 James N. Cobley1*
James N. Cobley1* Nikos V. Margaritelis2
Nikos V. Margaritelis2 James P. Morton3
James P. Morton3 Graeme L. Close3
Graeme L. Close3 Michalis G. Nikolaidis2
Michalis G. Nikolaidis2 John K. Malone1
John K. Malone1- 1Division of Sport and Exercise Sciences, Abertay University, Dundee, UK
- 2Exercise Physiology and Biochemistry Laboratory, School of Physical Education and Sport Sciences at Serres, Aristotle University of Thessaloniki, Serres, Greece
- 3Muscle Metabolism Research Group, Research Institute for Sport and Exercise Science, Liverpool John Moores University, Liverpool, UK
Acute exercise increases reactive oxygen and nitrogen species generation. This phenomenon is associated with two major outcomes: (1) redox signaling and (2) macromolecule damage. Mechanistic knowledge of how exercise-induced redox signaling and macromolecule damage are interlinked is limited. This review focuses on the interplay between exercise-induced redox signaling and DNA damage, using hydroxyl radical (·OH) and hydrogen peroxide (H2O2) as exemplars. It is postulated that the biological fate of H2O2 links the two processes and thus represents a bifurcation point between redox signaling and damage. Indeed, H2O2 can participate in two electron signaling reactions but its diffusion and chemical properties permit DNA oxidation following reaction with transition metals and ·OH generation. It is also considered that the sensing of DNA oxidation by repair proteins constitutes a non-canonical redox signaling mechanism. Further layers of interaction are provided by the redox regulation of DNA repair proteins and their capacity to modulate intracellular H2O2 levels. Overall, exercise-induced redox signaling and DNA damage may be interlinked to a greater extent than was previously thought but this requires further investigation.
Introduction
Acute exercise disrupts homeostasis, imposing a transient stress that inducts beneficial cyto-protective responses and adaptations with repeated bouts (Cobley et al., 2012; Egan and Zierath, 2013; Hawley et al., 2014). One key homeostatic perturbation is the exercise-induced increase in reactive oxygen (ROS) and nitrogen (RNS) species generation (Cobley et al., 2014). The exercise-induced increase in ROS/RNS generation is bi-functional causing cellular damage and inducting redox signaling (Powers and Jackson, 2008; Cobley et al., 2015a,b). This bi-functionality explains how acute exercise can cause nuclear and mitochondrial DNA oxidation, a mutagenic and damaging event, but exercise training up-regulates DNA repair providing protection against exercise-induced genomic stress (Radak et al., 2011a; Cobley et al., 2013). This synergy between exercise-induced DNA damage and redox signaling whilst conceptually obvious is mechanistically ill-understood. This is compounded by the rare delineation of the species responsible for each outcome in the exercise literature. The purpose of this review and indeed its principal novel feature is to use two exemplar ROS, hydroxyl radical (·OH) and hydrogen peroxide (H2O2), to illustrate the basic chemistry of exercise-induced DNA damage and redox signaling before considering mechanisms that link the two processes together.
Exercise-induced DNA Damage: The Key Role of Hydroxyl Radical
From a chemical perspective, superoxide (O2.−), and nitric oxide (NO), two parent radicals formed during exercise (Sakellariou et al., 2014), do not directly damage DNA (Dizdaroglu and Jaruga, 2012). Analogously, H2O2 does not directly damage DNA (Halliwell and Gutteridge, 2007). Instead, DNA damage is mediated by ·OH and other species capable of modifying DNA including inter alia: peroxynitrite, carbonate radical, and nitrogen trioxide (Cadet et al., 2012). In particular, ·OH rapidly reacts with DNA bases and the ribose sugar at diffusion-controlled rates (e.g., guanine: k ~ 5–8 × 109 M−1 s−1, Chatgilialoglu et al., 2011). The chemistry of ·OH mediated DNA damage is complex but the salient points are: (1) ·OH reacts with DNA indiscriminately via addition (k ~ 4–9 × 109 M−1 s−1) or hydrogen abstraction reactions (k ~ 2 × 109 M−1 s−1; Von Sonntag, 2006) and (2) the resultant radical products can then react with other radicals (e.g., O2.− and NO) or O2 to generate a modified DNA adduct (Dizdaroglu, 2012; Dizdaroglu and Jaruga, 2012). It follows that the chemical identity of the product (s) formed varies according to (1) the type of base modified (2) the nature of the initial reaction (addition or hydrogen abstraction) and (3) levels of secondary reactants (e.g., O2.−). Accordingly, ·OH-DNA reactions yield a multitude of end-products. The oxidation of guanine alone can generate ≥20 end-products, with 8-oxo-7,8-dihydroguanine (8-oxoG) being a frequently assayed end-product owing to its mutagenicity (Radak et al., 2011b).
·OH can be generated by the reaction of H2O2 with transition metals (see below).
