Wong and Martin (2010), where human mutated SOD1 expression driven from the HSA promoter, resulted in pathologic phenotypes in both muscle and motor neurons reminiscent of ALS (Wong and Martin, 2010). The reasons for this discrepancy is unclear although it may be due to the fact the animals used were of a different age, being significantly younger in the work from Dobrowolny et al. (2008). Regardless of this, together these studies provide strong evidence that mutant SOD1 is toxic to skeletal muscle and challenged the accepted dogma that motor neuron degeneration, caused by the overexpression of mutant SOD1, is the principle driver of muscle atrophy.
Skeletal muscle mitochondrial defects have been reported in MLC/SOD1G93A mice as well as in other mouse models of ALS (Dupuis et al., 2003, 2004; Dobrowolny et al., 2008). Previous studies have suggested that mitochondrial defects may lead to motor neuron degeneration in the context of ALS. This notion is supported by the study of Dupuis et al. (2009) in which they demonstrate that muscle mitochondria uncoupling leads to muscle denervation and motor neuron degeneration. Furthermore, muscle mitochondria uncoupling exacerbates disease progression and survival in a mouse model of ALS (Dupuis et al., 2009).
Myogenesis in ALS
Altered expression of the myogenic program has previously been reported in a mouse model of ALS. Widespread differences in the transcript and protein levels of Pax7 and myogenic regulatory factors were reported at disease onset (Manzano et al., 2011). The up-regulation of these proteins may reflect an increase in satellite cell activation following myofiber degeneration that may occur at time of disease onset. The levels of myogenic regulatory factors were not differentially expressed in early and late pre-symptomatic ALS mice compared to controls. This result suggests that the aberrant expression of the myogenic program is unlikely to be a triggering event leading to skeletal muscle defects in ALS model mice. In a separate study, however, primary myoblasts isolated from ALS patients and induced to differentiate into myotubes showed decreased expression of MHC, and displayed fusion defects (Pradat et al., 2011). These results are reminiscent of what was observed in SMA and SBMA cell culture studies (Arnold et al., 2004; Shafey et al., 2005; Malena et al., 2013).
Mechanisms of Atrophy in ALS
The most overt symptom in ALS patients is muscle weakness, which ultimately leads to death. Understanding mechanisms of muscle atrophy in ALS, i.e., whether muscle atrophy in ALS is solely due to denervation or whether it is intrinsic, may offer potential therapeutic avenues able to alleviate atrophy-induced weakness. Léger et al. (2006) studied signaling pathways contributing to skeletal muscle atrophy using ALS human samples as well as ALS SOD1G93A model mice. A dramatic increase in the expression of the E3 ubiquitin ligase atrogin 1 was detected in both mouse and human ALS samples. No changes were observed in the expression of MuRF1 (Table 1) (Léger et al., 2006). Changes upstream of atrogenes have also been reported. IGF-1 levels were decreased and the expression of activated Akt was down-regulated suggesting that muscle atrophy was associated in part to intrinsic defects not associated with myogenin-induced atrophy during denervation (Léger et al., 2006; Lunetta et al., 2012). Further evidence demonstrating that muscle atrophy is not entirely neurogenic in nature comes from studies with MLC/SOD1G93A mouse models of ALS in which restricted mutated SOD1 expression to skeletal muscle leads to atrophy (Dobrowolny et al., 2008). Skeletal muscle atrophy in these mice is initiated by the Akt pathway which suppresses protein synthesis and induces FoxO3 mediated expression of atrogenes (e.g., atrogin1 and MuRF1) (Dobrowolny et al., 2011). Furthermore, transcript levels of genes in the autophagic pathway such as LC3, Bnip3, and CathepsinL (Table 1) are up-regulated in muscles from MLC/SOD1G93A animals and may contribute to decreased myofiber size while caspases are likely end stage contributors to atrophy in these mice (Dobrowolny et al., 2008, 2011). These pathways altered in ALS conditional mice differ from the myogenin-induced atrophy observed following denervation. This supports the idea that intrinsic atrophy mechanisms are contributing to decreased myofiber size in ALS and that therapeutic approaches to reverse atrophy and increase myofiber size may prove beneficial in this disease. Based on these studies, it is difficult to overlook the importance of skeletal muscle as a contributor to the pathogenesis of ALS.
Skeletal Muscle IGF-1 as a Therapeutic Approach in ALS
The muscle-specific overexpression of IGF-1 has led to remarkable improvement in ALS model mice. Using the muscle MLC promoter, Dobrowolny et al. (2005) overexpressed IGF-1 onto the SOD1G93A mouse model of ALS (SOD1G93A/mIgf-1). This delayed disease onset and increased survival. Muscle atrophy was attenuated and satelitte cell activation was enhanced in SOD1G93A/mIgf-1 mice (Dobrowolny et al., 2005). Importantly, IGF-1 overexpression in muscle also preserved neuromuscular junction integrity and protected motor neurons from degeneration.
It has also been reported that viral delivery of IGF-1 to skeletal muscles leads to increased survival and protected motor neurons in ALS model mice (Kaspar et al., 2003). Interestingly, muscle viral delivery of IGF-1 in ALS model mice combined with exercise has a remarkable synergistic effect leading to an increase in survival beyond what was obseved following gene therarpy or exercise alone (Kaspar et al., 2005). It remains to been seen whether the adminstration of IGF-1 gene therapy combined with treatment of exercise mimetics such as AICAR and GW501516, can yeild similar results since exercise is likely not a feasable longterm option in severe ALS patients. Despite significant improvements in ALS model mice achieved by genetic or viral overexpression of IGF-1, to date efficacy studies of recombiant IGF-1 have yeilded positvie results in two human trials while a third trial reported no improvements in muscle strength. However, no trial has demonstated any imporovments regarding patient survival (Beauverd et al., 2012).
