 Julie B. McLean
Julie B. McLean Jennifer S. Moylan
Jennifer S. Moylan Francisco H. Andrade
Francisco H. Andrade- 1Department of Physiology, University of Kentucky, Lexington, KY, USA
- 2Center for Muscle Biology, University of Kentucky, Lexington, KY, USA
Aims: Cancer cachexia is a syndrome which results in severe loss of muscle mass and marked fatigue. Conditioned media from cachexia-inducing cancer cells triggers metabolic dysfunction in skeletal muscle, including decreased mitochondrial respiration, which may contribute to fatigue. We hypothesized that Lewis lung carcinoma conditioned medium (LCM) would impair the mitochondrial electron transport chain (ETC) and increase production of reactive oxygen species, ultimately leading to decreased mitochondrial respiration. We incubated C2C12 myotubes with LCM for 30 min, 2, 4, 24 or 48 h. We measured protein content by western blot; oxidant production by 2′,7′-dichlorofluorescin diacetate (DCF), 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF), and MitoSox; cytochrome c oxidase activity by oxidation of cytochrome c substrate; and oxygen consumption rate (OCR) of intact myotubes by Seahorse XF Analyzer. Results: LCM treatment for 2 or 24 h decreased basal OCR and ATP-related OCR, but did not alter the content of mitochondrial complexes I, III, IV and V. LCM treatment caused a transient rise in reactive oxygen species (ROS). In particular, mitochondrial superoxide (MitoSOX) was elevated at 2 h. 4-Hydroxynonenal, a marker of oxidative stress, was elevated in both cytosolic and mitochondrial fractions of cell lysates after LCM treatment. Conclusion: These data show that lung cancer-conditioned media alters electron flow in the ETC and increases mitochondrial ROS production, both of which may ultimately impair aerobic metabolism and decrease muscle endurance.
Introduction
Cancer kills nearly 600,000 Americans annually, equating to approximately 1500 deaths every day (Society, 2012). Fifty percent of all cancer patients experience cachexia, a severe wasting syndrome that includes loss of muscle mass, weakness and fatigue (Tan and Fearon, 2008; Fearon et al., 2011). Cachexia is unresponsive to nutritional interventions, and limits the response to cancer treatments (Fearon et al., 2013). Eighty percent of patients with advanced lung, prostate, colon, and pancreatic cancers present with cachexia (Tisdale, 2009; Von Haehling et al., 2010; Fearon et al., 2011; Johns et al., 2013). Although studies are ongoing, we currently know that malignant tumors alter their surrounding environment via tumor-derived and host-derived paracrine factors, which promote cachexia and may also lead to mitochondrial dysfunction. Some recent studies found evidence of mitochondrial dysfunction in cachexia, which may contribute to muscle pathology (Tan and Fearon, 2008; Constantinou et al., 2011; Fearon et al., 2011; Julienne et al., 2012, 2014; Wang et al., 2012; White et al., 2012; Dumas et al., 2013; Fontes-Oliveira et al., 2013; Tzika et al., 2013).
Mitochondria are multi-functional organelles that provide a majority of ATP to cells, are the major source of reactive oxygen species (ROS), and participate in multiple signaling cascades, including apoptosis. Reactive oxygen species play an important role in maintenance and adaptation of skeletal muscle (Irrcher et al., 2009; Merry et al., 2010; Dutka et al., 2012; Luo et al., 2013; Michaelson et al., 2013; Prosser et al., 2013). At low concentrations, ROS function in homeostatic signaling cascades, while at high concentrations they cause damage by oxidizing DNA, proteins, and lipids (Lee et al., 2012); in skeletal muscle, excessive oxidant production affects skeletal muscle mass and function in ways consistent with cachexia (Andrade et al., 1998, 2001; Reid et al., 2005; Hardin et al., 2008; Gilliam et al., 2011).
In this study, we examined the effect of Lewis lung carcinoma condition media (LCM) on mitochondrial function, protein content, and ROS production in C2C12 skeletal muscle myotubes. C2C12s are an immortalized cell line of mouse skeletal muscle that fuse and differentiate into myotubes under low-serum conditions (Yaffe and Saxel, 1977). They are a useful model is studying signaling pathways in skeletal muscle under controlled conditions. We chose Lewis lung carcinoma as our cancer model because it is known to induce cachexia and because lung cancer accounts for 23% of all cancer deaths worldwide (Carbo et al., 2004; Argiles et al., 2008; Jemal et al., 2011; Puppa et al., 2014). Then, we exposed C2C12 myotubes to LCM for 30 min, 2, 4, 24, or 48 h. We hypothesized that LCM would impair mitochondrial respiration, increase ROS production, and lead to oxidative stress. To test our hypothesis, we assessed the following in myotubes: mitochondrial oxygen consumption, content of mitochondrial complexes, voltage-dependent anion channel (VDAC), 4-hydroxynonenal (4HNE), and uncoupling protein 3 (UCP3), the activity of cytochrome c oxidase, and oxidant production via DCF, DAF, and Mitosox.
Materials and Methods
Myotubes
C2C12 myoblasts (American Type Culture Collection) were plated at a density of 10,000 cells/cm2 in growth medium [Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum, 1.6 g/L sodium bicarbonate, and 100U/ml PenStrep (Invitrogen)] and cultured at 37°C in 5% CO2. Cells reached ~90% confluence after 3 days, at which time cells were serum restricted in differentiation media (DMEM as above with 2% horse serum replacing fetal bovine serum). After 4 days of serum restriction, multinucleated myotubes were ready for treatment. Fresh medium was added every 2 days (Moylan et al., 2014).
