 Francisco Westermeier1
Francisco Westermeier1 Jaime A. Riquelme1
Jaime A. Riquelme1 Mario Pavez1
Mario Pavez1 Valeria Garrido1
Valeria Garrido1 Ariel Díaz1
Ariel Díaz1 Hugo E. Verdejo2,3Pablo F. Castro2,3
Hugo E. Verdejo2,3Pablo F. Castro2,3 Lorena García1
Lorena García1 Sergio Lavandero1,4*
Sergio Lavandero1,4*- 1Faculty of Chemical and Pharmaceutical Sciences and Faculty of Medicine, Advanced Center for Chronic Diseases, University of Chile, Santiago, Chile
- 2Faculty of Medicine, Advanced Center for Chronic Diseases, Pontifical Catholic University of Chile, Santiago, Chile
- 3Division of Cardiovascular Diseases, Faculty of Medicine, Pontifical Catholic University of Chile, Santiago, Chile
- 4Department of Internal Medicine (Division of Cardiology), University of Texas Southwestern Medical Center, Dallas, TX, USA
Type 2 diabetes mellitus (T2DM) is a highly prevalent disease worldwide. Cardiovascular disorders generated as a consequence of T2DM are a major cause of death related to this disease. Diabetic cardiomyopathy (DCM) is characterized by the morphological, functional and metabolic changes in the heart produced as a complication of T2DM. This cardiac disorder is characterized by constant high blood glucose and lipids levels which eventually generate oxidative stress, defective calcium handling, altered mitochondrial function, inflammation and fibrosis. In this context, insulin is of paramount importance for cardiac contractility, growth and metabolism and therefore, an impaired insulin signaling plays a critical role in the DCM development. However, the exact pathophysiological mechanisms leading to DCM are still a matter of study. Despite the numerous questions raised in the study of DCM, there have also been important findings, such as the role of micro-RNAs (miRNAs), which can not only have the potential of being important biomarkers, but also therapeutic targets. Furthermore, exosomes also arise as an interesting variable to consider, since they represent an important inter-cellular communication mechanism and therefore, they may explain many aspects of the pathophysiology of DCM and their study may lead to the development of therapeutic agents capable of improving insulin signaling. In addition, adenosine and adenosine receptors (ARs) may also play an important role in DCM. Moreover, the possible cross-talk between insulin and ARs may provide new strategies to reverse its defective signaling in the diabetic heart. This review focuses on DCM, the role of insulin in this pathology and the discussion of new molecular insights which may help to understand its underlying mechanisms and generate possible new therapeutic strategies.
Diabetic Cardiomyopathy: Pathophysiology and Clinical Features
Over the past two decades, the prevalence of type 2 diabetes mellitus (T2DM) has steadily increased as a consequence of higher rates of obesity, an increase in sedentary lifestyle, and changes in the environment. If unaddressed, nearly half a billion people will be burdened by this chronic disease in 2030 (Shaw et al., 2010). At one time, whether T2DM contributed to cardiac dysfunction in diabetic patients was a matter of debate (Rubler et al., 1972). However, diabetic cardiomyopathy (DCM)—with a prevalence of 12% in T2DM patients (Bertoni et al., 2004)—is now recognized as a serious complication of T2DM resulting from sustained hyperglycaemia and hyperlipidemia that leads to increases in cardiac oxidative stress, inflammation, and myocardial fibrosis, as well as detrimental changes in Ca2+ handling and mitochondrial function (Bugger and Abel, 2014; Jia et al., 2016). Several signaling pathways converge in the development of DCM, and the goal of finding suitable therapeutic targets remains a challenge. From a clinical standpoint, DCM is defined as the presence of left ventricular (LV) dysfunction in patients with T2DM in the absence of arterial hypertension, coronary artery disease (CAD) or evidence of any other structural cardiac disease (Battiprolu et al., 2010, 2013; Maisch et al., 2011; Adeghate and Singh, 2014; Isfort et al., 2014). Its clinical presentation ranges from early-stage DCM, which is characterized by abnormal myocardial energy metabolism, diastolic dysfunction and reduced LV strain to overt heart failure (HF), associated with cardiomyocyte hypertrophy, myocardial fibrosis and cardiomyocyte death (Voulgari et al., 2010; Falcao-Pires and Leite-Moreira, 2012; Battiprolu et al., 2013; Dhalla et al., 2014; Go et al., 2014; Isfort et al., 2014).
The first stage of DCM is clinically asymptomatic, but is evidenced by a characteristic diastolic dysfunction (increased ventricular stiffness, left atrial enlargement, and elevated LV end-diastolic pressure) (Battiprolu et al., 2010). The prevalence of diastolic dysfunction is several times higher in T2DM patients than in sex- and age-matched populations and can impact up to 40% of T2DM patients (Poulsen et al., 2010). Several mechanisms contribute to ventricular stiffness in T2DM: for instance, sustained hyperglycemia promotes formation of advanced glycation end products (AGEs), which are particularly prone to cross-linking with extracellular proteins, such as collagen and elastin, altering their structural properties and impairing cardiac relaxation (Murarka and Movahed, 2010; Jia et al., 2016).
The second stage of DCM is characterized by cardiac remodeling, LV hypertrophy (LVH), and the emergence of clinical indications of HF (Ozasa et al., 2008); however, whether LV systolic dysfunction ultimately develops is a matter of debate. Although some authors have suggested that there is an association between systolic dysfunction and long-standing diabetes (Mbanya et al., 2001), most publications indicate that resting systolic function is normal in the majority of T2DM patients, as assessed by LV ejection fraction (LVEF), although some subjects may exhibit systolic impairment during exercise (Vered et al., 1984). However, LVEF can be a poor approximation of cardiac contractility and may not detect minor changes in myocardial function (Sonoyama et al., 2007). These abnormalities in myocardial function correlate closely with changes in Ca2+ handling, caused in part by AGEs that decrease mechanical efficiency of the sarcomere and impair electromechanical coupling, ultimately leading to reduced contractility (Bugger and Abel, 2014). In addition, the activity of the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2), the ryanodine receptor (RyR) and Na+/Ca2+ exchanger (NCX1) are markedly reduced in DCM, leading to diminished release of Ca2+ from endoplasmic reticulum stores and slowed replenishment of these stores during relaxation. These changes, in turn, lead to elevated end-diastolic Ca2+ levels and a progressive decrease in the frequency of Ca2+ transients (Zhao et al., 2014). There is evidence that hyperglycemia may activate caspase-3 directly (Cai et al., 2002) causing apoptosis of cardiomyocytes and increased interstitial fibrosis (Fiordaliso et al., 2000). Thus, increased cardiomyocyte apoptosis may be a critical step in the transition from compensated to decompensated HF (Frustaci et al., 2000). It has also been proposed that insulin-resistance in DCM may be related to increased risk of cardiac LVH or HF (Abel et al., 2012). Altered myocardial insulin signaling is a hallmark of DCM (Battiprolu et al., 2012; Bugger and Abel, 2014; Jia et al., 2016); however, the molecular mechanisms that have been proposed to contribute to the development of insulin resistance in DCM are not fully understood. In this review, we summarize the current knowledge about cardiac insulin signaling, with emphasis on new molecular insights regarding its role in DCM.
Insulin Signaling in the Normal Heart
An appreciation for the role of insulin in glucose homeostasis began with its discovery in 1922 (Banting et al., 1922). The ability of insulin to induce glucose uptake (Levine et al., 1949) by activating specific cell surface receptors on target tissues (Kahn et al., 1974; Ebina et al., 1985; Ullrich et al., 1985), thus, triggering the translocation of the glucose transporter 4 (GLUT4) to the plasma membrane (Cushman and Wardzala, 1980; Suzuki and Kono, 1980), is well documented. However, much remains to be learned regarding both the physiological and pathophysiological impact of insulin signaling in the diversity of tissues, such as adipose tissue and skeletal muscle, upon which it acts (Leto and Saltiel, 2012). Although a comprehensive understanding of the mechanisms underlying insulin action in the heart is only just emerging, there is abundant evidence that insulin impacts cardiac metabolism by influencing a wide range of cellular processes, such as glucose transport, glycolysis, glycogen synthesis (Abel, 2004), cardiac hypertrophy, protein synthesis, lipid metabolism (Belke et al., 2002). Moreover, insulin also participates in myocardial contractility (Fu et al., 2014), protection against ischemia-induced necrotic death (Diaz et al., 2015), autophagy (Riehle et al., 2013), and cell survival, either directly or through the closely associated insulin-like growth factor-1 (IGF-1) (Troncoso et al., 2014).