Reaction 1: H2O2 + Fe2+ → Fe3+ + −OH + ·OH (k ~ 76 M−1 s−1; Halliwell and Gutteridge, 2007)
Reaction 2: H2O2 + Cu+ → Cu2+ + −OH + ·OH (k ~ 4.7 × 103 M−1 s−1; Halliwell and Gutteridge, 2007)
Acute exercise increases proxies of ·OH generation (Close et al., 2005) likely owing to increased H2O2 generation and disrupted transition metal handling, resulting in increased labile iron and copper (Cobley et al., 2015a). Only vicinal ·OH has capacity to damage mitochondrial and nuclear DNA owing to its diffusion-controlled reactivity with cellular biomolecules (Halliwell, 2012). A resultant mechanistic requirement exists for ·OH to be generated proximal to DNA and existence of conditions that promote reactions 1 and 2 (e.g., nuclear H2O2 diffusion) for exercise-induced ·OH mediated DNA damage to occur. An additional mechanistic point worthy of consideration is the origin of nuclear and mitochondrial H2O2 and thus ·OH. Mitochondria have a considerably greater capacity to generate H2O2 internally compared to nuclei, owing to localized O2.− generation and subsequent intra-mitochondrial O2.− to H2O2 dismutation capacity (Murphy, 2009, 2012). Exercise-induced nuclear DNA damage likely requires nuclear H2O2 diffusion from other intracellular sources (e.g., endoplasmic reticulum). The nature of this diffusion is poorly understood yet it is tempting to speculate the existence of retrograde H2O2 mitochondria-nuclei signaling and damage pathways (see Box 1). It should be noted that present assays, notably assessment of global 8-oxoG levels post-exercise, provide little mechanistic information, owing to the existence of DNA repair processes (Murphy et al., 2011).
Box 1. A modified mitochondrial bifurcation hypothesis.
In considering mitochondrial DNA oxidation, the bifurcation hypothesis becomes more complex. Mitochondrial O2.− originates from several sources, notably ETC complexes I and III (Murphy, 2009; Finkel, 2011; Goncalves et al., 2015). One fate of mitochondrial O2.− is reaction with SOD isoforms to generate H2O2 (McCord and Fridovich, 1969). The fates of H2O2 thereafter are numerous and include inter alia reaction with: (1) peroxiredoxin 3 and 5 (2) glutathione peroxidase 1 (3) protein thiols (4) labile transition metals or centered proteins (e.g., aconitase) and (5) diffusion out of the mitochondrion (Murphy, 2012). Fates 4 and 5 permit proximal and distal ·OH generation and thus damage to mitochondrial and nuclear DNA, respectively. Mitochondria manufacture hem and iron sulfur centered proteins (Collins et al., 2012), which could promote fate 4 and consequent mitochondrial DNA damage. Complexity is added by the O2.− source-function-energetic state relationship (Bleier et al., 2015). It is conceivable that some O2.− sources promote damage and others signaling, and that the outcome changes depending upon mitochondrial energetic state and the levels and functional state of O2.− and H2O2 reactants. Concordantly, Bleier et al. (2015) documented a differential pattern of target protein thiol modification when ETC I O2.− production was induced compared to ETC III. That is, the identity of the protein thiol modified depends on the site of O2.− generation. Analogously, O2.− and subsequent H2O2 generated by one source may damage mitochondrial DNA whereas another may not. Elucidating the site (s) that damage mitochondrial DNA would significantly advance current understanding. Fate 5 is intriguing because H2O2 diffusing out of the mitochondrion could function as a retrograde nuclear signal (Balaban et al., 2005; Murphy, 2009, 2012). This could involve direct or indirect (redox relay) H2O2 induced modification of redox-sensitive transcription factors and resultant regulation of nuclear transcription (Murphy, 2009). It could equally involve nuclear ·OH generation and consequent DNA damage, the sensing of which could also alter cell signaling processes (see main text).
Exercise-induced Redox Signaling: The Key Role of Hydrogen Peroxide
Exercise-induced ·OH mediated DNA damage proceeds in a random and indiscriminate chemical manner, exemplified by a myriad of guanine oxidation products (Radak et al., 2011b). Exercise-induced redox signaling, however, depends on the transduction of specific, reversible and compartmentalized chemical signals (Cobley et al., 2015a). ·OH is chemically unable to transmit a signal in this conventional manner (Holmstrom and Finkel, 2014). Hence, exercise-induced DNA damage and redox signaling are not necessarily mediated by the same species. Mechanistic knowledge of exercise-induced redox signaling is fragmentary (Cobley et al., 2015a). For instance, reversible cysteine oxidation is a key feature of redox signaling (Go and Jones, 2013). The proteome contains 214,000 cysteine residues, with ~21,000–40,000 modified following addition of oxidizing stimuli (Jones, 2008). If one assumes that a lower limit of ~21,000 cysteine residues are exercise-responsive then ≤1% of the exercise-responsive cysteine proteome has been mapped to date. Indeed, we are unaware of any cysteine based exercise proteomics study. Insights from the parent discipline (i.e., redox biology) are therefore, utilized to provide a brief chemical synopsis of the likely nature of exercise-induced redox signaling.
Current redox signaling paradigms are defined by specific and reversible target protein cysteine modifications that alter protein activity, partner binding and location (Janssen-Heininger et al., 2008; Winterbourn, 2008; Forman et al., 2014a,b). Notable modifications include disulfide, mixed disulfide formation and S-Nitrosylation (Gallogy and Mieyal, 2007; Benhar et al., 2009; Poole, 2015). Redox signaling is thought to be predominately mediated by two electron oxidants, with H2O2 considered to be one of the few species capable of selective reaction with reactive cysteine residues on target proteins (Forman et al., 2010). The direct reaction of H2O2 with often low abundant signaling proteins (e.g., KEAP1 estimated: k ~ 140 M−1 s−1; Marhino et al., 2014) must compete against the rapid reaction of H2O2 with several highly abundant redox enzymes (e.g., peroxiredoxins [Prx]: k ~ 105–108 M−1 s−1; Brigelius-Flohe and Flohe, 2011; Karplus, 2015). Further, acute exercise increases the activity of redox enzymes, notably catalase (Powers and Jackson, 2008). How H2O2 overcomes this kinetic bottleneck is a matter of debate, but could involve redox relays, wherein oxidizing equivalents are transferred from the antioxidant enzyme to the signaling protein (Marhino et al., 2014). For instance, the reaction of H2O2 with Prx II has recently been shown to be coupled to the oxidation of STAT3 (Sobatto et al., 2015). High local H2O2 gradients in specific cellular compartments (co-localization of target and source) allied to sequestration and/or inactivation of antioxidant enzymes may also facilitate direct H2O2 signaling (Woo et al., 2010; Marhino et al., 2014). Analogous to exercise, many unanswered questions remain regarding the precise chemical nature and spatiotemporal regulation of redox signaling (Brigelius-Flohe and Flohe, 2011; Levonen et al., 2014). It follows that one cannot fully appraise the interplay between exercise-induced redox signaling and DNA damage, since many mechanistic details have yet to be elucidated.
