 Pietro Mesirca
Pietro Mesirca Angelo G. Torrente
Angelo G. Torrente Matteo E. Mangoni
Matteo E. Mangoni- 1Laboratory of Excellence in Ion Channel Science and Therapeutics, Département de Physiologie, Institut de Génomique Fonctionnelle, Montpellier, France
- 2UMR-5203, Centre National de la Recherche Scientifique, Universités de Montpellier 1 and 2, Montpellier, France
- 3INSERM U 1191, Département de Physiologie, Universités de Montpellier 1 and 2, Montpellier, France
Pacemaker activity of automatic cardiac myocytes controls the heartbeat in everyday life. Cardiac automaticity is under the control of several neurotransmitters and hormones and is constantly regulated by the autonomic nervous system to match the physiological needs of the organism. Several classes of ion channels and proteins involved in intracellular Ca2+ dynamics contribute to pacemaker activity. The functional role of voltage-gated calcium channels (VGCCs) in heart automaticity and impulse conduction has been matter of debate for 30 years. However, growing evidence shows that VGCCs are important regulators of the pacemaker mechanisms and play also a major role in atrio-ventricular impulse conduction. Incidentally, studies performed in genetically modified mice lacking L-type Cav1.3 (Cav1.3−/−) or T-type Cav3.1 (Cav3.1−/−) channels show that genetic inactivation of these channels strongly impacts pacemaking. In cardiac pacemaker cells, VGCCs activate at negative voltages at the beginning of the diastolic depolarization and importantly contribute to this phase by supplying inward current. Loss-of-function of these channels also impairs atrio-ventricular conduction. Furthermore, inactivation of Cav1.3 channels promotes also atrial fibrillation and flutter in knockout mice suggesting that these channels can play a role in stabilizing atrial rhythm. Genomic analysis demonstrated that Cav1.3 and Cav3.1 channels are widely expressed in pacemaker tissue of mice, rabbits and humans. Importantly, human diseases of pacemaker activity such as congenital bradycardia and heart block have been attributed to loss-of-function of Cav1.3 and Cav3.1 channels. In this article, we will review the current knowledge on the role of VGCCs in the generation and regulation of heart rate and rhythm. We will discuss also how loss of Ca2+ entry through VGCCs could influence intracellular Ca2+ handling and promote atrial arrhythmias.
Introduction
Pacemaker activity in the heart is generated by specialized myocytes, able to generate periodical oscillations of their membrane potential. These cells are thus called “pacemaker” cells (Mangoni and Nargeot, 2008). Pacemaker cells are localized in the sino-atrial node (SAN), which is a thin tissue located in the right atrium (for anatomical description see Dobrzynski et al., 2005). Under physiological conditions the cardiac impulse has origin in the SAN. The pacemaker impulse spreads from the SAN to the cardiac conduction system (composed by the atrioventricular node and Purkinje fibers network), driving the contraction of the whole working myocardium. In comparison to the rest of the conduction system, the SAN generates the fastest intrinsic automaticity, thereby inhibiting pacemaking in the atrioventricular node (AVN) and the Purkinje fibers network. Nevertheless, in case of SAN failure, the AVN can take over as dominant pacemaker center. Under conditions of atrioventricular block, Purkinje fibers are able to generate viable rhythm, even if at relatively low rates (James, 2003; Dobrzynski et al., 2013).
The generation of the automaticity in cardiac pacemaker cells is due to the diastolic depolarization, a spontaneous slowly depolarizing phase of the action potential cycle. During this phase the membrane potential progressively becomes less negative until it reaches the threshold for triggering a new action potential. The SAN action potential cycle length determines the heart rate. At the level of the individual SAN cell, different classes of ion channels of the plasma membrane, the sarcoplasmic reticulum (SR) and mytochondria contribute to the generation and regulation of automaticity, but their respective functional roles and interactions are still not fully understood.
In the recent past, two distinct, but not mutually exclusively, hypotheses were proposed to explain the mechanism underling the cardiac automaticity: the so-called “membrane clock” model of pacemaking, which considers the “funny” current (If), an inward Na+/K+ current activated by membrane hyperpolarization at negative voltages (Brown et al., 1979) and regulated directly by cAMP (Difrancesco and Tortora, 1991) as the key initiator of the diastolic depolarization (Difrancesco, 1991). In the “calcium clock” model of pacemaking the key mechanism in the diastolic depolarization is a spontaneous rhythmic phenomenon of Ca2+ release from the SR activating the Na+/Ca2+ exchanger (NCX) in forward mode. This NCX mediated inward current is able to depolarize the membrane voltage to the threshold of the following action potential (Bogdanov et al., 2001; Vinogradova et al., 2002). Recently, the Ca2+ clock view of pacemaking has been updated into the “coupled-clock” model (Lakatta et al., 2010). In the coupled-clock model of pacemaking, the activity of membrane ion channels and spontaneous Ca2+ release mutually entrain to generate pacemaking (Lakatta et al., 2010; Monfredi et al., 2013). For a more extended description of the issues raised by the If - based and the Ca2+ or coupled-clock models of pacemaking, the reader is referred to recent review by the principal authors (Difrancesco, 2010; Lakatta et al., 2010; Monfredi et al., 2013).
However, either the If -based or the coupled clock models of pacemaking do not fully appreciate the role of VGCCs in pacemaking. Indeed, in the If -based model of pacemaking the L-type Ca2+ current (ICa,L) is considered only as a determinant of the action potential upstroke and duration (Difrancesco, 1993, 2010). In the coupled-clock model of pacemaking, ICa,L is considered as a major mechanism to replenish SR Ca2+ content at each pacemaker cycle (Vinogradova et al., 2002). Finally, both the If -based and the coupled-clock models grant only a limited role to T-type VGCCs (Vinogradova et al., 2002).
However, during the last 10 years evidence accumulated showing that VGCCs contribute directly to pacemaking by carrying inward current during the diastolic depolarization phase (Zhang et al., 2002, 2011; Mangoni et al., 2003, 2006b; Marger et al., 2011a) or by stimulating the NCX activated by subsarcolemmal Ca2+ release during the diastolic depolarization (Lakatta et al., 2010). VGCCs also participate to the upstroke phase of the action potential (Hagiwara et al., 1988; Doerr et al., 1989; Marger et al., 2011a). Here we will focus on two distinct families of VGCCs, the L-type and the T-type Ca2+ channels. L-type VGCCs are expressed throughout the myocardium and are sensitive to antagonist and agonist dihydropyridines (DHPs) such as nifedipine and BAY K 8644 and are stimulated by PKA-dependent phosporylation (Striessnig, 1999; van der Heyden et al., 2005). In comparison with T-type channels, L-type VGCCs activate upon membrane depolarization at more positive potential, have Ca2+ and voltage dependent inactivation, as well as a higher single channel conductance (Perez-Reyes, 2003). T-type VGCCs are activated at more negative potentials than L-type VGCCs. The kinetic hallmark of native and heterogously expressed T-type mediated Ca2+ current is slow criss-crossing activation and fast voltage dependent inactivation (Carbone and Lux, 1987). Table 1 summarizes the main characteristics of the L- and T-type VGCCs isoforms involved in cardiac automaticity. The elucidation of the functional role of the cardiac VGCCs can give important insights into the mechanisms underlying different SAN and conduction system pathologies. Indeed, failure of generating the cardiac impulse underlies SAN bradycardia and rhythmic disease. Diseases of the sinus node account for more than 1,000,000 electronic pacemaker implantations each year. SAN disease is characterized by various symptoms including severe sinus bradycardia, sinus pauses or arrest, chronotropic incompetence, sinus node exit block (Dobrzynski et al., 2007). Heart failure, cardiomyopathy, administration of antiarrhythmic drugs and other acquired cardiac conditions can induce SAN dysfunction. Nevertheless, in a significant number of patients, SAN dysfunction shows inherited features (Sarachek and Leonard, 1972; Lehmann and Klein, 1978; Mackintosh and Chamberlain, 1979; Dobrzynski et al., 2007; Sanders et al., 2014). Mutations in genes regulating L-type VDCCs involved in SAN automaticity such as L-type Cav1.3 (Mangoni et al., 2003; Baig et al., 2011) and T-type Cav3.1 (Marger et al., 2011a; Strandberg et al., 2013) are associated with various forms of previously unexplained tachy-brady syndromes and conduction defects (Mangoni and Nargeot, 2008; Pfeufer et al., 2010).
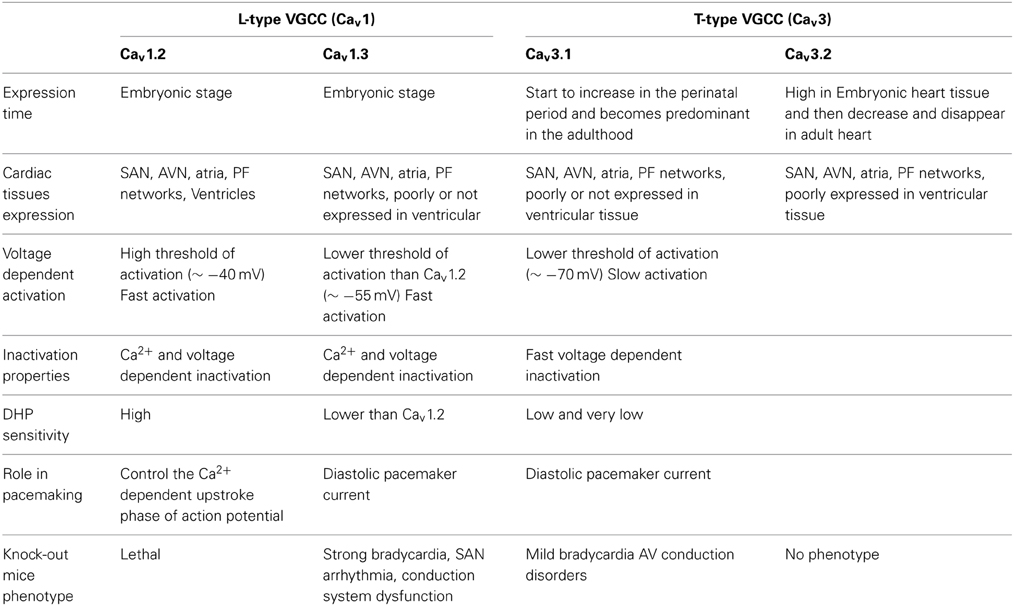
Table 1. Characteristics of the L- and T-type VGCCs isoforms involved in cardiac automaticity.