Cardiac Metabolism
Insulin-stimulated glucose uptake in cardiomyocytes is mediated primarily through mobilization of glucose transporter 4 (GLUT4). Basal cardiac glucose uptake is mediated by glucose transporter 1 (GLUT1), however, contraction-mediated activation of GLUT4 translocation may also contribute significantly to myocardial glucose uptake (Abel, 2004). We have reported that GLUT4-mediated glucose uptake in cardiomyocytes can be activated through a G protein, phosphatidylinositol 3-kinase (PI3K)γ and 1,4,5-inositol-triphosphate (IP3) receptor (IP3R) signaling axis, leading to generation of a cytoplasmic Ca+2 signal that mobilizes GLUT4 (Contreras-Ferrat et al., 2010; Figure 1A). Although the mechanisms of insulin regulation of cardiac GLUT4 are not yet fully defined, recent data show that constitutive activation of PI3K and AKT can reduce glucose uptake in mouse cardiomyocytes through a mechanism that does not involve GLUT4 translocation, suggesting that insulin impacts GLUT4 activity beyond the ability to regulate translocation to the plasma membrane (Zhu et al., 2013a). Moreover, in the heart of mice with a cardiomyocyte-selective deletion of GLUT4 (G4H−∕−), there was a reduction in the levels of insulin-responsive aminopeptidase (IRAP) which was related to impaired GLUT4 endosomal trafficking (Abel et al., 2004), further suggesting that GLUT4 may play a role in regulating glycolysis beyond its ability to transport glucose (Tian and Abel, 2001), evidence that highlight the critical role of GLUT4 and GLUT1 in the insulin-mediated glucose transport on cardiac metabolism.
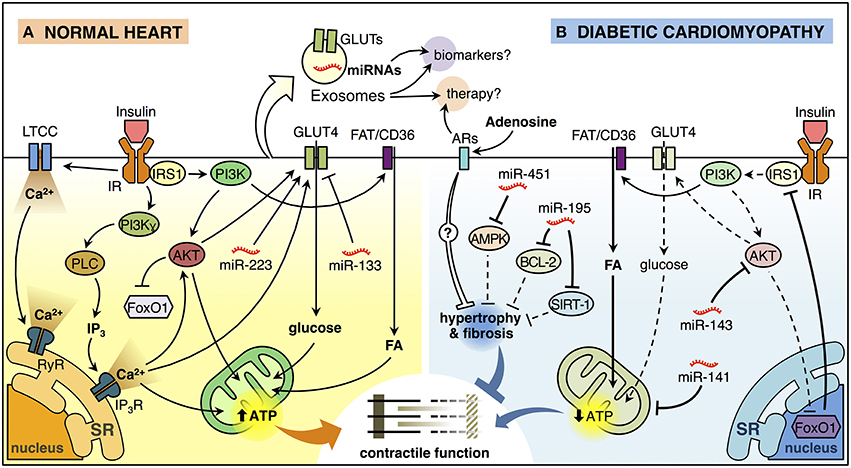
Figure 1. Insulin signaling in the normal heart and diabetic cardiomyopathy. (A) Insulin binding to its receptor (insulin receptor, IR) promotes a biphasic [Ca2+]i response in cardiomyocytes. The first phase of insulin-dependent [Ca2+]i increase involves extracellular Ca2+ influx through L-type calcium channel (LTCC) and activation of ryanodine receptor (RyR) Ca2+ channel which in turn releases Ca2+ from the sarcoendoplasmic reticulum (SR). The second phase involves a non-inhibitory G protein coupled receptor (Gβγ subunits) that activates downstream effectors, including phosphatidylinositol 3-kinase (PI3K)γ and phospholipase C (PLC). PLC generates inositol-1,4,5-trisphosphate (IP3) that opens the IP3-ligated Ca2+channels in the SR. These mechanisms trigger the translocation of glucose transporter 4 (GLUT4) from an intracellular store to the plasma membrane (PM) and increase glucose uptake. Insulin also induces the translocation of the fatty acid (FA) transporter FAT/CD36 to the PM trough PI3K activation promoting the FA uptake. Increased lipid and glucose uptake increase mitochondrial oxidative metabolism generating adenosine triphosphate (ATP) that supports myocardial contractile function. Insulin activation of AKT, downstream of PI3K, inactivates the transcription factor FoxO1. In addition, miR-223 stimulates and miR-133 reduces GLUT4-dependent glucose uptake. (B) Insulin signaling is deficient in diabetic cardiomyopathy (DCM). FoxO1-dependent downregulation of insulin receptor substrate 1 (IRS1) and AKT are associated with reduced insulin-induced GLUT4 translocation to the PM and lower glucose uptake. At the same time, cardiomyocytes accumulate lipids, reducing mitochondrial oxidative metabolism and promoting mitochondrial uncoupling, which in turn, affects cardiomyocyte function. Also, miR-143 inhibits insulin signaling, whereas miR-141 may impact mitochondrial function and the production of ATP. On the other hand, miR-451 is associated with inhibition of AMPK, while miR-195 inhibits the expression of BCL-2 and SIRT-1. Both of these miRNAs are associated with hypertrophy, which will consequently generate contractile dysfunction. Adenosine may have a paracrine effect on cardiomyocytes, which could reduce hypertrophy and myocardial fibrosis through the activation of adenosine receptors (ARs). Exosomes can ferry miRNAs and proteins such as GLUT4 from one cell to another and therefore, they can be used as potential biomarkers or therapeutic agents/targets for DCM.
Cardiac Contractility
Several animal studies have shown the cardioprotective effects of insulin (Das, 2003). In addition, clinical studies have suggested an association between insulin resistance and HF (Arnlov et al., 2001; Ingelsson et al., 2005). However, excessive cardiac insulin signaling can exacerbate systolic dysfunction induced by pressure overload in mice (Shimizu et al., 2010). Cardiac specific insulin receptor knockout (CIRKO) hearts show a mild reduction in cardiac function (Belke et al., 2002) and developed more severe contractile dysfunction compared with control hearts in response to isoproterenol exposure, a combined β1- and β2-adrenergic receptor (βAR) agonist (McQueen et al., 2005). Fu et al. proposed an interesting model for crosstalk between the insulin receptor (IR) and β2AR signaling in the heart (complex IR/β2AR) that could impact cardiac contractility (Fu et al., 2014). In their model, increased IR signaling promotes dissociation of the IR/β2AR complex, leading to an impaired contractile response to β2AR subsequent activation. This effect was seen in isolated neonatal and adult cardiomyocytes, as well as Langendorff-perfused hearts (Fu et al., 2014). Moreover, CIRKO mice, which are deficient for IR signaling, display defective electrical impulse propagation, elevated K+ extracellular concentrations (Punske et al., 2004), and diminished levels of several key K+ channel components involved in ventricular repolarization (Lopez-Izquierdo et al., 2014). Moreover, insulin can diminish markers of cytosolic oxidative stress (Li et al., 2012) and improve impaired cardiac electrical activity during hypoxia in G4H−∕− hearts (Sohn et al., 2013). Altogether, this evidence suggests a link between insulin signaling and the electrical function in the heart independent of its activation of GLUT4-dependent glucose uptake.
Cardiac Growth
In the adult heart, myocardial growth is achieved primarily through hypertrophic expansion of individual cardiomyocytes (Pasumarthi and Field, 2002). Heart size increases during post-natal development in response to several stimuli (Olson and Schneider, 2003), and these responses can be classified either as “pathological” or “physiological” (Richey and Brown, 1998). Insulin is considered an anabolic hormone that promotes protein synthesis, cell growth and hypertrophy (Shiojima et al., 2002). CIRKO mice are characterized by a 30% reduction in heart size (Belke et al., 2002), consistent with the effects observed in cardiomyocytes with reduced PI3K/AKT signaling, where insulin-stimulated hypertrophy is blunted (Shioi et al., 2000; Shiojima et al., 2002). G4H−∕− mice developed modest cardiac hypertrophy, with preserved contractile function, that is associated with an increase in cardiomyocyte size (Abel et al., 1999). Another study conducted by O'Neill et al. suggests that PI3K activates AKT-independent signaling pathways that mediate metabolic and mitochondrial adaptations during physiological cardiac hypertrophy (O'Neill et al., 2007). Cardiomyocyte-specific overexpression of constitutively activated mechanistic target of rapamycin (mTOR), an important target and modulator of the insulin-stimulated AKT pathway, did not elicit a significant increase in adult heart weight (Shen et al., 2008), even though mTOR is essential for cardiac development and growth during embryogenesis (Zhu et al., 2013b). Cardiomyocytes treated with the peroxisome proliferator-activated receptor (PPAR)γ agonist troglitazone also respond by increasing protein content (Bell and Mcdermott, 2000); however, the hypertrophic response of cardiomyocytes to PPARγ agonists does not appear to be mediated by changes in myocardial insulin signaling (Sena et al., 2007).
