 Daniel Zielonka1*
Daniel Zielonka1* Michal Mielcarek
Michal Mielcarek- 1Department of Social Medicine, Poznan University of Medical Sciences, Poznan, Poland
- 2MRC National Institute for Medical Research, London, UK
- 3Department of Medical and Molecular Genetics, King's College London, London, UK
Huntington's disease (HD) is a hereditary neurodegenerative disorder caused by the expansion of a polyglutamine stretch within the huntingtin protein (HTT). The neurological symptoms, that involve motor, cognitive and psychiatric disturbances, are caused by neurodegeneration that is particularly widespread in the basal ganglia and cereberal cortex. HTT is ubiquitously expressed and in recent years it has become apparent that HD patients experience a wide array of peripheral organ dysfunction including severe metabolic phenotype, weight loss, HD-related cardiomyopathy and skeletal muscle wasting. Although skeletal muscles pathology became a hallmark of HD, the mechanisms underlying muscular atrophy in this disorder are unknown. Skeletal muscles account for approximately 40% of body mass and are highly adaptive to physiological and pathological conditions that may result in muscle hypertrophy (due to increased mechanical load) or atrophy (inactivity, chronic disease states). The atrophy is caused by degeneration of myofibers and their replacement by fibrotic tissue is the major pathological feature in many genetic muscle disorders. Under normal physiological conditions the muscle function is orchestrated by a network of intrinsic hypertrophic and atrophic signals linked to the functional properties of the motor units that are likely to be imbalanced in HD. In this article, we highlight the emerging field of research with particular focus on the recent studies of the skeletal muscle pathology and the identification of new disease-modifying treatments.
Introduction
Huntington's disease (HD) is neurodegenerative disorder caused by the expansion of polyglutamine stretch within the huntingtin protein (HTT) (Gusella et al., 1993; Vonsattel and DiFiglia, 1998; Novak and Tabrizi, 2010). The disease is caused by the expansion of a CAG repeat to over 35 CAG repeats in exon1 of the huntingtin (HTT) gene which are normally observed in healthy objects. Neurodegeneration, particularly widespread in the striatal nuclei, basal ganglia and cereberal cortex, is a source of neurological symptoms that involve motor, cognitive and psychiatric disturbances (Novak and Tabrizi, 2010). This leads to a wide-range of clinical features including personality changes, motor impairment, dementia and weight loss that are likely to progress over the course of 15–20 years to death (HDRG, 1993; Walker, 2007). In mammals HTT is expressed in many tissues and organs (Hoogeveen et al., 1993; Strong et al., 1993; Trottier et al., 1995). HTT has been identified to be involved in many critical cellular processes like transcription, protein trafficking and vesicle transport (Li and Li, 2004). In mice HTT deletion is embryonically lethal, leading to defects in all germ layers (Zeitlin et al., 1995). It has been established that HTT is affecting organelles and functional systems that are essential for all type cells i.e., mitochondria, ubuquitin-proteasome system and this phenomena is not tissue specific (Li and Li, 2004; Sassone et al., 2009; van der Burg et al., 2009; Zielonka et al., 2014). A recent study of the heart function in two HD mouse models identified pronounced contractile heart dysfunction, which might be a part of dilatated cardiomyopathy (DCM). This was accompanied by the re-expression of fetal genes, apoptotic cardiomyocyte loss and a moderate degree of interstitial fibrosis (Mielcarek et al., 2014). Therefore it is likely that the peripheral pathology of HD, such as weight loss and severe skeletal muscle atrophy, might have a significant input to the disease progression.