Cardiac Voltage Gated Ca2+ Channels: Molecular Determinants and Expression
VGCCs are an important pathway for Ca2+ entry in pacemaker cells. In the mammalian heart, L- and T-type mediated Ca2+ currents are expressed in SAN, AVN, and Purkinje Fibers network and they have been consistently recorded in pacemaker SAN and AVN cells (Tseng and Boyden, 1989; Mangoni et al., 2003, 2006b; Marger et al., 2011a). Hagiwara et al. (1988) were the first to report the expression of ICa,L in isolated SAN pacemaker cells and to describe its kinetic and pharmacologic properties. In particular, they defined ICa,L as a “high”-threshold Ca2+ current activated from about -30 mV and distinguished from T-type mediated Ca2+ current (ICa,T), a “low” threshold Ca2+ current activated at −50 mV, suggesting that both currents participate the latter half of the slow diastolic depolarization (Hagiwara et al., 1988).
L-type VGCCs are hetero-oligomeric complexes constituted by a voltage sensitive pore, the so–called α1-subunits, together with different accessory subunits (α2δ, β, and γ) (Striessnig, 1999) and they are highly sensitive to DHP Ca2+ channels modulators. Four α1-subunits have been cloned and classified for the L- type Ca2+ channel, namely Cav1.1, Cav1.2 Cav1.3, and Cav1.4 (Catterall et al., 2003). Cav1.1 subunits are expressed in the skeletal muscle, where they couple membrane excitation to contraction (Tanabe et al., 1988; Tuluc et al., 2009), Cav1.4 expression is predominant in the retina, spinal cord and immune cells (McRory et al., 2004; Striessnig and Koschak, 2008). Cav1.2 and Cav1.3 are expressed in neurons, as well as in cells from the neuroendocrine and cardiovascular systems (Catterall, 2000). Cav1.2 is expressed in the whole heart but predominantly in atria and ventricles; Cav1.3 expression is predominant in the supraventricular regions with higher amounts of Cav1.3 in the rhythmogenic centers (Marionneau et al., 2005). Electrophysiological measurements showed clear differences between Cav1.3 and Cav1.2 mediated ICa,L. Cav1.3-mediated ICa,L activates at more negative voltages and displays slower current inactivation during depolarization allowing these channels to mediate long lasting Ca2+ influx during weak depolarization (Platzer et al., 2000; Koschak et al., 2001). As showed by Hagiwara (Hagiwara et al., 1988), T-type VGCCs are activated at more negative potentials than L-type VGCCs. Moreover, ICa,T have faster voltage-dependent inactivation and inactivation is complete at more negative membrane potentials than ICa,L (Perez-Reyes, 2003) (Table 1).
Three genes encoding for T-type α-subunits have been cloned and named Cav3.1, Cav3.2, and Cav3.3. While the Cav3.3 isoform is not present in the heart, the expression of Cav3.1 and Cav3.2 isoforms in the myocardium varies according to the developmental status of the tissue. Cav3.2 constitutes the predominant T-type isoform in embryonic heart tissue (Ferron et al., 2002); Cav3.1 channels expression increases during perinatal period and reaches its maximal in adulthood (Marshall et al., 1993). In adult SAN Cav3.1 expression is higher than Cav3.2 (Bohn et al., 2000). Contrary to the Cav1 family, the Cav3 family is almost insensible to DHPs and at present, no selective inhibitor to discriminate the contribution of Cav3.1 and Cav3.2 channels to the total ICa,T is available.
Regulation of L- and T-type Ca2+ Channels in Cardiac Tissues
Cardiac VGCCs are subject of multiple regulatory mechanisms involving both intramolecular regulatory sites and interactions with cellular second messengers and kinases.
Ca2+ influx through VGCCs can “auto-regulate” the channel activity in a negative (CDI, Ca2+-dependent inactivation) or positive (CDF, Ca2+-dependent facilitation) manner. L-type Ca2+ channels undergo calmoduline-mediated CDI or calmoduline kinase II (CaMKII)-mediated CDF (Christel and Lee, 2012). On the contrary only CaMKII-mediated CDF has been described for T-type Ca2+ channels (Christel and Lee, 2012).
It has been shown that SAN L-type Ca2+ channels undergo voltage-dependent inactivation (VDI) and facilitation (VDF) (Mangoni et al., 2000; Christel et al., 2012). Christel et al. (2012) showed that Cav1.2-mediated ICa,L undergoes stronger VDI than Cav1.3-mediated ICa,L and that Cav1.3-mediated ICa,L exhibited stronger VDF than Cav1.2 -mediated ICa,L. Numerical modeling simulations predicted that VDF was responsible of 25% increase in Cav1.3-mediated ICa,L which, as a consequence, induced a small positive chronotropic effect. These data further support the importance of Cav1.3 Ca2+ channels regulation in cardiac pacemaker activity.
L-type Ca2+ channels are also potently regulated by cAMP-dependent protein kinase A (De Jongh et al., 1996; Ramadan et al., 2009). Regulation of T-type Ca2+ channels by cAMP dependent protein kinase A is still controversial (Chemin et al., 2006), however, in a recent work Li et al. (2012) found that in cardiac myocytes the activity of Cav3.1 T-type VGCCs was significantly increased by isoproterenol, a β-adrenergic agonist, and that this regulation was strictly connected to the adenylate cyclase/cAMP/PKA machinery similar to L-Type Ca2+ channels. One of the most important differences in the pharmacologic modulation of T- and L-type Ca2+ channels rises from their different sensitivity to DHPs. DHPs are known to act on ICa,L without affecting ICa,T (Hagiwara et al., 1988). Nevertheless, this concept has been challenged by different studies showing an effect of certain types of DHPs also on different subunits of T-type VGCCs (Bladen et al., 2014a,b). L-type Ca2+ channel voltage-dependence and expression are potently regulated by β subunits (see Buraei and Yang, 2010, for recent review). In the SAN, the predominant β subunit isoform expressed appears to be β2 (Marionneau et al., 2005). Co-expression of the β2 subunit with the Cav1.2 α1 subunit induces slowing of the voltage dependent inactivation of ICa,L (Cens et al., 1996). It has been proposed that β subunits regulate α1 protein trafficking (Buraei and Yang, 2013). It will be interesting to investigate whether β2 subunits regulates L-type Ca2+ channel trafficking in the cardiac conduction system. It has been showed that L-type Ca2+ channels, even to a lesser extent than other VGCCs such as T-type Ca2+ channels, are regulated also by phospholipids (Suh and Hille, 2005).
Functional Role of L-type and T-type Ca2+ Channels in Cardiac Automaticity
L-type Ca2+ Channels
Evidence for the importance of L-type Ca2+ current in SAN pacemaking have been reported by different studies (see Mangoni et al., 2006a, for review). Kodama et al. (1997) showed that blocking ICa,L by 2 μM nifedipine abolished the action potential in the primary central pacemaker area in rabbit SAN but not in spontaneously beating tissue from the periphery of the SAN. In contrast, they showed that tetrodotoxin 20 μM had no effect on electrical activity in the primary central pacemaker area, but depolarized the takeoff potential, decreased the upstroke velocity and slowed the spontaneous activity in nodal tissue from SAN periphery. These results are in line with the view that the rabbit pacemaker action potential strongly depends from ICa,L in the central area of SAN but not in the periphery, where it is more sensitive to Na+ current (INa). The heterogeneity of sensitivity to ICa,L in pacemaker cells highlights the problem of isolating the contribution of ICa,L to the diastolic depolarization phase from its contribution to the upstroke phase of the cardiac action potential.
Doerr et al. (1989) tried to overcome this major problem using the action potential clamp technique to evaluate the contribution of ICa,L in the pacemaker cycles in rabbit isolated SAN cells. They reported a net methoxyverapamil (L-type Ca2+ channels blocker)-sensitive current measurable during the early diastolic depolarization as well a long lasting component during the plateau phase. Verheijck et al. (1999) have recorded the net nifedipine-sensitive ICa,L at different times during action potential cycle. Notably, they provided direct evidence that ICa,L can be activated at potential as negative as −60 mV, typical of the early diastolic depolarization phase, and then increases up to the threshold potential supplying inward current during the entire diastolic depolarization. In comparison to the previous study by Hagiwara et al. (1988), Verheijck and co-workers recorded ICa,L starting from negative holding potentials (−90 mV), preventing partial steady-state inactivation of ICa,L at negative voltages. Second, Verheijck and co-workers employed a recording protocol combining current clamp, to let the cell to depolarize and repolarize spontaneously, with voltage clamping at discrete voltages spanning the diastolic depolarization to record ICa,L. This strategy allowed accurate measurement of the ICa,L current density in this phase.