Interplay between Exercise-induced DNA Damage and Redox Signaling: A Nuanced View
Chemical delineation of the species and reactions responsible for DNA damage and redox signaling is necessary from a mechanistic perspective (Buettner, 2015; Forman et al., 2015) but one should not view the two processes as wholly discrete and independent. Although redox signaling can occur without oxidative damage and vice versa (Jones, 2008; Jones and Go, 2010) the two processes can be interlinked in several hitherto underappreciated ways, in an exercise setting. Possible points of interaction will be appraised herein.
H2O2: An Upstream Bifurcation Point
We hypothesize that H2O2 acts as a bifurcation point between exercise-induced redox signaling and damage. A key feature of our hypothesis is that the H2O2 “interactome” does not trap all of the H2O2 generated at signaling sites during exercise, permitting distal diffusion and the induction of DNA damage following reaction with transition metals (Reactions 1, 2). In this model, ·OH mediated DNA damage is a by-product of exercise-induced redox signaling. To illustrate this model an exemplar scenario is next considered (see Box 1 for a mitochondrial scenario).
NADPH oxidase (NOX) isoforms appear to be the principal intracellular source of O2.− during exercise (Sakellariou et al., 2013). Prototypical models of redox signaling are defined by activation of plasma membrane bound NOX, secondary to Rac1 recruitment, and growth factor stimulation (Brandes et al., 2014; Holmstrom and Finkel, 2014). Note NOXs are likely activated by different stimuli (e.g., Ca2+ fluxes) during exercise (Sakellariou et al., 2014) and are redox regulated (Brandes et al., 2014). NOX generate extracellular O2.− (Reaction 3) which can spontaneously dismutate to H2O2 (k ~ 105 M−1 s−1; Forman et al., 2010) or be converted to H2O2 enzymatically in a reaction catalyzed by superoxide dismutase (SOD) isoforms (k ~ 109 M−1 s−1; Reaction 4, McCord and Fridovich, 1969). H2O2 can re-enter the cell via diffusion or though specialized aquaporin/peroxiporin channels (Bienert and Chamont, 2014; Sies, 2014). Thereafter, the topology of the plasma membrane permits spatial gating with lipid rafts and caveolae providing a means of regulating and channeling signals in discrete membrane domains (Patel and Insel, 2009). The relatively slow reaction of H2O2 with even highly reactive cysteine residues on target signaling proteins (k ~ 1–10 M−1 s−1; Winterbourn and Hampton, 2008) could therefore, be compensated for by “insulating” the signal against other reactants and increasing local concentrations of target and reactant (Forman et al., 2014b). Intricate spatial regulation is demonstrated by the observation that redox signaling proceeds with modification of only a small protein sub-population (Sobatto et al., 2015). Evidenced in the H2O2 → Prx II → STAT3 redox relay wherein only a fraction of total STAT3 is modified and thus exhibits reduced transcriptional activity (Sobatto et al., 2015). This microenvironment may facilitate proximal trapping of signal and target, perhaps by lipid barriers restricting say the lateral diffusion of H2O2. Nevertheless, some H2O2 diffusion is likely and this might have unwanted distal effects (e.g., DNA oxidation, see Figure 1). DNA oxidation may, therefore, be one of the biological “costs” of H2O2 signaling and may explain why H2O2 is toxic to cells, at even micromolar concentrations (Nakamura et al., 2003). Alternatively, DNA oxidation secondary to signaling may not be “costly” at all, but rather an indirect redox-sensing mechanism, providing information on cellular redox state via sensing of a terminal product (discussed below).
Reaction 3, NOX reaction: 2O2 + NADPH → NADP+ + H+ + 2O2.−
Reaction 4, SOD reaction: 2O2.− + 2H+ → H2O2 + O2
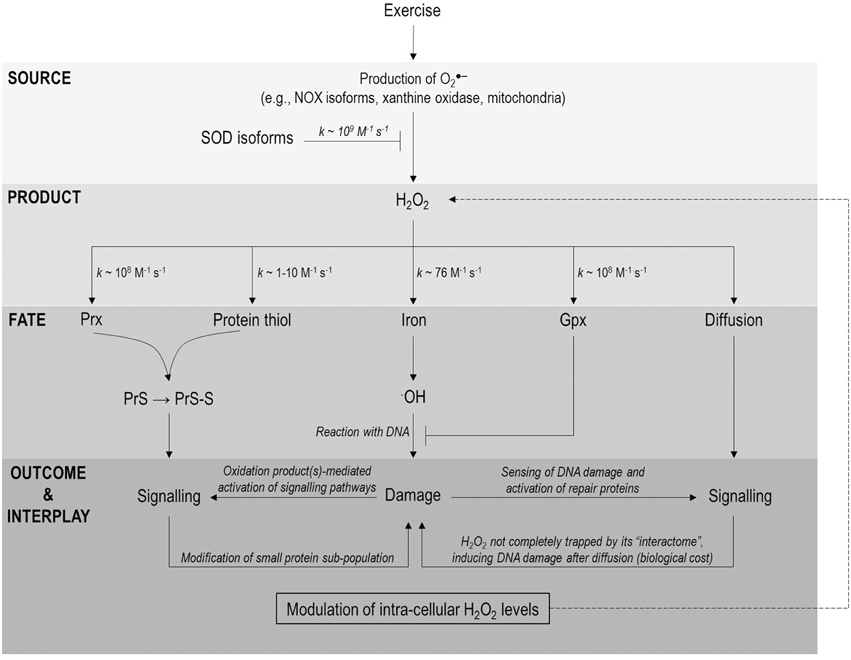
Figure 1. A H2O2 biological fate flow chart highlighting possible points of interaction between exercise-induced DNA damage and redox signaling. Exercise increases O2.− generation from a variety of sources (e.g., NOX isoforms, xanthine oxidase and mitochondria) which can be converted to H2O2 in a reaction catalyzed by SOD isoforms. Once formed H2O2 has 5 principal fates (1) reaction with Prx isoforms and oxidation of a signaling protein via a redox relay (2) direct reaction with a signaling protein (3) reaction with iron (other metals not shown for clarity) and ·OH generation (4) reaction with a protective redox enzyme (e.g., Gpx1) and (5) diffusion from source (permitting any of fates 1–4). Note the protective function of Prx and indeed catalase are not shown for the purposes of clarity. Fates 1–2 can lead to target protein modification and thus redox signaling whereas fate 3 can underpin DNA damage following the indiscriminate, diffusion-controlled ·OH-DNA reaction. Signaling and damage may be linked in two important ways from there (1) modification of redox repair enzymes and (2) modulation of H2O2 levels by redox repair enzymes (depicted a dashed line). Note many of these links have yet to be documented in an exercise setting and are thus speculative at present.
