 Rebeca C. Kalamgi
Rebeca C. Kalamgi Lars Larsson
Lars Larsson- 1Basic and Clinical Muscle Biology, Department of Physiology and Pharmacology, Karolinska Institutet, Stockholm, Sweden
- 2Department of Clinical Neuroscience, Clinical Neurophysiology, Karolinska Institutet, Stockholm, Sweden
The complete loss of mechanical stimuli of skeletal muscles, i.e., the loss of external strain, related to weight bearing, and internal strain, related to the contraction of muscle cells, is uniquely observed in pharmacologically paralyzed or deeply sedated mechanically ventilated intensive care unit (ICU) patients. The preferential loss of myosin and myosin associated proteins in limb and trunk muscles is a significant characteristic of critical illness myopathy (CIM) which separates CIM from other types of acquired muscle weaknesses in ICU patients. Mechanical silencing is an important factor triggering CIM. Microgravity or ground based microgravity models form the basis of research on the effect of muscle unloading-reloading, but the mechanisms and effects may differ from the ICU conditions. In order to understand how mechanical tension regulates muscle mass, it is critical to know how muscles sense mechanical information and convert stimulus to intracellular biochemical actions and changes in gene expression, a process called cellular mechanotransduction. In adult skeletal muscles and muscle fibers, this process may differ, the same stimulus can cause divergent response and the same fiber type may undergo opposite changes in different muscles. Skeletal muscle contains multiple types of mechano-sensors and numerous structures that can be affected differently and hence respond differently in distinct muscles.
Introduction
The complete loss of mechanical stimuli in skeletal muscles, i.e., the loss of external strain, related to weight bearing, and internal strain, related to the contraction of muscle cells, is uniquely observed in pharmacologically paralyzed or deeply sedated mechanically ventilated intensive care unit (ICU) patients. This mechanical silencing is an important factor triggering the specific critical illness myopathy (CIM; Ochala et al., 2011a; Llano-Diez et al., 2012; Renaud et al., 2013). Acquired muscle weaknesses in the ICU is a major complication that occurs in severely ill patients and has significant impact on the immune system, amino acid reserves, and temperature regulation (Puthucheary et al., 2010). Muscle wasting and weakness in the ICU is increasingly being recognized and the increasing prevalence of diagnosed ICU acquired weakness is both due to a growing awareness and improved survival of patients with prolonged organ failure (Puthucheary et al., 2010). For these reasons, a thorough understanding of the molecular mechanisms that regulate skeletal muscle mass is important for development of effective rehabilitation programs and possible pharmacological interventions that can prevent or alleviate the loss of skeletal muscle mass and function (Isaacson and Brotto, 2014).
CIM affects limb and trunk skeletal muscles resulting in quadriplegia due to a preferential myosin loss, muscle atrophy, reduced muscle membrane excitability, and unbalanced muscle metabolism (MacFarlane and Rosenthal, 1977; Rich et al., 1997, 1998a,b; Larsson et al., 2000; Sander et al., 2002). As a consequence, the illness often complicates weaning from the ventilator and increases the length of the stay in the ICU. This results in prolonged immobility, need for intensive rehabilitation and increased risk for pneumonia, morbidity, mortality, and negative economic consequences for health careproviders. Muscle atrophy occurs within 10–21 days of muscle disuse when healthy older adults are bound to bedrest. Similar adverse skeletal muscle changes happen as early as day 5 to 7 after ICU admission in critically ill patients (Puthucheary et al., 2010).
Many triggering factors have been suggested for CIM and muscle wasting in the ICU including sepsis, systemic inflammatory response syndrome (SIRS) and multiple organ failure. Animal models have, however, allowed the separation of sepsis and other confounding risk factors, e.g., mechanical ventilation and muscle unloading, which are usually present in ICU patients (Banduseela et al., 2009; Ochala et al., 2011b; Aare et al., 2013). Sepsis on its own has not been able to replicate the CIM phenotype (Friedrich et al., 2015). Other potential triggering factors evolve from the interventions used in modern anesthesiology and in the ICU, that is; prolonged mechanical ventilation, neuromuscular blockers (NMB), systemic corticosteroid hormone treatment, and muscle unloading (Larsson, 2007).
Although the muscle impairment associated with CIM is being described with increasing accuracy as a result of advances in genetic and proteomic science, the exact cause and underlying mechanisms of the disease remain unknown. The loss of skeletal muscle mass is a complex process that occurs as a consequence of a variety of stressors. Atrophy includes the reduction of muscle fiber cross sectional area (CSA) due to a net loss of proteins, organelles and cytoplasm. This occurs as a result of alterations in the balance between anabolic and catabolic processes, with the net result being a loss of muscle mass when protein breakdown exceeds protein synthesis (Bodine and Baehr, 2014). Acute muscle atrophy occurs in many pathological conditions, including neural inactivity, mechanical unloading, inflammation, metabolic stress, and elevated glucocorticoids levels. Acute muscle atrophy is then due to hyper-activation of the main cellular degradation pathways like the ubiquitin-proteasome system (UPS) and autophagy lysosome pathways (Sandri, 2013; Schiaffino et al., 2013). However, it is also well established that muscle unloading induces a rapid decrease in protein synthesis which contributes to the loss of muscle mass (Thomason and Booth, 1990; Han et al., 2007).
Muscle Unloading and Cellular Mechanotransduction
In order to understand how mechanical tension regulates muscle mass, it is critical to know how muscles sense mechanical information and convert stimulus to intracellular biochemical actions and changes in gene expression, a process called cellular mechanotransduction (Goldberg, 1968; Goldberg et al., 1975; Vandenburgh, 1987; Ingber et al., 2014). Skeletal muscle contains multiple types of mechano-sensors with diverse responses to changes in tension (Gautel, 2011a). Structures involved in mechanotransduction include integrins, sarcolemmal structure proteins, caveolae, cytoskeletal proteins, mitochondria, and sarcomeric proteins spanning from the Z-disc in to the M-line in the center of the sarcomere (Figure 1).