Lung Cancer Cells
Lewis lung carcinoma cancer cells (LL/2: American Type Culture Collection) were seeded at a density of 6000/cm2 in 100 mm cell culture plates in growth medium, as above. After 2 days, additional growth media was added to each plate. LL/2 cells are a heterogeneous mix of floating and adherent cells. After 4 days, we removed growth medium and harvested floating cells by centrifugation at 500 × g, 5 min. Pelleted cells and 10 mL differentiation media were added back to the adherent cells. After 2 days, we collected conditioned medium, removed cells and debris by centrifugation (500 × g, 5 min), and froze aliquots in liquid nitrogen for later use. For treatments, we used a 1 in 4 dilution of conditioned media mixed with fresh differentiation media.
Western Blot
C2C12 myotubes were homogenized in 2X protein loading buffer (120 mM Tris pH 7.5, 4% SDS, 200 mM DTT, 20% glycerol, 0.002% bromphenol blue). We separated proteins in equal volumes of lysates by SDS-PAGE (4–15% Criterion, BioRad) and determined relative total protein by scanning (Odyssey Infrared Imaging, LI-COR) stained gels (Simply Blue, Invitrogen). Fluorescence intensity data were used to normalize total protein for equal loading. Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF membranes for western blot using the Odyssey System (Moylan et al., 2014). For preparation of mitochondrial and cytosolic fractions, mitochondria were isolated as in Lemire et al., and western blots were performed as above (Lemire et al., 2008).
Antibodies
Primary antibodies were rabbit anti-VDAC and rabbit anti-4 HNE (Abcam), mouse anti-Complex I NDUFA9, mouse anti-Complex III, subunit core I, mouse anti-Complex IV, subunit IV (Novex), and mouse anti-ATP synthase α (Mitosciences). Secondary antibodies included anti-mouse IRDye 800CW and anti-rabbit IRDye 800CW (LI-COR).
Oxidant Assays and Antioxidant Treatments
To measure cytosolic oxidant activity, we aspirated off the cell culture media, replaced it with pre-warmed PBS supplemented with glucose (D-PBS) and 2′,7′-dichlorofluorescein diacetate (DCFH-DA 10 μM, Molecular Probes). We incubated C2C12 myotubes in DCFH-DA for 30 min, 37°C, 5% CO2. After incubation, we washed the cells twice with D-PBS, and added fresh D-PBS. To measure nitric oxide, we followed the same procedure as above using DAF-FM diacetate (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate) (DAF-FM 5 μM, Molecular Probes). We used a spectrofluorometer (Synergy H1, BioTek) to measure fluorescence of the oxidized derivatives, DCF (excitation: 480 nm, emission 520 nm) and DAF-FM (excitation: 495 nm, emission 515 nm), respectively. Antioxidant treatments included either 100 μM SS31, a mitochondrial-targeted antioxidant, or 5 mmol N-Acetylcysteine, a general antioxidant (Cho et al., 2007; Whiteman et al., 2008; Manczak et al., 2010; Powers et al., 2011). Both were administered simultaneously with LCM treatment.
Fluorescence Microscopy
We incubated C2C12 myotubes with MitoSox (5 μM, Molecular Probes), a mitochondrial superoxide anion specific indicator, for 30 min. Cells were rinsed twice with pre-warmed 1X PBS supplemented with glucose and sodium pyruvate. We captured live cell fluorescence images (excitation: 510 nm, emission 580 nm) using a CCD camera (CoolSNAP-ES, Roper Scientific Photometrics) attached to a Nikon TE2000 microscope with NIS Elements image acquisition software (Nikon).
Oxygen Consumption Rate
We cultured C2C12 myotubes as above in XFe24 Analyzer plates (Seahorse Bioscience). Before running the assay, we replaced differentiation media with Seahorse assay medium supplemented with 10 mM sodium pyruvate and 10 mM glucose, and we adjusted the pH to 7.4. Seahorse assay medium is non-buffered for measuring proton production. Plates were equilibrated in this medium for 1 h at 37°C with no CO2. We used the same media as vehicle for our injection compounds. The analyzer sequentially injected the following compounds: oligomycin (1 μM final concentration), which blocks ATP synthase and shows the OCR dedicated to ATP production; carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP: 4 μM final concentration), which permeabilizes the inner mitochondrial membrane and shows maximal OCR; and rotenone (1 μM final concentration), which blocks complex I and shows NADH-driven OCR. There were 3–4 wells per treatment group, and 4 plates in total. We measured OCR at three time points during basal respiration and after each injection using a Seahorse XFe24 Flux Analyzer (Seahorse Biosciences). We normalized OCR measurements to VDAC content in each well determined via western blot.
Cytochrome C Oxidase Activity
We measured mitochondrial complex IV activity using a Cytochrome Oxidase assay kit (Biovision) per manufacturer's protocol. Briefly, we lysed C2C12 myotubes by 3 freeze-thaw cycles in 20 mM hypotonic potassium phosphate buffer pH 7.5 supplemented with protease and phosphatase inhibitor cocktails (Sigma). In a 96 well plate, we added Cytochrome c and Cytochrome Oxidase Assay Buffer (Biovision) to 2 μg total protein and recorded the decrease in optical density (OD) at 550 nm over 45 min using a spectrophotometer (Synergy H1, BioTek). The decrease in optical density is proportional to an increase in oxidized cytochrome c. We derived activity from the reaction rate in the linear phase, i.e., cytochrome oxidase activity (Units/mg) = [(OD1-OD2)/(t1-t2)]/(ε x mg protein), where ε is the molar extinction coefficient of cytochrome c at 550 nm (7.04 mM−1cm−1) and one unit oxidizes one μmole reduced cytochrome c per minute.