The explanatory power of the bifurcation hypothesis, whilst cogent is conceptual in an exercise setting at present. It is, therefore, necessary to consider evidence for any distal reactions that could damage DNA. Measuring and deciphering the origin of nuclear ·OH is complicated by several technical issues (Halliwell and Whiteman, 2004). Although, direct evidence is lacking, redox signaling is commonly associated with the oxidation of the intracellular DCFH probe to DCF (Winterbourn, 2015). We emphasize that the DCFH assay does not measure H2O2 (no direct H2O2-DCFH reaction) and is oxidized by one electron mechanisms (Wardman, 2007; Winterbourn, 2014). Further the assay is prone to several artifacts, notably spurious amplification of the signal, via rapid reaction of the DCF radical intermediate with O2 (k ~ 108 M−1 s−1) to yield O2.− (Kalyanaraman et al., 2012; Winterbourn, 2014). Despite several caveats Kalyanaraman et al. (2012) note that the DCFH assay can provide valuable information on redox dependent iron signaling (Tampo et al., 2003). The temporal association of redox signaling and DCFH oxidation could, in part, reflect the efflux of H2O2 from signaling microdomains, subsequent reaction with iron and thus DNA oxidation. It could equally reflect an underappreciated role of free radicals in redox signaling, likely via the generation of thiyl radicals (Winterbourn and Hampton, 2008; Winterbourn, 2015). In any event, we postulate that exercise-induced redox perturbations are bi-functional, resulting in signaling and damage, and that the fate of H2O2 can, in part, regulate the extent of each outcome.
Sensing DNA Damage: Implications for Redox Signaling
Conceptually, DNA oxidation may influence redox signaling directly through DNA oxidation product (s) and/or indirectly through the redox regulation of proteins implicated in DNA repair (Radak et al., 2013). Each possibility will next be considered in turn with exemplars provided.
Oxidized macromolecule adducts are not chemically inert (Niki, 2009) and are thus not a passive end-point of exercise-induced reactive species generation. Indeed, many oxidized macromolecule adducts are biologically active and can influence cell signaling processes (Brigelius-Flohe and Flohe, 2011). A proof-of-concept example is the participation of lipid peroxidation products in Nrf-2-KEAP-1 signaling (Niki, 2012). The Nrf-2-KEAP-1 pathway regulates the expression of cyto-protective genes (e.g., hem oxygenase; Kasper et al., 2009). In the inactive state, KEAP-1 sequesters Nrf-2 in the cytoplasm, facilitating the E3 ubiquitin ligase cullin-3 mediated Nrf-2 proteolysis (Levonen et al., 2014). This inhibition can be relieved by S-alkylation and the subsequent degradation of KEAP-1, enabling the nuclear translocation of Nrf-2 (Forman et al., 2014a). KEAP-1 S-alkylation can be mediated by lipid peroxidation products, such as 4-hydroxy-2-noneneal (Chen et al., 2005, 2006). In considering DNA oxidation products, application of exogenous 8-oxo-G to cells alters signaling (Aguilera-Aguirre et al., 2015). This is, however, likely mediated by the binding of 8-oxo-G to DNA repair proteins and not post-translational modification (Aguilera-Aguirre et al., 2015). Indeed, to the best of our knowledge there are no examples of DNA oxidation products directly altering the post-translational modification state of signaling proteins. This notwithstanding, free 8-oxo-G can be oxidized to a hydroperoxide like derivative that could signal, but this remains speculative at present (Hajas et al., 2012). Altogether, the possibility that DNA damage products directly participate in signaling reactions is not excluded but is not presently an example of the interplay between exercise-induced oxidative damage and signaling and is thus not considered further.
