 Erick O. Hernández-Ochoa
Erick O. Hernández-Ochoa Stephen J. P. Pratt
Stephen J. P. Pratt Richard M. Lovering
Richard M. Lovering Martin F. Schneider1
Martin F. Schneider1- 1Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD, USA
- 2Department of Orthopaedics, University of Maryland School of Medicine, Baltimore, MD, USA
The skeletal muscle Ca2+ release channel, also known as ryanodine receptor type 1 (RyR1), is the largest ion channel protein known and is crucial for effective skeletal muscle contractile activation. RyR1 function is controlled by Cav1.1, a voltage gated Ca2+ channel that works mainly as a voltage sensor for RyR1 activity during skeletal muscle contraction and is also fine-tuned by Ca2+, several intracellular compounds (e.g., ATP), and modulatory proteins (e.g., calmodulin). Dominant and recessive mutations in RyR1, as well as acquired channel alterations, are the underlying cause of various skeletal muscle diseases. The aim of this mini review is to summarize several current aspects of RyR1 function, structure, regulation, and to describe the most common diseases caused by hereditary or acquired RyR1 malfunction.
Introduction to Excitation-Contraction Coupling
The process of voluntary muscle contraction starts with the intention to move, as generated in the frontal lobe and motor cortical regions of the brain (Dum and Strick, 2002). These cortical areas extend neuronal projections to the spinal cord (Rizzolatti and Luppino, 2001). The motoneurons' dendritic arbors and cell bodies serve as the final processing station for corticospinal signals; their axons then leave the spinal cord forming motor nerves, which subsequently deviate from the nerve as it finally approaches the target muscle. Single axons then branch to innervate and control a fixed number of muscle fibers, collectively known as the motor unit. The action potential ultimately reaches the neuromuscular junction, a specialized synapse between the motoneuron and its target muscle fiber, where acetylcholine (ACh) is released from the presynaptic membrane (Hubbard, 1973; Katz, 1996) and binds to postsynaptic ACh nicotinic receptors (Marques et al., 2000; Wu et al., 2010). Activation of ACh nicotinic receptors induces an end-plate potential and this electrical impulse continues along the muscle fiber as an action potential. The process by which this electrical impulse of muscle fibers (aka myofibers) initiates muscle contraction is termed excitation-contraction coupling (ECC; Sandow, 1952).
Each action potential is propagated both axially along the myofiber and radially into the myofiber (Figures 1A,B; Huxley and Taylor, 1958; Adrian et al., 1969; Adrian and Peachey, 1973). The action potential depolarization is generated primarily by current through Nav1.4 sodium channels, the skeletal muscle voltage-gated sodium channel, localized at the sarcolemma and through the transverse (T)-tubule system of the myofiber (Figure 1B). T-tubules are extensions of the sarcolemma that form a branched network of radial invaginations of the surface membrane, penetrating the myofiber to surround each myofibril, this occurs at the level of the A-I junction in each mammalian muscle sarcomere (Adrian et al., 1970; Adrian and Marshall, 1977; Jurkat-Rott et al., 2006). Depolarization of the T-tubule membrane induces conformational changes in the T-tubule EC coupling voltage sensor, the voltage-gated Ca2+ channel (Cav1.1; Figures 1B,C), also known as the dihydropyridine receptor (DHPR; Rios and Brum, 1987). In skeletal muscle, the DHPR is mechanically coupled to the ryanodine receptor Ca2+ release channel type 1 (RyR1), which in contrast to the DHPR, rests in the sarcoplasmic reticulum (SR; Figures 1B,C; Takeshima et al., 1989). The Cav1.1-coupled RyR1 channels mediate, by a mechanism poorly understood, rapid Ca2+ release from the SR into the cytosol in response to the muscle action potential in the T-tubules, leading to Ca2+ binding to thin filament troponin C and activation for contraction (Schneider and Chandler, 1973; Block et al., 1988). The activity of RyR1 is also regulated by Ca2+ (Meissner et al., 1986; Endo, 2006), therefore Ca2+ released through molecularly coupled RyR1 could activate Cav1.1-uncoupled RyR1s in close proximity by a process known as Ca2+-induced Ca2+ release (CICR; Endo, 2009) or via allosteric interactions between adjacent RyR1s (Marx et al., 1998). The contribution of CICR and allosteric coupled RyR1 gating during physiological skeletal muscle activity remains controversial (Endo, 2009; Figueroa et al., 2012). In the process of muscle activation, the sequence of communication from Cav1.1 to RyR is called orthograde signaling (Beam and Horowicz, 2004). The opposite sequence of coupling, from RyR1 to Cav1.1 is known as retrograde signaling (Nakai et al., 1996; Dirksen, 2002). Retrograde signaling causes an enhancement in the activity of Cav1.1 in response to the activation of RyR1 induced by Cav1.1 (Dirksen, 2002). Membrane depolarization of the sarcolemma and T-tubule system also activates Ca2+ influx through Cav1.1 (Stanfield, 1977; Sanchez and Stefani, 1978). While this Cav1.1-dependent Ca2+ flux appears to be important for activation of Ca2+-dependent signaling pathways, SR refilling during sustained activity and modulation of energy utilization, it is not essential for active ECC in adult skeletal muscle (Georgiou et al., 2015; Lee et al., 2015).
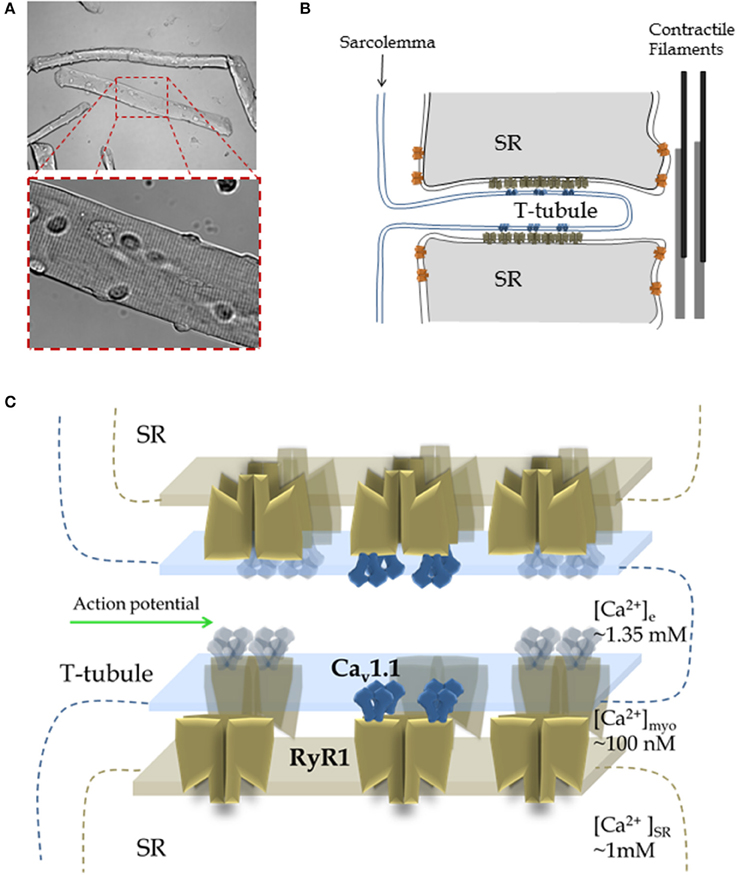
Figure 1. Muscle structure and function: from the myofiber to RyR1 Ca2+ release complex. (A) Morphology of skeletal muscle fibers. Top Panel: Transmitted light image of isolated muscle fibers from mouse flexor digitorum brevis via enzymatic dissociation; image was acquired using low magnification. Lower Panel: A higher magnification image of the fiber segment enclosed by a dashed rectangle. Note the characteristic striated pattern of muscle fibers, which results from highly organized array between sarcolemma, sarcoplasmic reticulum (SR), contractile elements and cytoarchitecture of the fibers. (B) Structure of the triad. In most cases the myoplasmic side of the junctional SR is closely apposed either to T-tubule or surface membrane, forming different junctions called dyads, triads, or peripheral coupling. The cartoon depicts a longitudinal section of the T-tubule axis, but a cross-section of the triad, a specialized array formed by the T-tubule and two segments of the terminal junctional SR (aka the terminal cisternae). The T-tubules are invaginations of the sarcolemma that propagate the action potential and possess the Cav1.1 voltage sensors that initiate the early steps of ECC. The voltage dependent Ca2+ channels, Cav1.1 (blue, aka DHPR), are positioned in both the T-tubule and sarcolemma. The SR Ca2+ release channel, RyR1 (brown), is located on the junctional domain of the SR surface, facing the T-tubules, and is also known as the junctional SR face membrane. Some RyRs may be present in adjacent parajunctional SR domains (orange). (C) Detailed architecture of the Cav1.1-RyR1 complex shown in (B). About half of the total RyR1s do not associate with Cav1.1, resulting in an alternating pattern of “free” and Cav1.1-associated RyR1s. Note: In addition to Cav1.1 and RyR1, many other proteins form part of the T-tubule- junctional SR complex (e.g., FKPB12, triadin, junctin, Casq1) and are not shown here. Panels (B,C) are based on references: (Franzini-Armstrong and Porter, 1964; Franzini-Armstrong and Nunzi, 1983; Block et al., 1988; Franzini-Armstrong and Jorgensen, 1994; Franzini-Armstrong and Kish, 1995; Franzini-Armstrong and Protasi, 1997).