Moreover, they demonstrated the presence of a “low-voltage”-activated ICa,L component, pharmacologically distinct from ICa,T, in the diastolic depolarization range, opening the way to the description of the functional role of Cav1.3 L-type channels in pacemaking.
The first in vivo evidence of the contribution of ICa,L in cardiac pacemaker was provided by Lande et al. (2001); they recorded DHP-induced bradycardia in anesthetized mice. Subsequently, the unexpected result that electrocardiogram (ECG) recordings revealed SAN dysfunction (bradycardia and arrhythmia) in mice lacking L-type Cav1.3 channels was the first genetic evidence of their importance in heart automaticity (Platzer et al., 2000). Two independent studies showed that Cav1.3 channels have a key role in automaticity both in vitro (Figure 1A) and in vivo (Figures 1B,C) (Zhang et al., 2002; Mangoni et al., 2003) also unmasking important differences between Cav1.3-mediated and Cav1.2-mediated ICa,L (Figure 2A). The heart chambers histology and thickness as well SAN and AVN structure did not show any differences between Cav1.3−/− and the wild type mice, suggesting that inactivation of Cav1.3 channels has no effect on heart structure (Matthes et al., 2004). Inactivation of Cav1.3-mediated ICa,L impairs pacemaking and atrioventricular conduction, but has no effect on myocardial contractility (Matthes et al., 2004). Zhang et al. (2005) showed that intracardiac atrial stimulation induced atrial fibrillation and atrial flutter in Cav1.3−/− mice but not in wild-type littermates even in the absence of vagal stimulation with carbachol, a muscarinic agonist which is known to induce atrial fibrillation in control mice (Kovoor et al., 2001). In contrast, no ventricular arrhythmias were induced in either the wild-type or mutant mice (Zhang et al., 2005). These data further support the view of an important functional role of Cav1.3 in the atria.
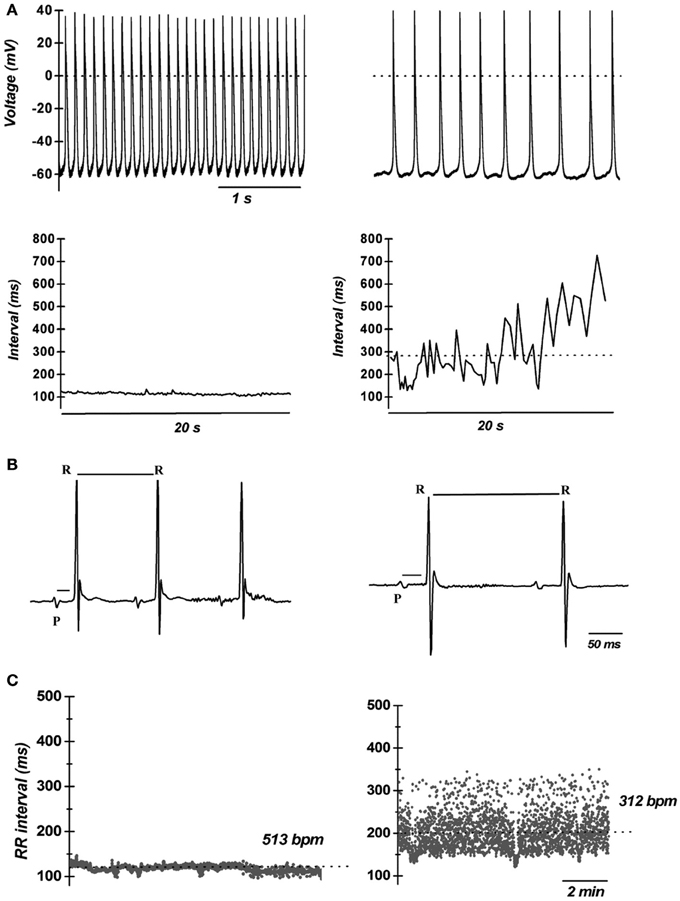
Figure 1. Importance of L-type VGCCs in cardiac automaticity. (A) Representative recordings of consecutive action potentials recorded in pacemaker cells from wild-type (top left panel) and Cav1.3−/− mice (top right panel). Cellular arrhythmia is evident as irregular cycle length duration in Cav1.3−/− cells (bottom right panel) compared with wild-type cells (bottom left panel). Dotted lines indicate the zero voltage level (Data from Mangoni et al., 2003). (B) Telemetric ECGs showing prolongation of RR interval PQ interval in Cav1.3−/− mice (top right panel) with respect to wild type littermates (top left panel). (C) Dot plot of beat to beat variability in wild-type (left panel) and Cav1.3−/− mice (right panel) observed during 10 min recordings. Note the dispersion of the RR intervals in Cav1.3 knockout mice, revealing strong sinus arrhythmia. The dotted lines indicate the average heart rate as the number of beats per minutes (bpm) (reprinted from Mangoni et al., 2006a, with permission from Elsevier).
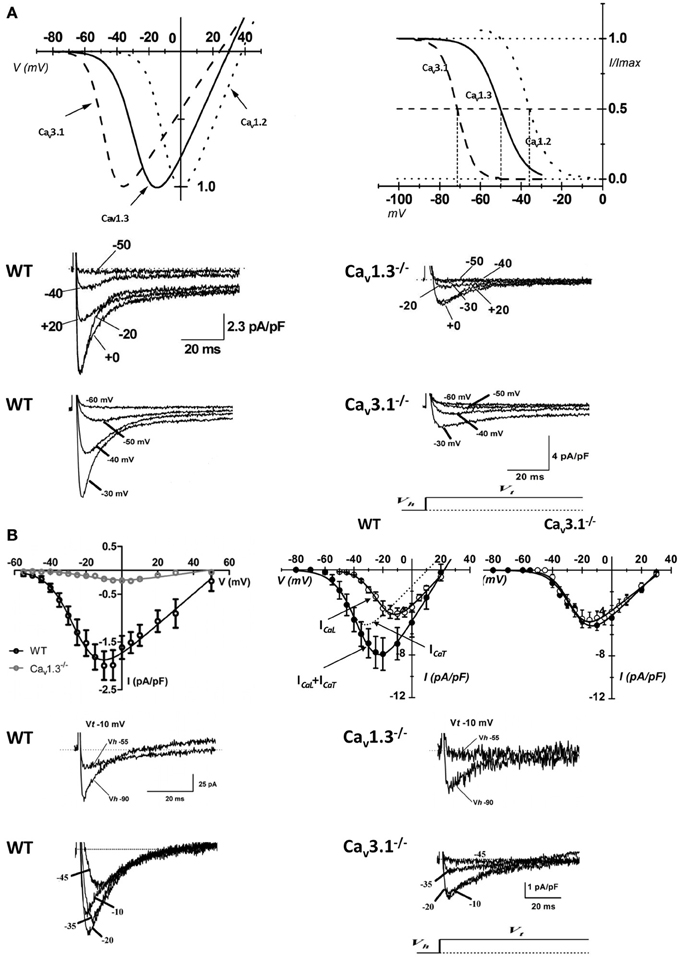
Figure 2. Properties of VGCCs in cardiac pacemaker cells. (A) I–V curve (top left panel) and steady-state inactivation (top right panel) of native SAN Cav3.1 (dashed curve), Cav1.3 (solid curve), and Cav1.2 (dotted curve) channels (reprinted from Mangoni et al., 2006a, with permission from Elsevier). Examples of voltage dependent calcium currents recorded in pacemakers cells from WT (middle and bottom left panel), Cav1.3−/− (middle right panel) and Cav3.1−/− mice (bottom right panel). (B) Top left panel: I–V curve of L-type Ca2+ channels obtained from WT (black open circles) and Cav1.3−/− (gray open circles) isolated AVN cells. Top right panels: current to voltage relationship in isolated AVN cells from WT and Cav3.1−/− mice. Sample traces of ICa,L (middle panels) and ICa,T (bottom panels) recorded in isolated AVN cells from WT, Cav1.3−/− and Cav3.1−/− mice. For ICa,L recordings the holding potential (Vh) was set at −55 mV, for Cav3.1 at −90 mV. Test potential (Vt) is reported near the trace (reprinted from Mangoni et al., 2006b with permission from Wolters Kluwer Health).
Using a knock-in mouse strain in which the DHP sensitivity in Cav1.2 α1 subunits was eliminated (Cav 1.2DHP−/−), without affecting channel function and expression, it has been possible to separate the DHP effects of Cav1.2 from those of Cav1.3 and other L-Type Ca2+ channels.
The heart rate reducing effect induced by DHP (isradipine) in Cav1.2DHP−/− mice demonstrated that Cav1.3 is the major L-Type Ca2+ channel controlling diastolic depolarization (Sinnegger-Brauns et al., 2004).