The consequences of oxidative damage, genomic rearrangements, and strand breaks, are sensed by repair proteins (Dizdaroglu, 2012). For example, 8-oxoG is excised by OGG1 a key component of the mitochondrial and nuclear base excision repair pathway (Radak et al., 2011b). OGG1 is regulated by a plethora of post-translational modifications, including redox-regulated disulfide formation, which is associated with reduced OGG1 activity (Bravard et al., 2006). Indeed, several other proteins implicated in DNA repair including but not limited to SIRT1 (Hwang et al., 2013), SIRT6 (Hu et al., 2015), and p53 (Malliet and Pervaiz, 2012) are also redox regulated. Redox signaling has the capacity therefore, to influence DNA repair and could constitute one convergence point between exercise-induced DNA damage and redox signaling. Another point is provided by the interaction of OGG1 with Rac1 (Hajas et al., 2013). Rac1 is a small GTPase that is regulated by GTP loading, being active in the GTP but not the GDP bound state (Bosco et al., 2009). Rac1 GTP loading is redox-sensitive being regulated by cys18 oxidation and S-glutathionylation (Heo and Campbell, 2005; Hobbs et al., 2015). Rac1, in turn, regulates cellular redox state in several ways, notably through binding and activating NOX isoforms (Leto et al., 2009; Nauseef, 2014). Interestingly, 8-oxoG bound OGG1 complexes can bind Rac1 and promote GTP loading and subsequent NOX4 activation following a rise in 8-oxo-G levels (Hajas et al., 2013). This is associated with increased intracellular H2O2 probe and general redox indicator probe DCFH oxidation (Hajas et al., 2013). This interaction provides a mechanistic link between redox signaling and damage that may constitute a feedback loop. The functionality of this feedback loop is, however, unclear and remains to be investigated in an exercise setting. Overall, two points of interplay are apparent: (1) the redox regulation of DNA repair proteins and (2) the capacity of DNA repair proteins to modulate intracellular H2O2 levels and perhaps redox signaling and damage.
Concluding Perspectives
The terminal reactions that define exercise-induced ·OH mediated DNA damage and H2O2 mediated signaling are chemically distinct yet we have delineated possible points of interaction between the two processes. Of course, redox signaling can proceed without DNA damage and vice versa (Jones, 2006, 2008). Indeed, this paradigm is well-established in many settings and likely occurs with the nanomolar (~10–100 nM; Levonen et al., 2014) H2O2 fluxes that define growth factor signaling in the resting state (Rhee, 2006). Exercise-induced quantal H2O2 yields are likely in the micromolar range (~1 μM; Palomero et al., 2008) and in this situation DNA damage and redox signaling are unlikely to be mutually exclusive. We postulate that the biological fate of H2O2 represents a bifurcation point that, in part, delineates the extent of exercise-induced DNA damage and signaling. In this scenario, crosstalk between redox signaling and DNA damage is facilitated by (1) the redox regulation of DNA repair proteins (e.g., OGG1; Bravard et al., 2006) and (2) the capacity of DNA repair proteins to modulate intracellular H2O2 levels (OGG1-Rac1-Nox4) axis (Hajas et al., 2013). Perhaps exercise-induced H2O2 levels define an interface between redox responses that are typically, but not always specific “on/off switches” (e.g., kinase activation) and general rheostats (e.g., repair processes that “sense” DNA damage). This hypothesis may have considerable explanatory power. The dual functionality of exercise-induced H2O2 fluxes is consistent with the temporal co-incidence of the redox-regulation of signaling proteins (e.g., PGC-1α, Kang et al., 2009) and generalized signaling responses to macromolecule damage (e.g., apoptosis secondary to DNA oxidation, Winterbourn, personal communication). Saliently, a generalized signaling response need not require reactive species to signal in a classical way, akin to a phosphorylation cascade. Rather, it simply requires the sensing of a redox perturbation at a critical juncture: DNA oxidation. This discourse may provide a mechanistic framework to further explain how acute exercise-induced DNA damage acts as an adaptive signal to stimulate protection against exercise-induced genomic stress. It is emphasized that other points of interaction may exist but were not considered owing to space constraints. Indeed, the biological fate of peroxynitrite may constitute another salient bifurcation point. Ultimately, this dialog is intended to stimulate further investigation into the mechanisms regulating exercise-induced redox signaling and damage.
Author Contributions
JC conceived, drafted and edited the manuscript. NM and MN conceived the figures and drafted sub-sections of the manuscript. All authors (JC, NM, JPM, GC, MN, and JKM) critically edited and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize for any works we were unable to cite in this review owing to space constraints. Prof Christine Winterbourn (University of Otago) is thanked for useful scientific discussions. JC gratefully acknowledges the generous financial support provided by the Carnegie Trust (R70103).