Spinal and Bulbar Muscular Atrophy (SBMA)
Disease Characteristics
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease, is an adult onset neuromuscular disease characterized by the degeneration and loss of lower motor neurons leading to muscle wasting. The initial phase of the disease presents as muscle cramping, hand tremors and fatigue, with muscle weakness considered a late manifestation of disease (Sambataro and Pennuto, 2012). In addition to muscle weakness, SBMA patients display other symptoms such as fasciculations that are especially prevalent in the face, neck and tongue. The disease affects an estimated 1–2 per 100,000 people (Katsuno et al., 2012).
At the molecular level, SBMA is caused by the expansion of a polyglutamine (polyQ)-encoding CAG trinucleotide repeat in the first exon of the gene coding for the androgen receptor (AR). These repeats are toxic and lead to motor neuron death causing respiratory weakness in SBMA patients (Bricceno et al., 2012a). Motor neurons express high levels of the AR relative to other neuronal populations, and the loss of AR function attributed to the expanded polyQ tract is believed to contribute to SBMA pathogenesis. However, the predominant disease mechanism involves a gain-of-toxic function that accompanies the expanded polyQ tract AR in motor neurons (Katsuno et al., 2012).
A correlation exists between the number of repeats and the age at onset of muscle weakness (Igarashi et al., 1992). Full disease manifestations are observed in men only while heterozygous females are mostly asymptomatic and women homozygous for the mutation are rare and show only mild symptoms (Schmidt et al., 2002). The AR gene is located on the proximal arm of chromosome Xq11-12 (Katsuno et al., 2012). The AR is a nuclear receptor and is part of the steroid/thyroid hormone receptor family. Upon binding to its natural ligands testosterone and dihydrotestosterone, the AR is translocated to the nucleus where it binds DNA as well as transcriptional co-regulators to control the expression of a subset of genes. However, pathological CAG triplet repeats in the AR lead to the nuclear accumulation of the receptor and this disrupts its transactivation domain's ability to interact with transcriptional co-activators. The AR is required for androgen dependent changes including proper male pubertal sexual development. SBMA patients demonstrate subtle androgen sensitivity highlighted by gynecomastia, fertility complications and atrophy of the gonads (Sambataro and Pennuto, 2012). Castration of SBMA model mice prevents any phenotype while treating female animals with testosterone exacerbates the phenotype (Katsuno et al., 2002). These results demonstrate that disease manifestations are androgen-dependent and this explains the gender bias observed in SBMA patients.
Muscle Defects in SBMA Patients
Although SBMA is believed to be primarily a motor neuron disease, increasing evidence suggests that myogenic defects contribute significantly to the disease pathogenesis. Studies performed on SBMA patients report very high levels of serum creatine kinase (CK), which are often 10 times higher than normal (Lee et al., 2005). It has been demonstrated that CK levels are higher in SBMA than any other motor neuron or myopathic disease and the increase can be detected prior to the onset of SBMA clinical symptoms (Chahin and Sorenson, 2009). Elevated serum CK levels are indicative of muscle damage and often seen in muscular dystrophy patients.
Histological studies performed on SBMA patients' muscle biopsies have revealed the presence of both neurogenic and primary myogenic defects. With muscle samples from SBMA patients, Sorarù et al. (2008), demonstrate the presence of fiber type grouping, atrophic fibers and angulated fibers, which are defects observed following chronic denervation. Necrotic myofibers as well as myofibers with centrally located nuclei, which are indicative of regenerating fibers, are also observed in SBMA muscle biopsies and are associated with primary myogenic changes (Sorarù et al., 2008; Chahin and Sorenson, 2009).
Studies using primary myoblasts isolated from SBMA patients have demonstrated that these cells proliferate and differentiate normally (Malena et al., 2013). Furthermore, primary SBMA myoblasts express myogenic regulatory factors such as MyoD and myogenin at levels comparable to controls. These results are androgen-dependent and suggest that early myogenesis is not affected in SBMA. However, defects such as cytoskeletal perturbations as well as aberrant myotube fusion were observed in primary myoblasts isolated from SBMA patients.
Muscle Defects in Mouse Models of SBMA
The use of mouse models has significantly contributed to our understanding of the myogenic defects in the SBMA phenotype. Yu et al. (2006) used gene targeting to generate a mouse model in which they converted the mouse AR sequence to the human sequence and in the process introduced 113 CAG repeats to recapitulate the human phenotype. These knock-in mice, designated AR113Q, are smaller, weaker compared to control counterparts, and die at 2–4 months of age. Histologically, muscles from AR113Q mice have atrophic and angulated fibers. At the molecular level, myogenin and acetylcholine receptor expression were up-regulated in AR113Q mice while MyoD expression was unchanged compared to controls. These morphological and gene expression changes are reflective of those observed following denervation (Moresi et al., 2010). Furthermore, myogenin was not mis-regulated in primary SBMA myoblasts supporting the idea that myogenin-dependent atrophy is present in AR113Q mice (Malena et al., 2013). However, the skeletal muscle pathology in AR113Q animals was thought to be cell-autonomous because it was detected prior to overt polyQ tract-induced pathology in the spinal cord. These results raise the possibility that the AR toxicity occurs in muscle prior to motor neurons, and may be the initiating factor leading to denervation-induced atrophy.
Other histological changes such as myofibers with centrally located nuclei present in AR113Q mice are more reflective of intrinsic myopathic defects. The expression of genes important for muscle function, such as the muscle chloride channel 1 (CLCN-1) and the muscle-specific sodium channel SCN4A, were decreased in AR113Q animals. The levels of CLCN-1 and SCN4A have previously been shown to decrease following experimental denervation (Kallen et al., 1993; Klocke et al., 1994). However, subsequent analyses in AR113Q animals demonstrated that the mis-regulation of CLCN-1 and SCN4A was not due to denervation (Yu et al., 2006). Furthermore, the down-regulation of CLCN-1 is attributed to the mis-splicing of the gene, which is reminiscent of what is observed in the muscle disease myotonic dystrophy (Yu et al., 2009). Therefore, SBMA model mice show muscle defects that are both intrinsic and motor neuron dependent. The denervation-like defects in AR113Q skeletal muscles can either be associated with intrinsic muscle toxicity-induced denervation, or to a secondary consequence linked to muscle denervation since the innervation status of skeletal muscles was not directly assessed.
