 Marco Falasca
Marco Falasca Tania Maffucci
Tania Maffucci- Inositide Signalling Group, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, UK
Phosphoinositide 3-kinases (PI3Ks) regulate several cellular functions that are critical for cancer progression and development, including cell survival, proliferation and migration. Three classes of PI3Ks exist with the class I PI3K encompassing four isoforms of the catalytic subunit known as p110α, p110β, p110γ, and p110δ. Although for many years attention has been mainly focused on p110α recent evidence supports the conclusion that p110β, p110γ, and p110δ can also have a role in cancer. Amongst these, accumulating evidence now indicates that p110γ is involved in several cellular processes associated with cancer and indeed this specific isoform has emerged as a novel important player in cancer progression. Studies from our laboratory have identified a specific overexpression of p110γ in human pancreatic ductal adenocarcinoma (PDAC) and in hepatocellular carcinoma (HCC) tissues compared to their normal counterparts. Our data have further established that selective inhibition of p110γ is able to block PDAC and HCC cell proliferation, strongly suggesting that pharmacological inhibition of this enzyme can directly affect growth of these tumors. Furthermore, increasing evidence suggests that p110γ plays also a key role in the interactions between cancer cells and tumor microenvironment and in particular in tumor-associated immune response. It has also been reported that p110γ can regulate invasion of myeloid cells into tumors and tumor angiogenesis. Finally p110γ has also been directly involved in regulation of cancer cell migration. Taken together these data indicate that p110γ plays multiple roles in regulation of several processes that are critical for tumor progression and metastasis. This review will discuss the role of p110γ in gastrointestinal tumor development and progression and how targeting this enzyme might represent a way to target very aggressive tumors such as pancreatic and liver cancer on multiple fronts.
Introduction
Gastrointestinal cancers comprise a group of cancers that affect the gastrointestinal tract and include esophageal, stomach, liver, pancreatic and colorectal cancer. Gastrointestinal cancers have one of the poorest prognoses among all cancers partly because of their silent nature and tendency for late discovery but also because of their peculiar resistance to chemotherapy and radiation therapy (Lockhart et al., 2005; Schneider et al., 2005; Thrumurthy et al., 2013; Zhang, 2013; Brenner et al., 2014; Singh et al., 2014). The prospect for patients with liver and pancreatic cancer is particularly dismal. For instance, the 5 year survival rate for pancreatic ductal adenocarcinomas (PDAC) is the lowest among all cancers (Li et al., 2004). Similarly in 2011 hepatocellular carcinoma (HCC) was estimated as the second and sixth leading cause of cancer-related death in men and women respectively (Jemal et al., 2011). There have been great advances in survival rates for many types of cancers over the past few decades but hardly any change for PDAC and HCC. There are very few treatments for PDAC, most of them just palliative. Similarly only a small percentage of patients with HCC are eligible for surgery since the majority of patients present with advanced or unresectable disease (Kuper et al., 2000; Thomas et al., 2010; Singh et al., 2014). For many years systemic chemotherapy also proved to be only minimally effective (Ryder, 2003; Thomas et al., 2008) and currently only the multikinase inhibitor sorafenib is approved for advanced HCC patients (Thomas et al., 2010). There is therefore an urgent need to better understand the mechanisms underlying progression of these cancer types in order to develop novel potential chemotherapeutic agents.
It is well known that mutations in K-Ras oncogene occur in 75–90% of PDAC and accumulate early in the disease progression (Moskaluk et al., 1997; Hruban et al., 2000). On the other hand, metastatic colorectal cancer represents one of the largest hurdles in cancer treatment and metastatic colorectal tumors with a mutation in K-Ras do not respond to available treatments such as anti-epidermal growth factor receptor monoclonal antibodies (Brand and Wheeler, 2012). K-Ras signaling promotes the neoplastic phenotype via activation of downstream targets that control membrane trafficking, cellular proliferation, differentiation and cytoskeleton organization (Lockhart et al., 2005). A key downstream target of the Ras family is phosphoinositide 3-kinase (PI3K), the enzyme responsible for generation of 3-phosphorylated phosphoinositides and activation of the protein kinase B/Akt (Kodaki et al., 1994; Chang et al., 1997; Khwaja et al., 1997; Luo et al., 2003). Indeed activation of Akt has been observed in PDAC and represents a biological indicator of the aggressiveness of the disease (Yamamoto et al., 2004). Akt and in particular its downstream effector mechanistic target of rapamycin (mTOR) have also been established as key molecular targets in HCC and inhibitors of these molecules have been tested in clinical trials (Shen et al., 2013). Although eight distinct PI3K isoforms exist most of the studies on PI3K and cancer have been focused so far on one specific isoform, p110α that has been found to be mutated in several cancer types. Only recently increasing evidence has suggested that other PI3K isoforms may also play a non-redundant role in different tumor settings. The aim of this review is to summarize the evidence indicating that the PI3K isoform p110γ plays a key role in gastrointestinal cancers.
Phosphoinositide 3-Kinases
PI3Ks catalyze the phosphorylation of lipids known as phosphoinositides in position 3 of their inositol rings (Falasca and Maffucci, 2012). Phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3], originally identified in activated neutrophils from human donors (Traynor-Kaplan et al., 1988), is the best characterized of the PI3K lipid products and it plays a key role in activation of several signaling molecules. These include Akt, 3 phosphoinositide dependent protein kinase 1 and their effector proteins such as mTOR that in turn regulate signaling cascades involved in cell growth, survival, proliferation, motility and morphology. It is now well-established that the PI3K/Akt pathway plays a pivotal role in several processes that are critical for cancer development and progression including inhibition of apoptosis, stimulation of cell proliferation and drug resistance (Luo et al., 2003; Takeda et al., 2004). Indeed it has been estimated that at least 50% of all cancer types present some deregulation of this signaling pathway (Yuan and Cantley, 2008). In particular, the PI3K/Akt pathway is activated in almost 60% of PDAC (Bondar et al., 2002; Schlieman et al., 2003) and it plays a critical role in HCC (Zhou et al., 2011).
Eight PI3K isoforms exist in mammalian cells and they have been grouped into three classes according to their structure and substrate specificity (Vanhaesebroeck et al., 2001; Falasca and Maffucci, 2007, 2012) with the PI3K class I encompassing four isoforms of the catalytic subunit known as p110α, p110β, p110γ, and p110δ. Amongst these, p110α has a well-established role in cancer and gain of function of this isoform due to mutation of its gene PIK3CA is common in several human cancers (Samuels et al., 2004; Zhao and Vogt, 2008). Whilst mutations to PI3K are commonly associated with PIK3CA it is important to notice that mutations have actually been found in all PI3K isoforms, although their prevalence and functional relevance in disease is considered limited. An overview of these mutations can be found on the COSMIC website (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Although somatic mutations of the genes encoding the other isoforms are less frequent, accumulating data now suggest that p110β, p110γ, and p110δ can also have a role in cancer.
It was previously reported that while overexpression of wild-type p110α does not have transforming potential, overexpression of the wild-type catalytic subunits p110β, p110γ, and p110δ is sufficient to induce an oncogenic phenotype in cultured cells (Kang et al., 2006). These data suggested that increased expression levels of the “non α” catalytic subunits rather than gain of function mutations can be relevant in cancer development and progression. Indeed increased levels of both p110β and p110δ have been observed in glioblastoma and in some colon and bladder tumors (Bénistant et al., 2000; Knobbe and Reifenberger, 2003). p110β has been shown to stimulate cell proliferation and invasive cell growth (Czauderna et al., 2003) whereas p110δ controls proliferation in acute myeloid leukemia (Sujobert et al., 2005) and migration of breast cancer cells (Sawyer et al., 2003). More recently it has been reported that inactivation of p110δ in mice models inhibits different cancer types and induces tumor regression (Ali et al., 2014). Interestingly, amongst the distinct cancer models investigated in this study, the authors also reported that treatment with PI-3065, a small molecule inhibitor with selectivity for p110δ, prolonged survival and reduced the incidence of macroscopic metastases in the LSL KRasG12D/+; p53R172H/+; PdxCretg/+ model of PDAC (Ali et al., 2014).