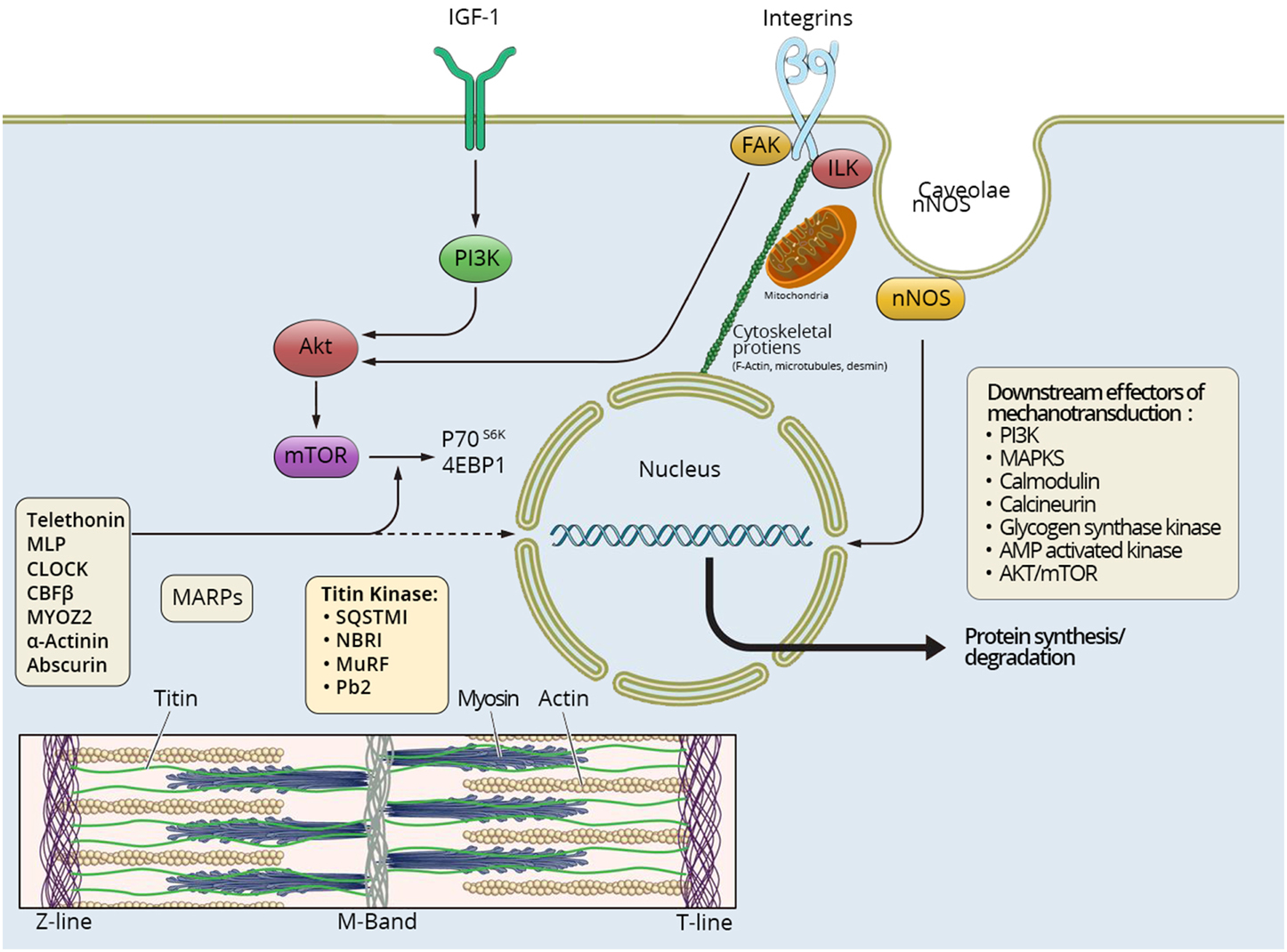
Figure 1. Mechanosensing in skeletal muscle. Different pathways involved in mechanosensing and tensegrity in skeletal muscle are briefly summarized. Multiple signaling pathways influence protein synthesis and degradation in the muscle fiber spanning from the muscle membrane and extracellular matrix to the M-band in the center of the sarcomere. IGF-1 has been suggested to play an important role for the muscle hypertrophy induced by mechanical overload. In addition caveolae, respond to cell stress and stretch-induced signaling, and many different proteins involved in cell signaling bind to caveolins, such as neural nitric oxide synthase (nNOS), tyrosine kinases, small GTPases, and growth receptors. Integrins spanning from the extacellular matrix to the interior of the muscle cell, linked to cytoskeletal actin, directly connect to the nuclei and mitochondria, thus allowing a “hard-wired” and rapid signal propagation to nuclear and mitochondrial DNA. A number of sarcomeric proteins are involved in mechanosensing, and there is emerging evidence of a very dynamic exchange of multiple sarcomeric proteins to the cytoplasmic pool affecting muscle gene expression in response to mechanical load from the Z-line to the center of the sarcomere in the M-band. Multiple major signaling cascades are downstream effectors of mechanosensing, such as PI-3K, MAPKs, calmodulin, calcineurin, glycogen synthase kinase, AMP activated kinase, and Akt/mTOR.
Mechanotransduction Pathways
Integrins are a large family of cell surface receptors that links the extra cellular matrix (ECM) to the cytoskeleton, regulating many cellular functions including cell proliferation, migration, and differentiation (Ingber, 1991; Giancotti and Ruoslahti, 1999). Integrins work both as mechanotransducers and adhesive receptors that can signal trough the cell membrane in both directions. ECM proteins can bind to integrin receptors and then form a cluster at the level of the cell membrane. The cytoplasmic tail of integrins can further associate with various intracellular proteins which then physically link the integrins to the internal cytoskeleton. The anchorage complex of ECM proteins, integrins, and cytoskeletal proteins is known as focal adhesion that ranges from one side of the cell membrane to the other (Wary et al., 1996; Giancotti and Ruoslahti, 1999; Wei et al., 1999; Graham et al., 2015). By forming focal adhesions, integrins allow mechanical forces to be transmitted to membrane proteins. Integrins also interact with mechano sensitive ion channels in focal adhesions (Martinac, 2014).
Caveolae are 60–80 nm cup-shaped invaginations of plasma membrane formed by caveolins. They are present in many cells but are particularly abundant in muscle cells, adipocytes, and endothelial cells. Sinha and colleges established that cells respond to an acute mechanical stress through rapid flattening of caveolae into the plasma membrane (Sinha et al., 2011). Stress-induced flattening of caveolae is strictly passive whereas the formation of invaginated caveolae is energetically favored through interactions with cortical actin in the presence of ATP. The caveolae invaginations are supported by extracellular matrix (ECM), which serves as external support, and the cytoskeleton, whose contraction and expansion locally regulate the mechanosensitivity of mechanosensitive channels in animal and human cells.
Wang and co-workers, were able to demonstrate that mitochondria also play an essential role in mechanotransduction. By confocal microscopy they confirmed that mitochondria associate directly with microtubules and distribute throughout the entire cytoplasm (Wang et al., 2001). In addition to microtubules, other cytoskeletal proteins, such as the intermediate filament desmin, are connected to mitochondria to provide a coordinated movement and alignment throughout the depth of the cell (Rappaport et al., 1998; Milner et al., 2000). When a mechanical pulling stress is applied to integrins, mitochondria show movement near the surface of the nucleus. These results demonstrate that the strains transported across integrins to microfilaments are passed via load-bearing interconnections to microtubules on which the mitochondria anchor, indicating the essential role of mitochondria in mechano-sensing.
The cytoskeleton is the core molecular structure of an active cell and actin microfilaments, microtubules, and intermediate filaments comprise the three major filamentous components of this structure. These filaments link to themselves, to form larger fibrils, and to each other to form structurally coupled networks (Ingber et al., 2014). The fundamental idea of cellular tensegrity is that the cytoskeleton is stabilized by an elastic prestress that is generated and maintained through a complementary force balance between contractile actomyosin filaments and by intracellular structures, such as microtubules, and extracellular binding sites to the extracellular matrix and to other cells (Ingber et al., 1981, 1985; Ingber, 1993, 1997, 2003, 2006). Actin is also the major protein component of the thin filaments in the sarcomere and when myosin, the motor protein binds to actin, substantial mechanical stress is generated on various structures of the sarcomere (Gautel, 2011b). The sarcomeres in striated muscle represent the basic molecular unit for the contractile force. Actin in the thin filaments are crosslinked at the Z-disc, whereas the thick filaments, mainly composed of myosin, are attached to the M-band of the sarcomere that can deliver cross stabilization. The distance between the myosin and actin cross-links, at the M-band and Z-disc, is determined by the giant sarcomeric scaffold titin (aka. connectin). In addition to their mechanical function, these sub-sarcomeric structures and their molecular components are greatly involved in cellular mechanotransduction (Hoshijima, 2006; Linke, 2008). The sarcomeric Z-disc, as well as the M-band, has increasingly been recognized as hubs for signaling pathways that mediate diverse intracellular processes including cell growth and differentiation, as well as protein turnover and gene expression (Hoshijima, 2006; Durieux et al., 2007).