Perhaps more compelling evidence supporting muscle contributions to SBMA pathogenesis comes from a mouse model in which a wild type AR was overexpressed in skeletal muscle and recapitulated the phenotypic features observed in other mouse models of SBMA (Monks et al., 2007). In this model, the AR overexpression in skeletal muscle was achieved using the HSA promoter (AR-HSA). AR-HSA animals showed muscle pathology and molecular changes such as increased levels of myogenin and AChR that are reminiscent of those observed following denervation. Interestingly, AR-HSA transgenic mice did not display motor neuron cell body loss but rather exhibited axonopathy. Again, these data suggest that the AR muscle toxicity may lead to motor neuron axon loss that in turn leads to denervation-induced pathology in skeletal muscles. Moreover, this result indicates that motor neuron defects occur distally first and eventually lead to cell body loss, a dying back phenomena, and may therefore explain the phenotype observed in the AR113Q mouse model of SBMA. The authors demonstrate that the toxicity to muscle occurred upon the activation of the AR-HSA by its ligand rather than the overexpression of the AR-HSA per se. However, it is difficult to conclude that the results observed from the AR-HSA mouse model can be directly generalized to SBMA, especially considering that the molecular pathogenesis of the AR-HSA model, which lacks the expanded polyQ tract, is very different.
Skeletal Muscle as a Therapeutic Target in SBMA
It has long been known that AR translocation to the nucleus is ligand-mediated and that this process is intensified in men given that the AR ligand is testosterone. Modifying testosterone levels in SBMA model mice is a ligand-targeted therapy that has proven very successful. In a mouse model of SBMA in which 97 CAG repeats were introduced into the AR gene (AR97Q), reduction of testosterone by way of castration prevented the appearance of the phenotype in male mice (Katsuno et al., 2002). In AR97Q female animals, administration of testosterone induced aberrant motor phenotypes and neuropathology. Treatment of AR-HSA female mice with testosterone provoked denervation-like symptoms similar to AR-HSA males. However, upon testosterone treatment cessation, AR-HSA females recovered normal motor function and expressed muscle genes at usual levels (Johansen et al., 2009). In agreement with these findings, survival and muscle pathology of AR-HSA mice is significantly ameliorated by prenatal treatment with flutamide, an AR antagonist. These data reveal that the AR expression restricted to muscle alone can lead to SBMA symptoms in mice and that these defects can be improved by blocking androgen binding. Thus, the SBMA phenotype strongly correlates with androgen expression and skeletal muscle may be an important target for SBMA treatment.
Although the use of anti-androgens is effective in treating mouse models of SBMA, it may prove to have unwanted side effects in humans. In an attempt to better understand how the AR is endogenously regulated, Palazzolo et al. (2007) uncovered a role for Akt in regulating AR function. Phosphorylation of the AR by Akt represses the AR activity through a ligand-dependent manner, thus preventing androgen from activating the AR. Modulation of Akt in cell culture models of SBMA rescued the polyQ associated cell toxicity indicating that manipulating the Akt pathway might provide phenotypic improvements in SBMA model mice (Palazzolo et al., 2007). Indeed, skeletal muscle overexpression of IGF-1, a protein that acts upstream of Akt, delayed disease onset and extended survival in SBMA model mice (Palazzolo et al., 2009). IGF-1 activates Akt and thus increases AR phosphorylation, which in turn reduces AR nuclear accumulation in muscles from SBMA model mice. A striking observation was that overexpression of IGF-1 in skeletal muscle improved motor behavior and reduced motor neuron cell loss in SBMA model mice. IGF-1 overexpression in skeletal muscle ameliorated both myofiber and motor neuron health by reducing AR nuclear aggregates. IGF-1 has previously been shown to benefit motor neurons by promoting sprouting and axonal growth, therefore IGF-1 may improve motor neuron health via its trophic properties in addition to its effects on mutant AR (Caroni and Grandes, 1990). Furthermore, overexpression of IGF-1 in skeletal muscle of wild type mice leads to gross muscle hypertrophy (Musaro et al., 2001). Therefore, IGF-1 might ameliorate muscle pathology independently of AR even though gene array data suggest that the IGF-1 signaling pathway is largely unaffected in skeletal muscle of multiple mouse models of SBMA (Mo et al., 2010).
Taken together, muscle defects are evident in SBMA and for the most part, were believed to be a secondary response to muscle denervation. However, a growing body of evidence supports the notion that muscle is the primary target of polyQ-AR toxicity and that muscle defects lead to motor neuron abnormalities. Results from various mouse models and muscle-specific targeted therapies support this idea.
Conclusion
Although considered motor neuron diseases, the use of conditional mouse models as well cell culture systems have highlighted the contributions of intrinsic skeletal muscle defects in SMA, ALS and SBMA (Table 2). The nature of the skeletal muscle defects differs between these three diseases. For instance, the most notable muscle defects in SMA are associated primarily with impaired development but defects neurogenic in origin are also observed. While in ALS, muscle defects are due to intrinsic and neurogenic factors, and appear to be present in mature myofibers rather than in developing muscle. In SBMA, cell-autonomous muscle degeneration is an overt contributor to the phenotype as are denervation-induced defects. However, results from ALS and SBMA mouse models suggest that in these diseases, skeletal muscle defects can initiate motor neuron defects. Such evidence to support this notion in SMA is currently lacking, indeed the complete absence of Smn in skeletal muscle was not associated with motor neuron defects (Cifuentes-Diaz et al., 2001). The use of appropriate conditional animals models will serve to explore this possibility further.