Studies of mice with cardiomyocyte-specific deletion of either insulin receptor substrate 1 (IRS1) (CIRS1KO mice) or IRS2 (CIRS2KO mice) reported that at baseline, adult CIRS1KO hearts were reduced in size, whereas CIRS2KO hearts exhibited cardiac hypertrophy. However, following a swim training exercise regimen, mitochondrial capacity and substrate oxidation rates increased in wild-type controls, but not in IRS-deficient hearts, suggesting that although different IRS isoforms may perform different roles during resting conditions, both are required for the hypertrophic and metabolic adaptation to exercise (Riehle et al., 2014). In summary, these studies suggest an important role for insulin signaling in physiological cardiac growth, however studies by Ikeda et al. indicated that in response to exercise IGF-1 may actually activate both the IGF-1 receptor (IGF-1R) and IR in the heart (Ikeda et al., 2009). Thus, deciphering the crosstalk between the IGF-1 and insulin signaling pathways will be crucial to understanding the role of IR in exercise-induced cardiac hypertrophy.
Insulin Signaling in Diabetic Cardiomyopathy
Defective insulin signaling in DCM has been described in both animal models (Wright et al., 2009) and humans (Cook et al., 2010), primarily observed as reduced GLUT4 translocation to the plasma membrane. Cardiac dysfunction induced by pressure overload in mice is associated with insulin resistance, hyperinsulinemia and increased AKT activation (Shimizu et al., 2010). Hearts from obese and insulin-resistant rodent models (ob/ob and db/db mice) exhibit higher rates of fatty acid (FA) oxidation and MVO2, but lower rates of glucose oxidation and cardiac efficiency, which precede the development of hyperglycemia (Mazumder et al., 2004; Buchanan et al., 2005). Moreover, CIRKO mice exhibit decreased LV function associated with mitochondrial dysfunction and reduced mitochondrial FA oxidative capacity following a myocardial infarct (Sena et al., 2009), as well as an enhanced decline in cardiac function when treated with streptozotocin (STZ) compared to wild-type mice (Bugger et al., 2012). In addition, these animals have defects in FA and pyruvate metabolism and reduced tricarboxylic acid flux associated with mitochondrial uncoupling. Thus, altered insulin signaling in the heart may contribute directly to mitochondrial dysfunction in the setting of obesity and T2DM (Boudina et al., 2009). In contrast, the type 1 diabetic Akita mouse model (Akita) is characterized by normal cardiac function and preserved myocardial insulin sensitivity, and does not exhibit FA-induced mitochondrial uncoupling (Bugger et al., 2008). These results suggest that the activity of pathways controlling cardiac mitochondrial function may be different in insulin-responsive type 1 and insulin-resistant T2DM hearts.
Interestingly, FoxO (forkhead box-containing protein, O subfamily) proteins have been described as important regulators of several cellular processes in the myocardium such as cardiac growth (Skurk et al., 2005; Ni et al., 2006), and autophagy (Sengupta et al., 2009; Schips et al., 2011). In addition, Ni et al. (2007) showed that FoxO activation leads to altered insulin signaling and decreased glucose uptake in cardiac myocytes (Ni et al., 2007). In this sense, activation of FoxO1 in response to the HFD was directly related to down-regulation of IRS1 activity, decreased AKT signaling, and insulin resistance, suggesting that FoxO1 might be a key step in the development of DCM preventing cardiac dysfunction by restoring insulin responsiveness (Battiprolu et al., 2012; Figure 1B). Recently, it has been shown that impaired cardiac insulin signaling FoxO1-mediated stimulates the β-myosin heavy chain (β-MHC) expression promoting cardiac dysfunction (Qi et al., 2015). Thus, FoxO1 plays a key role in the control of cardiac insulin signaling both in physiological and pathophysiological conditions, and it might prove a novel therapeutic or preventive strategy for treating individuals with DCM.
On the other hand, FAs are the major substrate for ATP synthesis in the heart; nevertheless, excess lipid accumulation is associated with decreased cardiac function in obesity and diabetes (Finck et al., 2002, 2003). In this context, insulin is also able to induce FA uptake in cardiomyocytes, promoting the translocation of the FA transporter FAT/CD36 (Glatz et al., 2006; Coort et al., 2007) to the plasma membrane mediated by PI3K activation (Luiken et al., 2002; Figure 1A). Overexpression of PPARγ led to cardiac lipid accumulation and cardiomyopathy associated with increased FAT/CD36 mRNA, but this effect was antagonized by WY-14,643, a potent PPARα agonist (Son et al., 2010). Nevertheless, comprehensive understanding of the relationship between PPARs and insulin in DCM are not yet fully understood (Jia et al., 2016). The heart can metabolize either non-esterified FA (bound to albumin) or FA obtained from lipolysis of lipoprotein-bound triglycerides (TG). Hearts from mice with cardiomyocyte-specific knockout of lipoprotein lipase (LpL) showed reduced FA oxidation and increased glucose oxidation, associated with increased expression of GLUT4, and reduced expression of pyruvate dehydrogenase kinase 4 (PDK4), suggesting that heart is able to adapt by balancing between the use of glucose and FA (Augustus et al., 2006). However, recent studies indicate that deletion of mTOR impairs myocardial palmitate oxidation and reduces expression of several genes associated with FA metabolism while also reducing insulin-stimulated AKT Ser473 phosphorylation (Zhu et al., 2013c). Thus, intact mTOR signaling may be essential for maintaining appropriate control over substrate usage. In addition, cardiomyocytes from HFD-fed mice exhibit increased FA oxidation rates and myocardial oxygen consumption (MVO2), correlating with reduced GLUT4 content and impaired GLUT4 translocation in response to insulin (Wright et al., 2009; Figure 1B) and consistent with earlier studies involving the response of cardiomyocytes to the adipocytokine resistin (Graveleau et al., 2005). In summary, cardiac lipotoxicity involves detrimental effects that may adversely impact cardiomyocyte function and participate in progression of DCM.
New Insights
miRNAs
miRNAs are small, single-strand non-coding RNA, that are transcribed by RNA polymerase II (Pol II), generating a primary miRNA (pri-miRNA). In the nucleus, the RNase III endonuclease Drosha and the double-stranded RNA-binding domain protein DGCR8/Pasha cleave the pri-miRNA to produce the precursor miRNA (pre-miRNA) of ~70-nt. Exportin-5 transports the pre-miRNA into the cytoplasm, where it is cleaved by another RNase III endonuclease, Dicer, together with the protein TRBP/Loquacious, releasing miRNA containing a 21- to 23-nt (Bushati and Cohen, 2007; Pasquinelli, 2012). They serve as important post-transcriptional regulators that down-regulate the expression of their target genes (Pritchard et al., 2012) The exact and detailed role of miRNAs in the heart is far from being completely understood, but there is mounting evidence showing that alterations in many miRNAs correlate with the development and progression of human diseases, including cardiovascular diseases, such as DCM (Asrih and Steffens, 2013). In this context, Zheng et al. (2015) found that miR-195 is up-regulated in the hearts of C57BL/6 mice treated with STZ and this was associated with a decrease expression of its target proteins B cell leukemia/lymphoma 2 (BCL-2) and sirtuin 1 (SIRT-1) (Zheng et al., 2015). Moreover, inhibition of miR-195 inhibited apoptosis, reduced oxidative stress and hypertrophy in diabetic hearts. Additionally, cardiac function and coronary blood flow was improved and the expression of BCL-2 and SIRT-1 was increased, suggesting that targeting miR-195 may attenuate DCM (Zheng et al., 2015). In order to further elaborate on the subject, we briefly discuss about a few miRNAs and their potential role in DCM. The models used, beneficial or detrimental effect on cardiac function and possible mechanisms suggested so far are summarized in Table 1.