Statistics
Prism 5.0b (GraphPad Software) and SAS served as our statistics software. We used t-test, One-Way ANOVA and post-hoc Bonferroni test, or repeated measures ANOVA as appropriate. Data are presented as mean ± SE. Statistical significance was defined as p < 0.05.
Results
Pre-Treatment with LCM Decreases Mitochondrial Respiration
To assess how LCM affects mitochondrial function in skeletal muscle, we examined C2C12 myotubes after 30 min, 2, or 24 h in LCM. LCM treatment altered respiration in a time-dependent manner (Figure 1A). Western blots showed that VDAC content is unaffected by LCM treatment; therefore, we normalized OCR to VDAC content (Figure 1B). Thirty minutes of LCM treatment did not affect electron transport chain (ETC) activity; however, 2 or 24 h of LCM treatment resulted in depressed basal respiration (Figure 1C). The difference between basal respiration and oligomycin-sensitive respiration represents the OCR dedicated to ATP production. Compared to control myotubes, myotubes exposed to 2 or 24 h of LCM had significantly less ATP-related OCR (Figure 1D).
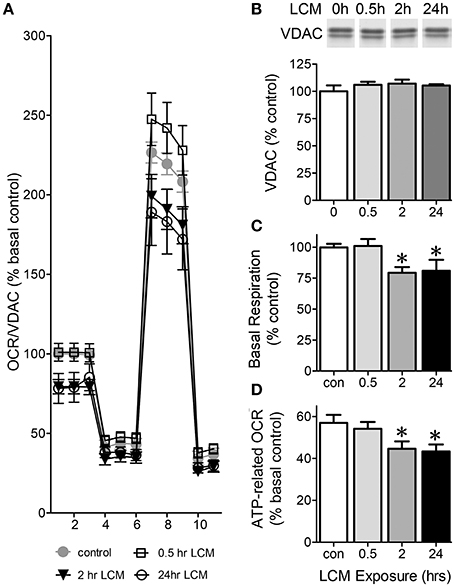
Figure 1. Oxygen consumption rate (OCR) in C2C12 myotubes. Eleven total OCR measurements were taken over 2 h: 3 basal respiration, 3 Oligomycin-sensitive respiration, 3 maximal respiratory capacity, and 2 non-mitochondrial respiration. The x-axis in (A) describes the measurement number. (A) OCR normalized to VDAC content presented at % basal control in control myotubes vs. myotubes treated for 30 min, 2 or 24 h with LCM (n = 12, *P < 0.01 for treatment effect, repeated measures ANOVA). (B) VDAC content in myotubes treated with LCM, represented as percent control. (C) Change basal respiration in myotubes treated with LCM, expressed as percent of control respiration (n = 12, *P < 0.01, ANOVA, Bonferroni post-hoc). (D) ATP-related OCR/VDAC in myotubes treated with LCM expressed as percent basal control (n = 12, *P < 0.05, ANOVA, Bonferroni post-hoc test).
Protein Content of Mitochondrial Complexes in C2C12 Myotubes Treated with LCM
To test whether the decreased mitochondrial respiration in LCM-treated myotubes was caused by changes in ETC complexes, we measured their content by western blot. Each complex in the ETC has multiple subunits, and we measured a representative subunit for each complex as follows: mitochondrial complex I NDUFA9; complex III subunit core I; complex IV subunit IV; and complex V, subunit α. LCM treatment did not alter the content of any ETC complex subunit measured (Figure 2).
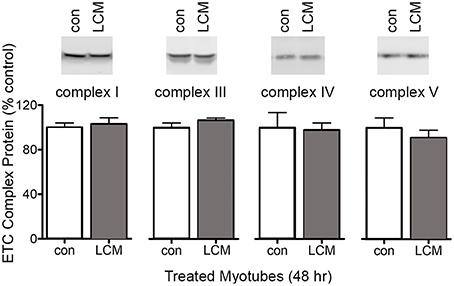
Figure 2. Protein content of electron transport chain complexes. Samples were equal protein loaded, which was determined via SDS-PAGE and staining with Simply Blue. Western blot membranes showed no changes in protein content for mitochondrial complex I NDUFA9, complex III subunit core I, complex IV subunit IV, and complex V, subunit α (n = 6).
Effect of LCM Treatment on Cytochrome C Oxidase Activity
Cytochrome c oxidase is the last complex in the electron transport chain. It transfers four electrons to molecular oxygen. A decrease in cytochrome c absorbance indicates an increase in cytochrome c oxidase activity. Cytochrome c oxidase activity in myotubes treated with LCM for 2 h increased compared to control myotubes (Figure 3). These results suggest that there is an early burst of cytochrome c oxidase activity upon exposure to LCM.