Via the sarcomeric protein telethonin located in the Z-disc, the Z-disc cross talks to growth factors. Further, it has also been shown that telethonin interacts with several other proteins outside the Z-disk and relocalizes in response to stress or atrophy signals (Gautel, 2008). The sarcomeric M-band has also been shown to play a critical role in mechanotransduction and an M-band signaling complex, consisting of titin, Nbr1, p62, and MURF2 mediate gene expression and muscle protein turnover in response to biomechanical stress via the nuclear translocation of the transcription factor serum response factor (SRF) (Lange et al., 2005; Peng et al., 2005; Will et al., 2010). Another member of the M-band proteins, Myosin-interacting M-band-associated stress-responsive protein (myosap, aka. leucine rich repeat containing 39 protein, LRRC39) binds to the myosin tail domain. Will and co-workers demonstrated that myosin is involved in mechanically activated gene transcription via SRF in cardiomyocytes (Will et al., 2010). This indicates that the M-band may contain several mechanostransducers that can control gene expression (Gautel, 2008). Additionally, titin has a kinase domain that is linked to the control of muscle gene expression and protein turnover, located at the M-band of the sarcomere (Lange et al., 2005). Titin was long considered a simple molecular spring, controlling the resting length of sarcomeres. However, it has been established that titin is involved in mechanical signaling pathways in heart muscle (Krüger and Linke, 2009) and several ubiquitin-linked signaling proteins are targeted to, and around the protein kinase domain of titin (Witt et al., 2005; Gautel, 2008).
Preferential Myosin Loss in Muscle Unloading Associated with CIM
To date, most studies on the effects of muscle unloading have focused on the effects of removing the external load induced by weight bearing in response to hind limb suspension or microgravity. In immobilized ICU patients, in addition to the removal of the external load by weight bearing, the internal strain in response to activation of contractile proteins is also missing. This results in the complete mechanical silencing, unique for mechanically ventilated, and deeply sedated or pharmacologically paralyzed ICU patients. Microgravity or ground based microgravity models form the basis of research on the effect of muscle unloading-reloading, but the mechanisms and effects may differ from the ICU conditions. The preferential loss of myosin and myosin associated proteins in limb and trunk muscles is a significant characteristic of CIM which separates CIM from other types of acquired muscle weaknesses in ICU patients (Larsson et al., 2000). This phenotype is reproduced in a unique experimental rat ICU model, used in our research group, allowing long-term mechanical ventilation in a deeply sedated, extensively monitored, and pharmacologically paralyzed rats by post synaptic neuromuscular blockade (Norman et al., 2006; Larsson, 2007). This model is not limited by the early mortality (within 1–3 days) observed when commercially available ventilators are used and the longest duration a rat has been continuously monitored and mechanically ventilated to date is 3 months (Dworkin and Dworkin, 1990). In this experimental model, the complete mechanical silencing for long durations, typically longer than 1 week, results in a similar geno- and phenotype as in ICU patients with CIM, i.e., a preferential loss of myosin and myosin associated proteins in parallel with a transcriptional down-regulation of contractile proteins and activation of protein degradation pathways in a typical temporal pattern in limb and trunk muscles (Norman et al., 2006; Ochala et al., 2011a). In accordance with the tensegrity model, mild passive mechanical loading reduces the preferential myosin loss seen in both fast- and slow-twitch muscles as well as the loss of specific force at the single muscle fiber level (Renaud et al., 2013). Similar findings were also observed in response to passive loading in deeply sedated and immobilized ICU patients (Llano-Diez et al., 2012). The significant muscle sparing effect in response to loading is mediated via multiple mechanosensing pathways involving protein degradation pathways and mitochondria resulting in decreased protein degradation, reduced oxidative stress and improved function of mitochondria.
Protein Synthesis in Muscle Unloading
Removing the pre-stress generated by the complementary force balance, i.e., muscle unloading, induces a rapid decrease in protein synthesis which contributes to the loss of muscle mass (Thomason and Booth, 1990; Han et al., 2007). The primary effect of mechanical stimulation on protein synthesis appears to occur at the level of translational efficiency (Goldspink, 1977; Kimball et al., 2002) and components of the mammalian/mechanistic target of rapamycin (mTOR) play a critical role in the mechanical regulation of protein synthesis and skeletal muscle mass (Thomason and Booth, 1990; Jackman and Kandarian, 2004; Han et al., 2007; Hornberger, 2011). However, different types of mechanical loading/unloading regulates protein synthesis and muscle mass in a distinct manner (Isaacson and Brotto, 2014).
Initially mTOR was identified as a cytoplasmic protein associated with intracellular membranes in early non-muscle studies (Withers et al., 1997; Sabatini et al., 1999) and subsequently shown to associate with the endoplasmic reticulum (ER) and Golgi apparatus in different cell lines (Drenan et al., 2004). Additionally mTOR has been found to be associated with the outer mitochondrial membrane, suggesting a potential role in the regulation of energy metabolism and also demonstrating the involvement of mTOR in the process of mechanotransduction (Desai et al., 2002; Isaacson and Brotto, 2014).
mTOR occurs in two complexes; mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is the rapamycin sensitive complex that consist of the regulatory associated protein of mTOR (RAPTOR), mammalian lethal with SEC13 protein 8 (mLST8; a.k.a. GbL), the Dep domain-containing mTOR-interacting protein (DEPTOR) and the proline-rich Akt substrate of 40 kDa (PRAS40). In contrast, mTORC2 is the rapamycin-insensitive complex, composed of the rapamycin insensitive companion of mTOR (RICTOR), mLST8, DEPTOR, stress-activated protein kinase-interacting protein 1 (mSIN1) and the protein observed with RICTOR (PROTOR; Weigl, 2012).
Several studies have shown that mTORC1 signaling regulates initiation of translation through the phosphorylation of substrates such as eukaryotic initiation factor (eIF), 4E binding protein-1 (4EBP-1), ribosomal protein S6 (S6), and p70 ribosomal protein S6 kinase (S6K) (Heitman et al., 1991; Haghighat et al., 1995; Hara et al., 1997; Baar and Esser, 1999). These substrates are key regulators of protein synthesis, and are expressed in all cell types (Weigl, 2012). The efficiency of an mRNA is mainly controlled by the eukaryotic initiation factor 4E (eIF4E) which is also part of the eIF4F-complex, together with eIF4G and eIF4A. eIF4A is an RNA helicase which is able to unwind hairpin structures in the 5′untranslated region (UTR) of mRNAs, hence promoting the translational process (Ray et al., 1983; Rogers et al., 2001). 4E-BP1 is an inhibitory phospho-protein, that in the hypo-phosphorylated state, is bound to eIF4E and thereby hindering the association of the eIF4F-complex. When mTORC1 phosphorylates 4EBP-1, 4EBP-1 detaches from eIF4E, increases the formation of the eIF4F complex and improves protein synthesis rate (Weigl, 2012). S6K1 is a positive regulator of protein translation initiation. mTORC1 phosphorylates S6K which in turn phosphorylates several other proteins affecting translation initiation like S6 and eIF4B, another member of the initiation eIF4F-complex (Holz et al., 2005). Thus, the control of translation initiation by mTOR is a key regulator of protein synthesis in skeletal muscle (You et al., 2015).
The upstream regulators of mTORC1 are linked with growth factors, nutrients, energy and stress. Growth factors like insulin and insulin growth factor (IGF) stimulate phosphoinositide 3 kinase (PI3K), which leads to the activation of Akt (aka. protein kinase B, PKB). Activated Akt phosphorylates the tuberous sclerosis complex 2 (TSC2) which is a protein that forms the TSC1/2 complex, together with TSC1 (Inoki et al., 2002). Phosphorylation of TSC2, by Akt, prevent its inhibition of Rheb from promoting mTORC1 activity (Stocker et al., 2003; Weigl, 2012). In addition to Akt, mTORC1 responds to many upstream signals, including amino acids. Further, mTORC1 controls several cellular processes in addition to protein synthesis such as autophagy (Sandri, 2010). Rapamycin inhibits mechanically induced growth and mechanically induced changes in protein synthesis in skeletal muscles (Kubica et al., 2005; Hornberger et al., 2007), this together with studies using transgenic mice models of mTORC1 inactivation indicate that mechanical stimulation of skeletal muscles is sufficient to activate mTORC1, and that the activation of mTORC1 is the basis for the increased protein synthesis and muscle growth observed under mechanical stimulation (Weigl, 2012).