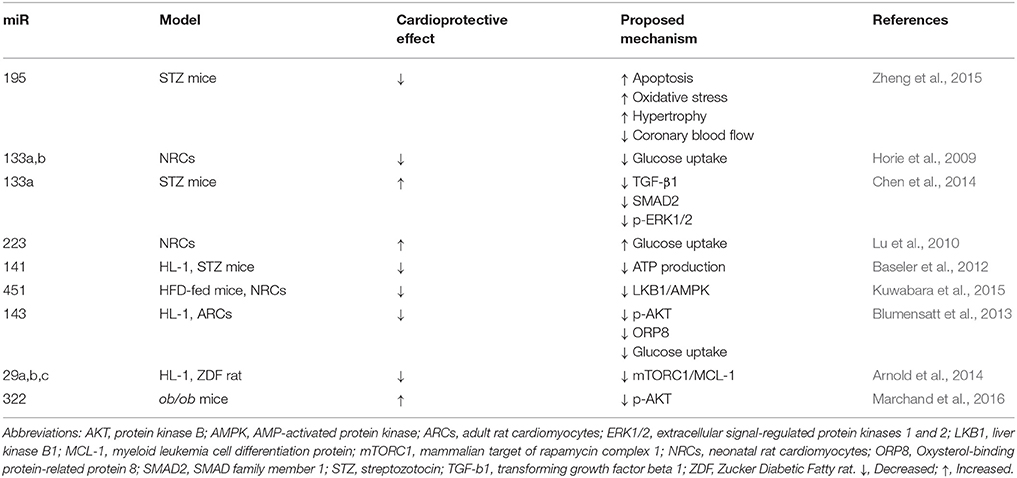
Table 1. Effects of microRNAs (miRs) on diabetic cardiomyopathy (DCM) models: beneficial or detrimental effect on cardiac function and possible mechanisms.
miR-133a,b expression have been shown to participate in metabolic control by reducing the expression of GLUT4 and insulin-mediated glucose uptake in neonatal rat cardiomyocytes (NRCs) (Horie et al., 2009). In contrast, Chen et al. (2014) found that miR-133a expression was reduced in the hearts of diabetic mice and this was associated with an increase in the expression of fibrosis markers. Moreover, they observed that overexpression of miR-133a in the heart attenuated cardiac fibrosis, which is one of the central causes for the HF generated by DCM (Chen et al., 2014). Considering the apparent contradiction between these two studies performed in different models, the results should be analyzed with precaution and further in vivo studies are required to fully clarify the role of miR-133a in DCM.
On the other hand, overexpression of miR-223 in NRCs significantly increased glucose uptake, in similar magnitude to that observed in cells stimulated with insulin. In vitro, overexpression of miR-223 in cardiomyocytes increased total GLUT4 level and induced GLUT4 translocation from the cytoplasmic compartment to the cell membrane. In vivo, inhibition of miR-223 in the heart resulted in a significant decrease in GLUT4 expression (Lu et al., 2010). miR-141 may be another miRNA of importance in DCM, since it was found to be significantly increased in the diabetic heart and its overexpression decreased the content of the inner mitochondrial membrane phosphate transporter, solute carrier family 25 member 3 (Slc25a3) in the mouse atrial cardiomyocyte cell line HL-1 (Baseler et al., 2012), suggesting that altered miR-141 expression may influence cardiac mitochondrial function and ATP generation (Baseler et al., 2012).
Further extending the evidence for the role of miRNA in the pathophysiology DCM, miR-451 levels were found to be markedly increased in palmitate-stimulated NRCs and DIO mouse heart (model of pre-T2DM and obesity with elevated blood glucose and impaired glucose tolerance). It was found that Cab39 (Calcium-binding protein 39, a scaffold protein of LKB1, a kinase upstream of AMPK) was a direct target to miR-451 in the heart. Furthermore, miR-451 knockdown partly rescued lipotoxicity in vitro, and HFD-induced cardiac hypertrophy was ameliorated in miR-451 KO mice through the LKB1/AMPK pathway (Kuwabara et al., 2015). Moreover, activin A released from epicardial adipose tissue in T2DM, inhibits insulin action via the induction of miR-143 in cardiomyocytes. This miRNA inhibits the AKT pathway through down-regulation of the novel regulator of insulin action, ORP8 (Blumensatt et al., 2013). ORP8 binds 25-OH-cholesterol, and this regulates AKT signaling in apoptosis through the induction of AKT degradation via proteasome and/or inhibition of PI3K in macrophages (Jordan et al., 2011). Moreover, activation of AKT in response to insulin and the consequent phosphorylation of glycogen synthase kinase 3β (GSK3β) were diminished in HepG2 cells depleted of ORP8 (Jordan et al., 2011). Future studies should confirm these results in the heart to further clarify the role of miR-143 and ORP8 in DCM.
Regarding insulin, it has been shown that miR-29, which downregulates the pro-survival protein myeloid cell leukemia 1 (MCL-1) (Roggli et al., 2012), may be involved in the development of DCM. Arnold et al., reported that insulin inhibited the expression of miR-29a, b and c and increased the mRNA levels of MCL-1 in HL-1 cells. Additionally, they observed that dysregulation of the miR-29-MCL-1 axis as a consequence of low levels of insulin or inhibition of mTORC1 in Zucker Diabetic Fatty (ZDF) rats was associated with structural damage of the diabetic myocardium (Arnold et al., 2014). In addition, Marchand et al. have recently reported that miR-322 modulates insulin pathway, protecting the heart against the cardiac dysfunction observed in C57Bl/6N mice fed with HFD, showing that miR-322 overexpression reduced AKT phosphorylation induced by insulin. Thus, miR-322 regulates insulin signaling and might protect the heart against consequences of hyperinsulinemia (Marchand et al., 2016). Moreover, Costantino et al. showed that altered miRs profile induced by hyperglycaemia in the diabetic heart is not reverted by the restoration of normal glycaemia levels with insulin (Costantino et al., 2016). This study also showed through ingenuity pathway analysis that the miRs increased in the diabetic heart were involved in myocardial cell signaling pathways associated with apoptosis, fibrosis, hypertrophic growth, autophagy, oxidative stress, and HF and this may explain why the detrimental effects of DCM persist even after the restoration of normal glucose levels (Costantino et al., 2016). Considering all the evidence shown above, miRNAs clearly participate in the pathophysiological mechanisms involved in DCM and more studies are required to fully understand and control their diagnostic and therapeutic potential.
Exosomes
Exosomes are nanosized extracellular vesicles ranging from 50 to 90 nm in diameter. These nanovesicles are formed in multivesicular bodies, which after fusing with the plasma membrane; release their content to the extracellular medium (Thery et al., 2006). Exosomes can carry proteins, RNA or miRNA from one cell to another through the bloodstream, protecting its content from enzymatic degradation and thus, these extracellular vesicles may be mediating intercellular communications (Yellon and Davidson, 2014). Moreover, de Jong et al reported that cellular stress may induce variations in the protein and mRNA content of endothelial cell-derived exosomes (De Jong et al., 2012), which suggests that exosomes may represent the physiological state of the cell (Yellon and Davidson, 2014). Therefore, exosomes are a very attractive target to develop possible biomarkers for different diseases.
The potential role of exosomes in cardiovascular diseases is starting to be explored. In this context, it has been described that exosomes exert a cardioprotective effect in the myocardium against ischemia-reperfusion (Giricz et al., 2014; Vicencio et al., 2015). It has also been reported that exosomes may play a role in septic cardiomyopathy, cardiac hypertrophy, and peripartum cardiomyopathy (Ailawadi et al., 2015).
Regarding DCM and the importance of miRNAs in this disease, exosomes arise as potential biomarkers for diagnosis. However, much remains to be elucidated about these nanovesicles and their role in DCM. In this context, it has been suggested that most of the plasma miRNAs are contained in exosomes, however, it has also been reported that plasma miRNAs can be bound to complexes of proteins like argonaute or high density lipoproteins (Yellon and Davidson, 2014) and therefore, further studies are required to clarify this point.