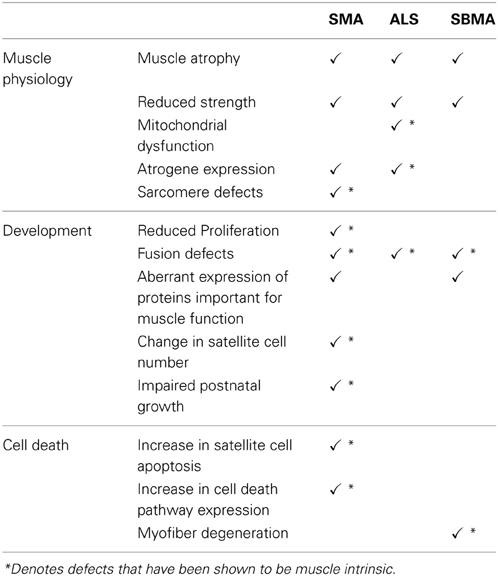
Table 2. Summary of defects observed in muscle in motor neuron diseases.
A common therapeutic approach used in all three diseases involves the muscle-specific overexpression of IGF-1. This approach has led to remarkable improvements in mouse models of ALS and SBMA, and to a lesser degree in SMA. The reason for this may be associated to the type of muscle defects in these diseases. The IGF-1 therapeutic approach may prove to be more beneficial in the context of mature muscle that is diseased, and may not be as efficient in SMA where muscle development is impaired.
It has always been assumed that muscle defects in SMA, ALS, and SBMA were a secondary consequence of motor neuron pathology. However, the studies described in this review shed light onto the importance of intrinsic muscle defects as a primary contributor to the pathogenesis of these diseases. As such, more research should be focused on treating intrinsic skeletal muscle defects. Indeed, regardless of whether muscle is a primary or secondary contributor to the pathogenesis of these diseases, muscle defects are present and therefore muscle is still an important therapeutic target to consider.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Lyndsay M. Murray for providing valuable comments during the preparation of the manuscript. Work in the Kothary laboratory is funded by grants from the Canadian Institutes of Health Research (CIHR). Justin G. Boyer was a recipient of a Frederick Banting and Charles Best CIHR Doctoral Research Award, Andrew Ferrier was supported by an Ontario Graduate Scholarship, and Rashmi Kothary is a recipient of a University Health Research Chair from the University of Ottawa.
References
Aguirre, T., Van Den Bosch, L., Goetschalckx, K., Tilkin, P., Mathijs, G., Cassiman, J. J. et al. (1998). Increased sensitivity of fibroblasts from amyotrophic lateral sclerosis patients to oxidative stress. Ann. Neurol. 43, 452–457. doi: 10.1002/ana.410430407
Arnold, A. S., Gueye, M., Guettier-Sigrist, S., Courdier-Fruh, I., Coupin, G., Poindron, P. et al. (2004). Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab. Invest. 84, 1271–1278. doi: 10.1038/labinvest.3700163
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067. doi: 10.1016/j.bbadis.2006.03.008
Beauverd, M., Mitchell, J. D., Wokke, J. H., and Borasio, G. D. (2012). Recombinant human insulin-like growth factor I (rhIGF-I) for the treatment of amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 11:CD002064. doi: 10.1002/14651858.CD002064.pub3
Biondi, O., Grondard, C., Lecolle, S., Deforges, S., Pariset, C., Lopes, P. et al. (2008). Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 28, 953–962. doi: 10.1523/JNEUROSCI.3237-07.2008
Bodine, S. C., Latres, E., Baumhueter, S., Lai, V. K., Nunez, L., Clarke, B. A. et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. doi: 10.1126/science.1065874
Boillee, S., Van de Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Bonaldo, P., and Sandri, M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 6, 25–39. doi: 10.1242/dmm.010389
Bosch-Marce, M., Wee, C. D., Martinez, T. L., Lipkes, C. E., Choe, D. W., Kong, L. et al. (2011). Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum. Mol. Genet. 20, 1844–1853. doi: 10.1093/hmg/ddr067
Bowerman, M., Murray, L. M., Beauvais, A., Pinheiro, B., and Kothary, R. (2012a). A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. 22, 263–276. doi: 10.1016/j.nmd.2011.09.007
Bowerman, M., Swoboda, K. J., Michalski, J. P., Wang, G. S., Reeks, C., Beauvais, A. et al. (2012b). Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 72, 256–268. doi: 10.1002/ana.23582
Boyer, J. G., Bowerman, M., and Kothary, R. (2010). The many faces of SMN: deciphering the function critical to spinal muscular atrophy pathogenesis. Future Neurol. 5, 873–890. doi: 10.2217/fnl.10.57
Boyer, J. G., Murray, L. M., Scott, K., De Repentigny, Y., Renaud, J. M., and Kothary, R. (2013). Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet. Muscle 3, 24. doi: 10.1186/2044-5040-3-24
Braun, S., Croizat, B., Lagrange, M. C., Warter, J. M., and Poindron, P. (1995). Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet 345, 694–695. doi: 10.1016/S0140-6736(95)90869-2
Bricceno, K. V., Fischbeck, K. H., and Burnett, B. G. (2012a). Neurogenic and myogenic contributions to hereditary motor neuron disease. Neurodegener. Dis. 9, 199–209. doi: 10.1159/000335311
Bricceno, K. V., Sampognaro, P. J., Van Meerbeke, J. P., Sumner, C. J., Fischbeck, K. H., and Burnett, B. G. (2012b). Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice. Hum. Mol. Genet. 21, 4448–4459. doi: 10.1093/hmg/dds286
Brzustowicz, L. M., Mérette, C., Kleyn, P. W., Lehner, T., Castilla, L. H., Penchaszadeh, G. K. et al. (1993). Assessment of nonallelic genetic heterogeneity of chronic (type II and III) spinal muscular atrophy. Hum. Hered. 43, 380–387. doi: 10.1159/000154164