Recently, Christel et al. (2012) showed a differential degree of co-localization between the ryanodine receptors (RYRs) of the SR and Cav1.3 or Cav1.2 channels in primary SAN pacemaker cells. The strong co-localization of Cav1.3 with RYR2 may be relevant for the functional role of RYR-mediated Ca2+ release in pacemaking (Vinogradova et al., 2002). During the late phase of the diastolic depolarization, RYR-mediated Ca2+ release promotes NCX activation, which accelerates reaching the threshold of the SAN action potential upstroke (Vinogradova et al., 2002). Close apposition of Cav1.3 with RYRs may facilitate SR Ca2+ release since ICa,L stimulates RYR open probability. In this respect, numerical simulations predicted that the slope of rise of diastolic RYR-dependent Ca2+ release increased as a function of Cav1.3-mediated ICa,L half-activation voltage (Christel et al., 2012). The coupling of this SR Ca2+ release to the depolarizing influence of NCX should accelerate attainment of the threshold for action potential firing of SAN cells (Vinogradova et al., 2002). L-type Ca2+ channels, and in particular Cav1.3 channels, have been shown to physically associate with RYR2 in the nervous system (Ouardouz et al., 2003; Kim et al., 2007). It will be interesting to investigate whether such a coupling also exists in SAN cells. As previously mentioned, Cav1.3−/− mice show slowing of atrioventricular conduction suggesting that these channels are important in AVN physiology (Figure 2B). It has been shown that Cav1.3 channels play a key role in pacemaking of AVN cells (Marger et al., 2011a; Zhang et al., 2011). In Cav1.3−/− AVN cells pacemaker activity is stopped and exhibited a depolarized membrane potential of -30 mV (Marger et al., 2011a) likely due the loss of crosstalk between Cav1.3 channels and SK2 K+ channels. Indeed, functional coupling between Cav1.3 and SK channels has been reported in mouse atrial myocytes (Lu et al., 2007), where Cav1.3 loss-of-function prolongs the action potential duration via reduction in the activity of SK channels. Interestingly, Zhang et al. (2008) showed that mice lacking SK2 channels exhibited significant sinus bradycardia with prolongation of the atrioventricular conduction intervals (PQ intervals), thus revealing a function role of these channels in AVN automaticity.
Saturating doses of the non-selective β-adrenergic agonist isoproterenol did not restore pacemaking in Cav1.3−/− AVN cells. Cellular automaticity could be initiated by injection of hyperpolarizing current to drive the membrane voltage to the maximum diastolic potential of −60 mV recorded in wild-type AVN cells. When this maximum diastolic potential voltage is maintained by constant hyperpolarizing current injection, AVN cells were able to fire spontaneous action potentials. However, this firing was very slow and arrhythmic. Furthermore, the slope of the diastolic depolarization in current injected Cav1.3−/− cells was very low. Indeed, during the diastolic phase, only sub-threshold low amplitude oscillations of the membrane potential were recorded. These oscillations occasionally met the threshold to evoke an action potential (Marger et al., 2011a). These data indicated that Cav1.3 channels have a key role in the generation of the diastolic depolarization in AVN pacemaker cells (Marger et al., 2011a). β-adrenergic stimulation induced by isoproterenol was able to increase the firing rate in current-injected Cav1.3−/− AVN cells. However, the firing rate of isoproterenol treated Cav1.3−/− myocytes did not reach the value of control AVN cells (Marger et al., 2011a).
The lack of spontaneous automaticity in Cav1.3−/− AVN cells in vitro does not imply un-excitability in vivo. A potential explication is that Cav1.3−/− cells embedded in tissue are kept at hyperpolarized membrane voltages by the electrotonic influence of the atrium (Verheijck et al., 2002), allowing the discharge of the INa dependent action potential in the presence of SAN impulse (Marger et al., 2011a). Incidentally, Marger et al. (2011b) showed that INa has an important role in the AVN automaticity as 20 μM TTX completely stop firing in AVN cells. Finally, it is well known that AVN is composed by different cell types, automatic and non-automatic, interacting each other and eventually implicated in different conduction pathways. These aspects too can explain the reason why Cav1.3−/− mice do not show complete atrioventricular block.
T-type Calcium Channels
ICa,T has been consistently found in all the three rhytmogenic centers of the heart: the SAN (Hagiwara et al., 1988; Fermini and Nathan, 1991), the AVN (Liu et al., 1993) and Purkinje fibers (Hirano et al., 1989; Tseng and Boyden, 1989) suggesting that T-type VGCCs may constitute a relevant mechanism in the generation of the diastolic depolarization.
Genetically modified mice with target inactivation of Cav3.2 and Cav3.1 subunit importantly helped to elucidate the role of T-type channel isoforms in cardiac pacemaking and impulse conduction (Figure 2A) (Chen et al., 2003; Mangoni et al., 2006b; Thuesen et al., 2014).
In comparison to wild-type littermates mice lacking Cav3.2 T-type channels do not show any significant differences in heart rate or the ECG waveform morphology; furthermore, no cardiac arrhythmias were observed in Cav3.2 deficient mice indicating that Cav3.2 mediated ICa,T do not contribute significantly to the generation and the conduction of the cardiac impulse (Chen et al., 2003). Contrary to what reported for Cav3.2 deficient mice, genetic inactivation of the Cav3.1 T-type Ca2+ channels in mice results in a moderate bradycardia and significant slowing of AV conduction. Moreover, SAN and AVN cells of Cav3.1−/− hearts do not show residual Cav3.2 mediated ICa,T. Niwa et al. (2004) and Ferron et al. (2002) showed that the embryonic myocardium express Cav3.2 channels, while the adult heart shows a higher expression of Cav3.1 channels. These results suggest that Cav3.2 underlies the functional T-type Ca2+ channels in the embryonic murine heart, and there is a subtype switching of transcripts from Cav3.2 to Cav3.1 in the perinatal period. As stated previously, ablation of Cav3.1 subunits causes heart rate reduction (around 10%) and prolongation of the PQ interval due to first-degree atrioventricular block (Mangoni et al., 2006b). Similar results are obtained in sedated Cav3.1−/− mice after autonomic blockade by atropine and propranolol indicating a direct impact of Cav3.1 subunits deletion in the SAN automaticity (Mangoni et al., 2006b) (Figure 3).
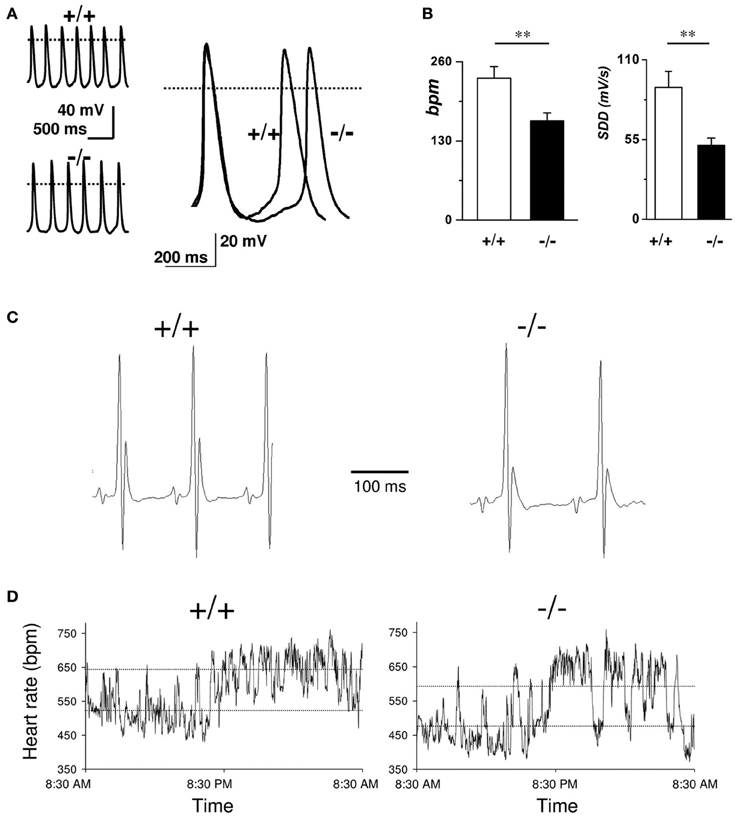
Figure 3. Role of T-type VGCCs in cardiac automaticity. (A) Representative sweeps of spontaneous action potentials obtained from SAN cells from WT (upper left trace) and Cav3.1−/− mice (lower left trace). Right panel: Superimposition of typical action potentials from a WT and from Cav3.1−/− SAN cell. (B) Histograms of the average bpm value and the slope of the diastolic depolarization (SDD). (C) Representative telemetric ECG recordings obtained on WT (left panel) and Cav3.1−/− animals (right panel). (D) Variation of heart rate (in bpm) in WT (left panel) and Cav3.1−/− mice (right panel) over a 24-h period. Dashed lines indicate mean day and night heart rates (reprinted from Mangoni et al., 2006b with permission from Wolters Kluwer Health).
In agreement with this observation in vivo, pacemaker activity in isolated SAN cells is slowed by about 30% (Mangoni et al., 2006b). The relatively lower impact of ICa,T loss of function on pacemaking in vivo compared to isolated cells can be explained supposing a shift of the leading pacemaker site in Cav3.1−/− SAN. This phenomenon is known as “pacemaker shift,” and can be observed when the SAN is challenged with neurotransmitters or pharmacologic agents that regulate pacemaker activity (Boyett et al., 2000; Lang et al., 2011; Inada et al., 2014). In the case of Cav3.1−/− hearts, it can be hypothesized that the leading pacemaker site of intact SAN is shifted to a location that is less sensitive to ICa,T than the leading site in wild-type hearts. In this respect, pacemaker shift can be viewed as a compensatory mechanism to keep SAN rate as high as possible in the absence of Cav3.1 channels. This hypothesis would need direct testing by employing optical or electrical mapping of pacemaking in wild-type and Cav3.1−/− SANs Similar hypothesis concerning pacemaker leading site shift have already been proposed to explain beat-to-beat variability, sinus node dysrhythmia and sinus pauses in mice lacking HCN1 channel (Fenske et al., 2013) or to partially explain the phenotype of human patients affected by “ankyrin B syndrome,” a disease characterized by sinus node dysfunction and increased susceptibility to spontaneous atrial fibrillation cause by Ankyrin-B dysfunction (Wolf et al., 2013). Another possible hypothesis to explain the difference in the pacing rate between isolated SAN cells and the heart rate in vivo in Cav3.1−/− mice would be the functional coupling between cardiac fibroblast and SAN myocytes. Indeed, it has been proposed that cardiac connective tissue facilitates impulse conduction in vivo (Camelliti et al., 2004; Kohl and Gourdie, 2014). Consequently, disruption of the electro-tonic coupling between fibroblast and Cav3.1−/− SAN cells during the cell isolation process could contribute reduce the pacing rate of isolated knockout myocytes. This hypothesis could also hold for other murine models lacking ion channels involved in pacemaker activity.