Application of a static stretch to skeletal muscle myotubes in vitro significantly increases amino acid transport into stretched cells (Vandenburgh and Kaufman, 1982) which may also activate mTORC1 in an Akt independent manner. Whether the increase in amino acid transport that results from mechanical activation of voltage-sensitive sodium channels contribute significantly to muscle growth or metabolism remains to be tested (Tidball, 2005).
Muscle Degradation in Muscle Unloading
Activation of proteolytic systems in cells is regulated at the gene level and the transcripts associated with muscle atrophy have been studied by gene expression profiling (Bodine et al., 2001; Gomes et al., 2001). A subset of genes that are commonly up- or down- regulated in muscle atrophy, irrespective of the underlying cause, have been identified and referred to as atrophy-related genes or “atrogenes” (Bodine et al., 2001; Gomes et al., 2001; Lecker et al., 2004; Sacheck et al., 2007). Among these atrogenes there are several transcripts associated with the ubiquitin proteasome system (UPS) and autophagy (Sandri, 2008).
Atrogenes and protein degradation are blocked by the activation of Akt. Akt phosphorylates Forkhead Box (Fox) O, thus inhibiting the UPS. Members of the FoxO family (FoxO1, 3, and 4), downstream Akt, have been identified as key transcription factors controlling the expression of MuRF1 and atrogin-1. Additionally it has been shown that FoxO3, specifically, also regulates autophagy (Sandri et al., 2004; Mammucari et al., 2007; Zhao et al., 2007). Recent work by Milan et al. (2015) demonstrates that muscle-specific deletion of FoxO members protects the muscle from atrophy, further demonstrating the role of FoxOs in the induction of the UPS and the autophagy lysosome pathway. Furthermore, it was demonstrated that FoxOs control several stress-response pathways such as the unfolded protein response, ROS detoxification, DNA repair and translation (Milan et al., 2015).
Ubiquitin Proteasome System
The UPS is an ATP-dependent proteolytic system that targets proteins with ubiquitin (Ub) molecule substrates, through a cascade of conjugating enzymes (ligases), for identification of degradation (Murton et al., 2008). The ubiquitin ligase enzymes (E3) bind the protein substrate and once a protein is ubiquitinated, it is unfolded and fed into the proteasome in an ATP-dependent process (Sandri, 2013). Two important atrogenes that have been found in several animal models of muscle atrophy including unloading are muscle-specific E3s; atrogin-1 and muscle RING finger-1 (MuRF1; Kisselev and Goldberg, 2001). These two atrogenes have been extensively studied in muscle protein degradation and their involvement in muscle atrophy is well known. Mice lacking atrogin-1 and MuRF1 are resistant to muscle atrophy in response to denervation and knockdown of atrogin-1 prevents muscle wasting induced by fasting (Bodine et al., 2001; Cong et al., 2011).
It has been shown that MuRF1 interacts with and controls the half-life of several essential muscle structural proteins like troponin I (Kedar et al., 2004), MyHC (Clarke et al., 2007; Fielitz et al., 2007), and actin (Polge et al., 2011). So far only a few muscle proteins have been identified as substrates for atrogin-1. The substrates found are however all related to growth-associated processes (Tintignac et al., 2005; Csibi et al., 2010).
Further, MuRF1 and atrogin-1 are also upregulated in limb muscles, in response to complete mechanical silencing and the upregulation of these E3 ligases precedes both the muscle atrophy and the preferential myosin loss in response to mechanical silencing (Ochala et al., 2011a; Renaud et al., 2013). In addition, a similar temporal up-regulation pattern of these two atrogenes is also observed in the diaphragm muscle in the experimental rat ICU model, used in our lab, but in the absence of the preferential myosin loss. Even though MuRF1 and atrogin-1 are significant Ub-ligases involved in muscle atrophy, there are likely to be other E3s related to muscle wasting in response to muscle mechanical silencing. Further, the preferential myosin loss in response to the ICU condition cannot be solely explained by the activation of these atrogenes. Particular Ub ligases can be involved in different muscle atrophy models and in different stages of the process. Recently a new set of Ub ligases have been discovered as significant atrogenes that are under the control of the FoxO-family. This set of Ub-ligases includes MUSA1, an E3, that has been found to be critical in muscle atrophy during denervation and fasting (Sartori et al., 2013), Fbxo31, an additional E3, Itch, a Ub-ligase that regulates the half-life of several transcription factors, and Fbxo21, a gene of unknown function but that contains an F-box motif and that has been given the name SMART (Specific of muscle atrophy and regulated by transcription). This novel set of E3s were all up-regulated in response to denervation induced atrophy (Milan et al., 2015).
Autophagy Lysosome Pathway
Autophagy is recognized as a crucial protein degradation mechanism in muscle in addition to the ubiquitin–proteasome system (Sandri, 2008), but is also emerging as a novel mechanism in regulating cellular signal transduction by removing activated signaling proteins (Gautel, 2008; Backues and Klionsky, 2011). Autophagy is a physiological process that is vital for the cells to eliminate damaged cell-components and to remodel the cellular architecture (Neel et al., 2013). There are three types of autophagy that all promote proteolytic degradation of cytosolic components; macro-autophagy, micro-autophagy, and chaperone mediated autophagy (CMA). Both macro- and micro-autophagy can ingest large structures through selective and non-selective mechanisms. Macro-autophagy transports cytoplasmic cargo to the lysosome through the intermediate of an autophagosome, a double membrane-bound vesicle. The autophagosome then fuses with the lysosome forming an autolysosome. In contrast, in micro-autophagy, the cytosolic components are taken up by the lysosome directly through invagination of the lysosomal membrane (Glick et al., 2010; Sandri, 2010). In the third type of CMA, proteins that have been targeted are translocated in a complex with chaperone proteins (such as heat shock proteins), and can cross the lysosomal membrane. This way the targeted proteins are recognized by the lysosomal membrane receptor, lysosomal-associated membrane protein 2A (LAMP-2A), and are subsequently unfolded and degraded (Saftig et al., 2008).
Autophagy is primarily considered to be a non-selective degradation pathway, but autophagy can trigger the selective elimination of specific organelles, such as mitochondria (via mitophagy). Parkin, PINK1, Bnip3, and Bnip3L are genes that have been identified in mammals to control mitophagy, inactivation of the genes encoding these proteins lead to abnormal mitochondria (Hara et al., 2006). During muscle wasting the mitochondrial network is drastically remodeled in response to fasting or denervation, and autophagy plays an important part in this process (Romanello et al., 2010; Romanello and Sandri, 2010). Alteration in mitochondrial dynamics is sufficient to cause muscle wasting in mice, indicating that disturbance of the mitochondrial network is essential for the muscle homeostasis (Romanello et al., 2010; Romanello and Sandri, 2010).
Muscle Specific Differences
Skeletal muscles are heterogeneous at the whole muscle, motor unit and individual muscle fiber levels. The different muscle properties decide the optimal function of each muscle, motor unit and muscle fiber. An important aspect of muscle diversity lies in their embryological origin (Sambasivan et al., 2009; Merrell and Kardon, 2013). Craniofacial muscles, are evolutionarily and developmentally distinct from trunk and limb muscles (Noden and Francis-West, 2006; Sambasivan et al., 2011). Trunk and limb muscles are derived from somites that originate from the paraxial mesoderm of the trunk, whereas the cranial mesoderm gives rise to head muscles like extraocular muscles and cheek muscles like the masseter and the buccinators (Grifone and Kelly, 2007; Nathan et al., 2008; Harel et al., 2009). MyoD, Myf5 and Mrf4 are myogenic regulatory factors that work together to control the entry to the myogenic differentiation program that applies to all skeletal muscles. However, the regulatory hierarchies that act upstream of the myogenic factors are diverse in somatic and cranial mesoderm (Czajkowski et al., 2014). Significant differences between muscles also lies in further development into mature skeletal muscles (Merrell and Kardon, 2013).