Caroni, P., and Grandes, P. (1990). Nerve sprouting in innervated adult skeletal muscle induced by exposure to elevated levels of insulin-like growth factors. J. Cell Biol. 110, 1307–1317. doi: 10.1083/jcb.110.4.1307
Carter, G. T., Joyce, N. C., Abresch, A. L., Smith, A. E., and Vandekeift, G. K. (2012). Using palliative care in progressive neuromuscular disease to maximize quality of life. Phys. Med. Rehabil. Clin. N. Am. 23, 903–909. doi: 10.1016/j.pmr.2012.08.002
Chahin, N., and Sorenson, E. J. (2009). Serum creatine kinase levels in spinobulbar muscular atrophy and amyotrophic lateral sclerosis. Muscle Nerve 40, 126–129. doi: 10.1002/mus.21310
Cifuentes-Diaz, C., Frugier, T., Tiziano, F. D., Lacene, E., Roblot, N., Joshi, V. et al. (2001). Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol. 152, 1107–1114. doi: 10.1083/jcb.152.5.1107
Cobb, M. S., Rose, F. F., Rindt, H., Glascock, J. J., Shababi, M., Miller, M. R. et al. (2013). Development and characterization of an SMN2-based intermediate mouse model of Spinal Muscular Atrophy. Hum. Mol. Genet. 22, 1843–1855. doi: 10.1093/hmg/ddt037
Coovert, D. D., Le, T. T., McAndrew, P. E., Strasswimmer, J., Crawford, T. O., Mendell, J. R. et al. (1997). The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 6, 1205–1214. doi: 10.1093/hmg/6.8.1205
Corti, S., Donadoni, C., Ronchi, D., Bordoni, A., Fortunato, F., Santoro, D. et al. (2009). Amyotrophic lateral sclerosis linked to a novel SOD1 mutation with muscle mitochondrial dysfunction. J. Neurol. Sci. 276, 170–174. doi: 10.1016/j.jns.2008.09.030
Cova, E., Cereda, C., Galli, A., Curti, D., Finotti, C., Di Poto, C. et al. (2006). Modified expression of Bcl-2 and SOD1 proteins in lymphocytes from sporadic ALS patients. Neurosci. Lett. 399, 186–190. doi: 10.1016/j.neulet.2006.01.057
Dachs, E., Hereu, M., Piedrafita, L., Casanovas, A., Caldero, J., and Esquerda, J. E. (2011). Defective neuromuscular junction organization and postnatal myogenesis in mice with severe spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 70, 444–461. doi: 10.1097/NEN.0b013e31821cbd8b
Derave, W., Van Den Bosch, L., Lemmens, G., Eijnde, B. O., Robberecht, W., and Hespel, P. (2003). Skeletal muscle properties in a transgenic mouse model for amyotrophic lateral sclerosis: effects of creatine treatment. Neurobiol. Dis. 13, 264–272. doi: 10.1016/S0969-9961(03)00041-X
Dobrowolny, G., Aucello, M., and Musaro, A. (2011). Muscle atrophy induced by SOD1G93A expression does not involve the activation of caspase in the absence of denervation. Skelet. Muscle 1, 3. doi: 10.1186/2044-5040-1-3
Dobrowolny, G., Aucello, M., Rizzuto, E., Beccafico, S., Mammucari, C., Boncompagni, S. et al. (2008). Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436. doi: 10.1016/j.cmet.2008.09.002
Dobrowolny, G., Giacinti, C., Pelosi, L., Nicoletti, C., Winn, N., Barberi, L. et al. (2005). Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 168, 193–199. doi: 10.1083/jcb.200407021
Dupuis, L., Di Scala, F., Rene, F., De Tapia, M., Oudart, H., Pradat, P. F. et al. (2003). Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J. 17, 2091–2093. doi: 10.1096/fj.02-1182fje
Dupuis, L., Gonzalez De Aguilar, J. L., Echaniz-Laguna, A., Eschbach, J., Rene, F., Oudart, H. et al. (2009). Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 4:e5390. doi: 10.1371/journal.pone.0005390
Dupuis, L., Gonzalez De Aguilar, J. L., Echaniz-Laguna, A., and Loeffler, J. P. (2006). Mitochondrial dysfunction in amyotrophic lateral sclerosis also affects skeletal muscle. Muscle Nerve 34, 253–254. doi: 10.1002/mus.20566
Dupuis, L., Oudart, H., Rene, F., Gonzalez De Aguilar, J. L., and Loeffler, J. P. (2004). Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. U.S.A. 101, 11159–11164. doi: 10.1073/pnas.0402026101
Echaniz-Laguna, A., Zoll, J., Ponsot, E., N'guessan, B., Tranchant, C., Loeffler, J. P. et al. (2006). Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp. Neurol. 198, 25–30. doi: 10.1016/j.expneurol.2005.07.020
Fuchs, C. S., Giovannucci, E. L., Colditz, G. A., Hunter, D. J., Speizer, F. E., and Willett, W. C. (1994). A prospective study of family history and the risk of colorectal cancer. N. Engl. J. Med. 331, 1669–1674. doi: 10.1056/NEJM199412223312501
Gavrilina, T. O., McGovern, V. L., Workman, E., Crawford, T. O., Gogliotti, R. G., Didonato, C. J. et al. (2008). Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 17, 1063–1075. doi: 10.1093/hmg/ddm379
Hamilton, G., and Gillingwater, T. H. (2013). Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 19, 40–50. doi: 10.1016/j.molmed.2012.11.002
Hammond, S. M., Gogliotti, R. G., Rao, V., Beauvais, A., Kothary, R., and Didonato, C. J. (2010). Mouse survival motor neuron alleles that mimic SMN2 splicing and are inducible rescue embryonic lethality early in development but not late. PLoS ONE 5:e15887. doi: 10.1371/journal.pone.0015887