Ca2+ release is subsequently decreased during depolarization by Ca2+-induced inactivation of RyR1, a negative feedback mechanism, and eventually terminated by membrane repolarization, which drives the return of Cav1.1 to a closed and resting state by reversing the activation of the Cav1.1 voltage sensor (Schneider and Hernández-Ochoa, 2012). Muscle relaxation then results from Ca2+ removal from myoplasm via transport back to the SR primarily via sarcoplasmic-endoplasmic reticulum ATPase (SERCA), the SR ATP-dependent Ca2+ pump (Hasselbach, 1964; Schneider and Simon, 1988; Schneider and Hernández-Ochoa, 2012). These processes prevent continuous Ca2+ influx, restore the initial resting state, and allow the ECC cycle to be repeated (Melzer et al., 1995; Berchtold et al., 2000).
RyR1 Function, Structure, and Regulation
The function of skeletal muscle relies on the movement of Ca2+ out of and back into the storage compartment, the SR (Green and MacLennan, 2002). Release of Ca2+ from the SR, via RyR1, results in muscle contraction. The RyR1 is a ~2.3 MDa assembly of four identical subunits (each subunit is formed by ~5000 amino acids; Imagawa et al., 1987; Inui et al., 1987). The subunit topology consists of a pore region formed by six transmembrane helices located within the C-terminal region, representing ca. 20% of total protein. The N-terminal region is a large cytoplasmic region that represents 80% of the total protein, known as the foot region (see Figure 2; Radermacher et al., 1992; Serysheva et al., 1995, 2005, 2008; Ludtke et al., 2005; Samso et al., 2005, 2006, 2009; Yan et al., 2015; Zalk et al., 2015). The myoplasmic portion of the channel (280 Å × 280 Å × 120 Å) is continuous with the transmembrane region (120 Å × 120 Å × 60 Å; Figure 2). The SR transmembranal region forms the Ca2+ release channel (Ludtke et al., 2005; Samso et al., 2005, 2006; Serysheva et al., 2005; Yan et al., 2015; Zalk et al., 2015).
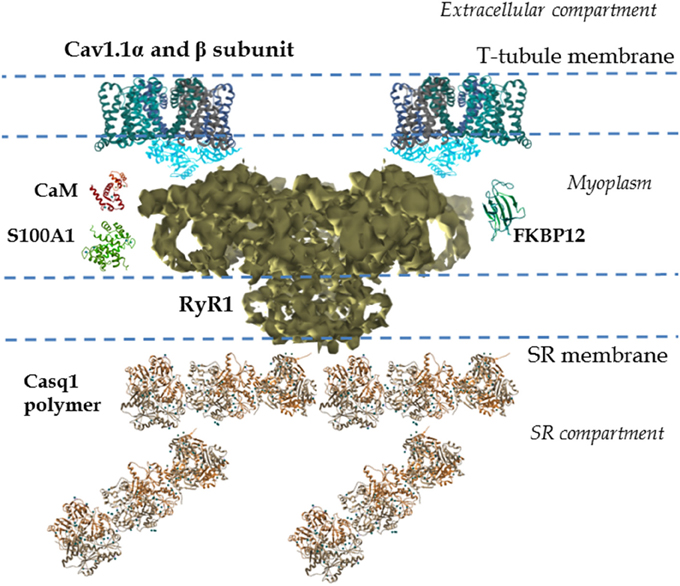
Figure 2. Cav1.1-RyR1 complex: molecular details. Shown is a reconstruction from cryo-electron microscopy of the mammalian RyR1 at 4.8 Å illustrating the different regions. This includes the large myoplasmic segment and its multiple cavities and processes, and the SR transmembranal segment, which forms the Ca2+ conduction pathway (Electron microscopy data bank entry 6106, Zalk et al., 2015). The above properties allow the RyR1 to interact with multiple SR luminal and cytosolic proteins. Several 3-D structures of proteins interacting with RyR1, as well as their relative locations, are also shown. EMData bank PDB entry and references: calsequestrin-1 (Casq1) polymer (3UOM, Sanchez et al., 2012), FKBP-12 (1D6O, Burkhard et al., 2000), S100A1 (2K2F; Wright et al., 2008), calmodulin (CaM; 1CLL, Chattopadhyaya et al., 1992), Cav1.1α subunit (4MS2, Tang et al., 2014; Hu et al., 2015), and Cav1.1 β subunit (1T0J, Van Petegem et al., 2004; Hu et al., 2015); only two alpha and two beta subunits are shown. In the muscle fiber Cav1.1 channels are clustered in groups of four (or tetrads) in the T-tubules. Cav1.1 channels in the tetrad interact with the four subunits of the RyR1, one Cav1.1 per each RyR1 subunit. All structures were generated using UCSF Chimera (Pettersen et al., 2004).
RyR1s are organized in regular checkered arrays within the terminal cisternae of the junctional SR (Franzini-Armstrong and Nunzi, 1983; Franzini-Armstrong et al., 1998). Cav1.1 channels are clustered in groups of four (or tetrads) in the T-tubule membrane portion that is in the vicinity of junctional SR (Block et al., 1988). Cav1.1 channels in the tetrad assume a coordinated location relative to the four subunits of the apposed RyR1 (see Figure 1C; Block et al., 1988), however these interactions only occur at every other RyR1. This results in half of RyR1s that are not associated with Cav1.1 (uncoupled RyR1; Franzini-Armstrong and Kish, 1995). The location of coupled and uncoupled RyR1 is dictated by the organization of the Cav1.1 molecules in the apposed T-tubule membrane, and thus mark a “checkerboard” array of coupled and uncoupled RyR1s that follows the Cav1.1 lattice organization (see Figure 1; Yin and Lai, 2000). Depolarization-induced activation of RyR1 is mediated via direct interactions with T-tubule voltage-sensitive Cav1.1 channels positioned in the abutted T-tubule membrane (Rios and Brum, 1987; Nakai et al., 1998a,b; Protasi et al., 2002). Exactly where and how these T-tubule voltage-dependent interactions between Cav1.1 and RyR1 occur remains elusive.
The RyR1 components, including SR luminal segments, transmembrane domains, large cytosolic domains, and interaction with the Cav1.1 channels, allow the RyR1s to be modulated and fine-tuned by multiple mechanisms (Serysheva et al., 2005; Samso et al., 2006; Yan et al., 2015; Zalk et al., 2015). Intracellular ions and components (e.g., cytosolic Ca2+, ATP, Mg2+, SR Ca2+ concentration, Meissner et al., 1986; Fill and Copello, 2002) and post-translational modifications (e.g., oxidation, S-nitrosylation, phosphorylation, Sonnleitner et al., 1997; Marengo et al., 1998; Eu et al., 2000; Sun et al., 2001; Fill and Copello, 2002; Aracena et al., 2003; Hidalgo et al., 2005) all regulate the activity of the RyR1. The RyR1 also interacts with numerous SR intraluminal proteins (e.g., calsequestrin, junctin, triadin; MacLennan and Wong, 1971; Brillantes et al., 1994; Glover et al., 2002; Beard et al., 2009; Treves et al., 2009), cytosolic proteins (e.g., FKBP12, calmodulin (CaM), S100A1; Fuentes et al., 1994; Buratti et al., 1995; Ikemoto et al., 1995; Tripathy et al., 1995; Ahern et al., 1997; Samso and Wagenknecht, 2002; Chelu et al., 2004; Samso et al., 2006; Prosser et al., 2008; Wright et al., 2009) and domains of the Cav1.1 α and β subunits (Nakai et al., 1994, 1998a,b; Gregg et al., 1996; Beurg et al., 1999a,b; Sheridan et al., 2006), located in the T-tubule and myoplasm, respectively. Further details on RyR1 structure-function relationship can be found in recent literature (Van Petegem et al., 2004; Lanner et al., 2010; Lanner, 2012; Van Petegem, 2015).
Given the critical role of RyR1s for skeletal muscle ECC, mutations in the RyR1s and the resulting Ca2+ dysregulation are a primary cause of several hereditary skeletal muscle myopathies. In addition, indirect or secondary RyR1 malfunction is also present in other myopathies not related to RyR1 mutations. Both congenital and acquired myopathies are a heterogeneous group of skeletal muscle weakness disorders caused by mutations or dysfunction in different structural, contractile, or regulatory muscle proteins. Several reviews have been published on various aspects of skeletal muscle myopathies (Betzenhauser and Marks, 2010; Lanner et al., 2010; Maclennan and Zvaritch, 2011; Lanner, 2012; Dowling et al., 2014; Vallejo-Illarramendi et al., 2014). Here we focus on primary RyR1 mutation-related myopathies, as well as considering Duchenne muscular dystrophy (DMD), a disease with a secondary involvement of RyR1.