Skeletal Muscle Pathology in HD Patients
A case-study report showed that a semi-professional marathon runner (43 CAGs) developed signs of a slowly progressing myopathy with elevated creatine kinase levels many years before first signs of chorea were detected. Muscle biopsy revealed a mild myopathy with mitochondrial pathology including a complex IV deficiency (Kosinski et al., 2007). The isometric muscle strength of 6 lower limb muscle groups was measured in 20 people with HD and matched healthy controls. HD patients had reduced muscle strength by 50% on average in comparison to healthy matched controls (Busse et al., 2008). Several studies have reported defects in the mitochondrial function of the central nervous system and skeletal muscles in HD patients (Reddy, 2014). For example, the non-invasive method of 31P-MRS (31P- Magnetic Resonance Spectroscopy) showed a reduced phosphocreatine to inorganic phosphate ratio in the symptomatic HD patients at rest. Muscle ATP/phosphocreatine and inorganic phosphate levels were significantly reduced in both symptomatic and presymptomatic HD subjects (Lodi et al., 2000). During recovery from exercise, the maximum rate of mitochondrial ATP production was reduced by 44% in the symptomatic HD patients and by 35% in the presymptomatic HD carriers. HD subjects showed also a deficit in the mitochondrial oxidative metabolism and that might support a role for mitochondrial dysfunction as a key factor involved in the HD-related muscle pathogenesis (Lodi et al., 2000; Saft et al., 2005). In addition, the total exercise capacity was normal in HD subjects but notably the presymptomatic HD patients had a lower anaerobic threshold and increased level of plasma lactate (Ciammola et al., 2011). In vitro muscle cell cultures revealed that HD cells produced more lactate and that might be indicative of a higher glycolysis level (Ciammola et al., 2011). Furthermore, muscle cultures showed cellular abnormalities including mitochondrial membrane potential, cytochrome c release, increased CASPASE-3, −8, and −9 levels and defective cell differentiation, likely due to the formation of HTT inclusions in differentiated myotubes (Ciammola et al., 2006). Finally, electron microscopy showed striking mitochondrial defects like abnormally elongated and swollen mitochondria with derangement of cristae and vacuoles (Ciammola et al., 2011).
On the Way to Understand Muscle Pathology in HD—an Animal Model
Recent years of research on HD pathogenesis resulted in a generation of many HD transgenic mouse models including mHTT N-terminal fragments and full-length murine or human mHTT (Crook and Housman, 2011; Lee et al., 2013; Rattray et al., 2013). R6/2 mice are transgenic for a mutated N-terminal exon 1 HTT fragment and are the most frequently used in pre-clinical settings. Behaviorally, R6/2 animals at first display a spatial learning deficit at 3–4 weeks of age (Lione et al., 1999). Attention learning deficits and abnormal performance in motor tests (swimming and high speed rotarod) appear at 5–6 weeks of age (Carter et al., 1999; Lione et al., 1999; Murphy et al., 2000), followed by development of a resting tremor, gait disturbances and visual learning deficits at 8–9 weeks of age (Carter et al., 1999; Murphy et al., 2000). There is support that mHTT may have detrimental effects in the skeletal muscles of R6/2 mice due to poly(Q) aggregate accumulation (Sathasivam et al., 1999; Moffitt et al., 2009) and formation of these inclusions in myoblasts and myotubes have been confirmed in vitro (Orth et al., 2003). Alternatively, the toxicity of triplet repeat-containing RNA and/or patially mis-spliced huntingtin gene (Htt) could be considered as an additional mechanism of HD pathology (Sathasivam et al., 2013).
Transcriptional deregulation is a typical feature of HD pathology in the brain (Luthi-Carter et al., 2002). A similar transcriptional profile in skeletal muscles (quadriceps) from R6/2 mice, HdhQ150 homozygous knock-in mice and HD patients has been identified and that was consistent with a transition from fast-twitch to slow-twitch muscle fiber types (Luthi-Carter et al., 2002). On the other hand, based on immunohistochemistry both type I and II muscles were atrophic. Although atrophy occurred in both type fibers, there was more type I fibers in the R6/2 skeletal muscles. Hence, there was a conversion of type II fibers to type I during the process of muscle atrophy (Ribchester et al., 2004). However, these findings in pre-clinical settings are inconsistent with an increased glycolysis observed in human patients (Ciammola et al., 2011). Metabolic adaptations similar to those induced by diabetes or fasting are also present in HD mouse models but neither metabolic disorder could explain the full phenotype of HD muscle (Strand et al., 2005). Consequently, at the ultrastructural level, the sciatic nerve displayed abnormalities in large myelinated fibers in the presymptomatic R6/2 mice. A significant