Hayhurst, M., Wagner, A. K., Cerletti, M., Wagers, A. J., and Rubin, L. L. (2012). A cell-autonomous defect in skeletal muscle satellite cells expressing low levels of survival of motor neuron protein. Dev. Biol. 368, 323–334. doi: 10.1016/j.ydbio.2012.05.037
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F. et al. (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. doi: 10.1038/nature10485
Igarashi, S., Tanno, Y., Onodera, O., Yamazaki, M., Sato, S., Ishikawa, A. et al. (1992). Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology 42, 2300–2302. doi: 10.1212/WNL.42.12.2300
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772. doi: 10.1083/jcb.200908164
Jaarsma, D., Teuling, E., Haasdijk, E. D., De Zeeuw, C. I., and Hoogenraad, C. C. (2008). Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J. Neurosci. 28, 2075–2088. doi: 10.1523/JNEUROSCI.5258-07.2008
Johansen, J. A., Yu, Z., Mo, K., Monks, D. A., Lieberman, A. P., Breedlove, S. M. et al. (2009). Recovery of function in a myogenic mouse model of spinal bulbar muscular atrophy. Neurobiol. Dis. 34, 113–120. doi: 10.1016/j.nbd.2008.12.009
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Van de Velde, C. et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kallen, R. G., Cohen, S. A., and Barchi, R. L. (1993). Structure, function and expression of voltage-dependent sodium channels. Mol. Neurobiol. 7, 383–428. doi: 10.1007/BF02769184
Kariya, S., Park, G. H., Maeno-Hikichi, Y., Leykekhman, O., Lutz, C., Arkovitz, M. S. et al. (2008). Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 17, 2552–2569. doi: 10.1093/hmg/ddn156
Kaspar, B. K., Frost, L. M., Christian, L., Umapathi, P., and Gage, F. H. (2005). Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann. Neurol. 57, 649–655. doi: 10.1002/ana.20451
Kaspar, B. K., Llado, J., Sherkat, N., Rothstein, J. D., and Gage, F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842. doi: 10.1126/science.1086137
Katsuno, M., Adachi, H., Kume, A., Li, M., Nakagomi, Y., Niwa, H. et al. (2002). Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–854. doi: 10.1016/S0896-6273(02)00834-6
Katsuno, M., Tanaka, F., Adachi, H., Banno, H., Suzuki, K., Watanabe, H. et al. (2012). Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog. Neurobiol. 99, 246–256. doi: 10.1016/j.pneurobio.2012.05.007
Klocke, R., Steinmeyer, K., Jentsch, T. J., and Jockusch, H. (1994). Role of innervation, excitability, and myogenic factors in the expression of the muscular chloride channel ClC-1. A study on normal and myotonic muscle. J. Biol. Chem. 269, 27635–27639.
Kong, L., Wang, X., Choe, D. W., Polley, M., Burnett, B. G., Bosch-Marce, M. et al. (2009). Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 29, 842–851. doi: 10.1523/JNEUROSCI.4434-08.2009
Krasnianski, A., Deschauer, M., Neudecker, S., Gellerich, F. N., Muller, T., Schoser, B. G. et al. (2005). Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain 128, 1870–1876. doi: 10.1093/brain/awh540
La Bella, V., Cisterni, C., Salaun, D., and Pettmann, B. (1998). Survival motor neuron (SMN) protein in rat is expressed as different molecular forms and is developmentally regulated. Eur. J. Neurosci. 10, 2913–2923. doi: 10.1111/j.1460-9568.1998.00298.x
Lai, K. M., Gonzalez, M., Poueymirou, W. T., Kline, W. O., Na, E., Zlotchenko, E. et al. (2004). Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell. Biol. 24, 9295–9304. doi: 10.1128/MCB.24.21.9295-9304.2004
Lee, J. H., Shin, J. H., Park, K. P., Kim, I. J., Kim, C. M., Lim, J. G. et al. (2005). Phenotypic variability in Kennedy's disease: implication of the early diagnostic features. Acta Neurol. Scand. 112, 57–63. doi: 10.1111/j.1600-0404.2005.00428.x
Lee, S. J., and McPherron, A. C. (2001). Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. U.S.A. 98, 9306–9311. doi: 10.1073/pnas.151270098
Lee, Y. I., Mikesh, M., Smith, I., Rimer, M., and Thompson, W. (2011). Muscles in a mouse model of spinal muscular atrophy show profound defects in neuromuscular development even in the absence of failure in neuromuscular transmission or loss of motor neurons. Dev. Biol. 356, 432–444. doi: 10.1016/j.ydbio.2011.05.667
Lefebvre, S., Burlen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L. et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A. et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269. doi: 10.1038/ng0797-265
Léger, B., Vergani, L., Soraru, G., Hespel, P., Derave, W., Gobelet, C. et al. (2006). Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 20, 583–585. doi: 10.1096/fj.05-5249fje
Lino, M. M., Schneider, C., and Caroni, P. (2002). Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832.
Liu, Q., and Dreyfuss, G. (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555–3565.