The prolongation of PQ interval in Cav3.1−/− mice suggested an important role of ICa,T in AVN pacemaker cells. No residual ICa,T was recorded in Cav3.1−/− AVN cells (Figure 2B) and the loss of Cav3.1 mediated ICa,T had remarkable effects on AVN cells automaticity. Pacemaker activity in Cav3.1−/− AVN isolated cells was irregular and slower (40%) than that of control cells (Marger et al., 2011a) suggesting that the relative importance of these channels in AVN automaticity may be even higher than that of SAN.
The importance of T-type channels in automaticity has been also investigated also in the ventricular conduction system. Le Quang et al. (2013) performed a clever study on the role of Cav3.1 T- type Ca2+ channels subunits in escape rhythms and in bradycardia induced ventricular tachyarrhythmia after atrioventricular block. Adult male mice lacking Cav3.1 alpha subunits after induction of complete atrioventricular block showed slower escape rhythms, greater mortality and higher frequency of torsades de pointes than control mice. This study suggests that Cav3.1 channels play an important role in infra-nodal escape automaticity. Loss of Cav3.1 channels also worsens bradycardia-related mortality, increases bradycardia-associated adverse remodeling, and enhances the risk of malignant ventricular tachyarrhythmia following atrioventricular block.
Although data from different studies show clearly the involvement of ICa,T in cardiac automaticity and impulse conduction, mechanistic description of how T channels contribute to the diastolic depolarization is still lacking. Protas et al. (2001) proposed, for rabbit SAN cells, the existence of T-type window current component in the diastolic depolarization, but such a window current was not recorded either in the original study by Hagiwara et al. (1988) or Mangoni et al. (2006b) leaving this aspect still controversial. A previous study by our group employing numerical modeling of pacemaker activity of SAN and AVN mouse cells suggested that about 25 pA/pF of Cav3.1-mediated ICa,T flows during the diastolic depolarization (Hagiwara et al., 1988; Mangoni et al., 2006b; Marger et al., 2011a). Such a current density would be in theory sufficient to functionally contribute to the diastolic depolarization, since previous modeling work indicated that given the very high membrane resistance of SAN pacemaker cells at the maximum diastolic potential, a net inward current density as low as 1 pA/pF could initiate the diastolic depolarization (Difrancesco, 1993; Verheijck et al., 1999). Another hypothesis on how T-type channels can contribute to the pacemaking has emerged from the study by Huser et al. (2000). The paper states that in cat SAN and latent atrial pacemaker cells, the activation of T-type calcium channels during the late phase of the depolarization triggers elementary Ca2+ release events (Ca2+ sparks) from the SR which in turns stimulate NCX current to depolarize the pacemaker potential to threshold. These data were confirmed using 40 μM nickel (blocker of low voltage activated ICa,T). Effectively, nickel reduced Ca2+ sparks and the slope of the diastolic depolarization, suggesting a functional coupling between T-type channels and SR (Lipsius et al., 2001), which could explain previous results showing that prevention of SR Ca2+ release with 10 μM ryanodine reduced T-type Ca2+ current (Li et al., 1997). However, Vinogradova et al. (2002) showed that nickel 50 μM slightly decreased the spontaneous cycle length of rabbit SAN cells and did not decrease the number of SR Ca2+ release suggesting a cell-type dependent role of ICa,T in beating rate, SR Ca2+ release and diastolic depolarization. Therefore, the fact that ICa,T appears to play a more important role in cat atrial latent pacemaker activity (Huser et al., 2000) than in primary pacemaker activity of rabbit SAN cells might be explained on the basis of a more negative maximum diastolic potential in atrial subsidiary vs. SAN cells (Vinogradova et al., 2002).
Diseases of Heart Rhythm and Cardiac VGCCs
During the last years, mutations in ion channels contributing to cardiac automaticity in humans have been described (Dobrzynski et al., 2007; Sanders et al., 2014). These mutations underlie complex arrhythmic profiles in affected patients. Typical clinical profiles include bradycardia due to sinus node dysfunction (Baig et al., 2011), atrioventricular block (Brucato et al., 2000) and ventricular tachycardia (Ueda et al., 2004). In particular, the discovery of two congenital pathologies of heart automaticity and atrioventricular conduction that could be attributed to a down regulation or loss-of-function of Cav1.3 and/or Cav3.1 channels highlights the physiological relevance of VGCCs in the determination of heart rate and atrioventricular conduction in humans. In this context genetically modified mice lacking Cav1.3 or Cav3.1 channels are important tools to test mechanistic hypothesis linking ion channel loss-of-function to bradycardia in affected subjects and for testing potential therapeutic strategies.
Mice lacking Cav1.3-mediated ICa,L are phenotypically characterized by bradycardia and deafness (Platzer et al., 2000; Mangoni et al., 2003). Similar dysfunctions were discovered in two consanguineous families from Pakistan (Baig et al., 2011). Deep hearing loss and SAN dysfunction in these individuals closely are reminiscent of the phenotype of Cav1.3−/− mice. Because of the association between deafness and bradycardia, this newly described disease was named Sino-atrial Node Dysfunction and Deafness syndrome (SANDD). Patients with SANDD present SAN bradycardia at rest and exhibit variable degree of atrioventricular block and dissociated rhythms. This last observation can be explained by a recent result showing that Cav1.3 is important for automaticity of mice AVN cells (Marger et al., 2011a) (Figure 4A). No short or long QT syndrome (LQTS) was reported in SANDD patients, indicating that Cav1.3 channels do not directly participate to ventricular repolarization in humans. On the other hand recent data indicate that mutations in genes affecting regulation of Cav1.2 channels can affect action potential duration. Particularly, mutations in calmodulin have been shown to be associated with cathecolaminergic polymorphic ventricular tachycardia (CPVT) and cardiac arrest (Nyegaard et al., 2012; Crotti et al., 2013). Limpitikul et al. (2014) showed that expression of mutated calmodulin suppressed Ca2+/calmodulin mediated CDI in native Cav1.2 channels of ventricular myocytes. Suppression of CDI increased action potential durations and augmented the SR Ca2+ content. These works indicate that alteration in Cav1.2 channels can induce LQTS (Limpitikul et al., 2014).
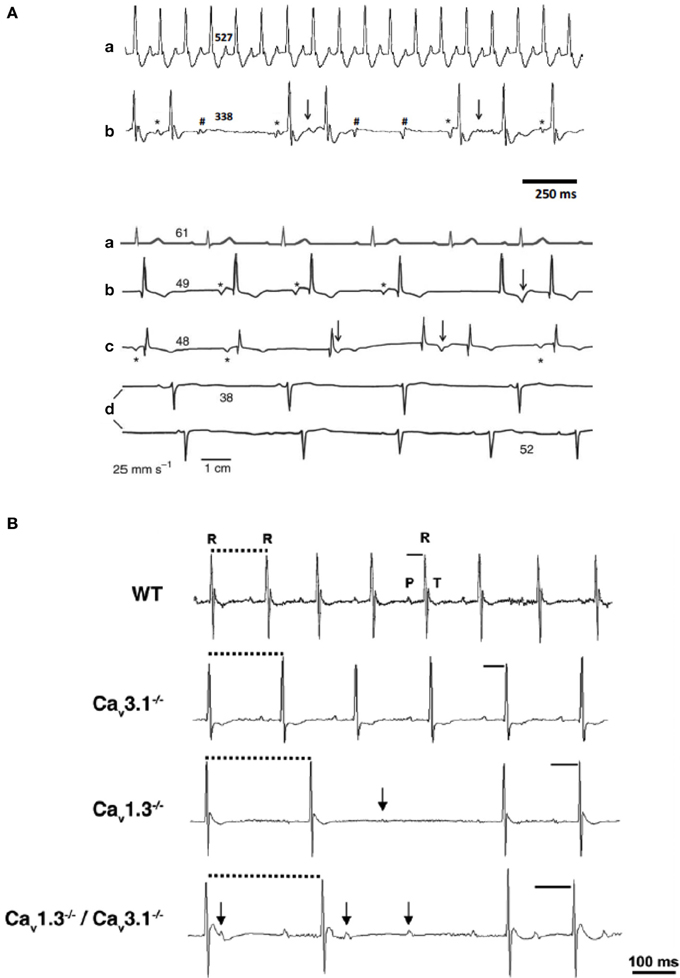
Figure 4. Cardiac VGCCs in cardiac automaticity pathology. (A) ECG sample recordings from WT (a) and Cav1.3−/− mice (b). (B) ECG recordings from a healthy person (a) and three individuals with SANDD syndrome (b–d). Asterisks mark P waves that precede QRS complexes; arrows indicate waveforms that suggest P waves coinciding with T waves; hashes indicate not conducted P waves. Numbers indicate heart rate (bpm) calculated from the corresponding beat-to-beat R-R interval (adapted from Baig et al., 2011). (B) Telemetric surface ECGs of freely moving WT, Cav3.1−/−, Cav1.3−/−, and Cav1.3−/−/Cav3.1−/− mice showed additive effect of Cav gene inactivation on atrioventricular conduction dysfunction. Solid bars indicate PQ interval, dotted bars indicate RR intervals and arrows indicate isolated P waves (reprinted from Marger et al., 2011a with permission from Taylor and Francis LLC http://www.tandfonline.com).