decrease in the axoplasm diameter of myelinated neurons and increased number of degenerating myelinated fibers were observed; although myelin thickness and unmyelinated fiber diameter were not affected (Wade et al., 2008). The synaptic transmission at the neuromuscular junction has also been studied in the R6/1 mouse model of HD. The morphological data suggest that the innervation pattern of the neuromuscular junctions in R6/1 muscles were normal in early symptomatic animals. However, the size and frequency of miniature endplate potentials were not changed in the R6/1 mice, while the amplitude of evoked endplate potentials increased. Consistent with a pre-synaptic increase of release probability, synaptic depression under high-frequency was higher in R6/1 mice. No changes were detected in size and dynamics of the recycling synaptic vesicle pool (Rozas et al., 2011). In contrast, it has been shown that skeletal muscles of R6/2 mice developed age-dependent denervation-like abnormalities, including reduced endplate area, supersensitivity to acetylcholine, decreased sensitivity to mu-conotoxin and anode-break action potentials (Ribchester et al., 2004). Moreover, the miniature endplate potential (mEPP) amplitude was notably increased while mEPP frequency was significantly reduced in R6/2 mice. Severely affected R6/2 mice developed a progressive increase in a number of motor endplates that fail to respond to nerve stimulation but there was no constitutive sprouting of motor neurons, even in severely atrophic muscles. In fact there was no age-dependent loss of regenerative capacity of motor neurons in R6/2 mice (Ribchester et al., 2004). Another group has studied the membrane properties of skeletal muscles that control contraction in the same HD mouse model. Adult skeletal muscle from R6/2 mice showed that the action potentials in diseased muscles were more easily triggered and prolonged than in wild type littermates. Furthermore, the expression of the muscle chloride channel (ClC-1) and Kcnj2 (Kir2.1 potassium channel) transcripts were significantly reduced and defects in mRNA processing were detected (Waters et al., 2013).
To better understand a mechanism underlying muscle wasting in the R6/2 mouse model, key pathways governing protein metabolism, apoptosis and autophagy were examined. R6/2 mice exhibited increased adiposity and elevated energy expenditure without altered food intake. A total protein synthesis was unexpectedly increased in the gastrocnemius muscle by 19%, which was associated with over-activation of rapamycin mTOR signaling (She et al., 2011). The transcript levels of androgens, like muscle ring finger-1 and atrophy F-box, were markedly attenuated during fasting and re-feeding. Additionally, the mRNA level of several caspase genes involved in both extrinsic and intrinsic apoptotic pathways, like CASPASE-3/7, −8, and −9, were elevated (She et al., 2011). Indeed, the CASPASE-6 up-regulation might be due to enhanced activity of the p53 in the muscles obtained from HD patients and from two different HD mouse models. It has been also shown that CASPASE-6 may target (cleave) laminin A (Ehrnhoefer et al., 2014). It was suggested that this phenomenon might be mitigated by a small molecule pifithrin-alpha, an inhibitor of p53 transcriptional activity (Ehrnhoefer et al., 2014).
Since mitochondrial dysfunction might play a crucial role in HD pathology (Quintanilla and Johnson, 2009), the role of PPAR γ coactivator 1α (PGC-1α) has been carefully assessed (Lin et al., 2005). Reduced levels of PGC-1α and its target genes in skeletal muscles of HD transgenic mice and HD subjects have been found. Treatment with guanidinopropionic acid (GPA) led to an increased expression level of AMPK, PGC-1α target genes and the genes characteristic for oxidative phosphorylation, electron transport chain and mitochondrial biogenesis. Oxygen consumption in response to GPA treatment was significantly reduced in myoblasts from HD patients (Chaturvedi et al., 2009). On the other hand, knockdown of mutant HTT resulted in increased PGC-1α expression in HD myoblast, while PGC-1α rescue led to increased expression of markers for oxidative muscle fibers and reversal of blunted response for GPA in HD mice (Chaturvedi et al., 2009). These findings showed that impaired function of PGC-1α plays a critical role in the skeletal muscles dysfunction in HD. Also, possible pharmacologic intervention with a small molecule could enhance PGC-1α function may exert therapeutic benefits (Chaturvedi et al., 2009). In addition, atrophic fibers of R6/2 mice showed increased fuchsinophilic aggregates and reduced cytochrome c oxidase by 15%. Complex I–dependent respiration of HD mitochondria showed more sensitivity to inhibition by Ca2+ than in wild-type mitochondria (Gizatullina et al., 2006). A summary of morphological and molecular characteristics of skeletal muscles in pre-clinical and clinical settings has been presented in Table 1.