Accumulating evidence from several groups indicates that the class IB isoform p110γ has a role in several cellular processes involved in tumor development and progression, including proliferation of pancreatic and liver cancer cells (Edling et al., 2010; Dituri et al., 2012), tumor angiogenesis (Hamada et al., 2005), drug resistance in chronic myeloid leukemia cells (Hickey and Cotter, 2006) and many more processes. This review will discuss the current evidence supporting the conclusion that inhibition of p110γ can represent an important strategy to target cancers on multiple fronts.
p110γ
The PI3K catalytic subunit p110γ is encoded by the PIK3CG gene, located on chromosome 7q22.3 and its activity is modulated via interaction with the p101 (PIK3R5) and p87 (PIK3R6) regulatory subunits (Fyffe et al., 2013). The original classification of this isoform into the class IB subgroup of PI3Ks mainly derived from the observation that p110γ can be activated downstream of G-protein coupled receptors (GPCRs). Evidence however suggests that tyrosine kinase receptors (RTKs) can also activate p110γ (Ptasznik et al., 1996; Vanhaesebroeck et al., 2010). Importantly, Ras has also a key role in p110γ activation.
p110γ is mainly expressed in hematopoietic cells and is involved in immune, inflammatory and allergic responses (Vanhaesebroeck et al., 2010). Several studies have indicated a key role for this isoform in migration of hematopoietic cell types. For instance it has been reported that p110γ modulates leukocyte chemotaxis to inflammatory sites and in response to chemoattractant agents and it is also involved in motility of dendritic cells (Del Prete et al., 2004). Both p110γ and p110δ are involved in natural killer cell development and migration toward the sites of inflammation and in T-lymphocyte migration and development (So and Fruman, 2012). In addition to a specific role in migration, p110γ can also regulate T-lymphocyte proliferation and cytokine production (So and Fruman, 2012). It has also been demonstrated that combined inactivation of p110γ and p110δ impairs B cell development and reduces B cell numbers to a greater extent than p110δ inactivation alone (Beer-Hammer et al., 2010). The lipid kinase activity of the enzyme seems to be important for regulation of these processes and indeed p110γ could play a role in leukocyte polarization and migration by regulating the spatial accumulation of PtdIns(3,4,5)P3, the organization of F-actin formation and integrin-based adhesion at the leading edge (Hirsch et al., 2000). Interestingly it has been demonstrated that p110γ also possesses a serine/threonine protein kinase activity that is independent from its lipid kinase activity (Hirsch et al., 2001). Specifically it has been reported that p110γ plays a key role in platelet aggregation and thrombosis by regulating αIIb/β3 integrin adhesive function in platelets downstream of P2Y12 through a mechanism that is independent from its lipid kinase activity (Hirsch et al., 2001). In addition it has been reported that p110γ can control cardiac contractility through formation of a multiprotein complex with PDE3B and independently from its kinase activity (Patrucco et al., 2004). Taken together these data indicate that p110γ can be involved in several cellular functions through distinct molecular mechanisms.
Role of p110γ in Cancer Cell Proliferation
Although p110γ is primarily expressed in leukocytes and has a well characterized role in immunity (Hirsch et al., 2014) evidence also indicates a role for this PI3K isoform in some cancer types. It must be noted that an original investigation reported that p110γ−/− mice on 129J background developed macroscopically visible tumors mainly at the proximal and distal parts of the large intestine (Sasaki et al., 2000). However, the authors did not observe the same tumor phenotype after backcrossing these mice onto a C57BL/6 background, neither they observed formation of tumors when they retargeted the allele in different ES cells using the same targeting construct. Therefore, the authors changed their original conclusion that inactivation of p110γ leads to development of invasive colorectal adenocarcinomas in mice (Sasaki et al., 2000) by stating that this inactivation does not in itself cause colon cancer (corrigenda Sasaki et al., 2003).
Although the prevalence and functional relevance of mutations in PI3Ks other than p110α are considered limited it is worth mentioning that significant recurrent mutations have been seen in both PIK3CG (9.7%) and PIK3C2B (12.9%), the gene encoding for the class II isoform PI3K-C2β, in lung cancer (Liu et al., 2012). Interestingly PIK3CG is located in a region of chromosome band 7q22 that is frequently deleted in myeloid malignancies.
Recently, new driver mutations in pancreatic cancer have been identified using cancer-specific high-throughput annotation of somatic mutations (Carter et al., 2010). Importantly, PIK3CG contains the second highest scoring predicted driver mutation among the set of genes not previously identified as a driver in pancreatic cancer. The specific PIK3CG mutation identified is R839C and the residue Arg 839 is located within the C-terminal catalytic domain. Although the authors suggest that this mutation may lead to a loss of function of the enzyme based on X-ray crystal structure (Carter et al., 2010) it actually remains to be established whether it indeed results in modulation of p110γ catalytic activity and the potential functional consequences on pancreatic cancer cells.
Recent studies in our laboratory have identified a selective accumulation of p110γ in specific cancer types. In particular, in an extensive investigation of the expression of all PI3Ks we detected a specific overexpression of p110γ in PDAC tissues compared to normal counterparts (Edling et al., 2010). Similarly immunohistochemistry analysis of PI3Ks in HCC and paired peritumoral human tissues showed p110γ expression in HCC tissues, in particular in epithelial cancer cells (Dituri et al., 2012). Importantly p110γ correlated with the proliferative marker Ki-67 in these tissues, indicating accumulation of this isoform in cancer cells with high proliferative index (Dituri et al., 2012). Consistent with this, our data demonstrated that p110γ is required for PDAC and HCC cell proliferation. Specifically we reported that chemical inhibition as well as selective siRNA-mediated downregulation of p110γ reduced pancreatic cancer cell growth without increasing cell apoptosis (Edling et al., 2010). Similarly we showed that downregulation of p110γ in HCC cell lines specifically induced arrest of cell cycle in the G2/M phase through modulation of p21 levels (Dituri et al., 2012). These data indicate that p110γ has a specific role in regulation of cancer cell proliferation and strongly suggest that overexpression of p110γ has a functional role in progression of PDAC and HCC. The molecular mechanisms regulating p110γ overexpression in specific cancer types are yet to be established.
Taken together these data suggest that inhibition of p110γ can inhibit proliferation of specific cancer types. In this respect we recently demonstrated that treatment with the caffeine analog CGS 15943 inhibited proliferation of HCC and PDAC cell lines (Edling et al., 2014). Interestingly, a kinase profiler analysis revealed that CGS 15943 is able to inhibit p110γ (Edling et al., 2014), further supporting the conclusion that targeting this PI3K isoform may prove beneficial to directly reduce PDAC and HCC growth (Falasca et al., 2011).