References
Aguilera-Aguirre, L., Hosoki, K., Bacsi, A., Radak, Z., Wood, T. G., Widen, S. G., et al. (2015). Whole transcriptome analysis reveals 8-oxoguanine DNA glycosylase-1-driven DNA repair dependent gene expression linked to essential biological processes. Free Radic. Biol. Med. 81, 107–118. doi: 10.1016/j.freeradbiomed.2015.01.004
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Benhar, M., Forrester, M. T., and Stamler, J. S. (2009). Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 10, 721–732. doi: 10.1038/nrm2764
Bienert, G. P., and Chamont, F. (2014). Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta. 1840, 1596–1604. doi: 10.1016/j.bbagen.2013.09.017
Bleier, L., Wittig, I., Heide, H., Steger, M., Brandt, U., and Drose, S. (2015). Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 78, 1–10. doi: 10.1016/j.freeradbiomed.2014.10.511
Bosco, E. E., Mulloy, J. C., and Zheng, Y. (2009). Rac1 GTPase: a Rac of all trades. Cell. Mol. Life Sci. 66, 370–374. doi: 10.1007/s00018-008-8552-x
Brandes, R. P., Weissman, N., and Schroder, K. (2014). Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic. Biol. Med. 76, 208–226. doi: 10.1016/j.freeradbiomed.2014.07.046
Bravard, A., Vacher, M., Gouget, B., Coutant, A., de Boisferon, F. H., Marsin, S., et al. (2006). Redox regulation of OGG1 activity in response to cellular oxidative stress. Mol. Cell. Biol. 26, 7430–7436. doi: 10.1128/MCB.00624-06
Brigelius-Flohe, R., and Flohe, L. (2011). Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 15, 2335–2381. doi: 10.1089/ars.2010.3534
Buettner, G. R. (2015). Moving free radical and redox biology ahead in the next decade(s). Free Radic. Biol. Med. 78, 236–238. doi: 10.1016/j.freeradbiomed.2014.10.578
Cadet, J., Ravanat, J., Taverna-Porro, M., Menoni, H., and Angelov, D. (2012). Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett. 327, 5–15. doi: 10.1016/j.canlet.2012.04.005
Chatgilialoglu, C., D'Angelantonio, M., Kciuk, G., and Bobrowski, K. (2011). New insights into the reaction paths of hydroxyl radicals with 2′-deoxyguanosine. Chem. Res. Toxicol. 24, 2200–2206. doi: 10.1021/tx2003245
Chen, Z. H., Saito, Y., Yoshida, Y., Serkine, A., Noguchi, N., and Niki, E. (2005). 4-Hydroxynonenal induces adaptive response and enhances PC12 cell tolerance primarily through induction of thioredoxin reductase 1 via activation of Nrf2. J. Biol. Chem. 280, 41921–41927. doi: 10.1074/jbc.M508556200
Chen, Z. H., Yoshida, Y., Saito, Y., Serkine, A., Noguchi, N., and Niki, E. (2006). Induction of adaptive response and enhancement of PC12 cell tolerance by 7-hydroxycholesterol and 15- deoxy-delta(12,14)-prostaglandin J2 through up-regulation of cellular glutathione via different mechanisms. J. Biol. Chem. 281, 14440–14445. doi: 10.1074/jbc.M600260200
Close, G. L., Ashton, T., McArdle, A., and Jackson, M. J. (2005). Microdialysis studies of extracellular reactive oxygen species production in skeletal muscle: factors influencing the reduction of cytochrome c and hydroxylation of salicylate. Free Radic. Biol. Med. 39, 1460–1467. doi: 10.1016/j.freeradbiomed.2005.07.009
Cobley, J. N., Bartlett, J. D., Kayani, A. C., Murray, S. W., Louhelainen, J., Donovan, T., et al. (2012). PGC-1α transcriptional response and mitochondrial adaptation to acute exercise is maintained in skeletal muscle of sedentary elderly males. Biogerontology 13, 621–631. doi: 10.1007/s10522-012-9408-1
Cobley, J. N., McHardy, H., Morton, J. P., Nikolaidis, M. G., and Close, G. L. (2015a). Influence of vitamin C and vitamin E on redox signalling: implications for exercise adaptation. Free Radic. Biol. Med. 84, 65–76. doi: 10.1016/j.freeradbiomed.2015.03.018
Cobley, J. N., Moult, P. R., Burniston, J. G., Morton, J. P., and Close, G. L. (2015b). Exercise improves mitochondrial and redox-regulated stress responses in the elderly: better late than never! Biogerontology 16, 249–264. doi: 10.1007/s10522-014-9546-8
Cobley, J. N., Sakellariou, G. K., Owens, D. J., Murray, S., Waldron, S., Gregson, W., et al. (2014). Lifelong training preserves some redox-regulated adaptive responses following an acute exercise stimulus in aged human skeletal muscle. Free Radic. Biol. Med. 70, 23–32. doi: 10.1016/j.freeradbiomed.2014.02.004
Cobley, J. N., Sakellariou, G. K., Waldron, S., Murray, G., Burniston, J. G., Morton, J. P., et al. (2013). Life-long training attenuates residual genotoxic stress in the elderly. Longev. Healthspan. 2:11. doi: 10.1186/2046-2395-2-11
Collins, Y., Chouchani, E. T., James, A. M., Menger, K. E., Cocheme, H. M., and Murphy, M. P. (2012). Mitochondrial redox signalling at a glance. J. Cell Sci. 125, 801–806. doi: 10.1242/jcs.098475
Dizdaroglu, M. (2012). Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett. 327, 26–47. doi: 10.1016/j.canlet.2012.01.016
Dizdaroglu, M., and Jaruga, P. (2012). Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 46, 381–419. doi: 10.3109/10715762.2011.653969
Egan, B., and Zierath, J. R. (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184. doi: 10.1016/j.cmet.2012.12.012
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Forman, H. J., Augusto, O., Brigelius-Flohe, R., Dennery, P. A., Kalyanaraman, B., Ishiropoulos, H., et al. (2015). A suggested guide to free radical research terminology and methodology. Free Radic. Biol. Med. 78, 233–235. doi: 10.1016/j.freeradbiomed.2014.10.504
Forman, H. J., Davies, K. J. A., and Ursini, F. (2014a). How do nutritional antioxidants really work: nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 66, 24–35. doi: 10.1016/j.freeradbiomed.2013.05.045
Forman, H. J., Maiorino, M., and Ursini, F. (2010). Signaling functions of reactive oxygen species. Biochemistry 49, 835–842. doi: 10.1021/bi9020378
Forman, H. J., Ursini, F., and Maiorino, M. (2014b). An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 73, 2–9. doi: 10.1016/j.yjmcc.2014.01.018
Gallogy, M. M., and Mieyal, J. J. (2007). Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7, 381–391. doi: 10.1016/j.coph.2007.06.003
Go, Y. M., and Jones, D. P. (2013). The redox proteome. J. Biol. Chem. 288, 26512–26520. doi: 10.1074/jbc.R113.464131
Goncalves, R. L. S., Quinlam, C. L., Perevoshchikova, I. V., Hey-Mogensen, M., and Brand, M. D. (2015). Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 290, 209–227. doi: 10.1074/jbc.M114.619072
Hajas, G., Bacsi, A., Aguilerra-Aguirre, L., German, P., Radak, Z., Sur, S., et al. (2012). Biochemical identification of a hydroperoxide derivative of the free 8-oxo-7,8-dihydroguanine base. Free Radic. Biol. Med. 52, 749–756. doi: 10.1016/j.freeradbiomed.2011.11.015
Hajas, G., Bacsi, A., Augilera-Aguirre, L., Hegde, M. L., Tapas, K. H., Sur, S., et al. (2013). 8-Oxoguanine DNA glycolyase-1 link DNA repair to cellular signaling via the activation of the small GTPase Rac1. Free Radic. Biol. Med. 61, 384–394. doi: 10.1016/j.freeradbiomed.2013.04.011
Halliwell, B. (2012). The antioxidant paradox. Br. J. Clin. Pharmacol. 75, 637–644. doi: 10.1111/j.1365-2125.2012.04272.x
Halliwell, B., and Gutteridge, J. M. G. (2007). Free Radicals in Biology and Medicine, 4th Edn. Oxford: Oxford University Press.