RyR1 Primary Mutation-Related Myopathies
General Implication of RyR1 Disease Mutations
Mutations in RyR1 can be grouped into four general categories according to their effects on RyR1 function (Treves et al., 2008; Loy et al., 2011). The first category of RyR1 mutations cause a higher probability of activation of RyR1 by muscle fiber electrical depolarization or by RyR1 activators, and are manifested in the malignant hyperthermia (MH) phenotype. In the second category, RyR1 mutations cause leaky channels leading to Ca2+ dysregulation and depletion of Ca2+ from SR, and result in central core disease (CCD). The third category of RyR1 mutations causes deficits on the Cav1.1-mediated voltage dependent activation of SR Ca2+ release, a process also known as excitation-contraction uncoupling, and result in certain forms of CCD. The fourth category of mutations are a consequence of wild type allele silencing, a process that mimics homozygosity, and causes a decrease of RyR1 channel expression, resulting in multi-minicore disease (MmD.) In the majority of these primary myopathies, mutations in the RyR1, result in gain of function and Ca2+ dysregulation. The consequences of these mutations on RyR1's activity are compound; RYR1-related MH and CCD produce hypersensitive and/or leaky RyR1 channels (Tong et al., 1997; Lynch et al., 1999; Yang et al., 2007). Some CCD present excitation-contraction uncoupling (Avila et al., 2001; Dirksen and Avila, 2002; Kraeva et al., 2013) or exhibit RyR1 leaky channels (Dirksen and Avila, 2002; Zvaritch et al., 2009), but the exact mechanisms are unclear (Dowling et al., 2014). Many RyR1 mutations are not so obviously correlated with alterations in Ca2+ release (Maclennan and Zvaritch, 2011; Dowling et al., 2014), and their pathogenicity is less clear.
Malignant Hyperthermia
Malignant hyperthermia is a subclinical, autosomal dominant, pharmacogenetic disorder that causes skeletal muscle intracellular Ca2+ dysregulation, characterized by a sudden and potentially fatal hypermetabolic adverse response to volatile anesthetics (e.g., isoflurane, sevoflurane, halothane) and/or the depolarizing muscle relaxant, succinylcholine (Denborough et al., 1962; Rosenberg et al., 2007; Maclennan and Zvaritch, 2011). MH manifests mostly in individuals carrying mutations in RyR1 (MacLennan et al., 1990), but some Cav1.1 mutations are also linked to MH (Rosenberg et al., 2007; Maclennan and Zvaritch, 2011). When MH-susceptible individuals are expose to trigger agent(s) (Denborough et al., 1962), they develop hypercarbia, tachycardia, muscle rigidity (some cases), rapid and severe hyperthermia (e.g., core body temperatures of 43°C), rhabdomyolysis and metabolic acidosis (Rosenberg et al., 2007; Larach et al., 2010; Maclennan and Zvaritch, 2011). MH may occur at any time during the anesthesia and in the postoperative period (Rosenberg et al., 2007). MH episodes account for many anesthetic-induced deaths in the operating room in otherwise healthy individuals (Maclennan and Zvaritch, 2011; Rosenberg et al., 2007). Untreated, episodes of MH are lethal in 90% of the cases. Consequences of MH include damage to brain, kidney and muscle tissues. In United States, MH complicates 1 out of 100,000 surgeries in adults and 1 out of 30,000 surgeries in children (Malignant Hyperthermia Association of United Sates; http://www.mhaus.org).
Different regions of RyR1 are targets of MH causing mutations (Manning et al., 1998; Barone et al., 1999; Brandt et al., 1999; Chamley et al., 2000; Rueffert et al., 2004; Betzenhauser and Marks, 2010). Dominant RyR1 mutations are the leading cause of MH. Studies on the Ca2+ conducting properties of RyR1 channels containing MH mutations expressed in myotubes (Dietze et al., 2000; Yang et al., 2003; Chelu et al., 2006; Bannister et al., 2010) and mature muscle fibers (Owen et al., 1997; Andronache et al., 2009), as well as animal models of MH (Mickelson and Louis, 1996; Chelu et al., 2006) and affected humans (Denborough et al., 1962; Melzer and Dietze, 2001), have concluded that MH mutations cause a channel gain of function, characterized by hyper-activation and hyper-sensitization to pharmacological activators. Current treatment includes immediate suspension of the triggering agent(s), cooling measures (hypothermic blankets), and 100% oxygen administration, as well as prompt treatment with dantrolene, a drug that acts as Ca2+ release inhibitor (Hainaut and Desmedt, 1974; Kobayashi et al., 2005), presumably acting on RyR1 (Fruen et al., 1997) and that requires of CaM for its effects (Oo et al., 2015). Ca2+ entry, via non-specific sarcolemmal channels, contributes to the pathogenesis of MH episodes (Eltit et al., 2013). Dantrolene also attenuates elevations on resting [Ca2+] that is dependent on Ca2+ entry (Cherednichenko et al., 2008; Eltit et al., 2013). Because dantrolene appears to suppress Ca2+ release and Ca2+ entry, it is not surprising that dantrolene could affect other proteins involved in ECC. Regardless of the action(s), dantrolene is currently the only available pharmacological treatment for MH (Mickelson and Louis, 1996; Lyfenko et al., 2004; Litman and Rosenberg, 2005) and reduces MH mortality significantly (Rosenberg et al., 2007; Schneiderbanger et al., 2014). Susceptibility to MH attacks can be detected prior to surgery using in-vitro assays that measure muscle contractile responses and halothane or caffeine sensitivity assays using muscle biopsies from individuals at risk or with family history of enhanced sensitivity to these agents, or in known individuals with MH-mutations (Rosenberg et al., 2007; Schneiderbanger et al., 2014).
The RyR1-Related Congenital Myopathies: Central Core Disease and Multi-Minicore Disease
These RyR1-related congenital myopathies are rare muscle disorders (incidence of 6 out of 100,000 births) that exhibit either autosomal dominant or recessive inheritance patterns. They are typically manifested at birth, or during childhood, and are characterized by generalized muscle weakness and low muscle tone (Magee and Shy, 1956; Shuaib et al., 1987; Jungbluth, 2007a,b). There are many forms of congenital myopathy, each defined by a specific pattern of histological abnormalities, and clinical course severity (Jungbluth, 2007a,b; Nance et al., 2012). Most individuals with congenital myopathies follow a relatively stable clinical course. Typically these patients present with ophthalmoparesis (eye muscle weakness), myasthenia (muscle weakness), and abnormal movement due to joint contractures. Patients may develop dysfunction of respiratory muscles, scoliosis, or difficulty swallowing (dysphagia; Jungbluth, 2007a,b). In comparison to MH episodes, the symptoms in CCD are not elicited by an external trigger. Currently, there is no cure for CCD; the treatment is supportive and aimed to maintain mobility and independence (Jungbluth, 2007a,b). At the cellular level, core myopathies are characterized by regions in muscle fibers devoid of mitochondria that appear as “cores” on oxidative stains (Dubowitz and Pearse, 1960; Gonatas et al., 1965). In CCD, cores are large and extend longitudinally. In MmD, the cores are short and vary in size and location (Sewry et al., 2002). Mutations in RYR1 are an important cause/factor of several forms of congenital myopathies, >60 mutations in RyR1 are linked to CCD (Quane et al., 1993; Zhang et al., 1993; Treves et al., 2008). Studies on the Ca2+ conducting properties of RyR1 channels containing CCD mutations expressed in myotubes concluded that CCD mutations cause SR Ca2+ leak due to hyperactive RyR1 channels (Tong et al., 1997; Lynch et al., 1999) or ECC uncoupling (Avila et al., 2001; Dirksen and Avila, 2002), and link these events to the development of cores and muscle weakness. The specific underlying process that causes the core formation is still unknown.
Secondary Involvement of RyR1 in Muscle Duchenne Muscular Dystrophy
DMD, a devastating and lethal inherited X-linked neuromuscular disease, is one of the most common genetic diseases (Mah et al., 2014). DMD almost exclusively affects boys, resulting in muscular dysfunction and premature death (Emery, 2002). The primary defect in DMD is the lack of dystrophin, a 427 kDa cytoskeletal protein that structurally links the contractile machinery to the dystrophin-glycoprotein complex and to components of the extracellular matrix (Hoffman et al., 1987; Campbell, 1995; Blake et al., 2002). Lack of dystrophin results in myofibers structurally defective and prone to damage. However, the absence of dystrophin alone cannot immediately account for the intricate clinical course and pathogenic mechanisms seen in DMD (Berchtold et al., 2000; Allen et al., 2010; Allen and Whitehead, 2011), and multiple hypotheses have been proposed as intervening events to explain the pathological alterations observed in DMD (Florence et al., 1985; McArdle et al., 1995; Tutdibi et al., 1999; Berchtold et al., 2000; Lovering et al., 2009a, 2013; Allen et al., 2010; Allen and Whitehead, 2011; Pratt et al., 2013, 2015; Xu et al., 2012; Mazala et al., 2015).