Role of p110γ in Cancer Metastasis
Metastasis, the ability of cancer cells to spread from a primary site and form tumors at distant sites, is the main cause of death of most cancer patients. Several steps regulate the development of metastasis including migration of cancer cells out of the primary tumor, local invasion, intravasation into the circulatory system, survival, extravasation, initiation and maintenance of micro-metastases at distant sites and vascularization of the resulting tumors (Nguyen and Massague, 2007). A peculiar feature of this process is the variability in metastatic tissue tropism shown by different types of cancer. Indeed, even though cancer cells can spread to almost every area of the body, each cancer type shows specific preference for common regions where cancer may spread to (Nguyen et al., 2009). Cancer cells with high metastatic potential are characterized by high proliferation and migration. The endogenous signaling pathways associated with high metastatic potential remain unclear.
Although it was well established that p110γ is involved in the regulation of migration of different cell types, including leukocytes and endothelial cells (ECs) (as discussed below) only recently a specific role for this isoform in regulation of cancer cell dissemination and metastasis has been suggested (Attoub et al., 2008; Brazzatti et al., 2012; Xie et al., 2013). Specifically it has been reported that shRNA-mediated downregulation of p110γ reduces the ability of breast cancer cells MDA-MB-231 to metastasize in vivo (Brazzatti et al., 2012). Similar data were obtained in the 4T1.2 mouse model of breast cancer (Brazzatti et al., 2012). Several mechanisms of p110γ-dependent regulation of metastasis formation have been proposed. For instance recent data indicate that p110γ exerts transforming functions via several mechanisms in human colon epithelial cancer cells, including alteration of homotypic cell–cell adhesion and induction of collagen type I invasion through canonical pro-invasive pathways (Attoub et al., 2008). The pro-metastatic activity of p110γ in metastatic epithelial carcinoma cells seems to involve its ability to prevent anoikis, an apoptotic process resulting from disruption of cell-matrix interaction-dependent survival (Brazzatti et al., 2012). Evidence also suggests that p110γ can be directly involved in cancer cell migration and invasion, as it has been shown in MDA-MB-231 (Brazzatti et al., 2012; Xie et al., 2013) and in melanoma cells (Monterrubio et al., 2009) in response to the chemokine CXCL12. In addition, p110γ is involved in lysophosphatidic acid (LPA) signaling (Edling et al., 2010). LPA signaling has been shown to play a role in cancer and in particular in cancer cell migration (Van Meeteren and Moolenaar, 2007). Both CXCL12 and LPA are key regulators of metastatic processes in several cancers including gastrointestinal cancers. Interestingly CXC chemokine receptor 4 plays an important role in pancreatic cancer metastasis, and initiates G-protein signaling when activated by its ligand CXCL12 (Marchesi et al., 2004).
Taken together these data support the conclusion that inhibition of p110γ can affect cancer cell migration and metastasis formation.
Roles of p110γ in Angiogenesis
A prerequisite for tumor growth is the supply of nutrients and oxygen that are provided by the formation of new blood vessels in a process named tumor angiogenesis (Welti et al., 2013). Several cancer types, including HCC, strongly rely on tumor angiogenesis and anti-angiogenic strategies are either in use or are currently being tested in clinical trials as anti-cancer strategies in several tumor settings.
Tumor angiogenesis involves the coordinated action of different cell types that are normally present or are recruited to the tumor site. In particular tumor angiogenesis is the results of proliferation, migration and remodeling of activated ECs that eventually form novel capillaries (Welti et al., 2013). Several transgenic mouse models have established the pivotal role for class I PI3K isoforms in EC functions. Specifically a central role for p110α has been demonstrated by the observation that knockout mice for this isoform die during embryogenesis (E9.5) due to defects in vasculogenesis and this was confirmed by knock-in strategies (Graupera et al., 2008). Transgenic mouse models further indicated specific roles for p110γ in ECs. Indeed, although knock out and knock in p110γ mice were mainly characterized by immunological and cardiac (in the case of p110γ−/− mice) defects (Morello et al., 2009) it was also observed that muscles from p110γ−/− mice presented reduced capillarization and arteriogenesis following unilateral limb ischemia compared to wild type mice which in turn resulted in delayed blood flow recovery (Madeddu et al., 2008). A role in proliferation and survival of ECs was suggested by the observation that the number of BrdU-positive capillaries was reduced in p110γ−/− ischemic gastrocnemius muscles compared to wild type whereas the percentage of apoptotic capillaries was increased. Interestingly, evidence also suggested that the lipid kinase activity of p110γ was not required in regulation of these cellular processes since post-ischemic neovascularization was not impaired and microvascular apoptosis was not increased in p110γKD/KD (kinase dead) mice. Furthermore, a defect in proliferation, survival and migration induced by stromal cell-derived factor-1 was detected in endothelial progenitor cells from p110γ−/− but not from p110γKD/KD mice, suggesting that p110γ may regulate endothelial progenitor cells function and muscular angiogenesis through a mechanism independent of its kinase activity. On the other hand both p110γ−/− and p110γKD/KD endothelial progenitor cells showed reduced integration into endothelial networks, indicating that the lipid kinase activity is required for some steps of capillary formation.
Evidence from several groups, including our own, has also identified a role for p110γ in regulation of EC migration. Specifically a previous study demonstrated that both p110β and p110γ are involved in sphingosine 1-phosphate (S1P)-mediated EC migration (Heller et al., 2008). We further reported that chemical inhibition and siRNA-mediated downregulation of p110γ reduced S1P-induced as well as high density lipoprotein 3-induced EC migration (Tibolla et al., 2013). Importantly downregulation of p110γ also inhibited S1P- and high density lipoprotein 3-induced remodeling of ECs on Matrigel indicating a key role for this isoform in regulation of capillary tube formation (Tibolla et al., 2013). Interestingly, analysis of cell motility further indicated a specific role for p110γ in regulation of cell speed and provided direct evidence that different PI3Ks are involved in EC tubule formation.
These data support the conclusion that inhibition of p110γ can reduce EC remodeling and formation of novel capillaries network and can therefore possibly represent a novel strategy to counteract tumor angiogenesis.
Roles of p110γ in Stroma Cells
Through generation of lipid second messengers and activation of several signaling molecules PI3Ks regulate several cellular functions, such as cell growth, survival, cytoskeletal remodeling and trafficking of intracellular organelles in many different cell types. The generation of transgenic mice has further revealed a critical role for some isoforms in regulation of the immune system in vivo in particular p110δ and p110γ that are preferentially expressed in immune cells.
It is now well-established that the immune system plays an important role during tumorigenesis (Sun and Karin, 2014). Deregulated cell proliferation and survival, genome destabilization and induction of migration and invasion (Elinav et al., 2013) as well as different steps involved in metastasis development (Antonioli et al., 2013) have all been associated with inflammation. For instance invasion and intravasation of cancer cells into blood and lymphatics vessels can be regulated by tumor-associated macrophages and inflammatory factors that can increase vascular permeability, prostaglandin production and matrix metalloproteinases-mediated tissue remodeling (Quail and Joyce, 2013).
Infiltrative macrophages and inflammatory cells have been detected in PDAC stroma which has been intensely investigated in recent years. Indeed PDAC is characterized by desmoplasia deriving from pancreatic stellate cells that proliferate and produce a dense extracellular matrix consisting of collagen, laminin and fibronectin (Apte et al., 2013). Although several lines of evidence support the conclusion that interaction between pancreatic stellate cells and PDAC cells plays a critical role in the disease (Haqq et al., 2014) this information has not been translated into efficient therapeutic strategies yet. In fact while preclinical studies indicated potential benefit in targeting the stroma-associated Hedgehog signaling pathway corresponding clinical trials yielded disappointing results. A renewed interest in understanding the cause of this failure has recently revealed a protective role for the stroma against PDAC (Lee et al., 2014; Özdemir et al., 2014; Rhim et al., 2014) indicating that alternative strategies must be investigated (Gore and Korc, 2014). HCC progression is also frequently associated with continuous hepatocyte death and inflammatory cell infiltration (He and Karin, 2011).