Lunetta, C., Serafini, M., Prelle, A., Magni, P., Dozio, E., Ruscica, M. et al. (2012). Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve 45, 200–208. doi: 10.1002/mus.22288
Mahoney, D. J., Kaczor, J. J., Bourgeois, J., Yasuda, N., and Tarnopolsky, M. A. (2006). Oxidative stress and antioxidant enzyme upregulation in SOD1-G93A mouse skeletal muscle. Muscle Nerve 33, 809–816. doi: 10.1002/mus.20542
Malena, A., Pennuto, M., Tezze, C., Querin, G., D'ascenzo, C., Silani, V. et al. (2013). Androgen-dependent impairment of myogenesis in spinal and bulbar muscular atrophy. Acta Neuropathol. 126, 109–121. doi: 10.1007/s00401-013-1122-9
Manzano, R., Toivonen, J. M., Olivan, S., Calvo, A. C., Moreno-Igoa, M., Munoz, M. J. et al. (2011). Altered expression of myogenic regulatory factors in the mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 8, 386–396. doi: 10.1159/000324159
Martinez, T. L., Kong, L., Wang, X., Osborne, M. A., Crowder, M. E., Van Meerbeke, J. P. et al. (2012). Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 32, 8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012
Martinez-Hernandez, R., Soler-Botija, C., Also, E., Alias, L., Caselles, L., Gich, I. et al. (2009). The developmental pattern of myotubes in spinal muscular atrophy indicates prenatal delay of muscle maturation. J. Neuropathol. Exp. Neurol. 68, 474–481. doi: 10.1097/NEN.0b013e3181a10ea1
McEachern, G., Kassovska-Bratinova, S., Raha, S., Tarnopolsky, M. A., Turnbull, J., Bourgeois, J. et al. (2000). Manganese superoxide dismutase levels are elevated in a proportion of amyotrophic lateral sclerosis patient cell lines. Biochem. Biophys. Res. Commun. 273, 359–363. doi: 10.1006/bbrc.2000.2933
Meister, G., Buhler, D., Laggerbauer, B., Zobawa, M., Lottspeich, F., and Fischer, U. (2000). Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum. Mol. Genet. 9, 1977–1986. doi: 10.1093/hmg/9.13.1977
Meister, G., Eggert, C., and Fischer, U. (2002). SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol. 12, 472–478. doi: 10.1016/S0962-8924(02)02371-1
Michaud, M., Arnoux, T., Bielli, S., Durand, E., Rotrou, Y., Jablonka, S. et al. (2010). Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis. 38, 125–135. doi: 10.1016/j.nbd.2010.01.006
Mo, K., Razak, Z., Rao, P., Yu, Z., Adachi, H., Katsuno, M. et al. (2010). Microarray analysis of gene expression by skeletal muscle of three mouse models of Kennedy disease/spinal bulbar muscular atrophy. PLoS ONE 5:e12922. doi: 10.1371/journal.pone.0012922
Monani, U. R., Sendtner, M., Coovert, D. D., Parsons, D. W., Andreassi, C., Le, T. T. et al. (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333–339. doi: 10.1093/hmg/9.3.333
Monks, D. A., Johansen, J. A., Mo, K., Rao, P., Eagleson, B., Yu, Z. et al. (2007). Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci. U.S.A. 104, 18259–18264. doi: 10.1073/pnas.0705501104
Moresi, V., Williams, A. H., Meadows, E., Flynn, J. M., Potthoff, M. J., McAnally, J. et al. (2010). Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143, 35–45. doi: 10.1016/j.cell.2010.09.004
Murdocca, M., Malgieri, A., Luchetti, A., Saieva, L., Dobrowolny, G., De Leonibus, E. et al. (2012). IPLEX administration improves motor neuron survival and ameliorates motor functions in a severe mouse model of spinal muscular atrophy. Mol. Med. 18, 1076–1085. doi: 10.2119/molmed.2012.00056
Musaro, A., McCullagh, K., Paul, A., Houghton, L., Dobrowolny, G., Molinaro, M. et al. (2001). Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 27, 195–200. doi: 10.1038/84839
Mutsaers, C. A., Wishart, T. M., Lamont, D. J., Riessland, M., Schreml, J., Comley, L. H. et al. (2011). Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum. Mol. Genet. 20, 4334–4344. doi: 10.1093/hmg/ddr360
Nicole, S., Desforges, B., Millet, G., Lesbordes, J., Cifuentes-Diaz, C., Vertes, D. et al. (2003). Intact satellite cells lead to remarkable protection against Smn gene defect in differentiated skeletal muscle. J. Cell Biol. 161, 571–582. doi: 10.1083/jcb.200210117
Ogawa, C., Usui, K., Aoki, M., Ito, F., Itoh, M., Kai, C. et al. (2007). Gemin2 plays an important role in stabilizing the survival of motor neuron complex. J. Biol. Chem. 282, 11122–11134. doi: 10.1074/jbc.M609297200
Osborne, M., Gomez, D., Feng, Z., McEwen, C., Beltran, J., Cirillo, K. et al. (2012). Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum. Mol. Genet. 21, 4431–4447. doi: 10.1093/hmg/dds285
Palazzolo, I., Burnett, B. G., Young, J. E., Brenne, P. L., La Spada, A. R., Fischbeck, K. H. et al. (2007). Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet. 16, 1593–1603. doi: 10.1093/hmg/ddm109
Palazzolo, I., Stack, C., Kong, L., Musaro, A., Adachi, H., Katsuno, M. et al. (2009). Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63, 316–328. doi: 10.1016/j.neuron.2009.07.019
Paushkin, S., Gubitz, A. K., Massenet, S., and Dreyfuss, G. (2002). The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 14, 305–312. doi: 10.1016/S0955-0674(02)00332-0
Pearn, J. (1978). Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 15, 409–413. doi: 10.1136/jmg.15.6.409
Pellizzoni, L., Charroux, B., Rappsilber, J., Mann, M., and Dreyfuss, G. (2001). A functional interaction between the survival motor neuron complex and RNA polymerase II. J. Cell Biol. 152, 75–85. doi: 10.1083/jcb.152.1.75
Pellizzoni, L., Yong, J., and Dreyfuss, G. (2002). Essential role for the SMN complex in the specificity of snRNP assembly. Science 298, 1775–1779. doi: 10.1126/science.1074962
Pradat, P. F., Barani, A., Wanschitz, J., Dubourg, O., Lombes, A., Bigot, A. et al. (2011). Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 264–271. doi: 10.3109/17482968.2011.566618
Pramatarova, A., Laganiere, J., Roussel, J., Brisebois, K., and Rouleau, G. A. (2001). Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 21, 3369–3374.