Regarding the involvement of RyR1 in muscular dystrophy, it is well established that dystrophin-deficient myofibers, especially those with malformed morphology (Lovering et al., 2009b; Hernández-Ochoa et al., 2015), have deficits in ECC (Collet et al., 1999; Woods et al., 2004, 2005; Hollingworth et al., 2008; Lovering et al., 2009b; Hernández-Ochoa et al., 2015), which is manifested as altered action potential-elicited RyR1 Ca2+ release from the SR (Woods et al., 2004). Several proposed mechanisms might explain the differences in ECC found between healthy and dystrophin-deficient myofibers, among them are alterations in the expression levels of Cav1.1 and RyR1, changes in Ca2+ buffering capacity (Doran et al., 2004; Dowling et al., 2004), dysfunction of membrane channels (Yeung et al., 2005), and differences in levels of ROS (Whitehead et al., 2010; Allen and Whitehead, 2011).
There is also evidence that increased S-nitrosylation of RyR1 in dystrophin deficient muscles, leads to FKBP12-RYR1 destabilization and increases RyR1 Ca2+ leak (channel opening at rest; Bellinger et al., 2009). It is likely that these modifications occur at intermediate or end stages of the transition from normal to apoptotic/necrotic fiber, where RyR1 leak will worsen Ca2+ dysregulation.
Thus, DMD appears to affect the activity of the RyR1 in two opposite ways: (1) First, in early stages of myofiber damage, or in pre-apoptotic phases, the overall function of RyR1 is reduced, leading to depressed action potential-induced RyR1 Ca2+ release (Collet et al., 1999; Woods et al., 2004, 2005; Hollingworth et al., 2008; Lovering et al., 2009b; Hernández-Ochoa et al., 2015). Since the magnitude of myofiber contraction and consequent force production are finely controlled by intracellular Ca2+ release (Chin, 2010), in DMD a gradual reduction RyR1 Ca2+ release contributes to the development of muscle weakness. (2) In advanced stages of the disease, RyR1 becomes hypersensitive to intracellular Ca2+ activation. Functional and post-translational modifications in RyR1 contribute to altered Ca2+ homeostasis in dystrophin-deficient muscles (Bellinger et al., 2009; Allen et al., 2010; Allen and Whitehead, 2011). Age-dependent S-nitrosylation of the RyR1 complex causes depletion of FKBP12 from the RyR1, resulting in a remodeling of the RyR1 channel complex, which in turn elicits intracellular SR Ca2+ leak and impaired contractility (Bellinger et al., 2009). The concurrent increased Ca2+ influx across the plasma membrane, due to inherent membrane fragility and consequent membrane damage, is thought to cause further membrane damage via multiple mechanisms including calpain activation (Berchtold et al., 2000; Allen et al., 2010; Allen and Whitehead, 2011).
Despite recent advances in understanding DMD, targeted pharmacological treatment for DMD is nearly non-existent (Andersson and Marks, 2010). In this regard, a new class of small-molecules “Rycals,” which reduce Ca2+ leak by stabilizing the FKBP12-RyR1 interaction, have been shown to improve contractility in skeletal muscle in the mdx mouse model of DMD (Bellinger et al., 2009). Rycal ARM210, has potential as a treatment for DMD; it has completed preclinical efficacy and safety studies, and has been selected for advancement to clinical development (Muscle Dystrophy Association; https://www.mda.org).
Given the critical role of RyR1 in Ca2+ release and Ca2+ signaling, any disease condition that affects RyR1 function via post-translational modifications could affect ECC, Ca2+-dependent metabolic and genetic programs, and untimely impinge on skeletal muscle function. In support of this view, recent reports have shown post-transcriptional alterations of RyR1 in muscle weakness related to arthritis (Yamada et al., 2016) and cancer (Waning et al., 2015) in murine models.
Conclusions
Many skeletal myopathies result from intracellular Ca2+ dysregulation and alterations in a myriad of Ca2+ dependent processes, including muscle contraction, signaling pathways, metabolism and gene regulation. Reduced SR Ca2+ release translates to deficient muscle contraction and muscle weakness. Sustained increases in cytoplasmic [Ca2+] or decreased SR intraluminal Ca2+ are key pathological events leading to muscle contractures (i.e., in MH episodes), apoptosis and necrotic muscle degeneration (i.e., dystrophinopathies; Maclennan and Zvaritch, 2011). In addition, many key channels, transporters, Ca2+ buffers and modulators of Ca2+ signaling contribute to abnormal Ca2+ handling in several muscle myopathies. Skeletal muscle research will continue to expand our understanding of these myopathies, as well as the function and dysfunction of the RyR1. Despite that the current near-atomic resolution RyR1 reconstructions are in the 4.8–3.8 Å range, more detailed RyR1 images are still needed, as well as specifics about RyR1's interactions with Cav1.1 and many other auxiliary and modulatory proteins (Van Petegem, 2015). Also, more in-depth function-structure studies (from molecular biophysics to systems physiology), increased sensitivity of structural biology methods and likely the development of new biochemical and physiological assays, are required. These studies will propel our knowledge about RyR1 function and dysfunction, and facilitate the development of new RyR1-selective drugs for RyR1-related myopathies.
Author Contributions
All authors contributed to this paper with literature review, manuscript drafting, editing, and final approval of the final version.
Funding
Grants to MS (R37-AR055099) and to RL (R01AR059179) from National Institutes of Health (NIH), National Institute of Arthritis and Musculoskeletal and Skin Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adrian, R. H., Chandler, W. K., and Hodgkin, A. L. (1970). Voltage clamp experiments in striated muscle fibres. J. Physiol. 208, 607–644. doi: 10.1113/jphysiol.1970.sp009139
Adrian, R. H., Costantin, L. L., and Peachey, L. D. (1969). Radial spread of contraction in frog muscle fibres. J. Physiol. 204, 231–257. doi: 10.1113/jphysiol.1969.sp008910
Adrian, R. H., and Marshall, M. W. (1977). Sodium currents in mammalian muscle. J. Physiol. 268, 223–250. doi: 10.1113/jphysiol.1977.sp011855
Adrian, R. H., and Peachey, L. D. (1973). Reconstruction of the action potential of frog sartorius muscle. J. Physiol. 235, 103–131. doi: 10.1113/jphysiol.1973.sp010380
Ahern, G. P., Junankar, P. R., and Dulhunty, A. F. (1997). Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys. J. 72, 146–162. doi: 10.1016/S0006-3495(97)78654-5
Allen, D. G., Gervasio, O. L., Yeung, E. W., and Whitehead, N. P. (2010). Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 88, 83–91. doi: 10.1139/Y09-058
Allen, D. G., and Whitehead, N. P. (2011). Duchenne muscular dystrophy–what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 43, 290–294. doi: 10.1016/j.biocel.2010.11.005
Andersson, D. C., and Marks, A. R. (2010). Fixing ryanodine receptor Ca leak - a novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov. Today Dis. Mech. 7, e151–e157. doi: 10.1016/j.ddmec.2010.09.009
Andronache, Z., Hamilton, S. L., Dirksen, R. T., and Melzer, W. (2009). A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2+ release in muscle fibers of Y522S RyR1 knock-in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 4531–4536. doi: 10.1073/pnas.0812661106
Aracena, P., Sanchez, G., Donoso, P., Hamilton, S. L., and Hidalgo, C. (2003). S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J. Biol. Chem. 278, 42927–42935. doi: 10.1074/jbc.M306969200
Avila, G., O'Brien, J. J., and Dirksen, R. T. (2001). Excitation–contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc. Natl. Acad. Sci. U.S.A. 98, 4215–4220. doi: 10.1073/pnas.071048198
Bannister, R. A., Esteve, E., Eltit, J. M., Pessah, I. N., Allen, P. D., Lopez, J. R., et al. (2010). A malignant hyperthermia-inducing mutation in RYR1 (R163C): consequent alterations in the functional properties of DHPR channels. J. Gen. Physiol. 135, 629–640. doi: 10.1085/jgp.200910329
Barone, V., Massa, O., Intravaia, E., Bracco, A., Di Martino, A., Tegazzin, V., et al. (1999). Mutation screening of the RYR1 gene and identification of two novel mutations in Italian malignant hyperthermia families. J. Med. Genet. 36, 115–118.
Beam, K. G., and Horowicz, P. (2004). Excitation-Contraction Coupling in Skeletal Muscle. New York, NY: McGraw-Hill.
Beard, N. A., Wei, L., and Dulhunty, A. F. (2009). Ca(2+) signaling in striated muscle: the elusive roles of triadin, junctin, and calsequestrin. Eur. Biophys. J. 39, 27–36. doi: 10.1007/s00249-009-0449-6
Bellinger, A. M., Reiken, S., Carlson, C., Mongillo, M., Liu, X., Rothman, L., et al. (2009). Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330. doi: 10.1038/nm.1916
Berchtold, M. W., Brinkmeier, H., and Muntener, M. (2000). Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 80, 1215–1265. Available online at: http://physrev.physiology.org/content/physrev/80/3/1215.full.pdf
Betzenhauser, M. J., and Marks, A. R. (2010). Ryanodine receptor channelopathies. Pflugers Arch. 460, 467–480. doi: 10.1007/s00424-010-0794-4
Beurg, M., Ahern, C. A., Vallejo, P., Conklin, M. W., Powers, P. A., Gregg, R. G., et al. (1999a). Involvement of the carboxy-terminus region of the dihydropyridine receptor beta1a subunit in excitation-contraction coupling of skeletal muscle. Biophys. J. 77, 2953–2967.