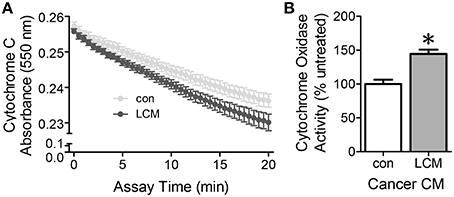
Figure 3. The effect of LCM treatment on cytochrome c oxidase activity. (A) Cytochrome C absorbance decreases after a 2 h exposure to LCM, indicating an increase in cytochrome c oxidase activity. (B) Cytochrome oxidase activity defined by the rate of change in the linear range per mg protein. LCM treatment for 2 h increased activity to 144% of control (n = 8, *P < 0.01, t-test).
LCM-Induced Changes in Oxidant Production
The ETC constitutively produces reactive oxygen species (ROS) during activity and is the greatest source of oxidants in skeletal muscle (Turrens, 2003). It is therefore likely that changes in ETC activity are reflected in alterations in ROS production. To test our hypothesis that LCM treatment would alter the level of oxidants, we examined general oxidant production, reactive nitrogen species (RNS) production, and mitochondrial superoxide anion production in myotubes treated with LCM for 30 min, 4, 6, and 24 h. DCF fluorescence measures overall cytosolic oxidant activity. Cytosolic oxidants increased at 30 min and 4 h compared to control myotubes (Figure 4A). However, after 24 h of LCM treatment, cytosolic oxidants decreased (Figure 4A). DAF fluorescence, which measures RNS production, was unaltered by LCM treatments (Figure 4B). These data indicated that RNS did not contribute to changes in overall cytosolic oxidant activity, as measured by DCF fluorescence. We hypothesized that some of the changes in DCF fluorescence might originate from the ETC. Using a CCD camera attached to a fluorescence microscope, we tested our hypothesis by measuring changes to the fluorescence intensity of mitochondrial superoxide indicator, MitoSox, in myotubes treated with LCM, LCM combined with mitochondrial-targeted antioxidant, SS31, and LCM combined with general antioxidant, N-Acetylcysteine (NAC). We found that after 2 h of LCM treatment, MitoSox fluorescence increased compared to control myotubes (Figures 4C,D). Myotubes treated for 2 h with LCM combined with mitochondrial antioxidant, SS31, had reduced MitoSox fluorescence compared to myotubes treated with LCM alone (Figure 4D). Myotubes treated with LCM combined with NAC had no significant alteration in MitoSox fluorescence (Figure 4D).
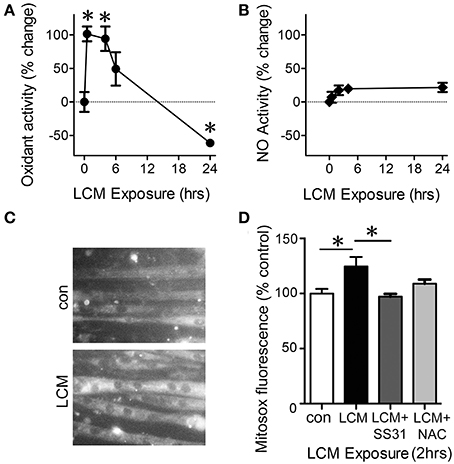
Figure 4. LCM treatment alters oxidant production. (A) DCFH fluorescence, a measurement of cytosolic oxidant levels, was increased by LCM treatment for 30 min and 4 h, but reduced by 24 h of treatment, represented as percent change (n = 10, *P < 0.05, ANOVA, Bonferroni post-hoc). (B) Unaltered DAF fluorescence in myotubes treated with LCM for 30 min, 2, and 24 h, represented as percent change (n = 20, non-significant, ANOVA, Bonferroni post-hoc). (C) Fluorescence images of control myotubes (top panel) and LCM-treated myotubes (bottom panel) treated with MitoSox. (D) 2 h of LCM treatment increased MitoSox fluorescence compared to control; 2 h of LCM treatment combined with SS31 decreased Mitosox fluorescence compared to LCM-treated myotubes; and 2 h of LCM treatment combined with NAC did not alter MitoSox fluorescence significantly (n = 4, *P < 0.05, Bonferroni post-hoc).
The Effect of LCM Treatment on UCP3 Content
The DCF results with 24 h of LCM treatment are consistent with ETC uncoupling. UCP3 is the most abundant uncoupling protein in skeletal muscle, and we speculated that its expression may be elevated with prolonged LCM exposure as a protection against LCM-induced oxidative damage by reducing ROS production (Maclellan et al., 2005; Toime and Brand, 2010). We examined the content of UCP3 via western blot and found that its expression was unaffected by LCM treatment (Figure 5).
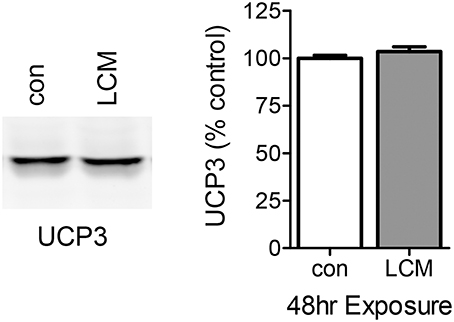
Figure 5. The effect of LCM treatment on UCP3 content. The left panel shows a representative western blot with anti-UCP3 in control and LCM-treated myotubes. The right panel shows the quantification of western blot represented as percent control (n = 6, t-test). 24 h of LCM treatment does not alter UCP3 content.