Recently, Neco et al. (2012), using a mouse model of CPVT carrying a mutation in RYR2 (RyR2R4496C), demonstrated a strong implication of SAN L-type channels in bradycardia associated with CPVT syndrome. RyR2R4496C mice manifested alteration in Ca2+ homeostasis together with SAN dysrhythmia (SAN pauses) and impaired positive chronotropic response to β-adrenergic stimulation. Isolated RyR2R4496C SAN cells showed Ca2+-dependent decrease of ICa,L density, together with depletion of SR Ca2+ load during the diastolic phase, two factors that impaired the generation of SAN action potential. Ca2+ dependent inactivation by excessive RYR dependent Ca2+ release provides a new mechanistic rationale of SAN dysfunction in CPVT disease. It has been shown that not only inherited, but also acquired cardiomyopathy can involve L-type Cav1.3. Rose et al. (2011) described a strong cardiac phenotype in a mouse model of chronic iron overload (CIO). SAN cells from CIO mice showed a strong decrease in Cav1.3-mediated ICa,L density. This decrease in ICa,L induced bradycardia, sinus pauses, prolonged PQ intervals and second degree heart block in vivo.
Congenital heart block (CHB) disease is another pathology in which cardiac VGCCs are strongly implicated. CHB disease affects fetuses and newborns. CHB is an acquired autoimmune disease that occurs during pregnancies of rheumatic mothers, but also in healthy mothers. CHB is usually diagnosed between weeks 18 and 24 of pregnancy by fetal echocardiography techniques. Although it may initially appear as a first- or second-degree atrioventricular block, most cases present with fetal bradycardia and complete third-degree atrioventricular block. Other arrhythmias, including sinus bradycardia, diverse atrial rhythms, and junctional ectopic and ventricular tachycardia, have also been reported in the context of CHB (Ambrosi et al., 2014). While the etiology of this disease has remained obscure for long time, there is now strong evidence that loss-of-function of Cav1.3 and Cav3.1 channels underlie this pathology (Strandberg et al., 2013). Hu et al. (2004) have reported inhibition of ICa,L and ICa,T by immunoglobulin G isolated from mothers having CHB-affected children. SAN bradycardia and CHB can be explained at least in part by down regulation of Cav1.3 and Cav3.1 channels by maternal antibodies (Hu et al., 2004) suggesting a strict correlation between loss of function of Cav1.3 and Cav3.1 VGCCs and CHB. Results published by our group (Marger et al., 2011a) further support this hypothesis. Indeed, we studied heart rate and atrioventricular conduction in mice with combined inactivation of Cav1.3 and Cav3.1 channels (Cav1.3−/−/Cav3.1−/−) showing that Cav3.1 and Cav1.3 inactivation have an additive effect on atrioventricular conduction (Figure 4B). Indeed, while inactivation of Cav3.1 channels alone causes moderate dysfunction of atrioventricular conduction, association with Cav1.3 inactivation induces severe atrioventricular block. Some Cav1.3−/−/Cav3.1−/− mice display complete block with dissociated atrial and ventricular. Disruption of both Cav1.3 and Cav3.1 subunits has also additive effects on AVN cells pacemaking. Cav1.3−/−/Cav3.1−/− AVN cells display poor or absent automaticity, thus stressing the importance of voltage-dependent Ca2+ channels in pacemaker activity of these cells. These results indicate that Cav1.3−/−/Cav3.1−/− mice constitute a faithful animal model of CHB and could be used for testing of new therapies (Marger et al., 2011a).
In conclusion, work on mouse models of SANDD and CIO (Cav1.3 channels), CPVT (ICa,L), and CHB (ICa,L and ICa,T) demonstrates that despite the differences between mouse and human cardiac rhythm the mouse is a valuable model for studying the role of ion channels in human pathologies of heart rhythm.
Concluding Remarks
The relevance of VGCCs in the generation and regulation of cardiac pacemaking, atrioventricular conduction and heart rate determination is now well established. Importantly, the functional role of VGCCs such as Cav1.3 and Cav3.1 channels seems to be conserved between rodents and humans. ICa,L and ICa,T play a major role in atrioventricular conduction as underscored by the presence of dissociated rhythms in Cav1.3−/− mice and SANDD patients (Baig et al., 2011) or in Cav1.3−/−/Cav3.1−/− mice and CHB patients (Marger et al., 2011a). Future studies will further address the role of VGCCs in pacemaker activity and in particular their importance in respect to other ion channels involved in automaticity such as HCN4 and RYRs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ambrosi, A., Sonesson, S. E., and Wahren-Herlenius, M. (2014). Molecular mechanisms of congenital heart block. Exp. Cell Res. 325, 2–9. doi: 10.1016/j.yexcr.2014.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baig, S. M., Koschak, A., Lieb, A., Gebhart, M., Dafinger, C., Nurnberg, G., et al. (2011). Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14, 77–84. doi: 10.1038/nn.2694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bladen, C., Gadotti, V. M., Gunduz, M. G., Berger, N. D., Simsek, R., Safak, C., et al. (2014a). 1,4-Dihydropyridine derivatives with T-type calcium channel blocking activity attenuate inflammatory and neuropathic pain. Pflugers Arch. doi: 10.1007/s00424-014-1566-3. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bladen, C., Gunduz, M. G., Simsek, R., Safak, C., and Zamponi, G. W. (2014b). Synthesis and evaluation of 1,4-dihydropyridine derivatives with calcium channel blocking activity. Pflugers Arch. 466, 1355–1363. doi: 10.1007/s00424-013-1376-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bogdanov, K. Y., Vinogradova, T. M., and Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ. Res. 88, 1254–1258. doi: 10.1161/hh1201.092095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohn, G., Moosmang, S., Conrad, H., Ludwig, A., Hofmann, F., and Klugbauer, N. (2000). Expression of T- and L-type calcium channel mRNA in murine sinoatrial node. FEBS Lett. 481, 73–76. doi: 10.1016/S0014-5793(00)01979-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyett, M. R., Honjo, H., and Kodama, I. (2000). The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 47, 658–687. doi: 10.1016/S0008-6363(00)00135-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, H., Difrancesco, D., and Noble, S. (1979). Cardiac pacemaker oscillation and its modulation by autonomic transmitters. J. Exp. Biol. 81, 175–204.
Brucato, A., Cimaz, R., Catelli, L., and Meroni, P. (2000). Anti-Ro-associated sinus bradycardia in newborns. Circulation 102, E88–E89. doi: 10.1161/01.CIR.102.11.e88
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buraei, Z., and Yang, J. (2010). The ss subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506. doi: 10.1152/physrev.00057.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buraei, Z., and Yang, J. (2013). Structure and function of the beta subunit of voltage-gated Ca(2) (+) channels. Biochim. Biophys. Acta 1828, 1530–1540. doi: 10.1016/j.bbamem.2012.08.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Camelliti, P., Green, C. R., Legrice, I., and Kohl, P. (2004). Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 94, 828–835. doi: 10.1161/01.RES.0000122382.19400.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carbone, E., and Lux, H. D. (1987). Kinetics and selectivity of a low-voltage-activated calcium current in chick and rat sensory neurons. J. Physiol. 386, 547–570. doi: 10.1113/jphysiol.1987.sp016551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555. doi: 10.1146/annurev.cellbio.16.1.521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catterall, W. A., Striessnig, J., Snutch, T. P., and Perez-Reyes, E. (2003). International union of pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol. Rev. 55, 579–581. doi: 10.1124/pr.55.4.8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cens, T., Mangoni, M. E., Richard, S., Nargeot, J., and Charnet, P. (1996). Coexpression of the beta2 subunit does not induce voltage-dependent facilitation of the class C L-type Ca channel. Pflugers Arch. 431, 771–774.