In adult skeletal muscles and muscle fibers, the same stimulus can cause divergent responses. The same fiber type may undergo opposite changes in different muscles. Even within the same muscle different fiber types react differently (Blaauw et al., 2013; Schiaffino et al., 2013). The nervous system utilizes the capacity of the muscles to generate force and movement for a variety of motor tasks. These tasks can roughly be divided into three main types; postural joint stabilization, long lasting repetitive activities (like respiration), and fast and powerful actions (such as jumping or kicking). In mammals the motor units are functionally organized into separate components, the motor units each consist of a motoneuron and the muscle fibers that it exclusively innervates. Muscle fibers attain a degree of specialized molecular structure and physiological parameters to perfectly suit the needs of the motor units (Blaauw et al., 2013). The heterogeneity of skeletal muscle fibers, therefore, mainly reflects an adaptation to the different patterns of activity; in addition it also indicates specialization in membrane properties, calcium shuttling mechanisms, contractile machinery, and the structure of the cytoskeleton (Polla et al., 2004; Schiaffino and Reggiani, 2011).
The diaphragm is a muscle engaged in continuous rhythmic activity and diaphragm muscle fibers are characterized by fatigue resistance (Polla et al., 2004; Merrell and Kardon, 2013). In addition to ventilation, the diaphragm muscle is also required for coughing, talking and singing, activities which are phasic and occasional. Diaphragm fibers generally have a smaller CSA than limb muscles. However, the number of capillary vessels surrounding each fiber is the same, the diffusion distance is reduced which makes the oxygen supply more efficient in the diaphragm than in other muscles. This might improve oxygen diffusion and contribute to the increased resistance of the diaphragm to fatigue. Respiratory muscle fibers do not undergo the same changes in response to training and inactivity as limb muscle fibers (Polla et al., 2004; Merrell and Kardon, 2013).
The diaphragm muscle is severely affected in many ICU patients exposed to mechanical ventilation resulting in the rapid development of diaphragmatic weakness due to both atrophy and contractile dysfunction. This harmful effect of prolonged MV has been named ventilator-induced diaphragmatic dysfunction (VIDD; Vassilakopoulos and Petrof, 2004; Powers et al., 2013). Approximately 25% of mechanically ventilated ICU patients experience difficult weaning (Daniel Martin et al., 2013) and the weaning procedures account for 40–60% of the total time on the ventilator (Esteban et al., 1994, 1995). This has significant negative consequences for patient quality of life with an increased risk for pneumonia, mortalaity, and morbidity as well as profound economic consequences for health care providers.
In spite of lack of a preferential myosin loss in the diaphragm, diaphragm muscle fiber function, i.e., fiber atrophy and maximum force normalized to muscle fiber cross-sectional area (specific force), was decreased by more than 85% after 10 days mechanical ventilation in the experimental rat ICU model (Corpeno et al., 2014). An early and maintained oxidative stress in response to controlled mechanical ventilation seems to be a key factor triggering post-translational protein modifications of the myosin molecule, impaired myofibrillar protein organization, and fiber atrophy (Corpeno et al., 2014). The temporal pattern, the phenotype, and mechanism underlying the loss of contractile proteins differ between the diaphragm and limb muscles in response to mechanical ventilation and may in part be related to the passive mechanical loading of the diaphragm by the ventilator, but is most probably also a consequence of other known and unknown factors such as muscle specific differences, oxidative stress, protein modifications and the release of specific factors from the respiratory system (Corpeno et al., 2014).
Conclusion
The uniquely observed mechanical silencing seen in pharmacologically paralyzed or deeply sedated mechanically ventilated ICU patients is an important factor triggering the specific CIM. Passive mechanical loading significantly alleviates these effects indicating the important role of cellular mechanotransduction in skeletal muscles. There is a growing body of evidence showing that mitochondrial activity plays an important role in the regulation of muscle size and there is emerging evidence demonstrating the important role of mitochondria in mechano signaling in skeletal muscle. The mechanisms controlling the effects of mechanosensation are part of an intricate biological system with important muscle specific differences that needs to be taken into account when designing strategies for reducing muscle wasting and weakness in immobilized and mechanically ventilated ICU patients.
Author Contributions
RCK wrote the manuscript with assistance of LL.
Funding
This study was financially supported by the Swedish Research Council (8651), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), the European Commission (MyoAge, EC Fp7 CT-223756 and COST CM1001), King Gustaf V and Queen Victoria's Foundation, and Karolinska Institutet to LL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aare, S., Radell, P., Eriksson, L. I., Akkad, H., Chen, Y. W., Hoffman, E. P., et al. (2013). Effects of corticosteroids in the development of limb muscle weakness in a porcine intensive care unit model. Physiol. Genomics 45, 312–320. doi: 10.1152/physiolgenomics.00123.2012
Baar, K., and Esser, K. (1999). Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am. J. Physiol. 276, C120–C127.
Backues, S. K., and Klionsky, D. J. (2011). Autophagy gets in on the regulatory act. J. Mol. Cell Biol. 3, 76–77. doi: 10.1093/jmcb/mjq033
Banduseela, V. C., Ochala, J., Chen, Y. W., Goransson, H., Norman, H., Radell, P., et al. (2009). Gene expression and muscle fiber function in a porcine ICU model. Physiol. Genomics 39, 141–159. doi: 10.1152/physiolgenomics.00026.2009
Blaauw, B., Schiaffino, S., and Reggiani, C. (2013). Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 3, 1645–1687. doi: 10.1002/cphy.c130009
Bodine, S. C., and Baehr, L. M. (2014). Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 307, 5. doi: 10.1152/ajpendo.00204.2014
Bodine, S. C., Latres, E., Baumhueter, S., Lai, V. K., Nunez, L., Clarke, B. A., et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. doi: 10.1126/science.1065874
Clarke, B. A., Drujan, D., Willis, M. S., Murphy, L. O., Corpina, R. A., Burova, E., et al. (2007). The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 6, 376–385. doi: 10.1016/j.cmet.2007.09.009
Cong, H., Sun, L., Liu, C., and Tien, P. (2011). Inhibition of atrogin-1/MAFbx expression by adenovirus-delivered small hairpin RNAs attenuates muscle atrophy in fasting mice. Hum. Gene Ther. 22, 313–324. doi: 10.1089/hum.2010.057
Corpeno, R., Dworkin, B., Cacciani, N., Salah, H., Bergman, H. M., Ravara, B., et al. (2014). Time course analysis of mechanical ventilation-induced diaphragm contractile muscle dysfunction in the rat. J. Physiol. 592, 3859–3880. doi: 10.1113/jphysiol.2014.277962