Effect of LCM Treatment on Protein Oxidation
LCM treatment caused a transient increase in ROS, potentially leading to oxidized proteins that may affect ETC activity (Figure 4). We isolated mitochondria from myotubes treated for 2 or 48 h with LCM. After 2 h of LCM treatment, 4-hydroxynonenal (HNE) was elevated in the cytosolic fraction, and after 48 h, HNE was elevated in both cytosolic and mitochondrial fractions (Figure 6). These data indicate that LCM oxidizes both cytosolic and mitochondrial proteins, which may result in alterations to their functions.
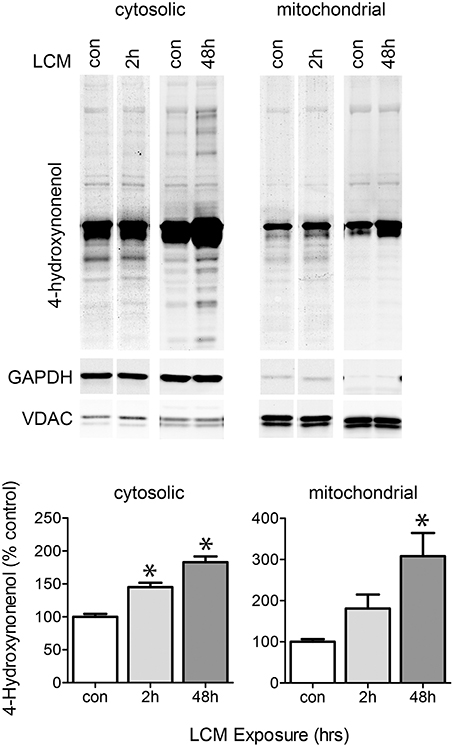
Figure 6. LCM treatment oxidizes proteins. The upper panel shows western blot for anti-4HNE in cytosolic and mitochondrial myotube fractions treated with LCM for 2 or 48 h, vs. control myotubes. The bottom panel shows the quantification anti-4HNE western blots (n = 4, *P < 0.05, ANOVA, Bonferroni post-hoc). LCM treatment for 2 and 48 h increased HNE content in both cytosolic and mitochondrial portions of cell lysates.
Discussion
Mitochondrial dysfunction has been reported in several models of cancer cachexia; however, there have been conflicting data regarding the nature of alterations to the ETC (Busquets et al., 2005; Constantinou et al., 2011; Julienne et al., 2012; Khamoui and Kim, 2012; Tzika et al., 2013). One primary feature of cachexia is that the remaining muscle mass becomes easily fatigued, suggesting possible ETC dysfunction. For example, alterations to the ETC could promote fatigue through loss of ATP production, increased production of ROS, or both (Roberts et al., 2013a,b). Previous studies have found that excessive ROS oxidize myofibrillar proteins in skeletal muscle, and it is reasonable to hypothesize that ROS may also contribute to cachectic fatigue in this manner (Bruton et al., 2008; Reid, 2008; Prochniewicz et al., 2008a,b; Fedorova et al., 2010). In this study, we used an in vitro model of cancer cachexia to focus on the direct action of cancer-conditioned media on mitochondrial function, protein content, ROS production, and oxidative stress in skeletal muscle.
Our results show that LCM changed ETC activity in C2C12 myotubes in a time-dependent manner (Figure 1A). ETC activity was altered by LCM treatment. LCM exposure of 2 and 24 h lead to depressed basal respiration and ATP-related OCR (Figures 1C,D). The altered ETC activity with longer treatment times could have several possible explanations: less mitochondria in LCM-treated cells, reduced content of mitochondrial complexes, or increased ROS production. We tested protein content of VDAC as a measurement of mitochondrial content and found it unaffected by LCM treatment (Figure 1B). With this finding, we used VDAC as a means of normalization in XF analyzer measurements of OCR. We also measured the protein content of ETC complexes I, III, IV, and V, and found that LCM treatment did not alter expression (Figure 2). With content of mitochondria and the complexes remaining intact, alterations to complex activity became a candidate to explain our OCR results. Complex IV, cytochrome c oxidase, is the last electron carrier of the ETC, passing four electrons from reduced cytochrome c to molecular oxygen to produce water. If complex IV activity is diminished, it suggests that electron transport is slowed. We found that 2 h of LCM treatment increased Complex IV activity, which seems to contradict our hypothesis (Figure 3). However, Turrens found that ETC-generated superoxide can reduce cytochrome c directly leading to increased Complex IV activity while bypassing complexes I and III (Turrens, 2003). In other words, the activity could increase if more reduced cytochrome c substrate was present due to an increase in superoxide production.
The complex IV activity data suggested the possibility that LCM treatment caused an increase in ROS production. Although normal physiological activity of the ETC constitutively produces some ROS as electrons slip off of complexes I and III, excessive oxidant production is known to cause oxidation damage to proteins, lipids, and DNA (St-Pierre et al., 2002; Turrens, 2003). We hypothesized that LCM treatment was inducing excessive oxidant production, leading to oxidative stress in our model. LCM treatment initially increased oxidant production, but by 24 h after treatment, oxidants were depressed below control conditions (Figure 4A). Lack of changes in DAF fluorescence eliminated RNS contributions in our system (Figure 4B). Increased MitoSox fluorescence with LCM treatment implicated mitochondrial superoxide as a source for the initial rise in oxidant production (Figure 4C). This hypothesis was supported by additional experiments in which we showed a decrease in MitoSox fluorescence when LCM treatment was combined with mitochondrial targeting antioxidant, SS31, but not with general antioxidant, NAC (Figures 4C,D) An increase in superoxide may also explain our increase in cytochrome c oxidase activity.