Halliwell, B., and Whiteman, M. (2004). Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol. 142, 231–255. doi: 10.1038/sj.bjp.0705776
Hawley, J. A., Hargreaves, M., Joyner, M. J., and Zierath, J. R. (2014). Integrative biology of exercise. Cell 159, 738–749. doi: 10.1016/j.cell.2014.10.029
Heo, J., and Campbell, S. L. (2005). Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J. Biol. Chem. 280, 31003–31010. doi: 10.1074/jbc.M504768200
Hobbs, A. G., Mitchell, L. E., Arrington, M. E., Gunawardena, H. P., DeCristo, M. J., Loeser, R. F., et al. (2015). Redox regulation of Rac1 by thiol oxidation. Free Radic. Biol. Med. 79, 237–250. doi: 10.1016/j.freeradbiomed.2014.09.027
Holmstrom, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421. doi: 10.1038/nrm3801
Hu, S., Liu, H., Ha, H., Luo, X., Motamedi, M., Gupta, M. P., et al. (2015). Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic. Biol. Med. 79, 176–185. doi: 10.1016/j.freeradbiomed.2014.11.011
Hwang, J. A., Yao, H., Caito, S., Sundar, I. K., and Rahman, I. (2013). Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 61, 95–110. doi: 10.1016/j.freeradbiomed.2013.03.015
Janssen-Heininger, Y. M., Mossman, B. T., Heintz, N. H., Forman, H. J., Kalyanaraman, B., Finkel, T., et al. (2008). Redox-based regulation of signal transduction: principles: pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17. doi: 10.1016/j.freeradbiomed.2008.03.011
Jones, D. P. (2006). Redefining oxidative stress. Antioxid. Redox Signal. 8, 1865–1879. doi: 10.1089/ars.2006.8.1865
Jones, D. P. (2008). Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 295, C849–C868. doi: 10.1152/ajpcell.00283.2008
Jones, D. P., and Go, Y. M. (2010). Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 12, 166–125. doi: 10.1111/j.1463-1326.2010.01266.x
Kalyanaraman, B., Usmar, V., Davies, K. J. A., Dennery, P. A., Forman, R. J., Grisham, M. B., et al. (2012). Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6. doi: 10.1016/j.freeradbiomed.2011.09.030
Kang, C., O'Moore, K., Dickman, J. R., and Ji, L. L. (2009). Exercise activation of muscle peroxisome proliferator activated receptor-γ co-activator-1α is redox sensitive. Free Radic. Biol. Med. 47, 1394–1400. doi: 10.1016/j.freeradbiomed.2009.08.007
Karplus, P. A. (2015). A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 80, 183–190. doi: 10.1016/j.freeradbiomed.2014.10.009
Kasper, J. W., Niture, S. K., and Jaiswal, A. K. (2009). Nrf2:INrf2 (Keap1) signalling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035
Leto, T. L., Morand, S., Hurt, D., and Ueyama, T. (2009). Targeting and regulation of reactive oxygen species by Nox family NADPH Oxidases. Antioxid. Redox Signal. 11, 2607–2619. doi: 10.1089/ars.2009.2637
Levonen, A. L., Hill, B. G., Kansanen, E., Xhang, J., and Darley-Usmar, V. M. (2014). Redox regulation of antioxidants, autophagy, and the response to stress: implications for electrophile therapeutics. Free Radic. Biol. Med. 71, 196, 207. doi: 10.1016/j.freeradbiomed.2014.03.025
Malliet, A., and Pervaiz, S. (2012). Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid. Redox Signal. 16, 1285–1294. doi: 10.1089/ars.2011.4434
Marhino, H. S., Real, C., Cryne, L., Soares, H., and Antunes, F. (2014). Hydrogen peroxide sensing, signalling and regulation of transcription factors. Redox Biol. 2, 535–562. doi: 10.1016/j.redox.2014.02.006
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase: an enzymatic function for erythrocuprein (Hemocuprein). J. Biol. Chem. 244, 6049–6055.