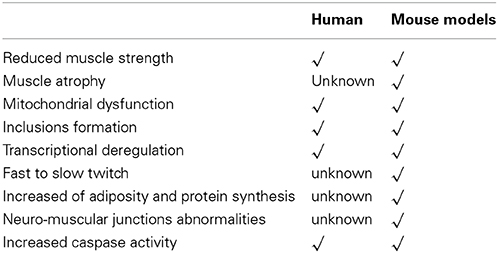
Table 1. Summary of defects observed in muscle in the pre-clinical and clinical HD settings.
Can we Delay HD Progression by Modulating Muscle Function?
As it has been mentioned in the previous paragraph, enhancing PGC-1α activity might be a good strategy to improve skeletal muscles function in HD. Indeed, pharmacologic treatment with the pan-PPAR agonist bezafibrate restored the PGC-1α, PPARs and downstream genes to wild type levels. It also prevented conversion of type I oxidative to type II glycolytic muscle fibers as well as increased muscle mitochondria numbers. Finally, bezafibrate rescued lipid accumulation and apparent vacuolization of brown adipose tissue in the HD mice (Johri et al., 2012).
The other strategy to improve muscle function in HD is based on the heat shock machinery modulation that could suppress mHTT aggregation (Labbadia and Morimoto, 2013). The R6/2 mice expressing an active heat shock transcription factor 1 (HSF1) isoform had reduced polyglutamine inclusion formation and improved body weight. Unexpectedly, the lifespan of R6/2:HSF1Tg mice were significantly improved despite the fact that active HSF1 was not expressed in the brain. These results indicated that active HSF1 has a strong inhibitory effect on polyglutamine aggregates formation in vivo (Fujimoto et al., 2005).
Recent studies also showed that HDAC4 function in the cytoplasm (Mielcarek et al., 2013a) and its reduction, delayed cytoplasmic aggregate formation and rescued neuronal and cortico-striatal synaptic function in HD mouse models. This was accompanied by an improvement in motor co-ordination, neurological phenotypes and increased lifespan (Mielcarek et al., 2013b). Given that HDAC4 has well-established functions in skeletal muscle, muscle atrophy is a major symptom of HD and that HDAC4 has been linked to disease progression in an ALS mouse model, it is likely that genetic reduction of HDAC4 in skeletal muscle was a contributing factor to the improved HD phenotypes (Bruneteau et al., 2013).
Summary
HD is a complex disease that has a peripheral component to its pathophysiology. Transcriptional changes in the HD skeletal muscles were comparable to those observed in the different brain regions and skeletal muscle wasting/atrophy is likely to be an important portion of HD pathogenesis. Some of the molecular and physiological changes in HD muscles can be detected, even in the pre-symptomatic HD individuals. On the molecular level, mitochondrial dysfunctions, PPAR alpha signaling and HSF1 activation were identified as major players in the muscle HD-related pathology. The major pathological pathways identified in skeletal muscles have been summarized in Figure 1. However, many aspects of HD neuromuscular transmission and muscle physiology remain unanswered and need to be studied more extensively. The proof of concept studies clearly showed that by improving muscle function in HD mouse models, the progression of disease onset could be delayed and the lifespan extended. Therefore this makes skeletal muscles an attractive target for future therapies. Two key signaling pathways, i.e., Insulin like growth Factor IGF and GDF-8/myostatin, have emerged in recent years to be potent regulators of skeletal muscle size. Moreover, several studies emphasized a role of hyperacetylation in muscle wasting. Therefore there is a need for more pre-clinical and clinical studies that will unravel the mechanism of HD skeletal muscles pathology, leading to potential therapies in HD. Further work is necessary in order to fully appreciate the complexity of the pathways that are affected during HD progression. Indeed, emerging evidence has clearly indicated that peripheral tissues are as much affected by the expression of the mutant huntingtin as the Central Nervous System. Furthermore, the possibility to test the effect of new drugs directly on human peripheral tissues is a new and exciting research area.