In the context of understanding the mechanisms involved in diabetes-induced ischemic cardiac disease (Ailawadi et al., 2015), Wang et al showed that cardiomyocyte-derived exosomes isolated from Goto-Kakizaki (GK) diabetic rats inhibited the proliferation and migration of endothelial cells. Interestingly, exosomes isolated from Wistar rat's cardiomyocytes generated the opposite effect in the endothelial cells, promoting migration and proliferation. In addition, the effects of both GK and Wistar rat's exosomes on endothelial cells were reversed by the exosome inhibitor GW4869. Moreover, GK-exosomes contained higher levels of miR-320 and lower levels of miR-126 compared to Wistar rat's cardiomyocyte-derived exosomes (Wang et al., 2014). Therefore, these results suggest that reduced angiogenesis in the diabetic myocardium may be mediated by exosomes with anti-angiogenic effects released from cardiomyocytes to the endothelium. These findings also suggest a possible role for exosomes for the treatment of DCM ischemic complications.
It has also been recently reported that cardiomyocyte-derived exosomes can regulate glucose transport in endothelial cells (Garcia et al., 2016). This study showed that under glucose deprivation conditions, the production of exosomes from cardiomyocytes was increased and these nanovesicles contained glucose transporters and enzymes involved in glycolysis, which in turn lead to an increase in glucose uptake, glycolysis and the production of pyruvate in the endothelial cells (Garcia et al., 2016). Taken the current evidence together, exosomes have massive potential to give new insights that could help to understand the pathophysiology of DCM and they can not only serve as potential biomarkers, but also act as therapeutic targets or agents that could reverse the impaired insulin signaling observed in DCM.
Adenosine
The purine nucleoside adenosine is an important paracrine/autocrine signaling molecule that modulates a wide range of biological processes activating four subtypes of G protein-coupled adenosine receptors (ARs), A1, A2A, A2B, and A3 (Fredholm, 2014). The ARs play a key role in tissue homeostasis both in physiological and pathophysiological conditions, such as inflammation, ischemia, hypoxia and cancer, regulating tissue homeostasis (Antonioli et al., 2013).
Interestingly, new evidence has emerged suggesting a link between adenosine signaling and T2DM (Antonioli et al., 2015). In this context, it has been described that adenosine modulates insulin secretion (Johansson et al., 2007; Yang et al., 2012; Ohtani et al., 2013) and also controls the proliferation and regeneration of β pancreatic cells (Ohtani et al., 2013; Andersson, 2014). Moreover, several studies have proposed that adenosine regulates the insulin signaling in muscle (Faulhaber-Walter et al., 2011; Figler et al., 2011; Johnston-Cox et al., 2012), liver (Figler et al., 2011; Johnston-Cox et al., 2012), and adipose tissue (Faulhaber-Walter et al., 2011; Figler et al., 2011; Johnston-Cox et al., 2012; Csoka et al., 2014).
In the cardiovascular system, the expression of ARs have been reported in vascular smooth muscle cells (Zhao et al., 1997), human umbilical vein endothelial cells (Wyatt et al., 2002), coronary artery endothelial cells (Olanrewaju et al., 2000), cardiac fibroblasts and cardiomyocytes (Sassi et al., 2014). In STZ-treated diabetic rats, cardiomyocytes exhibited increased levels of A1 and A3, reduced levels of A2A and unaltered levels of A2B (Grden et al., 2005); however, cardiac fibroblasts showed increased levels of A1 and A2A,reduced levels of A3 but did not change the levels of A2B (Grden et al., 2006). Thus, the evidence described above highlights the need to assess the functional role of ARs in DCM.
As we mentioned before, early-stage DCM is characterized by cardiomyocyte hypertrophy and myocardial fibrosis (Battiprolu et al., 2013; Go et al., 2014). In this context, Puhl et al. (2016) recently showed that A1 attenuates phenylephrine-mediated (α-1 adrenoceptor) cardiomyocyte hypertrophic and cardiac fibrosis, suggesting that A1 activation might be a novel therapeutic target to prevent cardiac remodeling (Puhl et al., 2016). In addition, cell-to-cell communication via cAMP/adenosine activating A1 and A2A/A2B in cardiomyocytes and fibroblast, respectively, may be also relevant in preventing isoproterenol-mediated (βAR agonist) cardiomyocyte hypertrophy and cardiac fibrosis, suggesting an interesting paracrine cardiac action of adenosine (Sassi et al., 2014). In summary, whether ARs participates in the molecular mechanisms of DCM, or whether they might be promoting the DCM-related defective insulin signaling are important issues that need to be addressed.
Concluding Remarks and Open Questions
The complete pathophysiology of DCM and the role of insulin in it are still being elucidated. Further studies are certainly required to clarify many aspects of this cardiovascular disease. However, there has also been emerging information on different mechanisms that may help answer the variety of questions raised in the study of DCM. Despite increasing amount of evidence that highlights the role of ARs in the regulation of insulin signaling in several tissues such as muscle, liver and adipose tissue, there are many open questions to deeply understand their roles both in the healthy heart and DCM. Thus, could insulin signaling in DCM be improved by activation of ARs? In addition, miRNAs may play a key role for early diagnosis of DCM and evidence such as the altered miRNA landscape in the diabetic heart suggests that new strategies are required to reverse the detrimental effects of DCM that persists despite normalization of blood glucose levels. In this context, is it possible to reverse the cardiovascular complications of DCM by targeting the miRNAs associated with these deleterious effects? Also, exosomes may also serve as potential biomarkers of DCM. These nanovesicles are a mechanism of inter-cellular communication and therefore, there is much excitement about the possibility of harnessing their power as therapeutic agents. Could normal exosomes isolated from healthy patients reverse the phenotype of patients with DCM? Is it possible to exogenously control the release of beneficial exosomes? Answers to these questions will help to elucidate the complex mechanisms underlying the defective insulin signaling in DCM, opening new fields for basic and clinical studies.
Author Contributions
FW and SL contributed to conception and design the manuscript. JR, MP, VG, AD, HV, PC, and LG, drafted and critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Comision Nacional de Ciencia y Tecnologia (CONICYT), Chile: FONDECYT 1141198 (PC), FONDAP 15130011 (HV, PC, LG, SL), FONDECYT 1161156 (SL), FONDECYT: 1110346 (LG), FONDECYT 3140532 (FW), FONDECYT 3160298 (JR), FONDECYT 1150359 (HV). MP, VG, AD, holds CONICYT PhD fellowship.
References
Abel, E. D., Graveleau, C., Betuing, S., Pham, M., Reay, P. A., Kandror, V., et al. (2004). Regulation of insulin-responsive aminopeptidase expression and targeting in the insulin-responsive vesicle compartment of glucose transporter isoform 4-deficient cardiomyocytes. Mol. Endocrinol. 18, 2491–2501. doi: 10.1210/me.2004-0175
Abel, E. D., Kaulbach, H. C., Tian, R., Hopkins, J. C., Duffy, J., Doetschman, T., et al. (1999). Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Invest. 104, 1703–1714. doi: 10.1172/JCI7605
Abel, E. D., O'Shea, K. M., and Ramasamy, R. (2012). Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 32, 2068–2076. doi: 10.1161/ATVBAHA.111.241984
Adeghate, E., and Singh, J. (2014). Structural changes in the myocardium during diabetes-induced cardiomyopathy. Heart Fail. Rev. 19, 15–23. doi: 10.1007/s10741-013-9388-5
Ailawadi, S., Wang, X., Gu, H., and Fan, G. C. (2015). Pathologic function and therapeutic potential of exosomes in cardiovascular disease. Biochim. Biophys. Acta 1852, 1–11. doi: 10.1016/j.bbadis.2014.10.008
Andersson, O. (2014). Role of adenosine signalling and metabolism in beta-cell regeneration. Exp. Cell Res. 321, 3–10. doi: 10.1016/j.yexcr.2013.11.019
Antonioli, L., Blandizzi, C., Csoka, B., Pacher, P., and Hasko, G. (2015). Adenosine signalling in diabetes mellitus–pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 11, 228–241. doi: 10.1038/nrendo.2015.10
Antonioli, L., Blandizzi, C., Pacher, P., and Hasko, G. (2013). Immunity, inflammation and cancer: a leading role for adenosine. Nat. Rev. Cancer 13, 842–857. doi: 10.1038/nrc3613
Arnlov, J., Lind, L., Zethelius, B., Andren, B., Hales, C. N., Vessby, B., et al. (2001). Several factors associated with the insulin resistance syndrome are predictors of left ventricular systolic dysfunction in a male population after 20 years of follow-up. Am. Heart J. 142, 720–724. doi: 10.1067/mhj.2001.116957
Arnold, N., Koppula, P. R., Gul, R., Luck, C., and Pulakat, L. (2014). Regulation of cardiac expression of the diabetic marker microRNA miR-29. PLoS ONE 9:e103284. doi: 10.1371/journal.pone.0103284
Asrih, M., and Steffens, S. (2013). Emerging role of epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc. Pathol. 22, 117–125. doi: 10.1016/j.carpath.2012.07.004
Augustus, A. S., Buchanan, J., Park, T. S., Hirata, K., Noh, H. L., Sun, J., et al. (2006). Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J. Biol. Chem. 281, 8716–8723. doi: 10.1074/jbc.M509890200
Banting, F. G., Best, C. H., Collip, J. B., Campbell, W. R., and Fletcher, A. A. (1922). Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J. 12, 141–146.