Depressed oxidant production after 24 h of LCM treatment suggests that uncoupling may occur in our model. Uncoupling increases in response to elevated ROS production as a means of reducing ROS and the potential damage they inflict (Maclellan et al., 2005; Toime and Brand, 2010). Mitochondria are considered uncoupled when electrons move through the inner mitochondrial membrane without participating in ATP production. Other studies on cachectic skeletal muscle have found both increased and unchanged expression of uncoupling protein three (UCP3), the main uncoupling protein in skeletal muscle (Vidal-Puig et al., 1997; Constantinou et al., 2011; Julienne et al., 2012; Antunes et al., 2014). We found no change in the expression of UCP3 despite the depression in ROS production (Figure 5). With UCP3 unchanged, we wondered if oxidative damage might affect ETC activity, leading to a subsequent decrease in ETC-generated ROS production over time.
We tested the effect of LCM treatment on oxidative stress. 4-hydroxynonenal is a product of lipid peroxidation, and acts as both a marker and inducer of oxidative stress. It is known to aggregate and cause oxidative damage to DNA and proteins (Chen and Niki, 2006). We tested for 4HNE-modified proteins in cytosolic and mitochondrial fractions of myotube lysates via anti-4HNE western blot. We found that LCM increased 4HNE modifications to proteins in both fractions after 48 h (Figure 6). These data show that LCM causes oxidative damage, perhaps affecting ETC activity.
Conclusions
Our results show that skeletal muscle cells treated with LCM experience altered ETC activity, including reduced basal respiration and reduced ATP-related OCR. LCM treatment initially induces a burst of ROS production, some of which is mitochondrial superoxide. Our 4HNE results show that LCM induced oxidative damage. 4HNE itself can act as a mediator of oxidative stress, and potentially creates a self-amplifying loop of oxidative stress. Both ROS and 4HNE can damage DNA, proteins, transcription factors, and lipids, resulting in widespread cellular damage. Future work should focus on discovering which LCM-derived paracrine factors are the upstream mediators in mitochondrial ETC alterations seen in cachexia.
Author Contributions
Julie B. McLean and Jennifer S. Moylan performed experiments and analyzed data; Jennifer S. Moylan and Julie B. McLean prepared figures, Julie B. McLean drafted manuscript, Julie B. McLean, Jennifer S. Moylan and Francisco H. Andrade edited and revised manuscript; Julie B. McLean, Jennifer S. Moylan and Francisco H. Andrade conceived experimental design; Julie B. McLean, Jennifer S. Moylan and Francisco H. Andrade approved final draft.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was supported by NIAMS/NIH R01 AR055974 to Francisco H. Andrade. We would like to thank Megan Endres, Ph.D., Department of Management, Eastern Michigan University, for her assistance in statistical tests for XFe24 analyzer data.
Abbreviations
ETC, electron transport chain; LCM, Lewis lung carcinoma conditioned medium; OCR, oxygen consumption rate; PAGE, polyacrylamide gel electrophoresis; VDAC, voltage dependent anion channel.
References
Andrade, F. H., Reid, M. B., Allen, D. G., and Westerblad, H. (1998). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 509 (Pt 2), 565–575.
Andrade, F. H., Reid, M. B., and Westerblad, H. (2001). Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox-modulation. FASEB J. 15, 309–311. doi: 10.1096/fj.00-0507fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Antunes, D., Padrao, A. I., Maciel, E., Santinha, D., Oliveira, P., Vitorino, R., et al. (2014). Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta 1841, 896–905. doi: 10.1016/j.bbalip.2014.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Argiles, J. M., Figueras, M., Ametller, E., Fuster, G., Olivan, M., De Oliveira, C. C., et al. (2008). Effects of CRF2R agonist on tumor growth and cachexia in mice implanted with Lewis lung carcinoma cells. Muscle Nerve 37, 190–195. doi: 10.1002/mus.20899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bruton, J. D., Place, N., Yamada, T., Silva, J. P., Andrade, F. H., Dahlstedt, A. J., et al. (2008). Reactive oxygen species and fatigue-induced prolonged low-frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J. Physiol. (Lond). 586, 175–184. doi: 10.1113/jphysiol.2007.147470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Busquets, S., Almendro, V., Barreiro, E., Figueras, M., Argiles, J. M., and Lopez-Soriano, F. J. (2005). Activation of UCPs gene expression in skeletal muscle can be independent on both circulating fatty acids and food intake. Involvement of ROS in a model of mouse cancer cachexia. FEBS Lett. 579, 717–722. doi: 10.1016/j.febslet.2004.12.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carbo, N., Costelli, P., Busquets, S., Lopez-Soriano, J., Lopez-Soriano, F. J., Baccino, F. M., et al. (2004). Effect of c-ski overexpression on the development of cachexia in mice bearing the Lewis lung carcinoma. Int. J. Mol. Med. 14, 719–723. doi: 10.3892/ijmm.14.4.719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Z. H., and Niki, E. (2006). 4-hydroxynonenal (4-HNE) has been widely accepted as an inducer of oxidative stress. Is this the whole truth about it or can 4-HNE also exert protective effects? IUBMB Life 58, 372–373. doi: 10.1080/15216540600686896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, S., Szeto, H. H., Kim, E., Kim, H., Tolhurst, A. T., and Pinto, J. T. (2007). A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J. Biol. Chem. 282, 4634–4642. doi: 10.1074/jbc.M609388200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Constantinou, C., Fontes De Oliveira, C. C., Mintzopoulos, D., Busquets, S., He, J., Kesarwani, M., et al. (2011). Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. Int. J. Mol. Med. 27, 15–24. doi: 10.3892/ijmm.2010.557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dumas, J. F., Peyta, L., Couet, C., and Servais, S. (2013). Implication of liver cardiolipins in mitochondrial energy metabolism disorder in cancer cachexia. Biochimie 95, 27–32. doi: 10.1016/j.biochi.2012.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dutka, T. L., Verburg, E., Larkins, N., Hortemo, K. H., Lunde, P. K., Sejersted, O. M., et al. (2012). ROS-mediated decline in maximum Ca2+-activated force in rat skeletal muscle fibers following in vitro and in vivo stimulation. PLoS ONE 7:e35226. doi: 10.1371/journal.pone.0035226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fearon, K., Arends, J., and Baracos, V. (2013). Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 10, 90–99. doi: 10.1038/nrclinonc.2012.209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fearon, K., Strasser, F., Anker, S. D., Bosaeus, I., Bruera, E., Fainsinger, R. L., et al. (2011). Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495. doi: 10.1016/S1470-2045(10)70218-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fedorova, M., Kuleva, N., and Hoffmann, R. (2010). Identification, quantification, and functional aspects of skeletal muscle protein-carbonylation in vivo during acute oxidative stress. J. Proteome Res. 9, 2516–2526. doi: 10.1021/pr901182r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fontes-Oliveira, C. C., Busquets, S., Toledo, M., Penna, F., Paz Aylwin, M., Sirisi, S., et al. (2013). Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim. Biophys. Acta 1830, 2770–2778. doi: 10.1016/j.bbagen.2012.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilliam, L. A., Moylan, J. S., Callahan, L. A., Sumandea, M. P., and Reid, M. B. (2011). Doxorubicin causes diaphragm weakness in murine models of cancer chemotherapy. Muscle Nerve 43, 94–102. doi: 10.1002/mus.21809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hardin, B. J., Campbell, K. S., Smith, J. D., Arbogast, S., Smith, J., Moylan, J. S., et al. (2008). TNF-alpha acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J. Appl. Physiol. (1985) 104, 694–699. doi: 10.1152/japplphysiol.00898.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Irrcher, I., Ljubicic, V., and Hood, D. A. (2009). Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 296, C116–C123. doi: 10.1152/ajpcell.00267.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. CA Cancer J. Clin. 61, 69–90. doi: 10.3322/caac.20107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johns, N., Stephens, N. A., and Fearon, K. C. (2013). Muscle wasting in cancer. Int. J. Biochem. Cell Biol. 45, 2215–2229. doi: 10.1016/j.biocel.2013.05.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Julienne, C. M., Dumas, J. F., Goupille, C., Pinault, M., Berri, C., Collin, A., et al. (2012). Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachexia Sarcopenia Muscle 3, 265–275. doi: 10.1007/s13539-012-0071-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Julienne, C. M., Tardieu, M., Chevalier, S., Pinault, M., Bougnoux, P., Labarthe, F., et al. (2014). Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer-induced cachexia without the involvement of adenine nucleotide translocase. Biochim. Biophys. Acta 1842, 726–733. doi: 10.1016/j.bbadis.2014.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khamoui, A. V., and Kim, J. S. (2012). Candidate mechanisms underlying effects of contractile activity on muscle morphology and energetics in cancer cachexia. Eur. J. Cancer Care (Engl). 21, 143–157. doi: 10.1111/j.1365-2354.2011.01287.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J., Giordano, S., and Zhang, J. (2012). Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 441, 523–540. doi: 10.1042/BJ20111451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemire, J., Mailloux, R. J., and Appanna, V. D. (2008). Mitochondrial lactate dehydrogenase is involved in oxidative-energy metabolism in human astrocytoma cells (CCF-STTG1). PLoS ONE 3:e1550. doi: 10.1371/journal.pone.0001550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, L., Sun, Z., Zhang, L., Li, X., Dong, Y., and Liu, T. C. (2013). Effects of low-level laser therapy on ROS homeostasis and expression of IGF-1 and TGF-beta1 in skeletal muscle during the repair process. Lasers Med. Sci. 28, 725–734. doi: 10.1007/s10103-012-1133-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maclellan, J. D., Gerrits, M. F., Gowing, A., Smith, P. J., Wheeler, M. B., and Harper, M. E. (2005). Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes 54, 2343–2350. doi: 10.2337/diabetes.54.8.2343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manczak, M., Mao, P., Calkins, M. J., Cornea, A., Reddy, A. P., Murphy, M. P., et al. (2010). Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J. Alzheimers Dis. 20(Suppl. 2), S609–S631. doi: 10.3233/JAD-2010-100564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merry, T. L., Steinberg, G. R., Lynch, G. S., and McConell, G. K. (2010). Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am. J. Physiol. Endocrinol. Metab. 298, E577–E585. doi: 10.1152/ajpendo.00239.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michaelson, L. P., Iler, C., and Ward, C. W. (2013). ROS and RNS signaling in skeletal muscle: critical signals and therapeutic targets. Annu. Rev. Nurs. Res. 31, 367–387. doi: 10.1891/0739-6686.31.367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moylan, J. S., Smith, J. D., Wolf Horrell, E. M., McLean, J. B., Deevska, G. M., Bonnell, M. R., et al. (2014). Neutral sphingomyelinase-3 mediates TNF-stimulated oxidant activity in skeletal muscle. Redox Biol. 2, 910–920. doi: 10.1016/j.redox.2014.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Powers, S. K., Hudson, M. B., Nelson, W. B., Talbert, E. E., Min, K., Szeto, H. H., et al. (2011). Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 39, 1749–1759. doi: 10.1097/CCM.0b013e3182190b62