Csibi, A., Cornille, K., Leibovitch, M. P., Poupon, A., Tintignac, L. A., Sanchez, A. M., et al. (2010). The translation regulatory subunit eIF3f controls the kinase-dependent mTOR signaling required for muscle differentiation and hypertrophy in mouse. PLoS ONE 5:0008994. doi: 10.1371/journal.pone.0008994
Czajkowski, M. T., Rassek, C., Lenhard, D. C., Brohl, D., and Birchmeier, C. (2014). Divergent and conserved roles of Dll1 signaling in development of craniofacial and trunk muscle. Dev. Biol. 395, 307–316. doi: 10.1016/j.ydbio.2014.09.005
Daniel Martin, A., Smith, B. K., and Gabrielli, A. (2013). Mechanical ventilation, diaphragm weakness and weaning: a rehabilitation perspective. Respir. Physiol. Neurobiol. 189, 377–383. doi: 10.1016/j.resp.2013.05.012
Desai, B. N., Myers, B. R., and Schreiber, S. L. (2002). FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. U.S.A. 99, 4319–4324. doi: 10.1073/pnas.261702698
Drenan, R. M., Liu, X., Bertram, P. G., and Zheng, X. F. (2004). FKBP12-rapamycin-associated protein or mammalian target of rapamycin (FRAP/mTOR) localization in the endoplasmic reticulum and the Golgi apparatus. J. Biol. Chem. 279, 772–778. doi: 10.1074/jbc.M305912200
Durieux, A. C., Desplanches, D., Freyssenet, D., and Fluck, M. (2007). Mechanotransduction in striated muscle via focal adhesion kinase. Biochem. Soc. Trans. 35, 1312–1313. doi: 10.1042/BST0351312
Dworkin, B. R., and Dworkin, S. (1990). Learning of physiological responses: I. Habituation, sensitization, and classical conditioning. Behav. Neurosci. 104, 298–319. doi: 10.1037/0735-7044.104.2.298
Esteban, A., Alia, I., Ibanez, J., Benito, S., and Tobin, M. J. (1994). Modes of mechanical ventilation and weaning. A national survey of Spanish hospitals. The Spanish Lung Failure Collaborative Group. Chest 106, 1188–1193. doi: 10.1378/chest.106.4.1188
Esteban, A., Frutos, F., Tobin, M. J., Alia, I., Solsona, J. F., Valverdu, I., et al. (1995). A comparison of four methods of weaning patients from mechanical ventilation. Spanish Lung Failure Collaborative Group. N. Engl. J. Med. 332, 345–350. doi: 10.1056/NEJM199502093320601
Fielitz, J., Kim, M. S., Shelton, J. M., Latif, S., Spencer, J. A., Glass, D. J., et al. (2007). Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Invest. 117, 2486–2495. doi: 10.1172/JCI32827
Friedrich, O., Reid, M. B., Van den Berghe, G., Vanhorebeek, I., Hermans, G., Rich, M. M., et al. (2015). The Sick and the weak: neuropathies/myopathies in the critically ill. Physiol. Rev. 95, 1025–1109. doi: 10.1152/physrev.00028.2014
Gautel, M. (2008). The sarcomere and the nucleus: functional links to hypertrophy, atrophy and sarcopenia. Adv. Exp. Med. Biol. 642, 176–191. doi: 10.1007/978-0-387-84847-1_13
Gautel, M. (2011a). Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch. 462, 119–134. doi: 10.1007/s00424-011-0946-1
Gautel, M. (2011b). The sarcomeric cytoskeleton: who picks up the strain? Curr. Opin. Cell Biol. 23, 39–46. doi: 10.1016/j.ceb.2010.12.001
Giancotti, F. G., and Ruoslahti, E. (1999). Integrin signaling. Science 285, 1028–1032. doi: 10.1126/science.285.5430.1028
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Goldberg, A. L. (1968). Protein synthesis during work-induced growth of skeletal muscle. J. Cell Biol. 36, 653–658. doi: 10.1083/jcb.36.3.653
Goldberg, A. L., Etlinger, J. D., Goldspink, D. F., and Jablecki, C. (1975). Mechanism of work-induced hypertrophy of skeletal-muscle. Med. Sci. Sports Exerc. 7, 248–261. doi: 10.1249/00005768-197500730-00016
Goldspink, D. F. (1977). The influence of denervation and stretch on the size and protein turnover of rat skeletal muscle [proceedings]. J Physiol. 269, 87P–88P.
Gomes, M. D., Lecker, S. H., Jagoe, R. T., Navon, A., and Goldberg, A. L. (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U.S.A. 98, 14440–14445. doi: 10.1073/pnas.251541198
Graham, Z. A., Gallagher, P. M., and Cardozo, C. P. (2015). Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil. 36, 305–315. doi: 10.1007/s10974-015-9415-3
Grifone, R., and Kelly, R. G. (2007). Heartening news for head muscle development. Trends Genet. 23, 365–369. doi: 10.1016/j.tig.2007.05.002
Haghighat, A., Mader, S., Pause, A., and Sonenberg, N. (1995). Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14, 5701–5709.
Han, B., Zhu, M. J., Ma, C., and Du, M. (2007). Rat hindlimb unloading down-regulates insulin like growth factor-1 signaling and AMP-activated protein kinase, and leads to severe atrophy of the soleus muscle. Appl. Physiol. Nutr. Metab. 32, 1115–1123. doi: 10.1139/H07-102
Hara, K., Yonezawa, K., Kozlowski, M. T., Sugimoto, T., Andrabi, K., Weng, Q. P., et al. (1997). Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 272, 26457–26463. doi: 10.1074/jbc.272.42.26457
Hara, Y., Hino, K., Okuda, M., Furutani, T., Hidaka, I., Yamaguchi, Y., et al. (2006). Hepatitis C virus core protein inhibits deoxycholic acid-mediated apoptosis despite generating mitochondrial reactive oxygen species. J. Gastroenterol. 41, 257–268. doi: 10.1007/s00535-005-1738-1
Harel, I., Nathan, E., Tirosh-Finkel, L., Zigdon, H., Guimarães-Camboa, N., Evans, S. M., et al. (2009). Distinct origins and genetic programs of head muscle satellite cells. Dev. Cell 16, 822–832. doi: 10.1016/j.devcel.2009.05.007
Heitman, J., Movva, N. R., and Hall, M. N. (1991). Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909. doi: 10.1126/science.1715094
Holz, M. K., Ballif, B. A., Gygi, S.P., and Blenis, J. (2005). mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 123, 569–580. doi: 10.1016/j.cell.2005.10.024
Hornberger, T. A. (2011). Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int. J. Biochem. Cell Biol. 43, 1267–1276. doi: 10.1016/j.biocel.2011.05.007
Hornberger, T. A., Sukhija, K. B., Wang, X. R., and Chien, S. (2007). mTOR is the rapamycin-sensitive kinase that confers mechanically-induced phosphorylation of the hydrophobic motif site Thr(389) in p70(S6k). FEBS Lett. 581, 4562–4566. doi: 10.1016/j.febslet.2007.08.045
Hoshijima, M. (2006). Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. Am. J. Physiol. Heart Circ. Physiol. 290, H1313–H1325. doi: 10.1152/ajpheart.00816.2005
Ingber, D. E. (1991). Control of capillary growth and differentiation by extracellular matrix. Use of a tensegrity (tensional integrity) mechanism for signal processing. Chest 99, 34s–40s. doi: 10.1378/chest.99.3_supplement.34s
Ingber, D. E. (1993). Cellular tensegrity: defining new rules of biological design that govern the cytoskeleton. J. Cell Sci. 104, 613–627.
Ingber, D. E. (1997). Tensegrity: the architectural basis of cellular mechanotransduction. Annu. Rev. Physiol. 59, 575–599. doi: 10.1146/annurev.physiol.59.1.575
Ingber, D. E. (2003). Tensegrity I. Cell structure and hierarchical systems biology. J. Cell Sci. 116, 1157–1173. doi: 10.1242/jcs.00359
Ingber, D. E. (2006). Cellular mechanotransduction: putting all the pieces together again. FASEB J. 20, 811–827. doi: 10.1096/fj.05-5424rev
Ingber, D. E., Madri, J. A., and Jamieson, J. D. (1981). Role of basal lamina in neoplastic disorganization of tissue architecture. Proc. Natl. Acad. Sci. U.S.A. 78, 3901–3905. doi: 10.1073/pnas.78.6.3901
Ingber, D. E., Madri, J. A., and Jamieson, J. D. (1985). Neoplastic disorganization of pancreatic epithelial cell-cell relations. Role of basement membrane. Am. J. Pathol. 121, 248–260.