Several lines of evidence suggest that p110γ plays a central role in cancer-associated inflammation. For instance, in a murine model of ulcerative colitis, it has been shown that p110γ regulates the innate immune system by controlling colon inflammation and tumor formation (Gonzalez-Garcia et al., 2010). In this model, p110γ-deficient mice and control mice were treated with dextran sulfate sodium to induce chronic colitis and colitis-associated cancer. The results showed that p110γ-deficient mice had a lower incidence of colitis-associated tumors as well as reduced tumor multiplicity. The reduced tumor development was a consequence of defective infiltration and activation of myeloid cells and defective recruitment of T cells to the colon and less colon inflammation.
It has been recently reported that a range of chemoattractants able to activate GPCRs, RTKs and Toll-like/IL-1 receptors unexpectedly initiate tumor inflammation by activating p110γ in Gr1+CD11b+ myeloid cells (Schmid et al., 2011). While GPCRs activate p110γ in a Ras/p101 dependent manner, RTKs and Toll-like/IL-1 receptors directly activate p110γ in a Ras/p87-dependent manner. Once activated, p110γ promotes inside-out activation of a single integrin, α4β 1, causing myeloid cell invasion in different tumor types including pancreatic cancer (Schmid et al., 2011). Pharmacological or genetic blockade of p110γ suppressed inflammation, growth and metastasis of implanted and spontaneous tumors, indicating that targeting p110γ can further affect tumor progression through its effect on stroma cells and reduction of inflammation.
Roles of p110γ in Cancer Metabolism
To meet their increasing demand of building block molecules, cancer cells switch to a heavily glucose-dependent metabolism (Ward and Thompson, 2012). Alteration of metabolic pathways is very common in cancer cells and indeed reprogramming of metabolism has been recently recognized as an emerging hallmark of cancer (Hanahan and Weinberg, 2011). In addition to altered glucose metabolism it is becoming increasingly evident that alteration of lipid metabolism plays also a critical role in cancer development. On the other hand, altered cell metabolism, as observed during obesity and insulin resistant conditions, is becoming increasingly associated with cancer development and progression. Indeed, accumulating evidence now suggests that obesity is associated with increased frequency of several cancer types, including prostate, kidney, esophagus, breast, endometrium cancers, as well as cancers of the stomach, colon, pancreas, gallbladder and liver (Møller et al., 1994; Wolk et al., 2001; Calle et al., 2003; Renehan et al., 2008; Basen-Engquist and Chang, 2011). For instance a recent analysis, with a follow up of 239,597 participants and 897 pancreatic cancer deaths, has indicated that obesity is independently associated with increased pancreatic cancer mortality in African Americans (Bethea et al., 2014). These results are consistent with a recent study of two large prospective cohorts comprising 902 patients with pancreatic cancer that also reported statistically significantly reduced survival in patients with higher body mass index before diagnosis (Yuan et al., 2013). Similarly several studies have indicated a link between obesity and increasing incidence of HCC (Møller et al., 1994; Wolk et al., 2001; Borena et al., 2012; Schlesinger et al., 2013; Turati et al., 2013; Karagozian et al., 2014). For instance a prospective cohort of the Cancer Prevention Study with more than 900,000 North American subjects revealed that men with body mass index ≥35 kg/m2 had a higher risk of dying from liver cancer (Calle et al., 2003). A similar study involving 362,552 Swedish men also indicated an increased risk of HCC in individuals with body mass index ≥30 kg/m2 (Samanic et al., 2006). The mechanisms responsible for this obesity-associated increase in cancer incidence are not completely known. Obesity is associated with a chronic low-grade inflammation, and specific anti-inflammatory interventions may be beneficial for the treatment of type 2 diabetes and other obesity-related diseases. On the other hand obesity is usually associated with insulin resistance and with increased levels of circulating insulin that can in turn promote cell proliferation and survival. Whether obesity per se or the associated insulin resistance is ultimately responsible for this increased cancer risk is still a matter of debate.
Consistent with its central role in leukocyte chemotaxis, mast cell degranulation, and EC activation, p110γ has been demonstrated to be critical for obesity-induced inflammation and insulin resistance. It has been shown that mice lacking functional p110γ were protected from insulin resistance, metabolic inflammation, and fatty liver largely because of their leaner phenotype (Becattini et al., 2011; Kobayashi et al., 2011). This phenotype appeared to be a consequence of decreased fat gain as a result of increased energy expenditure despite normal caloric intake. Indeed p110γ action on diet-induced obesity depends on p110γ activity within a non-hematopoietic compartment, where it promotes energetic efficiency for fat mass gain. Importantly, the metabolic modulation by p110γ depends on its lipid kinase activity but it might also involve kinase-independent signaling.
Apart from its role in control of weight gain, data also indicate that p110γ is directly involved in development of obesity-induced insulin resistance. By using murine models of both diet-induced and genetically induced obesity the role of p110γ in the accumulation of tissue macrophages and the development of obesity-induced insulin resistance was examined (Kobayashi et al., 2011). Mice lacking p110γ exhibited improved systemic insulin sensitivity with enhanced insulin signaling in the tissues of obese animals (Kobayashi et al., 2011). In adipose tissues and livers of obese p110γ−/− mice, the numbers of infiltrated pro-inflammatory macrophages were markedly reduced, leading to suppression of inflammatory reactions in these tissues. Furthermore, bone marrow-specific deletion and pharmacological blockade of p110γ also ameliorated obesity-induced macrophage infiltration and insulin resistance. These data suggest that p110γ plays a crucial role in the development of both obesity-induced inflammation and systemic insulin resistance. These data indicate that targeting p110γ can have additional beneficial effects in the context of obesity- and insulin resistance-associated cancers. Based on this evidence it is also tempting to speculate that p110γ inhibition may have a more generic role in modulation of cancer cell metabolism beyond the context of obesity and insulin resistant conditions.
p110γ Specific Inhibitors
Consistent with a key role for PI3Ks in cancer development, several inhibitors have been shown to possess anti-cancer activity in vitro and in vivo. Although PI3Ks and related signaling pathways have been recognized as important therapeutic targets, development of generic PI3K inhibitors has raised some concerns because of the large number of physiological functions that this family of enzymes controls. Many inhibitors of the PI3K pathway have been generated (Falasca, 2010, 2011) that can target either the enzymes themselves or their downstream effectors and these agents can be divided into four major classes: PI3K inhibitors, dual PI3K-mTOR inhibitors, Akt inhibitors and mTOR inhibitors.
PI3K inhibitors can be divided in pan-class I PI3K inhibitors, which target all class I PI3Ks, such as PI-103 and BEZ235 (Raynaud et al., 2007; Maira et al., 2008) and PI3K isoform-specific inhibitors, which specifically target a single PI3K isoform. Since p110δ and p110γ are highly enriched in leukocytes, they are particularly desirable targets for inhibition in the treatment of hematologic malignancies. Indeed among the first isoform-specific PI3K inhibitors developed, the specific p110δ inhibitor IC87114 was used to demonstrate that p110δ could play a role in the pathophysiology of acute myeloid leukemia (Sadhu et al., 2003). Subsequently, TGX-221, an analog of the generic PI3K inhibitor LY294002, and AS252424 were found to selectively inhibit p110β and p110γ respectively (Jackson et al., 2005; Pomel et al., 2006). In the last decade, a number of pharmaceutical companies have reported a wide variety of p110γ inhibitors (Venable et al., 2010), and several X-ray crystal structures with p110γ have been elucidated (Zvelebil et al., 2008). As a consequence, the efficacy of p110γ inhibitors generated has been demonstrated in different biological systems (Venable et al., 2010). However, one of the issues emerged from these studies is the selectivity of compounds tested. On the other hand, an increased interest has emerged in the development of dual p110δ and p110γ inhibitors (Randis et al., 2008). More recently a potent oral p110δ and p110γ inhibitor (IPI-145) has been characterized (Winkler et al., 2013). It has been demonstrated that IPI-145 exerts profound effects on adaptive and innate immunity by inhibiting B and T cell proliferation, blocking neutrophil migration, and inhibiting basophil activation. The therapeutic value of combined p110δ and p110γ blockade was explored, and IPI-145 showed potent activity in collagen-induced arthritis, ovalbumin-induced asthma, and systemic lupus erythematosus rodent models (Winkler et al., 2013). These findings support the hypothesis that inhibition of immune function can be achieved through p110δ and p110γ inhibition, potentially leading to significant therapeutic effects in multiple inflammatory, autoimmune, and hematologic diseases.