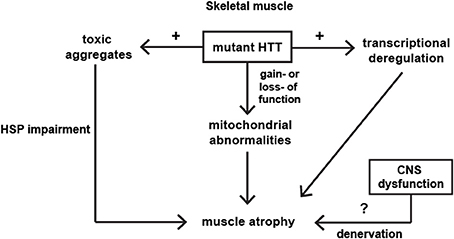
Figure 1. Summary of pathological events identified in the skeletal muscles in HD.
Conflict of Interest Statement
Conflict of Interest Statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bruneteau, G., Simonet, T., Bauche, S., Mandjee, N., Malfatti, E., Girard, E., et al. (2013). Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: potential role in reinnervation ability and disease progression. Brain 136, 2359–2368. doi: 10.1093/brain/awt164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Busse, M. E., Hughes, G., Wiles, C. M., and Rosser, A. E. (2008). Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington's disease. J. Neurol. 255, 1534–1540. doi: 10.1007/s00415-008-0964-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carter, R. J., Lione, L. A., Humby, T., Mangiarini, L., Mahal, A., Bates, G. P., et al. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J. Neurosci. 19, 3248–3257.
Chaturvedi, R. K., Adhihetty, P., Shukla, S., Hennessy, T., Calingasan, N., Yang, L., et al. (2009). Impaired PGC-1alpha function in muscle in Huntington's disease. Hum. Mol. Genet. 18, 3048–3065. doi: 10.1093/hmg/ddp243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ciammola, A., Sassone, J., Alberti, L., Meola, G., Mancinelli, E., Russo, M. A., et al. (2006). Increased apoptosis, Huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington's disease subjects. Cell Death Differ. 13, 2068–2078. doi: 10.1038/sj.cdd.4401967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ciammola, A., Sassone, J., Sciacco, M., Mencacci, N. E., Ripolone, M., Bizzi, C., et al. (2011). Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington's disease. Mov. Disord. 26, 130–137. doi: 10.1002/mds.23258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crook, Z. R., and Housman, D. (2011). Huntington's disease: can mice lead the way to treatment? Neuron 69, 423–435. doi: 10.1016/j.neuron.2010.12.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ehrnhoefer, D. E., Skotte, N. H., Ladha, S., Nguyen, Y. T., Qiu, X., Deng, Y., et al. (2014). p53 increases caspase-6 expression and activation in muscle tissue expressing mutant huntingtin. Hum. Mol. Genet. 23, 717–729. doi: 10.1093/hmg/ddt458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fujimoto, M., Takaki, E., Hayashi, T., Kitaura, Y., Tanaka, Y., Inouye, S., et al. (2005). Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 280, 34908–34916. doi: 10.1074/jbc.M506288200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gizatullina, Z. Z., Lindenberg, K. S., Harjes, P., Chen, Y., Kosinski, C. M., Landwehrmeyer, B. G., et al. (2006). Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann. Neurol. 59, 407–411. doi: 10.1002/ana.20754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gusella, J. F., MacDonald, M. E., Ambrose, C. M., and Duyao, M. P. (1993). Molecular genetics of Huntington's disease. Arch. Neurol. 50, 1157–1163. doi: 10.1001/archneur.1993.00540110037003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
HDRG. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72, 971–83. doi: 10.1016/0092-8674(93)90585-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoogeveen, A. T., Willemsen, R., Meyer, N., de Rooij, K. E., Roos, R. A., van Ommen, G. J., et al. (1993). Characterization and localization of the Huntington disease gene product. Hum. Mol. Genet. 2, 2069–2073. doi: 10.1093/hmg/2.12.2069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johri, A., Calingasan, N. Y., Hennessey, T. M., Sharma, A., Yang, L., Wille, E., et al. (2012). Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington's disease. Hum. Mol. Genet. 21, 1124–1137. doi: 10.1093/hmg/ddr541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kosinski, C. M., Schlangen, C., Gellerich, F. N., Gizatullina, Z., Deschauer, M., Schiefer, J., et al. (2007). Myopathy as a first symptom of Huntington's disease in a Marathon runner. Mov. Disord. 22, 1637–1640. doi: 10.1002/mds.21550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Labbadia, J., and Morimoto, R. I. (2013). Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 38, 378–385. doi: 10.1016/j.tibs.2013.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, C. Y., Cantle, J. P., and Yang, X. W. (2013). Genetic manipulations of mutant huntingtin in mice: new insights into Huntington's disease pathogenesis. FEBS J. 18, 4382–4394. doi: 10.1111/febs.12418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, S. H., and Li, X. J. (2004). Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet. 20, 146–154. doi: 10.1016/j.tig.2004.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, J., Handschin, C., and Spiegelman, B. M. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370. doi: 10.1016/j.cmet.2005.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lione, L. A., Carter, R. J., Hunt, M. J., Bates, G. P., Morton, A. J., and Dunnett, S. B. (1999). Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J. Neurosci. 19, 10428–10437.