Ingber, D. E., Wang, N., and Stamenovic, D. (2014). Tensegrity, cellular biophysics, and the mechanics of living systems. Reports on progress in physics. Phys. Soc. 77:46603. doi: 10.1088/0034-4885/77/4/046603
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K.-L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657. doi: 10.1038/ncb839
Isaacson, J., and Brotto, M. (2014). Physiology of mechanotransduction: how do muscle and bone “Talk” to one another? Clin. Rev. Bone Miner. Metab. 12, 77–85. doi: 10.1007/s12018-013-9152-3
Jackman, R. W., and Kandarian, S. C. (2004). The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 287, C834–C843. doi: 10.1152/ajpcell.00579.2003
Kedar, V., McDonough, H., Arya, R., Li, H. H., Rockman, H. A., and Patterson, C. (2004). Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. U.S.A. 101, 18135–18140. doi: 10.1073/pnas.0404341102
Kimball, S. R., Farrell, P. A., and Jefferson, L. S. (2002). Invited review: role of insulin in translational control of protein synthesis in skeletal muscle by amino acids or exercise. J. Appl. Physiol. (1985) 93, 1168–1180. doi: 10.1152/japplphysiol.00221.2002
Kisselev, A. F., and Goldberg, A. L. (2001). Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 8, 739–758. doi: 10.1016/S1074-5521(01)00056-4
Kruger, M., and Linke, W. A. (2009). Titin-based mechanical signalling in normal and failing myocardium. J. Mol. Cell. Cardiol. 46, 490–498. doi: 10.1016/j.yjmcc.2009.01.004
Kubica, N., Bolster, D. R., Farrell, P. A., Kimball, S. R., and Jefferson, L. S. (2005). Resistance exercise increases muscle protein synthesis and translation of eukaryotic initiation factor 2Bepsilon mRNA in a mammalian target of rapamycin-dependent manner. J. Biol. Chem. 280, 7570–7580. doi: 10.1074/jbc.M413732200
Lange, S., Xiang, F., Yakovenko, A., Vihola, A., Hackman, P., Rostkova, E., et al. (2005). The kinase domain of titin controls muscle gene expression and protein turnover. Science 308, 1599–1603. doi: 10.1126/science.1110463
Larsson, L. (2007). Experimental animal models of muscle wasting in intensive care unit patients. Crit. Care Med. 35, S484–S487. doi: 10.1097/01.CCM.0000278055.40121.54
Larsson, L., Li, X. P., Edström, L., Eriksson, L. I., Zackrisson, H., Argentini, C., et al. (2000). Acute quadriplegia and loss of muscle myosin in patients treated with nondepolarizing neuromuscular blocking agents and corticosteroids: mechanisms at the cellular and molecular levels. Crit. Care Med. 28, 34–45. doi: 10.1097/00003246-200001000-00006
Lecker, S. H., Jagoe, R. T., Gilbert, A., Gomes, M., Baracos, V., Bailey, J., et al. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 18, 39–51. doi: 10.1096/fj.03-0610com
Linke, W. A. (2008). Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 77, 637–648. doi: 10.1016/j.cardiores.2007.03.029
Llano-Diez, M., Renaud, G., Andersson, M., Marrero, H. G., Cacciani, N., Engquist, H., et al. (2012). Mechanisms underlying ICU muscle wasting and effects of passive mechanical loading. Crit Care 16:R209. doi: 10.1186/cc11841
MacFarlane, I. A., and Rosenthal, F. D. (1977). Severe myopathy after status asthmaticus. Lancet 2:615. doi: 10.1016/S0140-6736(77)91471-4
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Martinac, B. (2014). The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim. Biophys. Acta Biomembranes 1838, 682–691. doi: 10.1016/j.bbamem.2013.07.015
Merrell, A. J., and Kardon, G. (2013). Development of the diaphragm – a skeletal muscle essential for mammalian respiration. FEBS J. 280, 4026–4035. doi: 10.1111/febs.12274
Milan, G., Romanello, V., Pescatore, F., Armani, A., Paik, J.-H. H., Frasson, L., et al. (2015). Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6:6670. doi: 10.1038/ncomms7670
Milner, D. J., Mavroidis, M., Weisleder, N., and Capetanaki, Y. (2000). Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 150, 1283–1298. doi: 10.1083/jcb.150.6.1283
Murton, A. J., Constantin, D., and Greenhaff, P. L. (2008). The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim. Biophys. Acta 12, 730–743. doi: 10.1016/j.bbadis.2008.10.011
Nathan, E., Monovich, A., Tirosh-Finkel, L., Harrelson, Z., Rousso, T., Rinon, A., et al. (2008). The contribution of Islet1-expressing splanchnic mesoderm cells to distinct branchiomeric muscles reveals significant heterogeneity in head muscle development. Development 135, 647–657. doi: 10.1242/dev.007989
Neel, B. A., Lin, Y., and Pessin, J. E. (2013). Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol. Metab. 24, 635–643. doi: 10.1016/j.tem.2013.09.004
Noden, D. M., and Francis-West, P. (2006). The differentiation and morphogenesis of craniofacial muscles. Dev. Dyn. 235, 1194–1218. doi: 10.1002/dvdy.20697
Norman, H., Nordquist, J., Andersson, P., Ansved, T., Tang, X., Dworkin, B., et al. (2006). Impact of post-synaptic block of neuromuscular transmission, muscle unloading and mechanical ventilation on skeletal muscle protein and mRNA expression. Pflugers Arch. 453, 53–66. doi: 10.1007/s00424-006-0110-5
Ochala, J., Gustafson, A. M., Diez, M. L., Renaud, G., Li, M., Aare, S., et al. (2011a). Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J. Physiol. 589, 2007–2026. doi: 10.1113/jphysiol.2010.202044
Ochala, J., Renaud, G., Llano Diez, M., Banduseela, V. C., Aare, S., Ahlbeck, K., et al. (2011b). Diaphragm muscle weakness in an experimental porcine intensive care unit model. PLoS ONE 6:e20558. doi: 10.1371/journal.pone.0020558
Peng, J., Raddatz, K., Labeit, S., Granzier, H., and Gotthardt, M. (2005). Muscle atrophy in titin M-line deficient mice. J. Muscle Res. Cell Motil. 26, 381–388. doi: 10.1007/s10974-005-9020-y
Polge, C., Heng, A. E., Jarzaguet, M., Ventadour, S., Claustre, A., Combaret, L., et al. (2011). Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 25, 3790–3802. doi: 10.1096/fj.11-180968
Polla, B., D'Antona, G., Bottinelli, R., and Reggiani, C. (2004). Respiratory muscle fibres: specialisation and plasticity. Thorax 59, 808–817. doi: 10.1136/thx.2003.009894
Powers, S. K., Wiggs, M. P., Sollanek, K. J., and Smuder, A. J. (2013). Ventilator-induced diaphragm dysfunction: cause and effect. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R464–R477. doi: 10.1152/ajpregu.00231.2013
Puthucheary, Z., Montgomery, H., Moxham, J., Harridge, S., and Hart, N. (2010). Structure to function: muscle failure in critically ill patients. J. Physiol. 588, 4641–4648. doi: 10.1113/jphysiol.2010.197632
Rappaport, L., Oliviero, P., and Samuel, J. L. (1998). Cytoskeleton and mitochondrial morphology and function. Mol. Cell. Biochem. 184, 101–105. doi: 10.1023/A:1006843113166
Ray, B. K., Brendler, T. G., Adya, S., Daniels-McQueen, S., Miller, J. K., Hershey, J. W., et al. (1983). Role of mRNA competition in regulating translation: further characterization of mRNA discriminatory initiation factors. Proc. Natl. Acad. Sci. U.S.A. 80, 663–667. doi: 10.1073/pnas.80.3.663
Renaud, G., Llano-Diez, M., Ravara, B., Gorza, L., Feng, H. Z., Jin, J. P., et al. (2013). Sparing of muscle mass and function by passive loading in an experimental intensive care unit model. J. Physiol. 591, 1385–1402. doi: 10.1113/jphysiol.2012.248724
Rich, M. M., Bird, S. J., Raps, E. C., McCluskey, L. F., and Teener, J. W. (1997). Direct muscle stimulation in acute quadriplegic myopathy. Muscle Nerve 20, 665–673.