Preclinical studies have shown that PI3K inhibition is able to induce apoptosis and inhibit tumor growth of pancreatic cancer xenografts (Bondar et al., 2002). Our recent data have demonstrated that specific pharmacological inhibition of p110γ using selective inhibitors reduces proliferation of PDAC (Edling et al., 2010) and HCC cell lines (Dituri et al., 2012) in vitro. Interestingly we also reported that caffeine and its analog CGS 15943 block proliferation of HCC and PDAC cell lines by inhibiting the PI3K/Akt pathway (Edling et al., 2014). More specifically, a kinase profiling assay revealed that CGS 15943 targets p110γ therefore this study identified this compound as a promising lead compound to develop drugs that can specifically target this PI3K isoform in cancer (Edling et al., 2014).
Concluding Remarks
Given the predominant expression of p110γ in hematopoietic cells the interest of oncologists on this specific lipid kinase has been mainly focused on hematological malignancies. Nevertheless, the discovery of many solid cancers where p110γ seems to play a key role has clearly indicated that inhibition of this specific isoform can prove beneficial in a larger spectrum of cancer types. Furthermore, p110γ is a valid target in different tumor-related processes such as metastasis, angiogenesis and cancer associated inflammation (Figure 1). Therefore, this evidence underlines the potential of targeting p110γ in cancer, especially in gastrointestinal cancers. Several p110γ–specific inhibitors are available and they have been already used to alleviate symptoms in inflammatory chronic diseases such as rheumatoid arthritis and systemic lupus. Testing the effect of these inhibitors in solid cancers will represent an important future challenge. In particular, due to the central role of p110γ in regulation of the immune system it would be critical to assess whether such a strategy could result in potential side effects of immunosuppression. On the other hand small inhibitors designed to block the catalytic activity of the enzyme would not be beneficial to counteract its kinase-independent functions. Therefore, novel strategies should be developed to identify specific inhibitors that target the p110γ kinase-independent activity. We propose that certain gastrointestinal cancers represent the tumor types of election where the use of p110γ inhibitors can be particularly beneficial.
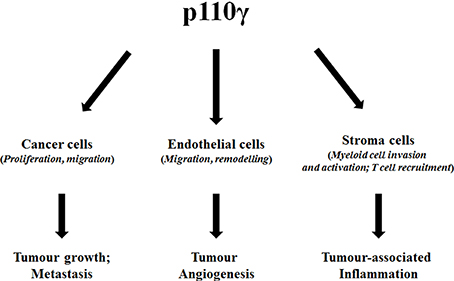
Figure 1. Diagram summarizing the effect of p110γ on different cell types and ultimately on cancer-associated processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in our laboratory is supported by Pancreatic Cancer Research Fund (to Marco Falasca) and Prostate Cancer UK (PG12-23 to Marco Falasca, PG13-029 to Tania Maffucci).
Abbreviations
EC, endothelial cells; GPCR, G-protein coupled receptor; HCC, hepatocellular carcinoma; LPA, lysophosphatidic acid; mTOR, mechanistic target of rapamycin; PDAC, pancreatic cancer adenocarcinoma; PI3K, phosphoinositide 3-kinase; RTK, receptor tyrosine kinase; S1P, sphingosine-1-phosphate.
References
Ali, K., Soond, D. R., Piñeiro, R., Hagemann, T., Pearce, W., Lim, E. L., et al. (2014). Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510, 407–411. doi: 10.1038/nature13444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Antonioli, L., Blandizzi, C., Pacher, P., and Haskó, G. (2013). Immunity, inflammation and cancer: a leading role for adenosine. Nat. Rev. Cancer 13, 842–857. doi: 10.1038/nrc3613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Apte, M. V., Wilson, J. S., Lugea, A., and Pandol, S. J. (2013). A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 144, 1210–1219. doi: 10.1053/j.gastro.2012.11.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Attoub, S., De Wever, O., Bruyneel, E., Mareel, M., and Gespach, C. (2008). The transforming functions of PI3-kinase-γ are linked to disruption of intercellular adhesion and promotion of cancer cell invasion. Ann. N.Y. Acad. Sci. 1138, 204–213. doi: 10.1196/annals.1414.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Basen-Engquist, K., and Chang, M. (2011). Obesity and cancer risk: recent review and evidence. Curr. Oncol. Rep. 13, 71–76. doi: 10.1007/s11912-010-0139-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becattini, B., Marone, R., Zani, F., Arsenijevic, D., Seydoux, J., Montani, J. P., et al. (2011). PI3Kγ within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, E854–E863. doi: 10.1073/pnas.1106698108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beer-Hammer, S., Zebedin, E., von Holleben, M., Alferink, J., Reis, B., Dresing, P., et al. (2010). The catalytic PI3K isoforms p110gamma and p110delta contribute to B cell development and maintenance, transformation, and proliferation. J. Leuk. Biol. 87, 1083–1095. doi: 10.1189/jlb.0809585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bénistant, C., Chapuis, H., and Roche, S. (2000). A specific function for phosphatidylinositol 3-kinase alpha (p85alpha-p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene 19, 5083–5090. doi: 10.1038/sj.onc.1203871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bethea, T. N., Kitahara, C. M., Sonderman, J. S., Patel, A. V., Harvey, C., Knutsen, S. F., et al. (2014). A pooled analysis of body mass index and pancreatic cancer mortality in African, Americans. Cancer Epidemiol. Biomarkers Prev. 23, 2119–2125. doi: 10.1158/1055-9965.EPI-14-0422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bondar, V. M., Sweeney-Gotsch, B., Andreeff, M., Mills, G. B., and McConkey, D. J. (2002). Inhibition of the phosphatidylinositol 3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol. Cancer Ther. 1, 989–997.