Baseler, W. A., Thapa, D., Jagannathan, R., Dabkowski, E. R., Croston, T. L., and Hollander, J. M. (2012). miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am. J. Physiol. Cell Physiol. 303, C1244–C1251. doi: 10.1152/ajpcell.00137.2012
Battiprolu, P. K., Gillette, T. G., Wang, Z. V., Lavandero, S., and Hill, J. A. (2010). Diabetic cardiomyopathy: mechanisms and therapeutic targets. Drug Discov. Today Dis. Mech. 7, e135–e143. doi: 10.1016/j.ddmec.2010.08.001
Battiprolu, P. K., Hojayev, B., Jiang, N., Wang, Z. V., Luo, X., Iglewski, M., et al. (2012). Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 122, 1109–1118. doi: 10.1172/JCI60329
Battiprolu, P. K., Lopez-Crisosto, C., Wang, Z. V., Nemchenko, A., Lavandero, S., and Hill, J. A. (2013). Diabetic cardiomyopathy and metabolic remodeling of the heart. Life Sci. 92, 609–615. doi: 10.1016/j.lfs.2012.10.011
Belke, D. D., Betuing, S., Tuttle, M. J., Graveleau, C., Young, M. E., Pham, M., et al. (2002). Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Invest. 109, 629–639. doi: 10.1172/JCI0213946
Bell, D., and Mcdermott, B. J. (2000). Troglitazone does not initiate hypertrophy but can sensitise cardiomyocytes to growth effects of serum. Eur. J. Pharmacol. 390, 237–244. doi: 10.1016/S0014-2999(99)00932-2
Bertoni, A. G., Hundley, W. G., Massing, M. W., Bonds, D. E., Burke, G. L., and Goff, D. C. Jr. (2004). Heart failure prevalence, incidence, and mortality in the elderly with diabetes. Diabetes Care 27, 699–703. doi: 10.2337/diacare.27.3.699
Blumensatt, M., Greulich, S., Herzfeld De Wiza, D., Mueller, H., Maxhera, B., Rabelink, M. J., et al. (2013). Activin A impairs insulin action in cardiomyocytes via up-regulation of miR-143. Cardiovasc. Res. 100, 201–210. doi: 10.1093/cvr/cvt173
Boudina, S., Bugger, H., Sena, S., O'Neill, B. T., Zaha, V. G., Ilkun, O., et al. (2009). Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119, 1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101
Buchanan, J., Mazumder, P. K., Hu, P., Chakrabarti, G., Roberts, M. W., Yun, U. J., et al. (2005). Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146, 5341–5349. doi: 10.1210/en.2005-0938
Bugger, H., and Abel, E. D. (2014). Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57, 660–671. doi: 10.1007/s00125-014-3171-6
Bugger, H., Boudina, S., Hu, X. X., Tuinei, J., Zaha, V. G., Theobald, H. A., et al. (2008). Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes 57, 2924–2932. doi: 10.2337/db08-0079
Bugger, H., Riehle, C., Jaishy, B., Wende, A. R., Tuinei, J., Chen, D., et al. (2012). Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J. Mol. Cell. Cardiol. 52, 1019–1026. doi: 10.1016/j.yjmcc.2012.02.001
Bushati, N., and Cohen, S. M. (2007). microRNA functions. Annu. Rev. Cell Dev. Biol. 23, 175–205. doi: 10.1146/annurev.cellbio.23.090506.123406
Cai, L., Li, W., Wang, G., Guo, L., Jiang, Y., and Kang, Y. J. (2002). Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51, 1938–1948. doi: 10.2337/diabetes.51.6.1938
Chen, S., Puthanveetil, P., Feng, B., Matkovich, S. J., Dorn, G. W. II., and Chakrabarti, S. (2014). Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell. Mol. Med. 18, 415–421. doi: 10.1111/jcmm.12218
Contreras-Ferrat, A. E., Toro, B., Bravo, R., Parra, V., Vasquez, C., Ibarra, C., et al. (2010). An inositol 1,4,5-triphosphate (IP3)-IP3 receptor pathway is required for insulin-stimulated glucose transporter 4 translocation and glucose uptake in cardiomyocytes. Endocrinology 151, 4665–4677. doi: 10.1210/en.2010-0116
Cook, S. A., Varela-Carver, A., Mongillo, M., Kleinert, C., Khan, M. T., Leccisotti, L., et al. (2010). Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Heart J. 31, 100–111. doi: 10.1093/eurheartj/ehp396
Coort, S. L., Bonen, A., Van Der Vusse, G. J., Glatz, J. F., and Luiken, J. J. (2007). Cardiac substrate uptake and metabolism in obesity and type-2 diabetes: role of sarcolemmal substrate transporters. Mol. Cell. Biochem. 299, 5–18. doi: 10.1007/s11010-005-9030-5
Costantino, S., Paneni, F., Lüscher, T. F., and Cosentino, F. (2016). MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J. 37, 572–576. doi: 10.1093/eurheartj/ehv599
Csoka, B., Koscso, B., Toro, G., Kokai, E., Virag, L., Nemeth, Z. H., et al. (2014). A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes 63, 850–866. doi: 10.2337/db13-0573
Cushman, S. W., and Wardzala, L. J. (1980). Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 255, 4758–4762.