Lodi, R., Schapira, A. H., Manners, D., Styles, P., Wood, N. W., Taylor, D. J., et al. (2000). Abnormal in vivo skeletal muscle energy metabolism in Huntington's disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 48, 72–76. doi: 10.1002/1531-8249(200007)48:1<72::AID-ANA11>3.0.CO;2-I
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luthi-Carter, R., Hanson, S. A., Strand, A. D., Bergstrom, D. A., Chun, W., Peters, N. L., et al. (2002). Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Genet. 11, 1911–1926. doi: 10.1093/hmg/11.17.1911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mielcarek, M., Inuabasi, L., Bondulich, M. K., Muller, T., Osborne, G. F., Franklin, S. A., et al. (2014). Dysfunction of the CNS-Heart Axis in Mouse Models of Huntington's Disease. PLoS Genet. 10:e1004550. doi: 10.1371/journal.pgen.1004550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mielcarek, M., Landles, C., Weiss, A., Bradaia, A., Seredenina, T., Inuabasi, L., et al. (2013b). HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 11:e1001717. doi: 10.1371/journal.pbio.1001717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mielcarek, M., Seredenina, T., Stokes, M. P., Osborne, G. F., Landles, C., Inuabasi, L., et al. (2013a). HDAC4 does not act as a protein deacetylase in the postnatal murine brain in vivo. PLoS ONE 8:e80849. doi: 10.1371/journal.pone.0080849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moffitt, H., McPhail, G. D., Woodman, B., Hobbs, C., and Bates, G. P. (2009). Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington's disease. PLoS ONE 4:e8025. doi: 10.1371/journal.pone.0008025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, K. P., Carter, R. J., Lione, L. A., Mangiarini, L., Mahal, A., Bates, G. P., et al. (2000). Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J. Neurosci. 20, 5115–5123.
Novak, M. J., and Tabrizi, S. J. (2010). Huntington's disease. BMJ 340:c3109. doi: 10.1136/bmj.c3109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orth, M., Cooper, J. M., Bates, G. P., and Schapira, A. H. (2003). Inclusion formation in Huntington's disease R6/2 mouse muscle cultures. J. Neurochem. 87, 1–6. doi: 10.1046/j.1471-4159.2003.02009.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quintanilla, R. A., and Johnson, G. V. W. (2009). Role of mitochondrial dysfunction in the pathogenesis of Huntington's disease. Brain Res. Bull. 80, 242–247. doi: 10.1016/j.brainresbull.2009.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rattray, I., Smith, E., Gale, R., Matsumoto, K., Bates, G. P., and Modo, M. (2013). Correlations of behavioral deficits with brain pathology assessed through longitudinal MRI and histopathology in the R6/2 mouse model of HD. PLoS ONE 8:e60012. doi: 10.1371/journal.pone.0060012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reddy, P. H. (2014). Increased mitochondrial fission and neuronal dysfunction in Huntington's disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov. Today 19, 951–955. doi: 10.1016/j.drudis.2014.03.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ribchester, R. R., Thomson, D., Wood, N. I., Hinks, T., Gillingwater, T. H., Wishart, T. M., et al. (2004). Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur. J. Neurosci. 20, 3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rozas, J. L., Gomez-Sanchez, L., Tomas-Zapico, C., Lucas, J. J., and Fernandez-Chacon, R. (2011). Increased neurotransmitter release at the neuromuscular junction in a mouse model of polyglutamine disease. J. neurosci. 31, 1106–1113. doi: 10.1523/JNEUROSCI.2011-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saft, C., Zange, J., Andrich, J., Muller, K., Lindenberg, K., Landwehrmeyer, B., et al. (2005). Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov. Disord. 20, 674–679. doi: 10.1002/mds.20373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sassone, J., Colciago, C., Cislaghi, G., Silani, V., and Ciammola, A. (2009). Huntington's disease: the current state of research with peripheral tissues. Exp. Neurol. 219, 385–397. doi: 10.1016/j.expneurol.2009.05.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sathasivam, K., Hobbs, C., Turmaine, M., Mangiarini, L., Mahal, A., Bertaux, F., et al. (1999). Formation of polyglutamine inclusions in non-CNS tissue. Hum. Mol. Genet. 8, 813–822. doi: 10.1093/hmg/8.5.813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sathasivam, K., Neueder, A., Gipson, T. A., Landles, C., Benjamin, A. C., Bondulich, M. K., et al. (2013). Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, 2366–2370. doi: 10.1073/pnas.1221891110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