Borena, W., Strohmaier, S., Lukanova, A., Bjørge, T., Lindkvist, B., Hallmans, G., et al. (2012). Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int. J. Cancer 131, 193–200. doi: 10.1002/ijc.26338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brand, T. M., and Wheeler, D. L. (2012). KRAS mutant colorectal tumors: past and present. Small GTPases 3, 34–39. doi: 10.4161/sgtp.18751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brazzatti, J. A., Klingler-Hoffmann, M., Haylock-Jacobs, S., Harata-Lee, Y., Niu, M., Higgins, M. D., et al. (2012). Differential roles for the p101 and p84 regulatory subunits of PI3Kγ in tumor growth and metastasis. Oncogene 31, 2350–2361. doi: 10.1038/onc.2011.414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brenner, H., Kloor, M., and Pox, C. P. (2014). Colorectal cancer. Lancet 383, 1490–1502. doi: 10.1016/S0140-6736(13)61649-9
Calle, E. E., Rodriguez, C., Walker-Thurmond, K., and Thun, M. J. (2003). Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 348, 1625–1638. doi: 10.1056/NEJMoa021423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carter, H., Samayoa, J., Hruban, R. H., and Karchin, R. (2010). Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM). Cancer Biol. Ther. 10, 582–587. doi: 10.4161/cbt.10.6.12537
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, H. W., Aoki, M., Fruman, D., Auger, K. R., Bellacosa, A., Tsichlis, P. N., et al. (1997). Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science 276, 1848–1850. doi: 10.1126/science.276.5320.1848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Czauderna, F., Fechtner, M., Aygün, H., Arnold, W., Klippel, A., Giese, K., et al. (2003). Functional studies of the PI(3)-kinase signalling pathway employing synthetic and expressed siRNA. Nucleic Acids Res. 31, 670–682. doi: 10.1093/nar/gkg141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Prete, A., Vermi, W., Dander, E., Otero, K., Barberis, L., Luini, W., et al. (2004). Defective dendritic cell migration and activation of adaptive immunity in P13Kgamma-deficient mice. Embo J. 23, 3505–3515. doi: 10.1038/sj.emboj.7600361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dituri, F., Mazzocca, A., Lupo, L., Edling, C. E., Azzariti, A., Antonaci, S., et al. (2012). PI3K class IB controls the cell cycle checkpoint promoting cell proliferation in hepatocellular carcinoma. Int. J. Cancer 130, 2505–2513. doi: 10.1002/ijc.26319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edling, C. E., Selvaggi, F., Buus, R., Maffucci, T., Di Sebastiano, P., Friess, H., et al. (2010). Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin. Cancer Res. 16, 4928–4937. doi: 10.1158/1078-0432.CCR-10-1210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edling, C. E., Selvaggi, F., Ghonaim, R., Maffucci, T., and Falasca, M. (2014). Caffeine and the analog CGS 15943 inhibit cancer cell growth by targeting the phosphoinositide 3-kinase/Akt pathway. Cancer Biol. Ther. 15, 524–532. doi: 10.4161/cbt.28018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elinav, E., Nowarski, R., Thaiss, C. A., Hu, B., Jin, C., and Flavell, R. A. (2013). Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer. 13, 759–771. doi: 10.1038/nrc3611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falasca, M. (2010). PI3K/Akt signalling pathway specific inhibitors: a novel strategy to sensitize cancer cells to anti-cancer drugs. Curr. Pharm. Des. 16, 1410–1416. doi: 10.2174/138161210791033950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falasca, M. (2011). Phosphoinositide 3-kinase pathway inhibitors: pharmacology, metabolism and drug development. Curr. Med. Chem. 18, 2673. doi: 10.2174/092986711796011210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falasca, M., and Maffucci, T. (2007). Role of class II phosphoinositide 3-kinase in cell signalling. Biochem. Soc. Trans. 35, 211–214. doi: 10.1042/BST0350211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falasca, M., and Maffucci, T. (2012). Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem J. 443, 587–601. doi: 10.1042/BJ20120008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Falasca, M., Selvaggi, F., Buus, R., Sulpizio, S., and Edling, C. E. (2011). Targeting phosphoinositide 3-kinase pathways in pancreatic cancer—from molecular signalling to clinical trials. Anticancer Agents Med. Chem. 11, 455–463. doi: 10.2174/187152011795677382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fyffe, C., Buus, R., and Falasca, M. (2013). Genetic and epigenetic regulation of phosphoinositide 3-kinase isoforms. Curr. Pharm. Des. 19, 680–686. doi: 10.2174/138161213804581873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Garcia, A., Sanchez-Ruiz, J., Flores, J. M., and Carrera, A. C. (2010). Phosphatidylinositol 3-kinase gamma inhibition ameliorates inflammation and tumor growth in a model of colitis associated cancer. Gastroenterology 138, 1374–1383. doi: 10.1053/j.gastro.2009.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gore, J., and Korc, M. (2014). Pancreatic cancer stroma: friend or foe? Cancer Cell 25, 711–712. doi: 10.1016/j.ccr.2014.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graupera, M., Guillermet-Guibert, J., Foukas, L. C., Phng, L. K., Cain, R. J., Salpekar, A., et al. (2008). Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453, 662–666. doi: 10.1038/nature06892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamada, K., Sasaki, T., Koni, P. A., Natsui, M., Kishimoto, H., Sasaki, J., et al. (2005). The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 19, 2054–2065. doi: 10.1101/gad.1308805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haqq, J., Howells, L. M., Garcea, G., Metcalfe, M. S., Steward, W. P., and Dennison, A. R. (2014). Pancreatic stellate cells and pancreas cancer: Current perspectives and future strategies. Eur. J. Cancer. 50, 2570–2582. doi: 10.1016/j.ejca.2014.06.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, G., and Karin, M. (2011). NF-κ B and STAT3—key players in liver inflammation and cancer. Cell Res. 21, 159–168. doi: 10.1038/cr.2010.183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heller, R., Chang, Q., Ehrlich, G., Hsieh, S. N., Schoenwaelder, S. M., Kuhlencordt, P. J., et al. (2008). Overlapping and distinct roles for PI3Kbeta and gamma isoforms in S1P-induced migration of human and mouse endothelial cells. Cardiovasc. Res. 80, 96–105. doi: 10.1093/cvr/cvn159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickey, F. B., and Cotter, T. G. (2006). BCR-ABL regulates phosphatidylinositol 3-kinase-p110gamma transcription and activation and is required for proliferation and drug resistance. J. Biol. Chem. 281, 2441–2450. doi: 10.1074/jbc.M511173200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirsch, E., Bosco, O., Tropel, P., Laffargue, M., Calvez, R., Altruda, F., et al. (2001). Resistence to thromboembolism in P13Kgamma-deficient mice. Faseb J. 15, 2019–2021.
Hirsch, E., Ciraolo, E., Franco, I., Ghigo, A., and Martini, M. (2014). PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene 33, 3083–3090. doi: 10.1038/onc.2013.265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirsch, E., Katanaev, V. L., Garlanda, C., Azzolino, O., Pirola, L., Silengo, L., et al. (2000). Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287, 1049–1053. doi: 10.1126/science.287.5455.1049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hruban, R. H., Goggins, M., Parsons, J., and Kern, S. E. (2000). Progression model for pancreatic cancer. Clin. Cancer Res. 6, 2969–7292.