Beurg, M., Sukhareva, M., Ahern, C. A., Conklin, M. W., Perez-Reyes, E., Powers, P. A., et al. (1999b). Differential regulation of skeletal muscle L-type Ca2+ current and excitation-contraction coupling by the dihydropyridine receptor beta subunit. Biophys. J. 76, 1744–1756. doi: 10.1016/S0006-3495(99)77336-4
Blake, D. J., Weir, A., Newey, S. E., and Davies, K. E. (2002). Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 82, 291–329. doi: 10.1152/physrev.00028.2001
Block, B. A., Imagawa, T., Campbell, K. P., and Franzini-Armstrong, C. (1988). Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107, 2587–2600. doi: 10.1083/jcb.107.6.2587
Brandt, A., Schleithoff, L., Jurkat-Rott, K., Klingler, W., Baur, C., and Lehmann-Horn, F. (1999). Screening of the ryanodine receptor gene in 105 malignant hyperthermia families: novel mutations and concordance with the in vitro contracture test. Hum. Mol. Genet. 8, 2055–2062. doi: 10.1093/hmg/8.11.2055
Brillantes, A. B., Ondrias, K., Scott, A., Kobrinsky, E., Ondriasova, E., Moschella, M. C., et al. (1994). Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77, 513–523. doi: 10.1016/0092-8674(94)90214-3
Buratti, R., Prestipino, G., Menegazzi, P., Treves, S., and Zorzato, F. (1995). Calcium dependent activation of skeletal muscle Ca2+ release channel (ryanodine receptor) by calmodulin. Biochem. Biophys. Res. Commun. 213, 1082–1090. doi: 10.1006/bbrc.1995.2238
Burkhard, P., Taylor, P., and Walkinshaw, M. D. (2000). X-ray structures of small ligand-FKBP complexes provide an estimate for hydrophobic interaction energies. J. Mol. Biol. 295, 953–962. doi: 10.1006/jmbi.1999.3411
Campbell, K. P. (1995). Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80, 675–679. doi: 10.1016/0092-8674(95)90344-5
Chamley, D., Pollock, N. A., Stowell, K. M., and Brown, R. L. (2000). Malignant hyperthermia in infancy and identification of novel RYR1 mutation. Br. J. Anaesth. 84, 500–504. doi: 10.1093/oxfordjournals.bja.a013478
Chattopadhyaya, R., Meador, W. E., Means, A. R., and Quiocho, F. A. (1992). Calmodulin structure refined at 1.7 A resolution. J. Mol. Biol. 228, 1177–1192. doi: 10.1016/0022-2836(92)90324-D
Chelu, M. G., Danila, C. I., Gilman, C. P., and Hamilton, S. L. (2004). Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc. Med. 14, 227–234. doi: 10.1016/j.tcm.2004.06.003
Chelu, M. G., Goonasekera, S. A., Durham, W. J., Tang, W., Lueck, J. D., Riehl, J., et al. (2006). Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 20, 329–330. doi: 10.1096/fj.05-4497fje
Cherednichenko, G., Ward, C. W., Feng, W., Cabrales, E., Michaelson, L., Samso, M., et al. (2008). Enhanced excitation-coupled calcium entry in myotubes expressing malignant hyperthermia mutation R163C is attenuated by dantrolene. Mol. Pharmacol. 73, 1203–1212. doi: 10.1124/mol.107.043299
Chin, E. R. (2010). Intracellular Ca2+ signaling in skeletal muscle: decoding a complex message. Exerc. Sport Sci. Rev. 38, 76–85. doi: 10.1097/JES.0b013e3181d495d2
Collet, C., Allard, B., Tourneur, Y., and Jacquemond, V. (1999). Intracellular calcium signals measured with indo-1 in isolated skeletal muscle fibres from control and mdx mice. J. Physiol. 520(Pt 2), 417–429. doi: 10.1111/j.1469-7793.1999.00417.x
Denborough, M. A., Forster, J. F., Lovell, R. R., Maplestone, P. A., and Villiers, J. D. (1962). Anaesthetic deaths in a family. Br. J. Anaesth. 34, 395–396. doi: 10.1093/bja/34.6.395
Dietze, B., Henke, J., Eichinger, H. M., Lehmann-Horn, F., and Melzer, W. (2000). Malignant hyperthermia mutation Arg615Cys in the porcine ryanodine receptor alters voltage dependence of Ca2+ release. J. Physiol. 526(Pt 3), 507–514. doi: 10.1111/j.1469-7793.2000.t01-1-00507.x
Dirksen, R. T. (2002). Bi-directional coupling between dihydropyridine receptors and ryanodine receptors. Front. Biosci. 7, d659–d670. doi: 10.2741/dirksen
Dirksen, R. T., and Avila, G. (2002). Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca(2+) release channels? Trends Cardiovasc. Med. 12, 189–197. doi: 10.1016/S1050-1738(02)00163-9
Doran, P., Dowling, P., Lohan, J., McDonnell, K., Poetsch, S., and Ohlendieck, K. (2004). Subproteomics analysis of Ca+-binding proteins demonstrates decreased calsequestrin expression in dystrophic mouse skeletal muscle. Eur. J. Biochem. 271, 3943–3952. doi: 10.1111/j.1432-1033.2004.04332.x
Dowling, J. J., Lawlor, M. W., and Dirksen, R. T. (2014). Triadopathies: an emerging class of skeletal muscle diseases. Neurotherapeutics 11, 773–785. doi: 10.1007/s13311-014-0300-3
Dowling, P., Doran, P., and Ohlendieck, K. (2004). Drastic reduction of sarcalumenin in Dp427 (dystrophin of 427 kDa)-deficient fibres indicates that abnormal calcium handling plays a key role in muscular dystrophy. Biochem. J. 379, 479–488. doi: 10.1042/bj20031311
Dubowitz, V., and Pearse, A. G. (1960). Oxidative enzymes and phosphorylase in central-core disease of muscle. Lancet 2, 23–24. doi: 10.1016/S0140-6736(60)92665-9
Dum, R. P., and Strick, P. L. (2002). Motor areas in the frontal lobe of the primate. Physiol. Behav. 77, 677–682. doi: 10.1016/S0031-9384(02)00929-0
Eltit, J. M., Ding, X., Pessah, I. N., Allen, P. D., and Lopez, J. R. (2013). Nonspecific sarcolemmal cation channels are critical for the pathogenesis of malignant hyperthermia. FASEB J. 27, 991–1000. doi: 10.1096/fj.12-218354
Emery, A. E. (2002). The muscular dystrophies. Lancet 359, 687–695. doi: 10.1016/S0140-6736(02)07815-7
Endo, M. (2006). Calcium ion as a second messenger with special reference to excitation-contraction coupling. J. Pharmacol. Sci. 100, 519–524. doi: 10.1254/jphs.CPJ06004X
Endo, M. (2009). Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 89, 1153–1176. doi: 10.1152/physrev.00040.2008
Eu, J. P., Sun, J., Xu, L., Stamler, J. S., and Meissner, G. (2000). The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell 102, 499–509. doi: 10.1016/S0092-8674(00)00054-4
Figueroa, L., Shkryl, V. M., Zhou, J., Manno, C., Momotake, A., Brum, G., et al. (2012). Synthetic localized calcium transients directly probe signalling mechanisms in skeletal muscle. J. Physiol. 590, 1389–1411. doi: 10.1113/jphysiol.2011.225854
Fill, M., and Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922. doi: 10.1152/physrev.00013.2002
Florence, J. M., Fox, P. T., Planer, G. J., and Brooke, M. H. (1985). Activity, creatine kinase, and myoglobin in Duchenne muscular dystrophy: a clue to etiology? Neurology 35, 758–761.
Franzini-Armstrong, C., and Jorgensen, A. O. (1994). Structure and development of E-C coupling units in skeletal muscle. Annu. Rev. Physiol. 56, 509–534. doi: 10.1146/annurev.ph.56.030194.002453
Franzini-Armstrong, C., and Kish, J. W. (1995). Alternate disposition of tetrads in peripheral couplings of skeletal muscle. J. Muscle Res. Cell Motil. 16, 319–324. doi: 10.1007/BF00121140
Franzini-Armstrong, C., and Nunzi, G. (1983). Junctional feet and particles in the triads of a fast-twitch muscle fibre. J. Muscle Res. Cell Motil. 4, 233–252. doi: 10.1007/BF00712033
Franzini-Armstrong, C., and Porter, K. R. (1964). Sarcolemmal Invaginations Constituting the T System in Fish Muscle Fibers. J. Cell Biol. 22, 675–696. doi: 10.1083/jcb.22.3.675
Franzini-Armstrong, C., and Protasi, F. (1997). Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77, 699–729.