Das, U. N. (2003). Insulin: an endogenous cardioprotector. Curr. Opin. Crit. Care 9, 375–383. doi: 10.1097/00075198-200310000-00007
De Jong, O. G., Verhaar, M. C., Chen, Y., Vader, P., Gremmels, H., Posthuma, G., et al. (2012). Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell Vesicles 1:18396. doi: 10.3402/jev.v1i0.18396
Dhalla, N. S., Takeda, N., Rodriguez-Leyva, D., and Elimban, V. (2014). Mechanisms of subcellular remodeling in heart failure due to diabetes. Heart Fail. Rev. 19, 87–99. doi: 10.1007/s10741-013-9385-8
Diaz, A., Humeres, C., Gonzalez, V., Gomez, M. T., Montt, N., Sanchez, G., et al. (2015). Insulin/NFkappaB protects against ischemia-induced necrotic cardiomyocyte death. Biochem. Biophys. Res. Commun. 467, 451–457. doi: 10.1016/j.bbrc.2015.09.171
Ebina, Y., Edery, M., Ellis, L., Standring, D., Beaudoin, J., Roth, R. A., et al. (1985). Expression of a functional human insulin receptor from a cloned cDNA in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. U.S.A. 82, 8014–8018. doi: 10.1073/pnas.82.23.8014
Falcao-Pires, I., and Leite-Moreira, A. F. (2012). Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail. Rev. 17, 325–344. doi: 10.1007/s10741-011-9257-z
Faulhaber-Walter, R., Jou, W., Mizel, D., Li, L., Zhang, J., Kim, S. M., et al. (2011). Impaired glucose tolerance in the absence of adenosine A1 receptor signaling. Diabetes 60, 2578–2587. doi: 10.2337/db11-0058
Figler, R. A., Wang, G., Srinivasan, S., Jung, D. Y., Zhang, Z., Pankow, J. S., et al. (2011). Links between insulin resistance, adenosine A2B receptors, and inflammatory markers in mice and humans. Diabetes 60, 669–679. doi: 10.2337/db10-1070
Finck, B. N., Han, X., Courtois, M., Aimond, F., Nerbonne, J. M., Kovacs, A., et al. (2003). A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc. Natl. Acad. Sci. U.S.A. 100, 1226–1231. doi: 10.1073/pnas.0336724100
Finck, B. N., Lehman, J. J., Leone, T. C., Welch, M. J., Bennett, M. J., Kovacs, A., et al. (2002). The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 109, 121–130. doi: 10.1172/JCI0214080
Fiordaliso, F., Li, B., Latini, R., Sonnenblick, E. H., Anversa, P., Leri, A., et al. (2000). Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II- dependent. Lab. Invest. 80, 513–527. doi: 10.1038/labinvest.3780057
Fredholm, B. B. (2014). Adenosine–a physiological or pathophysiological agent? J. Mol. Med. 92, 201–206. doi: 10.1007/s00109-013-1101-6
Frustaci, A., Kajstura, J., Chimenti, C., Jakoniuk, I., Leri, A., Maseri, A., et al. (2000). Myocardial cell death in human diabetes. Circ. Res. 87, 1123–1132. doi: 10.1161/01.RES.87.12.1123
Fu, Q., Xu, B., Liu, Y., Parikh, D., Li, J., Li, Y., et al. (2014). Insulin inhibits cardiac contractility by inducing a gi-biased beta2-adrenergic signaling in hearts. Diabetes 63, 2676–2689. doi: 10.2337/db13-1763
Garcia, N. A., Moncayo-Arlandi, J., Sepulveda, P., and Diez-Juan, A. (2016). Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc Res. 109, 397–408. doi: 10.1093/cvr/cvv260
Giricz, Z., Varga, Z. V., Baranyai, T., Sipos, P., Paloczi, K., Kittel, A., et al. (2014). Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J. Mol. Cell. Cardiol. 68, 75–78. doi: 10.1016/j.yjmcc.2014.01.004
Glatz, J. F., Bonen, A., Ouwens, D. M., and Luiken, J. J. (2006). Regulation of sarcolemmal transport of substrates in the healthy and diseased heart. Cardiovasc. Drugs Ther. 20, 471–476. doi: 10.1007/s10557-006-0582-8
Go, A. S., Mozaffarian, D., Roger, V. L., Benjamin, E. J., Berry, J. D., Blaha, M. J., et al. (2014). Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation 129, e28–e292. doi: 10.1161/01.cir.0000442015.53336.12
Graveleau, C., Zaha, V. G., Mohajer, A., Banerjee, R. R., Dudley-Rucker, N., Steppan, C. M., et al. (2005). Mouse and human resistins impair glucose transport in primary mouse cardiomyocytes, and oligomerization is required for this biological action. J. Biol. Chem. 280, 31679–31685. doi: 10.1074/jbc.M504008200
Grden, M., Podgorska, M., Kocbuch, K., Szutowicz, A., and Pawelczyk, T. (2006). Expression of adenosine receptors in cardiac fibroblasts as a function of insulin and glucose level. Arch. Biochem. Biophys. 455, 10–17. doi: 10.1016/j.abb.2006.08.022
Grden, M., Podgorska, M., Szutowicz, A., and Pawelczyk, T. (2005). Altered expression of adenosine receptors in heart of diabetic rat. J. Physiol. Pharmacol. 56, 587–597.
Horie, T., Ono, K., Nishi, H., Iwanaga, Y., Nagao, K., Kinoshita, M., et al. (2009). MicroRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochem. Biophys. Res. Commun. 389, 315–320. doi: 10.1016/j.bbrc.2009.08.136
Ikeda, H., Shiojima, I., Ozasa, Y., Yoshida, M., Holzenberger, M., Kahn, C. R., et al. (2009). Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 47, 664–675. doi: 10.1016/j.yjmcc.2009.08.028
Ingelsson, E., Sundstrom, J., Arnlov, J., Zethelius, B., and Lind, L. (2005). Insulin resistance and risk of congestive heart failure. JAMA 294, 334–341. doi: 10.1001/jama.294.3.334
Isfort, M., Stevens, S. C., Schaffer, S., Jong, C. J., and Wold, L. E. (2014). Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail. Rev. 19, 35–48. doi: 10.1007/s10741-013-9377-8
Jia, G., DeMarco, V. G., and Sowers, J. R. (2016). Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12, 144–153. doi: 10.1038/nrendo.2015.216
Johansson, S. M., Salehi, A., Sandstrom, M. E., Westerblad, H., Lundquist, I., Carlsson, P. O., et al. (2007). A1 receptor deficiency causes increased insulin and glucagon secretion in mice. Biochem. Pharmacol. 74, 1628–1635. doi: 10.1016/j.bcp.2007.08.006
Johnston-Cox, H., Koupenova, M., Yang, D., Corkey, B., Gokce, N., Farb, M. G., et al. (2012). The A2b adenosine receptor modulates glucose homeostasis and obesity. PLoS ONE 7:e40584. doi: 10.1371/journal.pone.0040584
Jordan, S. D., Kruger, M., Willmes, D. M., Redemann, N., Wunderlich, F. T., Bronneke, H. S., et al. (2011). Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 13, 434–446. doi: 10.1038/ncb2211
Kahn, C. R., Freychet, P., Roth, J., and Neville, D. M. Jr. (1974). Quantitative aspects of the insulin-receptor interaction in liver plasma membranes. J. Biol. Chem. 249, 2249–2257.
Kuwabara, Y., Horie, T., Baba, O., Watanabe, S., Nishiga, M., Usami, S., et al. (2015). MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ. Res. 116, 279–288. doi: 10.1161/CIRCRESAHA.116.304707
Leto, D., and Saltiel, A. R. (2012). Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383–396. doi: 10.1038/nrm3351
Levine, R., Goldstein, M., Klein, S., and Huddlestun, B. (1949). The action of insulin on the distribution of galactose in eviscerated nephrectomized dogs. J. Biol. Chem. 179, 985–986.
Li, Y., Wende, A. R., Nunthakungwan, O., Huang, Y., Hu, E., Jin, H., et al. (2012). Cytosolic, but not mitochondrial, oxidative stress is a likely contributor to cardiac hypertrophy resulting from cardiac specific GLUT4 deletion in mice. FEBS J. 279, 599–611. doi: 10.1111/j.1742-4658.2011.08450.x
Lopez-Izquierdo, A., Pereira, R. O., Wende, A. R., Punske, B. B., Abel, E. D., and Tristani-Firouzi, M. (2014). The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am. J. Physiol. Heart Circ. Physiol. 306, H747–H754. doi: 10.1152/ajpheart.00849.2013
Lu, H., Buchan, R. J., and Cook, S. A. (2010). MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc. Res. 86, 410–420. doi: 10.1093/cvr/cvq010
Luiken, J. J., Koonen, D. P., Willems, J., Zorzano, A., Becker, C., Fischer, Y., et al. (2002). Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 51, 3113–3119. doi: 10.2337/diabetes.51.10.3113
Maisch, B., Alter, P., and Pankuweit, S. (2011). Diabetic cardiomyopathy–fact or fiction? Herz 36, 102–115. doi: 10.1007/s00059-011-3429-4
Marchand, A., Atassi, F., Mougenot, N., Clergue, M., Codoni, V., Berthuin, J., et al. (2016). miR-322 regulates insulin signaling pathway and protects against metabolic syndrome-induced cardiac dysfunction in mice. Biochim. Biophys. Acta. 1862, 611–621. doi: 10.1016/j.bbadis.2016.01.010
Mazumder, P. K., O'Neill, B. T., Roberts, M. W., Buchanan, J., Yun, U. J., Cooksey, R. C., et al. (2004). Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53, 2366–2374. doi: 10.2337/diabetes.53.9.2366
Mbanya, J. C., Sobngwi, E., Mbanya, D. S., and Ngu, K. B. (2001). Left ventricular mass and systolic function in African diabetic patients: association with microalbuminuria. Diabetes Metab. 27, 378–382.