Jackson, S. P., Schoenwaelder, S. M., Goncalves, I., Nesbitt, W. S., Yap, C. L., Wright, C. E., et al. (2005). PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat. Med. 11, 507–514. doi: 10.1038/nm1232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. CA Cancer J. Clin. 61, 69–90. doi: 10.3322/caac.20107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, S., Denley, A., Vanhaesebroeck, B., and Vogt, P. K. (2006). Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 103, 1289–1294. doi: 10.1073/pnas.0510772103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karagozian, R., Derdák, Z., and Baffy, G. (2014). Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism 63, 607–617. doi: 10.1016/j.metabol.2014.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khwaja, A., Rodriguez-Viciana, P., Wennström, S., Warne, P. H., and Downward, J. (1997). Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 16, 2783–2793. doi: 10.1093/emboj/16.10.2783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knobbe, C. B., and Reifenberger, G. (2003). Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 13, 507–518. doi: 10.1111/j.1750-3639.2003.tb00481.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kobayashi, N., Ueki, K., Okazaki, Y., Iwane, A., Kubota, N., Ohsugi, M., et al. (2011). Blockade of class IB phosphoinositide-3 kinase ameliorates obesity-induced inflammation and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 5753–5758. doi: 10.1073/pnas.1016430108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodaki, T., Woscholski, R., Hallberg, B., Rodriguez-Viciana, P., Downward, J., and Parker, P. J. (1994). The activation of phosphatidylinositol 3-kinase by Ras. Curr. Biol. 4, 798–806. doi: 10.1016/S0960-9822(00)00177-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuper, H., Tzonou, A., Kaklamani, E., Hsieh, C. C., Lagiou, P., Adami, H. O., et al. (2000). Tobacco smoking, alcohol consumption and their interaction in the causation of hepatocellular carcinoma. Int. J. Cancer 85, 498–502. doi: 10.1002/(SICI)1097-0215(20000215)85:4<498::AID-IJC9>3.0.CO;2-F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J. J., Perera, R. M., Wang, H., Wu, D. C., Liu, X. S., Han, S., et al. (2014). Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. U.S.A. 111, E3091–E3100. doi: 10.1073/pnas.1411679111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, D., Xie, K., Wolff, R., and Abbruzzese, J. L. (2004). Pancreatic cancer. Lancet 363, 1049–1057. doi: 10.1016/S0140-6736(04)15841-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, P., Morrison, C., Wang, L., Xiong, D., Vedell, P., Cui, P., et al. (2012). Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 33, 1270–1276. doi: 10.1093/carcin/bgs148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lockhart, A. C., Rothenberg, M. L., and Berlin, J. D. (2005). Treatment for pancreatic cancer: current therapy and continued progress. Gastroenterology 128, 1642–1654. doi: 10.1053/j.gastro.2005.03.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, J., Manning, B. D., and Cantley, L. C. (2003). Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4, 257–262. doi: 10.1016/S1535-6108(03)00248-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madeddu, P., Kraenkel, N., Barcelos, L. S., Siragusa, M., Campagnolo, P., Oikawa, A., et al. (2008). Phosphoinositide 3-kinase gamma gene knockout impairs postischemic neovascularization and endothelial progenitor cell functions. Arterioscler. Thromb. Vasc. Biol. 28, 68–76. doi: 10.1161/ATVBAHA.107.145573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maira, S. M., Stauffer, F., Brueggen, J., Furet, P., Schnell, C., Fritsch, C., et al. (2008). Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863. doi: 10.1158/1535-7163.MCT-08-0017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchesi, F., Monti, P., Leone, B. E., Zerbi, A., Vecchi, A., Piemonti, L., et al. (2004). Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 64, 8420–8427. doi: 10.1158/0008-5472.CAN-04-1343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Møller, H., Mellemgaard, A., Lindvig, K., and Olsen, J. H. (1994). Obesity and cancer risk: a Danish record-linkage study. Eur. J. Cancer. 30A, 344–350. doi: 10.1016/0959-8049(94)90254-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monterrubio, M., Mellado, M., Carrera, A. C., and Rodríguez-Frade, J. M. (2009). PI3Kgamma activation by CXCL12 regulates tumor cell adhesion and invasion. Biochem. Biophys. Res. Commun. 388, 199–204. doi: 10.1016/j.bbrc.2009.07.153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morello, F., Perino, A., and Hirsch, E. (2009). Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 82, 261–271. doi: 10.1093/cvr/cvn325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moskaluk, C. A., Hruban, R. H., and Kern, S. E. (1997). p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 57, 2140–2143.
Nguyen, D. X., Bos, P. D., and Massagué, J. (2009). Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284. doi: 10.1038/nrc2622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, D. X., and Massague, J. (2007). Genetic determinants of cancer metastasis. Nat. Rev. Genet. 8, 341–352. doi: 10.1038/nrg2101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Özdemir, B. C., Pentcheva-Hoang, T., Carstens, J. L., Zheng, X., Wu, C. C., Simpson, T. R., et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. doi: 10.1016/j.ccr.2014.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patrucco, E., Notte, A., Barberis, L., Selvetella, G., Maffei, A., Brancaccio, M., et al. (2004). P13Kgamma modulates the cardiac response to chronic pressure overload by distinct Kinase-dependent and -independent effects. Cell 118, 375–387. doi: 10.1016/j.cell.2004.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pomel, V., Klicic, J., Covini, D., Church, D. D., Shaw, J. P., Roulin, K., et al. (2006). Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase gamma. J. Med. Chem. 49, 3857–3871. doi: 10.1021/jm0601598
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ptasznik, A., Prossnitz, E. R., Yoshikawa, D., Smrcka, A., Traynor-Kaplan, A. E., and Bokoch, G. M. (1996). A tyrosine kinase signaling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. J. Biol. Chem. 271, 25204–25207. doi: 10.1074/jbc.271.41.25204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quail, D. F., and Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. doi: 10.1038/nm.3394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Randis, T. M., Puri, K. D., Zhou, H., and Diacovo, T. G. (2008). Role of P13Kdelta and P13Kgamma in inflammatory arthritis and tissue localization of neutrophils. Eur. J. Immunol. 38, 1215–1224. doi: 10.1002/eji.200838266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raynaud, F. I., Eccles, S., Clarke, P. A., Hayes, A., Nutley, B., Alix, S., et al. (2007). Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 67, 5840–5850. doi: 10.1158/0008-5472.CAN-06-4615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Renehan, A. G., Tyson, M., Egger, M., Heller, R. F., and Zwahlen, M. (2008). Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371, 569–578. doi: 10.1016/S0140-6736(08)60269-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhim, A. D., Oberstein, P. E., Thomas, D. H., Mirek, E. T., Palermo, C. F., Sastra, S. A., et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. doi: 10.1016/j.ccr.2014.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ryder, S. D. (2003). Guidelines for the diagnosis and treatment of hepatocellular carcinoma (HCC) in adults. Gut 52(Suppl. 3), iii1–iii8. doi: 10.1136/gut.52.suppl_3.iii1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sadhu, C., Masinovsky, B., Dick, K., Sowell, C. G., and Staunton, D. E. (2003). Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J. Immunol. 170, 2647–2654. doi: 10.4049/jimmunol.170.5.2647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Samanic, C., Chow, W. H., Gridley, G., Jarvholm, B., and Fraumeni, J. F. Jr. (2006). Relation of body mass index to cancer risk in 362,552 Swedish men. Cancer Causes Control 17, 901–909. doi: 10.1007/s10552-006-0023-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Samuels, Y., Wang, Z., Bardelli, A., Silliman, N., Ptak, J., Szabo, S., et al. (2004). High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554. doi: 10.1126/science.1096502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sasaki, T., Irie-Sasaki, J., Horie, Y., Bachmaier, K., Fata, J. E., Li, M., et al. (2000). Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature 406, 897–902. doi: 10.1038/35022585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sasaki, T., Irie-Sasaki, J., Horie, Y., Bachmaier, K., Fata, J. E., Li, M., et al. (2003). Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature 426, 584. doi: 10.1038/nature02203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sawyer, C., Sturge, J., Bennett, D. C., O'Hare, M. J., Allen, W. E., Bain, J., et al. (2003). Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 63, 1667–1675.