Chemin, J., Traboulsie, A., and Lory, P. (2006). Molecular pathways underlying the modulation of T-type calcium channels by neurotransmitters and hormones. Cell Calcium 40, 121–134. doi: 10.1016/j.ceca.2006.04.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, C. C., Lamping, K. G., Nuno, D. W., Barresi, R., Prouty, S. J., Lavoie, J. L., et al. (2003). Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science 302, 1416–1418. doi: 10.1126/science.1089268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christel, C., and Lee, A. (2012). Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 1820, 1243–1252. doi: 10.1016/j.bbagen.2011.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christel, C. J., Cardona, N., Mesirca, P., Herrmann, S., Hofmann, F., Striessnig, J., et al. (2012). Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J. Physiol. 590, 6327–6342. doi: 10.1113/jphysiol.2012.239954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crotti, L., Johnson, C. N., Graf, E., De Ferrari, G. M., Cuneo, B. F., Ovadia, M., et al. (2013). Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Jongh, K. S., Murphy, B. J., Colvin, A. A., Hell, J. W., Takahashi, M., and Catterall, W. A. (1996). Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry 35, 10392–10402. doi: 10.1021/bi953023c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Difrancesco, D. (1991). The contribution of the ‘pacemaker’ current (if) to generation of spontaneous activity in rabbit sino-atrial node myocytes. J. Physiol. 434, 23–40. doi: 10.1113/jphysiol.1991.sp018457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Difrancesco, D. (1993). Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472. doi: 10.1146/annurev.ph.55.030193.002323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Difrancesco, D. (2010). The role of the funny current in pacemaker activity. Circ. Res. 106, 434–446. doi: 10.1161/CIRCRESAHA.109.208041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Difrancesco, D., and Tortora, P. (1991). Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351, 145–147. doi: 10.1038/351145a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobrzynski, H., Anderson, R. H., Atkinson, A., Borbas, Z., D'Souza, A., Fraser, J. F., et al. (2013). Structure, function and clinical relevance of the cardiac conduction system, including the atrioventricular ring and outflow tract tissues. Pharmacol. Ther. 139, 260–288. doi: 10.1016/j.pharmthera.2013.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobrzynski, H., Boyett, M. R., and Anderson, R. H. (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932. doi: 10.1161/CIRCULATIONAHA.106.616011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobrzynski, H., Li, J., Tellez, J., Greener, I. D., Nikolski, V. P., Wright, S. E., et al. (2005). Computer three-dimensional reconstruction of the sinoatrial node. Circulation 111, 846–854. doi: 10.1161/01.CIR.0000152100.04087.DB
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doerr, T., Denger, R., and Trautwein, W. (1989). Calcium currents in single SA nodal cells of the rabbit heart studied with action potential clamp. Pflugers Arch. 413, 599–603. doi: 10.1007/BF00581808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fenske, S., Krause, S. C., Hassan, S. I., Becirovic, E., Auer, F., Bernard, R., et al. (2013). Sick sinus syndrome in HCN1-deficient mice. Circulation 128, 2585–2594. doi: 10.1161/CIRCULATIONAHA.113.003712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fermini, B., and Nathan, R. D. (1991). Removal of sialic acid alters both T- and L-type calcium currents in cardiac myocytes. Am. J. Physiol. 260, H735–H743.
Ferron, L., Capuano, V., Deroubaix, E., Coulombe, A., and Renaud, J. F. (2002). Functional and molecular characterization of a T-type Ca(2+) channel during fetal and postnatal rat heart development. J. Mol. Cell. Cardiol. 34, 533–546. doi: 10.1006/jmcc.2002.1535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagiwara, N., Irisawa, H., and Kameyama, M. (1988). Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 395, 233–253. doi: 10.1113/jphysiol.1988.sp016916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirano, Y., Fozzard, H. A., and January, C. T. (1989). Characteristics of L- and T-type Ca2+ currents in canine cardiac Purkinje cells. Am. J. Physiol. 256, H1478–H1492.
Hu, K., Qu, Y., Yue, Y., and Boutjdir, M. (2004). Functional basis of sinus bradycardia in congenital heart block. Circ. Res. 94, e32–38. doi: 10.1161/01.RES.0000121566.01778.06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huser, J., Blatter, L. A., and Lipsius, S. L. (2000). Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. 524(Pt 2), 415–422. doi: 10.1111/j.1469-7793.2000.00415.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inada, S., Zhang, H., Tellez, J. O., Shibata, N., Nakazawa, K., Kamiya, K., et al. (2014). Importance of gradients in membrane properties and electrical coupling in sinoatrial node pacing. PLoS ONE 9:e94565. doi: 10.1371/journal.pone.0094565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
James, T. N. (2003). Structure and function of the sinus node, AV node and his bundle of the human heart: part II–function. Prog. Cardiovasc. Dis. 45, 327–360. doi: 10.1053/pcad.2003.1
Kim, S., Yun, H. M., Baik, J. H., Chung, K. C., Nah, S. Y., and Rhim, H. (2007). Functional interaction of neuronal Cav1.3 L-type calcium channel with ryanodine receptor type 2 in the rat hippocampus. J. Biol. Chem. 282, 32877–32889. doi: 10.1074/jbc.M701418200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodama, I., Nikmaram, M. R., Boyett, M. R., Suzuki, R., Honjo, H., and Owen, J. M. (1997). Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am. J. Physiol. 272, H2793–H2806.
Kohl, P., and Gourdie, R. G. (2014). Fibroblast-myocyte electrotonic coupling: does it occur in native cardiac tissue? J. Mol. Cell. Cardiol. 70, 37–46. doi: 10.1016/j.yjmcc.2013.12.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koschak, A., Reimer, D., Huber, I., Grabner, M., Glossmann, H., Engel, J., et al. (2001). alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 276, 22100–22106. doi: 10.1074/jbc.M101469200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kovoor, P., Wickman, K., Maguire, C. T., Pu, W., Gehrmann, J., Berul, C. I., et al. (2001). Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143. doi: 10.1016/S0735-1097(01)01304-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lakatta, E. G., Maltsev, V. A., and Vinogradova, T. M. (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ. Res. 106, 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lande, G., Demolombe, S., Bammert, A., Moorman, A., Charpentier, F., and Escande, D. (2001). Transgenic mice overexpressing human KvLQT1 dominant-negative isoform. Part II: pharmacological profile. Cardiovasc. Res. 50, 328–334. doi: 10.1016/S0008-6363(01)00232-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lang, D., Petrov, V., Lou, Q., Osipov, G., and Efimov, I. R. (2011). Spatiotemporal control of heart rate in a rabbit heart. J. Electrocardiol. 44, 626–634. doi: 10.1016/j.jelectrocard.2011.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lehmann, H., and Klein, U. E. (1978). Familial sinus node dysfunction with autosomal dominant inheritance. Br. Heart J. 40, 1314–1316. doi: 10.1136/hrt.40.11.1314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Le Quang, K., Benito, B., Naud, P., Qi, X. Y., Shi, Y. F., Tardif, J. C., et al. (2013). T-type calcium current contributes to escape automaticity and governs the occurrence of lethal arrhythmias after atrioventricular block in mice. Circ. Arrhythm. Electrophysiol. 6, 799–808. doi: 10.1161/CIRCEP.113.000407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, J., Qu, J., and Nathan, R. D. (1997). Ionic basis of ryanodine's negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am. J. Physiol. 273, H2481–H2489.
Li, Y., Wang, F., Zhang, X., Qi, Z., Tang, M., Szeto, C., et al. (2012). beta-Adrenergic stimulation increases Cav3.1 activity in cardiac myocytes through protein kinase A. PLoS ONE 7:e39965. doi: 10.1371/journal.pone.0039965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Limpitikul, W. B., Dick, I. E., Joshi-Mukherjee, R., Overgaard, M. T., George, A. L. Jr., and Yue, D. T. (2014). Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell. Cardiol. 74, 115–124. doi: 10.1016/j.yjmcc.2014.04.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipsius, S. L., Huser, J., and Blatter, L. A. (2001). Intracellular Ca2+ release sparks atrial pacemaker activity. News Physiol. Sci. 16, 101–106.
Liu, Y., Zeng, W., Delmar, M., and Jalife, J. (1993). Ionic mechanisms of electronic inhibition and concealed conduction in rabbit atrioventricular nodal myocytes. Circulation 88, 1634–1646. doi: 10.1161/01.CIR.88.4.1634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, L., Zhang, Q., Timofeyev, V., Zhang, Z., Young, J. N., Shin, H. S., et al. (2007). Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ. Res. 100, 112–120. doi: 10.1161/01.RES.0000253095.44186.72
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mackintosh, A. F., and Chamberlain, D. A. (1979). Sinus node disease affecting both parents and both children. Eur. J. Cardiol. 10, 117–122.