Franzini-Armstrong, C., Protasi, F., and Ramesh, V. (1998). Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann. N.Y. Acad. Sci. 853, 20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x
Fruen, B. R., Mickelson, J. R., and Louis, C. F. (1997). Dantrolene inhibition of sarcoplasmic reticulum Ca2+ release by direct and specific action at skeletal muscle ryanodine receptors. J. Biol. Chem. 272, 26965–26971. doi: 10.1074/jbc.272.43.26965
Fuentes, O., Valdivia, C., Vaughan, D., Coronado, R., and Valdivia, H. H. (1994). Calcium-dependent block of ryanodine receptor channel of swine skeletal muscle by direct binding of calmodulin. Cell Calcium 15, 305–316. doi: 10.1016/0143-4160(94)90070-1
Georgiou, D. K., Dagnino-Acosta, A., Lee, C. S., Griffin, D. M., Wang, H., Lagor, W. R., et al. (2015). Ca2+ binding/permeation via calcium channel, CaV1.1, regulates the intracellular distribution of the fatty acid transport protein, CD36, and fatty acid metabolism. J. Biol. Chem. 290, 23751–23765. doi: 10.1074/jbc.M115.643544
Glover, L., Quinn, S., Ryan, M., Pette, D., and Ohlendieck, K. (2002). Supramolecular calsequestrin complex. Eur. J. Biochem. 269, 4607–4616. doi: 10.1046/j.1432-1033.2002.03160.x
Gonatas, N. K., Perez, M. C., Shy, G. M., and Evangelista, I. (1965). Central “core” disease of skeletal muscle. ultrastructural and cytochemical observations in two cases. Am. J. Pathol. 47, 503–524.
Green, N. M., and MacLennan, D. H. (2002). Calcium callisthenics. Nature 418, 598–599. doi: 10.1038/418598b
Gregg, R. G., Messing, A., Strube, C., Beurg, M., Moss, R., Behan, M., et al. (1996). Absence of the beta subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the alpha 1 subunit and eliminates excitation-contraction coupling. Proc. Natl. Acad. Sci. U.S.A. 93, 13961–13966. doi: 10.1073/pnas.93.24.13961
Hainaut, K., and Desmedt, J. E. (1974). Effect of dantrolene sodium on calcium movements in single muscle fibres. Nature 252, 728–730. doi: 10.1038/252728a0
Hasselbach, W. (1964). ATP-driven active transport of calcium in the membranes of the sarcoplasmic reticulum. Proc. R. Soc. Lond. B Biol. Sci. 160, 501–504. doi: 10.1098/rspb.1964.0064
Hernández-Ochoa, E. O., Pratt, S. J., Garcia-Pelagio, K. P., Schneider, M. F., and Lovering, R. M. (2015). Disruption of action potential and calcium signaling properties in malformed myofibers from dystrophin-deficient mice. Physiol. Rep. 3:e12366. doi: 10.14814/phy2.12366
Hidalgo, C., Donoso, P., and Carrasco, M. A. (2005). The ryanodine receptors Ca2+ release channels: cellular redox sensors? IUBMB Life 57, 315–322. doi: 10.1080/15216540500092328
Hoffman, E. P., Brown, R. H. Jr., and Kunkel, L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. doi: 10.1016/0092-8674(87)90579-4
Hollingworth, S., Zeiger, U., and Baylor, S. M. (2008). Comparison of the myoplasmic calcium transient elicited by an action potential in intact fibres of mdx and normal mice. J. Physiol. 586, 5063–5075. doi: 10.1113/jphysiol.2008.160507
Hu, H., Wang, Z., Wei, R., Fan, G., Wang, Q., Zhang, K., et al. (2015). The molecular architecture of dihydropyrindine receptor/L-type Ca2+ channel complex. Sci. Rep. 5:8370. doi: 10.1038/srep08370
Hubbard, J. I. (1973). Microphysiology of vertebrate neuromuscular transmission. Physiol. Rev. 53, 674–723.
Huxley, A. F., and Taylor, R. E. (1958). Local activation of striated muscle fibres. J. Physiol. 144, 426–441. doi: 10.1113/jphysiol.1958.sp006111
Ikemoto, T., Iino, M., and Endo, M. (1995). Enhancing effect of calmodulin on Ca(2+)-induced Ca2+ release in the sarcoplasmic reticulum of rabbit skeletal muscle fibres. J. Physiol. 487(Pt 3), 573–582.
Imagawa, T., Smith, J. S., Coronado, R., and Campbell, K. P. (1987). Purified ryanodine receptor from skeletal muscle sarcoplasmic reticulum is the Ca2+-permeable pore of the calcium release channel. J. Biol. Chem. 262, 16636–16643.
Inui, M., Saito, A., and Fleischer, S. (1987). Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J. Biol. Chem. 262, 1740–1747.
Jungbluth, H. (2007a). Multi-minicore Disease. Orphanet J. Rare Dis. 2:31. doi: 10.1186/1750-1172-2-31
Jungbluth, H. (2007b). Central core disease. Orphanet J. Rare Dis. 2, 25. doi: 10.1186/1750-1172-2-25
Jurkat-Rott, K., Fauler, M., and Lehmann-Horn, F. (2006). Ion channels and ion transporters of the transverse tubular system of skeletal muscle. J. Muscle Res. Cell Motil. 27, 275–290. doi: 10.1007/s10974-006-9088-z
Katz, B. (1996). Neural transmitter release: from quantal secretion to exocytosis and beyond. The Fenn Lecture. J. Neurocytol. 25, 677–686. doi: 10.1007/BF02284834
Kobayashi, S., Bannister, M. L., Gangopadhyay, J. P., Hamada, T., Parness, J., and Ikemoto, N. (2005). Dantrolene stabilizes domain interactions within the ryanodine receptor. J. Biol. Chem. 280, 6580–6587. doi: 10.1074/jbc.M408375200
Kraeva, N., Zvaritch, E., Rossi, A. E., Goonasekera, S. A., Zaid, H., Frodis, W., et al. (2013). Novel excitation-contraction uncoupled RYR1 mutations in patients with central core disease. Neuromuscul. Disord. 23, 120–132. doi: 10.1016/j.nmd.2012.08.007
Lanner, J. T. (2012). Ryanodine receptor physiology and its role in disease. Adv. Exp. Med. Biol. 740, 217–234. doi: 10.1007/978-94-007-2888-2_9
Lanner, J. T., Georgiou, D. K., Joshi, A. D., and Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2:a003996. doi: 10.1101/cshperspect.a003996
Larach, M. G., Gronert, G. A., Allen, G. C., Brandom, B. W., and Lehman, E. B. (2010). Clinical presentation, treatment, and complications of malignant hyperthermia in North America from 1987 to 2006. Anesth. Analg. 110, 498–507. doi: 10.1213/ANE.0b013e3181c6b9b2
Lee, C. S., Dagnino-Acosta, A., Yarotskyy, V., Hanna, A., Lyfenko, A., Knoblauch, M., et al. (2015). Ca(2+) permeation and/or binding to CaV1.1 fine-tunes skeletal muscle Ca(2+) signaling to sustain muscle function. Skelet. Muscle 5, 4. doi: 10.1186/s13395-014-0027-1
Litman, R. S., and Rosenberg, H. (2005). Malignant hyperthermia: update on susceptibility testing. JAMA 293, 2918–2924. doi: 10.1001/jama.293.23.2918
Lovering, R. M., McMillan, A. B., and Gullapalli, R. P. (2009a). Location of myofiber damage in skeletal muscle after lengthening contractions. Muscle Nerve 40, 589–594. doi: 10.1002/mus.21389
Lovering, R. M., Michaelson, L., and Ward, C. W. (2009b). Malformed mdx myofibers have normal cytoskeletal architecture yet altered EC coupling and stress-induced Ca2+ signaling. Am. J. Physiol. Cell Physiol. 297, C571–C580. doi: 10.1152/ajpcell.00087.2009
Lovering, R. M., Shah, S. B., Pratt, S. J., Gong, W., and Chen, Y. (2013). Architecture of healthy and dystrophic muscles detected by optical coherence tomography. Muscle Nerve 47, 588–590. doi: 10.1002/mus.23711
Loy, R. E., Orynbayev, M., Xu, L., Andronache, Z., Apostol, S., Zvaritch, E., et al. (2011). Muscle weakness in Ryr1I4895T/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ ion permeation and release from the sarcoplasmic reticulum. J. Gen. Physiol. 137, 43–57. doi: 10.1085/jgp.201010523
Ludtke, S. J., Serysheva, I. I., Hamilton, S. L., and Chiu, W. (2005). The pore structure of the closed RyR1 channel. Structure 13, 1203–1211. doi: 10.1016/j.str.2005.06.005
Lyfenko, A. D., Goonasekera, S. A., and Dirksen, R. T. (2004). Dynamic alterations in myoplasmic Ca2+ in malignant hyperthermia and central core disease. Biochem. Biophys. Res. Commun. 322, 1256–1266. doi: 10.1016/j.bbrc.2004.08.031
Lynch, P. J., Tong, J., Lehane, M., Mallet, A., Giblin, L., Heffron, J. J., et al. (1999). A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. U.S.A. 96, 4164–4169. doi: 10.1073/pnas.96.7.4164
MacLennan, D. H., Duff, C., Zorzato, F., Fujii, J., Phillips, M., Korneluk, R. G., et al. (1990). Ryanodine receptor gene is a candidate for predisposition to malignant hyperthermia. Nature 343, 559–561. doi: 10.1038/343559a0
MacLennan, D. H., and Wong, P. T. (1971). Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 68, 1231–1235. doi: 10.1073/pnas.68.6.1231
Maclennan, D. H., and Zvaritch, E. (2011). Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim. Biophys. Acta 1813, 948–964. doi: 10.1016/j.bbamcr.2010.11.009
Magee, K. R., and Shy, G. M. (1956). A new congenital non-progressive myopathy. Brain 79, 610–621. doi: 10.1093/brain/79.4.610
Mah, J. K., Korngut, L., Dykeman, J., Day, L., Pringsheim, T., and Jette, N. (2014). A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 24, 482–491. doi: 10.1016/j.nmd.2014.03.008
Manning, B. M., Quane, K. A., Lynch, P. J., Urwyler, A., Tegazzin, V., Krivosic-Horber, R., et al. (1998). Novel mutations at a CpG dinucleotide in the ryanodine receptor in malignant hyperthermia. Hum. Mutat. 11, 45–50.