Schlesinger, S., Aleksandrova, K., Pischon, T., Fedirko, V., Jenab, M., Trepo, E., et al. (2013). Abdominal obesity, weight gain during adulthood and risk of liver and biliary tract cancer in a European cohort. Int. J. Cancer 132, 645–657. doi: 10.1002/ijc.27645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlieman, M. G., Fahy, B. N., Ramsamooj, R., Beckett, L., and Bold, R. J. (2003). Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer. 89, 2110–2115. doi: 10.1038/sj.bjc.6601396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmid, M. C., Avraamides, C. J., Dippold, H. C., Franco, I., Foubert, P., Ellies, L. G., et al. (2011). Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 19, 715–727. doi: 10.1016/j.ccr.2011.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, G., Siveke, J. T., Eckel, F., and Schmid, R. M. (2005). Pancreatic cancer: basic and clinical aspects. Gastroenterology 128, 1606–1625. doi: 10.1053/j.gastro.2005.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, Y. C., Lin, Z. Z., Hsu, C. H., Hsu, C., Shao, Y. Y., and Cheng, A. L. (2013). Clinical trials in hepatocellular carcinoma: an update. Liver Cancer 2, 345–364. doi: 10.1159/000343850
Singh, S., Singh, P. P., Roberts, L. R., and Sanchez, W. (2014). Chemopreventive strategies in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 11, 45–54. doi: 10.1038/nrgastro.2013.143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
So, L., and Fruman, D. A. (2012). PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem. J. 442, 465–481. doi: 10.1042/BJ20112092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sujobert, P., Bardet, V., Cornillet-Lefebvre, P., Hayflick, J. S., Prie, N., Verdier, F., et al. (2005). Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 106, 1063–1066. doi: 10.1182/blood-2004-08-3225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, B., and Karin, M. (2014). The therapeutic value of targeting inflammation in gastrointestinal cancers. Trends Pharmacol. Sci. 35, 349–357. doi: 10.1016/j.tips.2014.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takeda, A., Osaki, M., Adachi, K., Honjo, S., and Ito, H. (2004). Role of the phosphatidylinositol 3′-kinase-Akt signal pathway in the proliferation of human pancreatic ductal carcinoma cell lines. Pancreas 28, 353–358. doi: 10.1097/00006676-200404000-00026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, M. B., Jaffe, D., Choti, M. M., Belghiti, J., Curley, S., Fong, Y., et al. (2010). Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J. Clin. Oncol. 28, 3994–4005. doi: 10.1200/JCO.2010.28.7805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, M. B., O'Beirne, J. P., Furuse, J., Chan, A. T., Abou-Alfa, G., and Johnson, P. (2008). Systemic therapy for hepatocellular carcinoma: cytotoxic chemotherapy, targeted therapy and immunotherapy. Ann. Surg. Oncol. 15, 1008–1014. doi: 10.1245/s10434-007-9705-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thrumurthy, S. G., Chaudry, M. A., Hochhauser, D., and Mughal, M. (2013). The diagnosis and management of gastric cancer. BMJ 347, f6367. doi: 10.1136/bmj.f6367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tibolla, G., Piñeiro, R., Chiozzotto, D., Mavrommati, I., Wheeler, A. P., Norata, G. D., et al. (2013). Class II phosphoinositide 3-kinases contribute to endothelial cells morphogenesis. PLoS ONE 8:e53808. doi: 10.1371/journal.pone.0053808.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Traynor-Kaplan, A. E., Harris, A. L., Thompson, B. L., Taylor, P., and Sklar, L. A. (1988). An inositol tetrakisphosphate-containing phospholipid in activated neutrophils. Nature 334, 353–356. doi: 10.1038/334353a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turati, F., Talamini, R., Pelucchi, C., Polesel, J., Franceschi, S., Crispo, A., et al. (2013). Metabolic syndrome and hepatocellular carcinoma risk. Br. J. Cancer 108, 222–228. doi: 10.1038/bjc.2012.492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., and Bilanges, B. (2010). The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell. Biol. 11, 329–341. doi: 10.1038/nrm2882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vanhaesebroeck, B., Leevers, S. J., Ahmadi, K., Timms, J., Katso, R., Driscoll, P. C., et al. (2001). Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70, 535–602. doi: 10.1146/annurev.biochem.70.1.535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Meeteren, L. A., and Moolenaar, W. H. (2007). Regulation and biological activities of the autotaxin–LPA axis. Prog. Lipid Res. 46, 145–160. doi: 10.1016/j.plipres.2007.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Venable, J. D., Ameriks, M. K., Blevitt, J. M., Thurmond, R. L., and Fung-Leung, W. P. (2010). Phosphoinositide 3-kinase gamma (P13Kgamma) inhibitors for the treatment of inflammation and autoimmune disease. Recent Pat. Inflamm. Allergy Drug Discov. 4, 1–15. doi: 10.2174/187221310789895603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ward, P. S., and Thompson, C. B. (2012). Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308. doi: 10.1016/j.ccr.2012.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Welti, J., Loges, S., Dimmeler, S., and Carmeliet, P. (2013). Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J. Clin. Invest. 123, 3190–3200. doi: 10.1172/JCI70212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Winkler, D. G., Faia, K. L., DiNitto, J. P., Ali, J. A., White, K. F., Brophy, E. E., et al. (2013). PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem. Biol. 20, 1364–1374. doi: 10.1016/j.chembiol.2013.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolk, A., Gridley, G., Svensson, M., Nyrén, O., McLaughlin, J. K., Fraumeni, J. F., et al. (2001). A prospective study of obesity and cancer risk (Sweden). Cancer Causes Control. 12, 13–21. doi: 10.1023/A:1008995217664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xie, Y., Abel, P. W., Kirui, J. K., Deng, C., Sharma, P., Wolff, D. W., et al. (2013). Identification of upregulated phosphoinositide 3-kinase γ as a target to suppress breast cancer cell migration and invasion. Biochem. Pharmacol. 85, 1454–1462. doi: 10.1016/j.bcp.2013.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamamoto, S., Tomita, Y., Hoshida, Y., Morooka, T., Nagano, H., Dono, K., et al. (2004). Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 10, 2846–2850. doi: 10.1158/1078-0432.CCR-02-1441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, C., Bao, Y., Wu, C., Kraft, P., Ogino, S., Ng, K., et al. (2013). Prediagnostic body mass index and pancreatic cancer survival. J. Clin. Oncol. 31, 4229–4234. doi: 10.1200/JCO.2013.51.7532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, T. L., and Cantley, L. C. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510. doi: 10.1038/onc.2008.245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y. (2013). Epidemiology of esophageal cancer. World J. Gastroenterol. 19, 5598–5606. doi: 10.3748/wjg.v19.i34.5598
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, L., and Vogt, P. K. (2008). Class I PI3K in oncogenic cellular transformation. Oncogene 27, 5486–5496. doi: 10.1038/onc.2008.244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, Q., Lui, V. W., and Yeo, W. (2011). Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol. 7, 1149–1167. doi: 10.2217/fon.11.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zvelebil, M. J., Waterfield, M. D., and Shuttleworth, S. J. (2008). Structural analysis of P13-kinase isoforms: identification of residues enabling selective inhibition by small molecule ATP-competitive inhibitors. Arch. Biochem. Biophys. 477, 404–410. doi: 10.1016/j.abb.2008.06.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: angiogenesis, cancer, HCC, inflammation, metastasis, p110γ, PDAC, phosphoinositide 3-kinase
Citation: Falasca M and Maffucci T (2014) Targeting p110gamma in gastrointestinal cancers: attack on multiple fronts. Front. Physiol. 5:391. doi: 10.3389/fphys.2014.00391
Received: 20 August 2014; Paper pending published: 03 September 2014;
Accepted: 21 September 2014; Published online: 15 October 2014.
Edited by:
Andrzej Ptasznik, Cedars-Sinai Medical Center, USAReviewed by:
Klaus Okkenhaug, Babraham Institute, UKAlexis E. Traynor-Kaplan, ISM Therapeutics, USA
Andrzej Ptasznik, Cedars-Sinai Medical Center, USA
Copyright © 2014 Falasca and Maffucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Falasca, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, 4 Newark Street, London E1 2AT, UK e-mail: m.falasca@qmul.ac.uk