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prochniewicz, E., Lowe, D. A., Spakowicz, D. J., Higgins, L., O'Conor, K., Thompson, L. V., et al. (2008a). Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 294, C613–C626. doi: 10.1152/ajpcell.00232.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prochniewicz, E., Spakowicz, D., and Thomas, D. D. (2008b). Changes in actin structural transitions associated with oxidative inhibition of muscle contraction. Biochemistry 47, 11811–11817. doi: 10.1021/bi801080x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prosser, B. L., Khairallah, R. J., Ziman, A. P., Ward, C. W., and Lederer, W. J. (2013). X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca(2)(+)]i. J. Mol. Cell. Cardiol. 58, 172–181. doi: 10.1016/j.yjmcc.2012.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Puppa, M. J., Gao, S., Narsale, A. A., and Carson, J. A. (2014). Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma-induced cachexia. FASEB J. 28, 998–1009. doi: 10.1096/fj.13-240580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reid, M. B. (2008). Free radicals and muscle fatigue: of ROS, canaries, and the IOC. Free Radic. Biol. Med. 44, 169–179. doi: 10.1016/j.freeradbiomed.2007.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reid, M. B., Andrade, F. H., Balke, C. W., and Esser, K. A. (2005). Redox mechanisms of muscle dysfunction in inflammatory disease. Phys. Med. Rehabil. Clin. N. Am. 16, 925–949. doi: 10.1016/j.pmr.2005.08.016
Roberts, B. M., Ahn, B., Smuder, A. J., Al-Rajhi, M., Gill, L. C., Beharry, A. W., et al. (2013a). Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 27, 2600–2610. doi: 10.1096/fj.12-222844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roberts, B. M., Frye, G. S., Ahn, B., Ferreira, L. F., and Judge, A. R. (2013b). Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem. Biophys. Res. Commun. 435, 488–492. doi: 10.1016/j.bbrc.2013.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D. (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790. doi: 10.1074/jbc.M207217200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tan, B. H., and Fearon, K. C. (2008). Cachexia: prevalence and impact in medicine. Curr. Opin. Clin. Nutr. Metab. Care 11, 400–407. doi: 10.1097/MCO.0b013e328300ecc1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tisdale, M. J. (2009). Mechanisms of cancer cachexia. Physiol. Rev. 89, 381–410. doi: 10.1152/physrev.00016.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toime, L. J., and Brand, M. D. (2010). Uncoupling protein-3 lowers reactive oxygen species production in isolated mitochondria. Free Radic. Biol. Med. 49, 606–611. doi: 10.1016/j.freeradbiomed.2010.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. (Lond). 552, 335–344. doi: 10.1113/jphysiol.2003.049478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tzika, A. A., Fontes-Oliveira, C. C., Shestov, A. A., Constantinou, C., Psychogios, N., Righi, V., et al. (2013). Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 43, 886–894. doi: 10.3892/ijo.2013.1998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vidal-Puig, A., Solanes, G., Grujic, D., Flier, J. S., and Lowell, B. B. (1997). UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem. Biophys. Res. Commun. 235, 79–82. doi: 10.1006/bbrc.1997.6740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Von Haehling, S., Morley, J. E., Coats, A. J., and Anker, S. D. (2010). Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J. Cachexia Sarcopenia Muscle 1, 7–8. doi: 10.1007/s13539-010-0003-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Pickrell, A. M., Zimmers, T. A., and Moraes, C. T. (2012). Increase in muscle mitochondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer-induced cachexia. PLoS ONE 7:e33426. doi: 10.1371/journal.pone.0033426
White, J. P., Puppa, M. J., Sato, S., Gao, S., Price, R. L., Baynes, J. W., et al. (2012). IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2, 14. doi: 10.1186/2044-5040-2-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Whiteman, M., Spencer, J. P., Szeto, H. H., and Armstrong, J. S. (2008). Do mitochondriotropic antioxidants prevent chlorinative stress-induced mitochondrial and cellular injury? Antioxid. Redox Signal. 10, 641–650. doi: 10.1089/ars.2007.1879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cachexia, mitochondria, oxidants, electron transport chain, skeletal muscle
Citation: McLean JB, Moylan JS and Andrade FH (2014) Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Front. Physiol. 5:503. doi: 10.3389/fphys.2014.00503
Received: 08 October 2014; Accepted: 03 December 2014;
Published online: 18 December 2014.
Edited by:
Julio L. Vergara, University of California, Los Angeles, USAReviewed by:
Espen Spangenburg, University of Maryland, USARanganath Mamidi, Case Western Reserve University, USA
Copyright © 2014 McLean, Moylan and Andrade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco H. Andrade, Department of Physiology, University of Kentucky, 800 Rose St. MS-508, Lexington, KY 40536, USA e-mail: paco.andrade@uky.edu