Marengo, J. J., Hidalgo, C., and Bull, R. (1998). Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys. J. 74, 1263–1277. doi: 10.1016/S0006-3495(98)77840-3
Marques, M. J., Conchello, J. A., and Lichtman, J. W. (2000). From plaque to pretzel: fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J. Neurosci. 20, 3663–3675. Available online at: http://www.jneurosci.org/content/20/10/3663.full.pdf
Marx, S. O., Ondrias, K., and Marks, A. R. (1998). Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281, 818–821. doi: 10.1126/science.281.5378.818
Mazala, D. A., Pratt, S. J., Chen, D., Molkentin, J. D., Lovering, R. M., and Chin, E. R. (2015). SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol. 308, C699–C709. doi: 10.1152/ajpcell.00341.2014
McArdle, A., Edwards, R. H., and Jackson, M. J. (1995). How does dystrophin deficiency lead to muscle degeneration?–evidence from the mdx mouse. Neuromuscul. Disord. 5, 445–456. doi: 10.1016/0960-8966(95)00001-4
Meissner, G., Darling, E., and Eveleth, J. (1986). Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry 25, 236–244. doi: 10.1021/bi00349a033
Melzer, W., and Dietze, B. (2001). Malignant hyperthermia and excitation-contraction coupling. Acta Physiol. Scand. 171, 367–378. doi: 10.1046/j.1365-201x.2001.00840.x
Melzer, W., Herrmann-Frank, A., and Luttgau, H. C. (1995). The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta 1241, 59–116. doi: 10.1016/0304-4157(94)00014-5
Mickelson, J. R., and Louis, C. F. (1996). Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol. Rev. 76, 537–592.
Nakai, J., Adams, B. A., Imoto, K., and Beam, K. G. (1994). Critical roles of the S3 segment and S3-S4 linker of repeat I in activation of L-type calcium channels. Proc. Natl. Acad. Sci. U.S.A. 91, 1014–1018. doi: 10.1073/pnas.91.3.1014
Nakai, J., Dirksen, R. T., Nguyen, H. T., Pessah, I. N., Beam, K. G., and Allen, P. D. (1996). Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75. doi: 10.1038/380072a0
Nakai, J., Sekiguchi, N., Rando, T. A., Allen, P. D., and Beam, K. G. (1998a). Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J. Biol. Chem. 273, 13403–13406.
Nakai, J., Tanabe, T., Konno, T., Adams, B., and Beam, K. G. (1998b). Localization in the II-III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 273, 24983–24986.
Nance, J. R., Dowling, J. J., Gibbs, E. M., and Bonnemann, C. G. (2012). Congenital myopathies: an update. Curr. Neurol. Neurosci. Rep. 12, 165–174. doi: 10.1007/s11910-012-0255-x
Oo, Y. W., Gomez-Hurtado, N., Walweel, K., van Helden, D. F., Imtiaz, M. S., Knollmann, B. C., et al. (2015). Essential Role of Calmodulin in RyR Inhibition by Dantrolene. Mol. Pharmacol. 88, 57–63. doi: 10.1124/mol.115.097691
Owen, V. J., Taske, N. L., and Lamb, G. D. (1997). Reduced Mg2+ inhibition of Ca2+ release in muscle fibers of pigs susceptible to malignant hyperthermia. Am. J. Physiol. 272, C203–C211.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pratt, S. J., Shah, S. B., Ward, C. W., Inacio, M. P., Stains, J. P., and Lovering, R. M. (2013). Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J. Physiol. 591, 559–570. doi: 10.1113/jphysiol.2012.241679
Pratt, S. J., Valencia, A. P., Le, G. K., Shah, S. B., and Lovering, R. M. (2015). Pre- and postsynaptic changes in the neuromuscular junction in dystrophic mice. Front. Physiol. 6:252. doi: 10.3389/fphys.2015.00252
Prosser, B. L., Wright, N. T., Hernández-Ochoa, E. O., Varney, K. M., Liu, Y., Olojo, R. O., et al. (2008). S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J. Biol. Chem. 283, 5046–5057. doi: 10.1074/jbc.M709231200
Protasi, F., Paolini, C., Nakai, J., Beam, K. G., Franzini-Armstrong, C., and Allen, P. D. (2002). Multiple regions of RyR1 mediate functional and structural interactions with alpha(1S)-dihydropyridine receptors in skeletal muscle. Biophys. J. 83, 3230–3244. doi: 10.1016/S0006-3495(02)75325-3
Quane, K. A., Healy, J. M., Keating, K. E., Manning, B. M., Couch, F. J., Palmucci, L. M., et al. (1993). Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. 5, 51–55. doi: 10.1038/ng0993-51
Radermacher, M., Wagenknecht, T., Grassucci, R., Frank, J., Inui, M., Chadwick, C., et al. (1992). Cryo-EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys. J. 61, 936–940. doi: 10.1016/S0006-3495(92)81900-8
Rios, E., and Brum, G. (1987). Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature 325, 717–720. doi: 10.1038/325717a0
Rizzolatti, G., and Luppino, G. (2001). The cortical motor system. Neuron 31, 889–901. doi: 10.1016/S0896-6273(01)00423-8
Rosenberg, H., Davis, M., James, D., Pollock, N., and Stowell, K. (2007). Malignant hyperthermia. Orphanet J. Rare Dis. 2, 21. doi: 10.1186/1750-1172-2-21
Rueffert, H., Olthoff, D., Deutrich, C., Schober, R., and Froster, U. G. (2004). A new mutation in the skeletal ryanodine receptor gene (RYR1) is potentially causative of malignant hyperthermia, central core disease, and severe skeletal malformation. Am. J. Med. Genet. A 124A, 248–254. doi: 10.1002/ajmg.a.20404
Samso, M., Feng, W., Pessah, I. N., and Allen, P. D. (2009). Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7:e85. doi: 10.1371/journal.pbio.1000085
Samso, M., Shen, X., and Allen, P. D. (2006). Structural characterization of the RyR1-FKBP12 interaction. J. Mol. Biol. 356, 917–927. doi: 10.1016/j.jmb.2005.12.023
Samso, M., and Wagenknecht, T. (2002). Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 277, 1349–1353. doi: 10.1074/jbc.M109196200
Samso, M., Wagenknecht, T., and Allen, P. D. (2005). Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat. Struct. Mol. Biol. 12, 539–544. doi: 10.1038/nsmb938
Sanchez, E. J., Lewis, K. M., Danna, B. R., and Kang, C. (2012). High-capacity Ca2+ binding of human skeletal calsequestrin. J. Biol. Chem. 287, 11592–11601. doi: 10.1074/jbc.M111.335075
Sanchez, J. A., and Stefani, E. (1978). Inward calcium current in twitch muscle fibres of the frog. J. Physiol. 283, 197–209. doi: 10.1113/jphysiol.1978.sp012496
Sandow, A. (1952). Excitation-contraction coupling in muscular response. Yale J. Biol. Med. 25, 176–201.
Schneider, M. F., and Chandler, W. K. (1973). Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature 242, 244–246. doi: 10.1038/242244a0
Schneider, M. F., and Hernández-Ochoa, E. O. (2012). “Skeletal muscle excitation-contraction coupling,” in Muscle: Fundamental Biology and Mechanisms of Disease, eds J. A. Hill and E. Olson (London: Academic Press: Elsevier), 811–822.