Prior, T. W., Snyder, P. J., Rink, B. D., Pearl, D. K., Pyatt, R. E., Mihal, D. C. et al. (2010). Newborn and carrier screening for spinal muscular atrophy. Am. J. Med. Genet. A 152A, 1608–1616. doi: 10.1002/ajmg.a.33474
Rajendra, T. K., Gonsalvez, G. B., Walker, M. P., Shpargel, K. B., Salz, H. K., and Matera, A. G. (2007). A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J. Cell Biol. 176, 831–841. doi: 10.1083/jcb.200610053
Rindt, H., Buckley, D. M., Vale, S. M., Krogman, M., Rose, F. F. Jr., Garcia, M. L. et al. (2012). Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. 22, 277–285. doi: 10.1016/j.nmd.2011.10.012
Rochette, C. F., Gilbert, N., and Simard, L. R. (2001). SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 108, 255–266. doi: 10.1007/s004390100473
Rose, F. F. Jr., Mattis, V. B., Rindt, H., and Lorson, C. L. (2009). Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 18, 997–1005. doi: 10.1093/hmg/ddn426
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A. et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rossoll, W., Jablonka, S., Andreassi, C., Kroning, A. K., Karle, K., Monani, U. R. et al. (2003). Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 163, 801–812. doi: 10.1083/jcb.200304128
Rowland, L. P., and Shneider, N. A. (2001). Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700. doi: 10.1056/NEJM200105313442207
Sambataro, F., and Pennuto, M. (2012). Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases. Prog. Neurobiol. 97, 152–172. doi: 10.1016/j.pneurobio.2011.10.003
Schiaffino, S., and Mammucari, C. (2011). Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle 1, 4. doi: 10.1186/2044-5040-1-4
Schiaffino, S., and Reggiani, C. (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531. doi: 10.1152/physrev.00031.2010
Schmidt, B. J., Greenberg, C. R., Allingham-Hawkins, D. J., and Spriggs, E. L. (2002). Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology 59, 770–772. doi: 10.1212/WNL.59.5.770
Schrank, B., Gotz, R., Gunnersen, J. M., Ure, J. M., Toyka, K. V., Smith, A. G. et al. (1997). Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 94, 9920–9925. doi: 10.1073/pnas.94.18.9920
Shababi, M., Glascock, J., and Lorson, C. L. (2011). Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum. Gene Ther. 22, 135–144. doi: 10.1089/hum.2010.114
Shafey, D., Boyer, J. G., Bhanot, K., and Kothary, R. (2010). Identification of novel interacting protein partners of SMN using tandem affinity purification. J. Proteome Res. 9, 1659–1669. doi: 10.1021/pr9006987
Shafey, D., Cote, P. D., and Kothary, R. (2005). Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res. 311, 49–61. doi: 10.1016/j.yexcr.2005.08.019
Sorarù, G., D'ascenzo, C., Polo, A., Palmieri, A., Baggio, L., Vergani, L. et al. (2008). Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J. Neurol. Sci. 264, 100–105. doi: 10.1016/j.jns.2007.08.012
Sumner, C. J., Wee, C. D., Warsing, L. C., Choe, D. W., Ng, A. S., Lutz, C. et al. (2009). Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum. Mol. Genet. 18, 3145–3152. doi: 10.1093/hmg/ddp253
Tsai, L. K., Chen, Y. C., Cheng, W. C., Ting, C. H., Dodge, J. C., Hwu, W. L. et al. (2012). IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol. Dis. 45, 272–279. doi: 10.1016/j.nbd.2011.06.021
Vielhaber, S., Winkler, K., Kirches, E., Kunz, D., Buchner, M., Feistner, H. et al. (1999). Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 169, 133–139. doi: 10.1016/S0022-510X(99)00236-1
Walker, M. P., Rajendra, T. K., Saieva, L., Fuentes, J. L., Pellizzoni, L., and Matera, A. G. (2008). SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum. Mol. Genet. 17, 3399–3410. doi: 10.1093/hmg/ddn234
Wan, L., Battle, D. J., Yong, J., Gubitz, A. K., Kolb, S. J., Wang, J. et al. (2005). The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol. Cell. Biol. 25, 5543–5551. doi: 10.1128/MCB.25.13.5543-5551.2005
Wang, L., Sharma, K., Deng, H. X., Siddique, T., Grisotti, G., Liu, E. et al. (2008). Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol. Dis. 29, 400–408. doi: 10.1016/j.nbd.2007.10.004
Wiedemann, F. R., Winkler, K., Kuznetsov, A. V., Bartels, C., Vielhaber, S., Feistner, H. et al. (1998). Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 156, 65–72. doi: 10.1016/S0022-510X(98)00008-2
Will, C. L., and Luhrmann, R. (2001). Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13, 290–301. doi: 10.1016/S0955-0674(00)00211-8
Wong, M., and Martin, L. J. (2010). Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 19, 2284–2302. doi: 10.1093/hmg/ddq106
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A. et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116. doi: 10.1016/0896-6273(95)90259-7
Yu, Z., Dadgar, N., Albertelli, M., Gruis, K., Jordan, C., Robins, D. M. et al. (2006). Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Invest. 116, 2663–2672. doi: 10.1172/JCI28773
Keywords: mouse models, neuromuscular disease, myofiber degeneration, fusion defect, insulin-like growth factor 1
Citation: Boyer JG, Ferrier A and Kothary R (2013) More than a bystander: the contributions of intrinsic skeletal muscle defects in motor neuron diseases. Front. Physiol. 4:356. doi: 10.3389/fphys.2013.00356
Received: 30 October 2013; Paper pending published: 13 November 2013;
Accepted: 20 November 2013; Published online: 18 December 2013.
Edited by:
Lucas Guimarães-Ferreira, Federal University of Espirito Santo, BrazilReviewed by:
Julien Ochala, KIng's College London, UKRavindra N. Singh, Iowa State University, USA;
Christian Lorson, University of Missouri, USA
Copyright © 2013 Boyer, Ferrier and Kothary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rashmi Kothary, Ottawa Hospital Research Institute, 501 Smyth Road, Ottawa, ON K1H 8L6, Canada e-mail: rkothary@ohri.ca