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., Reimer, D., Striessnig, J., et al. (2003). Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. U.S.A. 100, 5543–5548. doi: 10.1073/pnas.0935295100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Couette, B., Marger, L., Bourinet, E., Striessnig, J., and Nargeot, J. (2006a). Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog. Biophys. Mol. Biol. 90, 38–63. doi: 10.1016/j.pbiomolbio.2005.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Fontanaud, P., Noble, P. J., Noble, D., Benkemoun, H., Nargeot, J., et al. (2000). Facilitation of the L-type calcium current in rabbit sino-atrial cells: effect on cardiac automaticity. Cardiovasc. Res. 48, 375–392. doi: 10.1016/S0008-6363(00)00182-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., and Nargeot, J. (2008). Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982. doi: 10.1152/physrev.00018.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Traboulsie, A., Leoni, A. L., Couette, B., Marger, L., Le Quang, K., et al. (2006b). Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 98, 1422–1430. doi: 10.1161/01.RES.0000225862.14314.49
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marger, L., Mesirca, P., Alig, J., Torrente, A., Dubel, S., Engeland, B., et al. (2011a). Functional roles of Ca(v)1.3, Ca(v)3.1 and HCN channels in automaticity of mouse atrioventricular cells: insights into the atrioventricular pacemaker mechanism. Channels (Austin) 5, 251–261. doi: 10.4161/chan.5.3.15266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marger, L., Mesirca, P., Alig, J., Torrente, A., Dubel, S., Engeland, B., et al. (2011b). Pacemaker activity and ionic currents in mouse atrioventricular node cells. Channels (Austin) 5, 241–250. doi: 10.4161/chan.5.3.15264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marionneau, C., Couette, B., Liu, J., Li, H., Mangoni, M. E., Nargeot, J., et al. (2005). Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 562, 223–234. doi: 10.1113/jphysiol.2004.074047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marshall, P. W., Rouse, W., Briggs, I., Hargreaves, R. B., Mills, S. D., and McLoughlin, B. J. (1993). ICI D7288, a novel sinoatrial node modulator. J. Cardiovasc. Pharmacol. 21, 902–906. doi: 10.1097/00005344-199306000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matthes, J., Yildirim, L., Wietzorrek, G., Reimer, D., Striessnig, J., and Herzig, S. (2004). Disturbed atrio-ventricular conduction and normal contractile function in isolated hearts from Cav1.3-knockout mice. Naunyn Schmiedebergs Arch. Pharmacol. 369, 554–562. doi: 10.1007/s00210-004-0940-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McRory, J. E., Hamid, J., Doering, C. J., Garcia, E., Parker, R., Hamming, K., et al. (2004). The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J. Neurosci. 24, 1707–1718. doi: 10.1523/JNEUROSCI.4846-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monfredi, O., Maltseva, L. A., Spurgeon, H. A., Boyett, M. R., Lakatta, E. G., and Maltsev, V. A. (2013). Beat-to-beat variation in periodicity of local calcium releases contributes to intrinsic variations of spontaneous cycle length in isolated single sinoatrial node cells. PLoS ONE 8:e67247. doi: 10.1371/journal.pone.0067247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neco, P., Torrente, A. G., Mesirca, P., Zorio, E., Liu, N., Priori, S. G., et al. (2012). Paradoxical effect of increased diastolic Ca2+ release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 126, 392–401. doi: 10.1161/CIRCULATIONAHA.111.075382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niwa, N., Yasui, K., Opthof, T., Takemura, H., Shimizu, A., Horiba, M., et al. (2004). Cav3.2 subunit underlies the functional T-type Ca2+ channel in murine hearts during the embryonic period. Am. J. Physiol. Heart Circ. Physiol. 286, H2257–H2263. doi: 10.1152/ajpheart.01043.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nyegaard, M., Overgaard, M. T., Sondergaard, M. T., Vranas, M., Behr, E. R., Hildebrandt, L. L., et al. (2012). Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 91, 703–712. doi: 10.1016/j.ajhg.2012.08.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ouardouz, M., Nikolaeva, M. A., Coderre, E., Zamponi, G. W., McRory, J. E., Trapp, B. D., et al. (2003). Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron 40, 53–63. doi: 10.1016/j.neuron.2003.08.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perez-Reyes, E. (2003). Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 83, 117–161. doi: 10.1152/physrev.00018.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeufer, A., van Noord, C., Marciante, K. D., Arking, D. E., Larson, M. G., Smith, A. V., et al. (2010). Genome-wide association study of PR interval. Nat. Genet. 42, 153–159. doi: 10.1038/ng.517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Platzer, J., Engel, J., Schrott-Fischer, A., Stephan, K., Bova, S., Chen, H., et al. (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102, 89–97. doi: 10.1016/S0092-8674(00)00013-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Protas, L., Difrancesco, D., and Robinson, R. B. (2001). L-type but not T-type calcium current changes during postnatal development in rabbit sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 281, H1252–H1259.
Ramadan, O., Qu, Y., Wadgaonkar, R., Baroudi, G., Karnabi, E., Chahine, M., et al. (2009). Phosphorylation of the consensus sites of protein kinase A on alpha1D L-type calcium channel. J. Biol. Chem. 284, 5042–5049. doi: 10.1074/jbc.M809132200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rose, R. A., Sellan, M., Simpson, J. A., Izaddoustdar, F., Cifelli, C., Panama, B. K., et al. (2011). Iron overload decreases CaV1.3-dependent L-type Ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ. Arrhythm. Electrophysiol. 4, 733–742. doi: 10.1161/CIRCEP.110.960401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanders, P., Lau, D. H., and Kalman, J. K. (2014). “Sinus node abnormalities,” in Cardiac Electrophysiology: from Cell to Bedside, 6th Edn., eds D. P. Zipes and J. Jalife (Philadelphia, PA: Elsevier Saunders), 691–696.
Sarachek, N. S., and Leonard, J. L. (1972). Familial heart block and sinus bradycardia. Classification and natural history. Am. J. Cardiol. 29, 451–458. doi: 10.1016/0002-9149(72)90432-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sinnegger-Brauns, M. J., Hetzenauer, A., Huber, I. G., Renstrom, E., Wietzorrek, G., Berjukov, S., et al. (2004). Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J. Clin. Invest. 113, 1430–1439. doi: 10.1172/JCI200420208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strandberg, L. S., Cui, X., Rath, A., Liu, J., Silverman, E. D., Liu, X., et al. (2013). Congenital heart block maternal sera autoantibodies target an extracellular epitope on the alpha1G T-type calcium channel in human fetal hearts. PLoS ONE 8:e72668. doi: 10.1371/journal.pone.0072668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Striessnig, J. (1999). Pharmacology, structure and function of cardiac L-type Ca(2+) channels. Cell. Physiol. Biochem. 9, 242–269. doi: 10.1159/000016320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Striessnig, J., and Koschak, A. (2008). Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2, 233–251. doi: 10.4161/chan.2.4.5847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suh, B. C., and Hille, B. (2005). Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 15, 370–378. doi: 10.1016/j.conb.2005.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanabe, T., Beam, K. G., Powell, J. A., and Numa, S. (1988). Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature 336, 134–139. doi: 10.1038/336134a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thuesen, A. D., Andersen, H., Cardel, M., Toft, A., Walter, S., Marcussen, N., et al. (2014). Differential effect of T-type voltage-gated Ca2+ channel disruption on renal plasma flow and glomerular filtration rate in vivo. Am. J. Physiol. Renal Physiol. 307, F445–F452. doi: 10.1152/ajprenal.00016.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tseng, G. N., and Boyden, P. A. (1989). Multiple types of Ca2+ currents in single canine Purkinje cells. Circ. Res. 65, 1735–1750. doi: 10.1161/01.RES.65.6.1735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tuluc, P., Molenda, N., Schlick, B., Obermair, G. J., Flucher, B. E., and Jurkat-Rott, K. (2009). A CaV1.1 Ca2+ channel splice variant with high conductance and voltage-sensitivity alters EC coupling in developing skeletal muscle. Biophys. J. 96, 35–44. doi: 10.1016/j.bpj.2008.09.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ueda, K., Nakamura, K., Hayashi, T., Inagaki, N., Takahashi, M., Arimura, T., et al. (2004). Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 279, 27194–27198. doi: 10.1074/jbc.M311953200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van der Heyden, M. A., Wijnhoven, T. J., and Opthof, T. (2005). Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc. Res. 65, 28–39. doi: 10.1016/j.cardiores.2004.09.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verheijck, E. E., van Ginneken, A. C., Wilders, R., and Bouman, L. N. (1999). Contribution of L-type Ca2+ current to electrical activity in sinoatrial nodal myocytes of rabbits. Am. J. Physiol. 276, H1064–H1077.
Verheijck, E. E., Wilders, R., and Bouman, L. N. (2002). Atrio-sinus interaction demonstrated by blockade of the rapid delayed rectifier current. Circulation 105, 880–885. doi: 10.1161/hc0702.104128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Bogdanov, K. Y., and Lakatta, E. G. (2002). beta-Adrenergic stimulation modulates ryanodine receptor Ca(2+) release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res. 90, 73–79. doi: 10.1161/hh0102.102271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolf, R. M., Glynn, P., Hashemi, S., Zarei, K., Mitchell, C. C., Anderson, M. E., et al. (2013). Atrial fibrillation and sinus node dysfunction in human ankyrin-B syndrome: a computational analysis. Am. J. Physiol. Heart Circ. Physiol. 304, H1253–H1266. doi: 10.1152/ajpheart.00734.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Q., Timofeyev, V., Lu, L., Li, N., Singapuri, A., Long, M. K., et al. (2008). Functional roles of a Ca2+-activated K+ channel in atrioventricular nodes. Circ. Res. 102, 465–471. doi: 10.1161/CIRCRESAHA.107.161778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Q., Timofeyev, V., Qiu, H., Lu, L., Li, N., Singapuri, A., et al. (2011). Expression and roles of Cav1.3 (alpha1D) L-type Ca(2)+ channel in atrioventricular node automaticity. J. Mol. Cell. Cardiol. 50, 194–202. doi: 10.1016/j.yjmcc.2010.10.002
Zhang, Z., He, Y., Tuteja, D., Xu, D., Timofeyev, V., Zhang, Q., et al. (2005). Functional roles of Cav1.3(alpha1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation 112, 1936–1944. doi: 10.1161/CIRCULATIONAHA.105.540070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Z., Xu, Y., Song, H., Rodriguez, J., Tuteja, D., Namkung, Y., et al. (2002). Functional Roles of Ca(v)1.3 (alpha(1D)) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ. Res. 90, 981–987. doi: 10.1161/01.RES.0000018003.14304.E2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: heart automaticity, L-type Ca2+ channel, T-type Ca2+ channels, sinoatrial node, atrioventricular node
Citation: Mesirca P, Torrente AG and Mangoni ME (2015) Functional role of voltage gated Ca2+ channels in heart automaticity. Front. Physiol. 6:19. doi: 10.3389/fphys.2015.00019
Received: 21 November 2014; Accepted: 12 January 2015;
Published online: 02 February 2015.
Edited by:
Ming Lei, University of Oxford, UKReviewed by:
Arie O. Verkerk, University of Amsterdam, NetherlandsYue-kun Ju, University of Sydney, Australia
Copyright © 2015 Mesirca, Torrente and Mangoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pietro Mesirca, Laboratory of Excellence in Ion Channel Science and Therapeutics, Département de Physiologie, Institut de Génomique Fonctionnelle;
UMR-5203, Centre National de la Recherche Scientifique, Universités de Montpellier 1 and 2;
INSERM U 1191, Département de Physiologie, Universités de Montpellier 1 and 2, 141 Rue de la Cardonille, 34094 Montpellier, France e-mail: pietro.mesirca@igf.cnrs.fr