Schneider, M. F., and Simon, B. J. (1988). Inactivation of calcium release from the sarcoplasmic reticulum in frog skeletal muscle. J. Physiol. 405, 727–745. doi: 10.1113/jphysiol.1988.sp017358
Schneiderbanger, D., Johannsen, S., Roewer, N., and Schuster, F. (2014). Management of malignant hyperthermia: diagnosis and treatment. Ther. Clin. Risk Manag. 10, 355–362. doi: 10.2147/TCRM.S47632
Serysheva, I. I., Hamilton, S. L., Chiu, W., and Ludtke, S. J. (2005). Structure of Ca2+ release channel at 14 A resolution. J. Mol. Biol. 345, 427–431. doi: 10.1016/j.jmb.2004.10.073
Serysheva, I. I., Ludtke, S. J., Baker, M. L., Cong, Y., Topf, M., Eramian, D., et al. (2008). Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proc. Natl. Acad. Sci. U.S.A. 105, 9610–9615. doi: 10.1073/pnas.0803189105
Serysheva, I. I., Orlova, E. V., Chiu, W., Sherman, M. B., Hamilton, S. L., and van Heel, M. (1995). Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat. Struct. Biol. 2, 18–24. doi: 10.1038/nsb0195-18
Sewry, C. A., Muller, C., Davis, M., Dwyer, J. S., Dove, J., Evans, G., et al. (2002). The spectrum of pathology in central core disease. Neuromuscul. Disord. 12, 930–938. doi: 10.1016/S0960-8966(02)00135-9
Sheridan, D. C., Takekura, H., Franzini-Armstrong, C., Beam, K. G., Allen, P. D., and Perez, C. F. (2006). Bidirectional signaling between calcium channels of skeletal muscle requires multiple direct and indirect interactions. Proc. Natl. Acad. Sci. U.S.A. 103, 19760–19765. doi: 10.1073/pnas.0609473103
Shuaib, A., Paasuke, R. T., and Brownell, K. W. (1987). Central core disease. Clinical features in 13 patients. Medicine (Baltimore) 66, 389–396. doi: 10.1097/00005792-198709000-00004
Sonnleitner, A., Fleischer, S., and Schindler, H. (1997). Gating of the skeletal calcium release channel by ATP is inhibited by protein phosphatase 1 but not by Mg2+. Cell Calcium 21, 283–290. doi: 10.1016/S0143-4160(97)90116-0
Stanfield, P. R. (1977). A calcium dependent inward current in frog skeletal muscle fibres. Pflugers Arch. 368, 267–270. doi: 10.1007/BF00585206
Sun, J., Xu, L., Eu, J. P., Stamler, J. S., and Meissner, G. (2001). Classes of thiols that influence the activity of the skeletal muscle calcium release channel. J. Biol. Chem. 276, 15625–15630. doi: 10.1074/jbc.M100083200
Takeshima, H., Nishimura, S., Matsumoto, T., Ishida, H., Kangawa, K., Minamino, N., et al. (1989). Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature 339, 439–445. doi: 10.1038/339439a0
Tang, L., Gamal El-Din, T. M., Payandeh, J., Martinez, G. Q., Heard, T. M., Scheuer, T., et al. (2014). Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 505, 56–61. doi: 10.1038/nature12775
Tong, J., Oyamada, H., Demaurex, N., Grinstein, S., McCarthy, T. V., and MacLennan, D. H. (1997). Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J. Biol. Chem. 272, 26332–26339. doi: 10.1074/jbc.272.42.26332
Treves, S., Jungbluth, H., Muntoni, F., and Zorzato, F. (2008). Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm. Curr. Opin. Pharmacol. 8, 319–326. doi: 10.1016/j.coph.2008.01.005
Treves, S., Vukcevic, M., Maj, M., Thurnheer, R., Mosca, B., and Zorzato, F. (2009). Minor sarcoplasmic reticulum membrane components that modulate excitation-contraction coupling in striated muscles. J. Physiol. 587, 3071–3079. doi: 10.1113/jphysiol.2009.171876
Tripathy, A., Xu, L., Mann, G., and Meissner, G. (1995). Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. 69, 106–119. doi: 10.1016/S0006-3495(95)79880-0
Tutdibi, O., Brinkmeier, H., Rudel, R., and Fohr, K. J. (1999). Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 515(Pt 3), 859–868.
Vallejo-Illarramendi, A., Toral-Ojeda, I., Aldanondo, G., and Lopez de Munain, A. (2014). Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 16:e16. doi: 10.1017/erm.2014.17
Van Petegem, F. (2015). Ryanodine receptors: allosteric ion channel giants. J. Mol. Biol. 427, 31–53. doi: 10.1016/j.jmb.2014.08.004
Van Petegem, F., Clark, K. A., Chatelain, F. C., and Minor, D. L. Jr. (2004). Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429, 671–675. doi: 10.1038/nature02588
Waning, D. L., Mohammad, K. S., Reiken, S., Xie, W., Andersson, D. C., John, S., et al. (2015). Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 21, 1262–1271. doi: 10.1038/nm.3961
Whitehead, N. P., Yeung, E. W., Froehner, S. C., and Allen, D. G. (2010). Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS ONE 5:e15354. doi: 10.1371/journal.pone.0015354
Woods, C. E., Novo, D., DiFranco, M., Capote, J., and Vergara, J. L. (2005). Propagation in the transverse tubular system and voltage dependence of calcium release in normal and mdx mouse muscle fibres. J. Physiol. 568, 867–880. doi: 10.1113/jphysiol.2005.089318
Woods, C. E., Novo, D., DiFranco, M., and Vergara, J. L. (2004). The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J. Physiol. 557, 59–75. doi: 10.1113/jphysiol.2004.061291
Wright, N. T., Cannon, B. R., Zimmer, D. B., and Weber, D. J. (2009). S100A1: structure, Function, and Therapeutic Potential. Curr. Chem. Biol. 3, 138–145. doi: 10.2174/2212796810903020138
Wright, N. T., Prosser, B. L., Varney, K. M., Zimmer, D. B., Schneider, M. F., and Weber, D. J. (2008). S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J. Biol. Chem. 283, 26676–26683. doi: 10.1074/jbc.M804432200
Wu, H., Xiong, W. C., and Mei, L. (2010). To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137, 1017–1033. doi: 10.1242/dev.038711
Xu, S., Pratt, S. J., Spangenburg, E. E., and Lovering, R. M. (2012). Early metabolic changes measured by 1H MRS in healthy and dystrophic muscle after injury. J. Appl. Physiol. (1985) 113, 808–816. doi: 10.1152/japplphysiol.00530.2012
Yamada, T., Fedotovskaya, O., Cheng, A. J., Cornachione, A. S., Minozzo, F. C., Aulin, C., et al. (2015). Nitrosative modifications of the Ca2+ release complex and actin underlie arthritis-induced muscle weakness. Ann. Rheum Dis. 74, 1907–1914. doi: 10.1136/annrheumdis-2013-205007
Yan, Z., Bai, X. C., Yan, C., Wu, J., Li, Z., Xie, T., et al. (2015). Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55. doi: 10.1038/nature14063
Yang, T., Esteve, E., Pessah, I. N., Molinski, T. F., Allen, P. D., and Lopez, J. R. (2007). Elevated resting [Ca(2+)](i) in myotubes expressing malignant hyperthermia RyR1 cDNAs is partially restored by modulation of passive calcium leak from the SR. Am. J. Physiol. Cell Physiol. 292, C1591–C1598. doi: 10.1152/ajpcell.00133.2006
Yang, T., Ta, T. A., Pessah, I. N., and Allen, P. D. (2003). Functional defects in six ryanodine receptor isoform-1 (RyR1) mutations associated with malignant hyperthermia and their impact on skeletal excitation-contraction coupling. J. Biol. Chem. 278, 25722–25730. doi: 10.1074/jbc.M302165200
Yeung, E. W., Whitehead, N. P., Suchyna, T. M., Gottlieb, P. A., Sachs, F., and Allen, D. G. (2005). Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 562, 367–380. doi: 10.1113/jphysiol.2004.075275
Yin, C. C., and Lai, F. A. (2000). Intrinsic lattice formation by the ryanodine receptor calcium-release channel. Nat. Cell Biol. 2, 669–671. doi: 10.1038/35023625
Zalk, R., Clarke, O. B., des Georges, A., Grassucci, R. A., Reiken, S., Mancia, F., et al. (2015). Structure of a mammalian ryanodine receptor. Nature 517, 44–49. doi: 10.1038/nature13950
Zhang, Y., Chen, H. S., Khanna, V. K., De Leon, S., Phillips, M. S., Schappert, K., et al. (1993). A mutation in the human ryanodine receptor gene associated with central core disease. Nat. Genet. 5, 46–50. doi: 10.1038/ng0993-46
Zvaritch, E., Kraeva, N., Bombardier, E., McCloy, R. A., Depreux, F., Holmyard, D., et al. (2009). Ca2+ dysregulation in Ryr1(I4895T/wt) mice causes congenital myopathy with progressive formation of minicores, cores, and nemaline rods. Proc. Natl. Acad. Sci. U.S.A. 106, 21813–21818. doi: 10.1073/pnas.0912126106
Keywords: skeletal muscle, excitation-contraction coupling, sarcolemma, sarcoplasmic reticulum, Ca2+ release channel, ryanodine receptor type 1, RyR1-related mutations, RyR1 dysfunction and disease
Citation: Hernández-Ochoa EO, Pratt SJP, Lovering RM and Schneider MF (2016) Critical Role of Intracellular RyR1 Calcium Release Channels in Skeletal Muscle Function and Disease. Front. Physiol. 6:420. doi: 10.3389/fphys.2015.00420
Received: 05 November 2015; Accepted: 21 December 2015;
Published: 12 January 2016.
Edited by:
Julio L. Vergara, University of California, Los Angeles, USAReviewed by:
Paul D. Allen, Harvard Medical School, USAFrancesco Zorzato, University of Ferrara, Italy
Cecilia Hidalgo, Universidad de Chile, Chile
Copyright © 2016 Hernández-Ochoa, Pratt, Lovering and Schneider. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erick O. Hernández-Ochoa, ehernandez-ochoa@som.umaryland.edu