She, P., Zhang, Z., Marchionini, D., Diaz, W. C., Jetton, T. J., Kimball, S. R., et al. (2011). Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington's disease. Am. J. Physiol. Endocrinol. Metab. 301, E49–E61. doi: 10.1152/ajpendo.00630.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strand, A. D., Aragaki, A. K., Shaw, D., Bird, T., Holton, J., Turner, C., et al. (2005). Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum. Mol. Genet. 14, 1863–1876. doi: 10.1093/hmg/ddi192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strong, T. V., Tagle, D. A., Valdes, J. M., Elmer, L. W., Boehm, K., Swaroop, M., et al. (1993). Widespread expression of the human and rat Huntington's disease gene in brain and nonneural tissues. Nat. Genet. 5, 259–265. doi: 10.1038/ng1193-259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trottier, Y., Devys, D., Imbert, G., Saudou, F., An, I., Lutz, Y., et al. (1995). Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat. Genet. 10, 104–110. doi: 10.1038/ng0595-104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van der Burg, J. M., Bjorkqvist, M., and Brundin, P. (2009). Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 8, 765–774. doi: 10.1016/S1474-4422(09)70178-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wade, A., Jacobs, P., and Morton, A. J. (2008). Atrophy and degeneration in sciatic nerve of presymptomatic mice carrying the Huntington's disease mutation. Brain Res. 1188, 61–68. doi: 10.1016/j.brainres.2007.06.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walker, F. O. (2007). Huntington's disease. Lancet 369, 218–228. doi: 10.1016/S0140-6736(07)60111-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waters, C. W., Varuzhanyan, G., Talmadge, R. J., and Voss, A. A. (2013). Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc. Natl. Acad. Sci. U.S.A. 110, 9160–9165. doi: 10.1073/pnas.1220068110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeitlin, S., Liu, J. P., Chapman, D. L., Papaioannou, V. E., and Efstratiadis, A. (1995). Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat. Genet. 11, 155–163. doi: 10.1038/ng1095-155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zielonka, D., Piotrowska, I., and Mielcarek, M. (2014). Cardiac Dysfunction in Huntington's Disease. Exp. Clin. Cardiol. 20, 2547–2554. Available online at: http://cardiologyacademicpress.com/soap/pdf/delme_1788_53e3f6e3313354.90986595.pdf
Keywords: Huntington's disease, peripheral pathology, skeletal muscle atrophy, disease modifying treatment
Citation: Zielonka D, Piotrowska I, Marcinkowski JT and Mielcarek M (2014) Skeletal muscle pathology in Huntington's disease. Front. Physiol. 5:380. doi: 10.3389/fphys.2014.00380
Received: 31 July 2014; Accepted: 13 September 2014;
Published online: 06 October 2014.
Edited by:
Julio L. Vergara, University of California, Los Angeles, USAReviewed by:
Seth L. Robia, Loyola University Chicago, USAAndrew Alvin Voss, Wright State University, USA
Copyright © 2014 Zielonka, Piotrowska, Marcinkowski and Mielcarek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Zielonka, Department of Social Medicine, Poznan University of Medical Sciences, Rokietnicka Str., No. 5 “C,” 60-806 Poznan, Poland e-mail: daniel.zielonka@gmail.com;
Michal Mielcarek, Department of Medical and Molecular Genetics, School of Medicine, King's College London, 8th Floor Tower Wing, Guy's Hospital Great Maze Pond, London, SE1 9RT, UK e-mail: michal.mielcarek@kcl.ac.uk