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Murphy, M. P. (2012). Mitochondrial thiols in antioxidant protection and redox signalling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495. doi: 10.1089/ars.2011.4289
Murphy, M. P., Holmgren, A., Larsson, N., Halliwell, B., Chang, C. J., Kalyanaraman, B., et al. (2011). Unravelling the biological roles of reactive oxygen species. Cell Metab. 13, 361–366. doi: 10.1016/j.cmet.2011.03.010
Nakamura, J., Purvis, E. R., and Swenberg, J. A. (2003). Micromolar concentrations of hydrogen peroxide induce oxidative DNA lesions more efficiently than millimolar concentrations in mammalian cells. Nucleic Acids Res. 31, 170–175. doi: 10.1093/nar/gkg263
Nauseef, W. M. (2014). Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. Biochim. Biophys. Acta. 1840, 757–767. doi: 10.1016/j.bbagen.2013.04.040
Niki, E. (2009). Lipid peroxidation: physiological levels and dual biological effects. Free Radic. Biol. Med. 47, 469–484. doi: 10.1016/j.freeradbiomed.2009.05.032
Niki, E. (2012). Do antioxidants impair signaling by reactive oxygen species and lipidoxidation products? FEBS Lett. 586, 3767–3770. doi: 10.1016/j.febslet.2012.09.025
Palomero, J., Pye, D., Kabayo, T., Spiller, D. G., and Jackson, M. J. (2008). In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibres by real-time fluorescence microscopy. Antioxid. Redox Signal. 10, 1463–1474. doi: 10.1089/ars.2007.2009
Patel, H. H., and Insel, P. A. (2009). Lipid rafts and caveolae and their role in compartmentation of redox signalling. Antioxid. Redox Signal. 11, 1357–1372. doi: 10.1089/ars.2008.2365
Poole, L. B. (2015). The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 80, 148–157. doi: 10.1016/j.freeradbiomed.2014.11.013
Powers, S. K., and Jackson, M. J. (2008). Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol. Rev. 88, 1243–1276. doi: 10.1152/physrev.00031.2007
Radak, Z., Bori, Z., Kolati, E., Fatourous, G. I., Jamaturas, A. Z., Douroudos, I. I., et al. (2011b). Age-dependent changes in 8-oxoguanine-DNA glyocloase activity are modulated by the adaptive response to physical exercise in human muscle. Free Radic. Biol. Med. 51, 417–423. doi: 10.1016/j.freeradbiomed.2011.04.018
Radak, Z., Zhao, Z., Goto, S., and Koltai, E. (2011a). Age-associated neurodegeneration and oxidative damage to lipids, proteins and DNA. Mol. Aspects Med. 32, 305–315. doi: 10.1016/j.mam.2011.10.010
Radak, Z., Zhao, Z., Kolati, E., Ohno, H., and Atalay, M. (2013). Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signalling. Antioxid. Redox Signal. 18, 1208–1246. doi: 10.1089/ars.2011.4498
Rhee, S. C. (2006). Cell signalling: H2O2, a necessary evil for cell signalling. Science 312, 1882–1883. doi: 10.1126/science.1130481
Sakellariou, G. K., Jackson, M. J., and Vasilaki, A. (2014). Redefining the major contributors to superoxide production in contracting skeletal muscle. Role of NAD(P)H oxidases. Free Radic. Res. 48, 12–29. doi: 10.3109/10715762.2013.830718
Sakellariou, G. K., Vasilaki, A., Palomero, J., Kayani, A., Zibrik, L., McArdle, A., et al. (2013). Studies of mitochondrial and non-mitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase (s) in the increase skeletal muscle superoxide generation that occurs following contractile activity. Antioxid. Redox Signal. 18, 603–621. doi: 10.1089/ars.2012.4623
Sies, H. (2014). Role of metabolic H2O2 generation: redox signalling and oxidative stress. J. Biol. Chem. 289, 8735–8741. doi: 10.1074/jbc.R113.544635
Sobatto, M. C., Liou, W., Stocker, S., Talwar, D., Oehler, M., Ruppert, T., et al. (2015). Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70. doi: 10.1038/nchembio.1695
Tampo, Y., Kotamraju, S., Chitambar, C. R., Kalivendi, S. V., Keszler, A., Joseph, J., et al. (2003). Oxidative stress-induced iron signaling is responsible for peroxide-dependent oxidation of dichlorodihydrofluorescein in endothelial cells: role of transferrin receptor-dependent iron uptake in apoptosis. Circ. Res. 92, 56–63. doi: 10.1161/01.RES.0000048195.15637.AC
Wardman, P. (2007). Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 43, 995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026
Winterbourn, C. C. (2008). Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286. doi: 10.1038/nchembio.85
Winterbourn, C. C. (2014). The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta. 1840, 730–738. doi: 10.1016/j.bbagen.2013.05.004
Winterbourn, C. C. (2015). Are free radicals involved in thiol-based redox signalling? Free Radic. Biol. Med. 80, 164–170. doi: 10.1016/j.freeradbiomed.2014.08.017
Winterbourn, C. C., and Hampton, M. B. (2008). Thiol chemistry and specificity in redox signalling. Free Radic. Biol. Med. 45, 549–561. doi: 10.1016/j.freeradbiomed.2008.05.004
Keywords: DNA damage, redox signaling, exercise, hydrogen peroxide, hydroxyl radical, mitochondrial DNA damage
Citation: Cobley JN, Margaritelis NV, Morton JP, Close GL, Nikolaidis MG and Malone JK (2015) The basic chemistry of exercise-induced DNA oxidation: oxidative damage, redox signaling, and their interplay. Front. Physiol. 6:182. doi: 10.3389/fphys.2015.00182
Received: 28 April 2015; Accepted: 02 June 2015;
Published: 17 June 2015.
Edited by:
Gareth Davison, University of Ulster, UKReviewed by:
Mariarosaria Santillo, University of Naples Federico II, ItalyMarcos Lopez, Fundación Cardiovascular de Colombia, Colombia
Sataro Goto, Juntendo University, Japan
Copyright © 2015 Cobley, Margaritelis, Morton, Close, Nikolaidis and Malone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James N. Cobley, Division of Sport and Exercise Sciences, Abertay University, Kydd Building, Bell Street, Dundee, DD1 1HG, UK, j.cobley@abertay.ac.uk