Rich, M. M., Pinter, M. J., Kraner, S. D., and Barchi, R. L. (1998a). Loss of electrical excitability in an animal model of acute quadriplegic myopathy. Ann. Neurol. 43, 171–179. doi: 10.1002/ana.410430207
Rich, M. M., Teener, J. W., Raps, E. C., and Bird, S. J. (1998b). Muscle inexcitability in patients with reversible paralysis following steroids and neuromuscular blockade. Muscle Nerve 21, 1231–1232.
Rogers, G. W., Richter, N. J., Lima, W. F., and Merrick, W. C. (2001). Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J. Biol. Chem. 276, 30914–30922. doi: 10.1074/jbc.m100157200
Romanello, V., Guadagnin, E., Gomes, L., Roder, I., Sandri, C., Petersen, Y., et al. (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785. doi: 10.1038/emboj.2010.60
Romanello, V., and Sandri, M. (2010). Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr. Hypertens. Rep. 12, 433–439. doi: 10.1007/s11906-010-0157-8
Sabatini, D. M., Barrow, R. K., Blackshaw, S., Burnett, P. E., Lai, M. M., Field, M. E., et al. (1999). Interaction of RAFT1 with gephyrin required for rapamycin-sensitive signaling. Science 284, 1161–1164. doi: 10.1126/science.284.5417.1161
Sacheck, J. M., Hyatt, J. P., Raffaello, A., Jagoe, R. T., Roy, R. R., Edgerton, V. R., et al. (2007). Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 21, 140–155. doi: 10.1096/fj.06-6604com
Saftig, P., Beertsen, W., and Eskelinen, E. L. (2008). LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy 4, 510–512. doi: 10.4161/auto.5724
Sambasivan, R., Gayraud-Morel, B., Dumas, G., Cimper, C., Paisant, S., Kelly, R. G., et al. (2009). Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev. Cell 16, 810–821. doi: 10.1016/j.devcel.2009.05.008
Sambasivan, R., Yao, R., Kissenpfennig, A., Van Wittenberghe, L., Paldi, A., Gayraud-Morel, B., et al. (2011). Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656. doi: 10.1242/dev.067587
Sander, H. W., Golden, M., and Danon, M. J. (2002). Quadriplegic areflexic ICU illness: selective thick filament loss and normal nerve histology. Muscle Nerve 26, 499–505. doi: 10.1002/mus.10233
Sandri, M. (2008). Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda). 23, 160–170. doi: 10.1152/physiol.00041.2007
Sandri, M. (2010). Autophagy in skeletal muscle. FEBS Lett. 584, 1411–1416. doi: 10.1016/j.febslet.2010.01.056
Sandri, M. (2013). Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 45, 2121–2129. doi: 10.1016/j.biocel.2013.04.023
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., et al. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412. doi: 10.1016/S0092-8674(04)00400-3
Sartori, R., Schirwis, E., Blaauw, B., Bortolanza, S., Zhao, J., Enzo, E., et al. (2013). BMP signaling controls muscle mass. Nat. Genet. 45, 1309–1318. doi: 10.1038/ng.2772
Schiaffino, S., Dyar, K. A., Ciciliot, S., Blaauw, B., and Sandri, M. (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294–4314. doi: 10.1111/febs.12253
Schiaffino, S., and Reggiani, C. (2011). Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531. doi: 10.1152/physrev.00031.2010
Sinha, B., Koster, D., Ruez, R., Gonnord, P., Bastiani, M., Abankwa, D., et al. (2011). Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144, 402–413. doi: 10.1016/j.cell.2010.12.031
Stocker, H., Radimerski, T., Schindelholz, B., Wittwer, F., Belawat, P., Daram, P., et al. (2003). Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat. Cell Biol. 5, 559–565. doi: 10.1038/ncb995
Thomason, D. B., and Booth, F. W. (1990). Atrophy of the soleus muscle by hindlimb unweighting. J. Appl. Physiol. (1985) 68, 1–12.
Tidball, J. G. (2005). Mechanical signal transduction in skeletal muscle growth and adaptation. J Appl Physiol (1985) 98, 1900–1908. doi: 10.1152/japplphysiol.01178.2004
Tintignac, L. A., Lagirand, J., Batonnet, S., Sirri, V., Leibovitch, M. P., and Leibovitch, S. A. (2005). Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 280, 2847–2856. doi: 10.1074/jbc.M411346200
Vandenburgh, H. H. (1987). Motion into mass: how does tension stimulate muscle growth? Med. Sci. Sports Exerc. 19, S142–S149.
Vandenburgh, H. H., and Kaufman, S. (1982). Coupling of voltage-sensitive sodium channel activity to stretch-induced amino acid transport in skeletal muscle in vitro. J. Biol. Chem. 257, 13448–13454.
Vassilakopoulos, T., and Petrof, B. J. (2004). Ventilator-induced diaphragmatic dysfunction. Am. J. Respir. Crit. Care Med. 169, 336–341. doi: 10.1164/rccm.200304-489CP
Wang, N., Naruse, K., Stamenovic, D., Fredberg, J. J., Mijailovich, S. M., Tolic-Norrelykke, I. M., et al. (2001). Mechanical behavior in living cells consistent with the tensegrity model. Proc. Natl. Acad. Sci. U.S.A. 98, 7765–7770. doi: 10.1073/pnas.141199598
Wary, K. K., Mainiero, F., Isakoff, S. J., Marcantonio, E. E., and Giancotti, F. G. (1996). The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 87, 733–743. doi: 10.1016/S0092-8674(00)81392-6
Wei, Y., Yang, X., Liu, Q., Wilkins, J. A., and Chapman, H. A. (1999). A role for caveolin and the urokinase receptor in integrin-mediated adhesion and signaling. J. Cell Biol. 144, 1285–1294. doi: 10.1083/jcb.144.6.1285
Weigl, L. G. (2012). Lost in translation: regulation of skeletal muscle protein synthesis. Curr. Opin. Pharmacol. 12, 377–382. doi: 10.1016/j.coph.2012.02.017
Will, R. D., Eden, M., Just, S., Hansen, A., Eder, A., Frank, D., et al. (2010). Myomasp/LRRC39, a heart- and muscle-specific protein, is a novel component of the sarcomeric M-band and is involved in stretch sensing. Circ. Res. 107, 1253–1264. doi: 10.1161/CIRCRESAHA.110.222372
Withers, D. J., Ouwens, D. M., Nave, B. T., van der Zon, G. C., Alarcon, C. M., Cardenas, M. E., et al. (1997). Expression, enzyme activity, and subcellular localization of mammalian target of rapamycin in insulin-responsive cells. Biochem. Biophys. Res. Commun. 241, 704–709. doi: 10.1006/bbrc.1997.7878
Witt, S. H., Granzier, H., Witt, C. C., and Labeit, S. (2005). MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 350, 713–722. doi: 10.1016/j.jmb.2005.05.021
You, J.-S. S., Anderson, G. B., Dooley, M. S., and Hornberger, T. A. (2015). The role of mTOR signaling in the regulation of protein synthesis and muscle mass during immobilization in mice. Dis. Model. Mech. 8, 1059–1069. doi: 10.1242/dmm.019414
Keywords: critical illness myopathy, mechanical silencing, mechanotransduction, sarcomere, mitochondria, mTORC1
Citation: Kalamgi RC and Larsson L (2016) Mechanical Signaling in the Pathophysiology of Critical Illness Myopathy. Front. Physiol. 7:23. doi: 10.3389/fphys.2016.00023
Received: 30 November 2015; Accepted: 18 January 2016;
Published: 04 February 2016.
Edited by:
Catherine Coirault, Institut National de la Santé et de la Recherche Médicale, FranceReviewed by:
Martina Krüger, Heinrich Heine University Düsseldorf, GermanyCharles S. Chung, Wayne State University, USA
Copyright © 2016 Kalamgi and Larsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebeca C. Kalamgi, r.corpeno@gmail.com