McQueen, A. P., Zhang, D., Hu, P., Swenson, L., Yang, Y., Zaha, V. G., et al. (2005). Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J. Mol. Cell. Cardiol. 39, 882–892. doi: 10.1016/j.yjmcc.2005.07.017
Murarka, S., and Movahed, M. R. (2010). Diabetic cardiomyopathy. J. Card. Fail. 16, 971–979. doi: 10.1016/j.cardfail.2010.07.249
Ni, Y. G., Berenji, K., Wang, N., Oh, M., Sachan, N., Dey, A., et al. (2006). Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168. doi: 10.1161/CIRCULATIONAHA.106.637124
Ni, Y. G., Wang, N., Cao, D. J., Sachan, N., Morris, D. J., Gerard, R. D., et al. (2007). FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc. Natl. Acad. Sci. U.S.A. 104, 20517–20522. doi: 10.1073/pnas.0610290104
Ohtani, M., Oka, T., and Ohura, K. (2013). Possible involvement of A(2)A and A(3) receptors in modulation of insulin secretion and beta-cell survival in mouse pancreatic islets. Gen. Comp. Endocrinol. 187, 86–94. doi: 10.1016/j.ygcen.2013.02.011
Olanrewaju, H. A., Qin, W., Feoktistov, I., Scemama, J. L., and Mustafa, S. J. (2000). Adenosine A(2A) and A(2B) receptors in cultured human and porcine coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 279, H650–H656.
Olson, E. N., and Schneider, M. D. (2003). Sizing up the heart: development redux in disease. Genes Dev. 17, 1937–1956. doi: 10.1101/gad.1110103
O'Neill, B. T., Kim, J., Wende, A. R., Theobald, H. A., Tuinei, J., Buchanan, J., et al. (2007). A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 6, 294–306. doi: 10.1016/j.cmet.2007.09.001
Ozasa, N., Furukawa, Y., Morimoto, T., Tadamura, E., Kita, T., and Kimura, T. (2008). Relation among left ventricular mass, insulin resistance, and hemodynamic parameters in type 2 diabetes. Hypertens. Res. 31, 425–432. doi: 10.1291/hypres.31.425
Pasquinelli, A. E. (2012). MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 13, 271–282. doi: 10.1038/nrg3162
Pasumarthi, K. B., and Field, L. J. (2002). Cardiomyocyte cell cycle regulation. Circ. Res. 90, 1044–1054. doi: 10.1161/01.RES.0000020201.44772.67
Poulsen, M. K., Henriksen, J. E., Dahl, J., Johansen, A., Gerke, O., Vach, W., et al. (2010). Left ventricular diastolic function in type 2 diabetes mellitus: prevalence and association with myocardial and vascular disease. Circ. Cardiovasc. Imaging 3, 24–31. doi: 10.1161/CIRCIMAGING.109.855510
Pritchard, C. C., Cheng, H. H., and Tewari, M. (2012). MicroRNA profiling: approaches and considerations. Nat. Rev. Genet. 13, 358–369. doi: 10.1038/nrg3198
Puhl, S. L., Kazakov, A., Müller, A., Fries, P., Wagner, D. R., Böhm, M., et al. (2016). A1 receptor activation attenuates cardiac hypertrophy and fibrosis in response to alpha1-adrenergic stimulation in vivo. Br. J. Pharmacol. 173, 88–102. doi: 10.1111/bph.13339
Punske, B. B., Rossi, S., Ershler, P., Rasmussen, I., and Abel, E. D. (2004). Optical mapping of propagation changes induced by elevated extracellular potassium ion concentration in genetically altered mouse hearts. J. Electrocardiol. 37(suppl.), 128–134. doi: 10.1016/j.jelectrocard.2004.08.037
Qi, Y., Zhu, Q., Zhang, K., Thomas, C., Wu, Y., Kumar, R., et al. (2015). Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and beta-myosin heavy chain gene expression. Circ. Heart Fail. 8, 198–208. doi: 10.1161/CIRCHEARTFAILURE.114.001457
Richey, P. A., and Brown, S. P. (1998). Pathological versus physiological left ventricular hypertrophy: a review. J. Sports Sci. 16, 129–141. doi: 10.1080/026404198366849
Riehle, C., Wende, A. R., Sena, S., Pires, K. M., Pereira, R. O., Zhu, Y., et al. (2013). Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J. Clin. Invest. 123, 5319–5333. doi: 10.1172/JCI71171
Riehle, C., Wende, A. R., Zhu, Y., Oliveira, K. J., Pereira, R. O., Jaishy, B. P., et al. (2014). Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol. Cell. Biol. 34, 3450–3460. doi: 10.1128/MCB.00426-14
Roggli, E., Gattesco, S., Caille, D., Briet, C., Boitard, C., Meda, P., et al. (2012). Changes in microRNA expression contribute to pancreatic beta-cell dysfunction in prediabetic NOD mice. Diabetes 61, 1742–1751. doi: 10.2337/db11-1086
Rubler, S., Dlugash, J., Yuceoglu, Y. Z., Kumral, T., Branwood, A. W., and Grishman, A. (1972). New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 30, 595–602. doi: 10.1016/0002-9149(72)90595-4
Sassi, Y., Ahles, A., Truong, D. J., Baqi, Y., Lee, S. Y., Husse, B., et al. (2014). Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J. Clin. Invest. 124, 5385–5397. doi: 10.1172/JCI74349
Schips, T. G., Wietelmann, A., Hohn, K., Schimanski, S., Walther, P., Braun, T., et al. (2011). FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc. Res. 91, 587–597. doi: 10.1093/cvr/cvr144
Sena, S., Hu, P., Zhang, D., Wang, X., Wayment, B., Olsen, C., et al. (2009). Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J. Mol. Cell. Cardiol. 46, 910–918. doi: 10.1016/j.yjmcc.2009.02.014
Sena, S., Rasmussen, I. R., Wende, A. R., McQueen, A. P., Theobald, H. A., Wilde, N., et al. (2007). Cardiac hypertrophy caused by peroxisome proliferator- activated receptor-gamma agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology 148, 6047–6053. doi: 10.1210/en.2006-1559
Sengupta, A., Molkentin, J. D., and Yutzey, K. E. (2009). FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 284, 28319–28331. doi: 10.1074/jbc.M109.024406
Shaw, J. E., Sicree, R. A., and Zimmet, P. Z. (2010). Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 87, 4–14. doi: 10.1016/j.diabres.2009.10.007
Shen, W. H., Chen, Z., Shi, S., Chen, H., Zhu, W., Penner, A., et al. (2008). Cardiac restricted overexpression of kinase-dead mammalian target of rapamycin (mTOR) mutant impairs the mTOR-mediated signaling and cardiac function. J. Biol. Chem. 283, 13842–13849. doi: 10.1074/jbc.M801510200
Shimizu, I., Minamino, T., Toko, H., Okada, S., Ikeda, H., Yasuda, N., et al. (2010). Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J. Clin. Invest. 120, 1506–1514. doi: 10.1172/JCI40096
Shioi, T., Kang, P. M., Douglas, P. S., Hampe, J., Yballe, C. M., Lawitts, J., et al. (2000). The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548. doi: 10.1093/emboj/19.11.2537
Shiojima, I., Yefremashvili, M., Luo, Z., Kureishi, Y., Takahashi, A., Tao, J., et al. (2002). Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J. Biol. Chem. 277, 37670–37677. doi: 10.1074/jbc.M204572200
Skurk, C., Izumiya, Y., Maatz, H., Razeghi, P., Shiojima, I., Sandri, M., et al. (2005). The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 280, 20814–20823. doi: 10.1074/jbc.M500528200
Sohn, K., Wende, A. R., Abel, E. D., Moreno, A. P., Sachse, F. B., and Punske, B. B. (2013). Absence of glucose transporter 4 diminishes electrical activity of mouse hearts during hypoxia. Exp. Physiol. 98, 746–757. doi: 10.1113/expphysiol.2012.070235
Son, N. H., Yu, S., Tuinei, J., Arai, K., Hamai, H., Homma, S., et al. (2010). PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARalpha deficiency despite increases in fatty acid oxidation. J. Clin. Invest. 120, 3443–3454. doi: 10.1172/JCI40905
Sonoyama, K., Greenstein, A., Price, A., Khavandi, K., and Heagerty, T. (2007). Vascular remodeling: implications for small artery function and target organ damage. Ther. Adv. Cardiovasc. Dis. 1, 129–137. doi: 10.1177/1753944707086358
Suzuki, K., and Kono, T. (1980). Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc. Natl. Acad. Sci. U.S.A. 77, 2542–2545. doi: 10.1073/pnas.77.5.2542
Thery, C., Amigorena, S., Raposo, G., and Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.22. doi: 10.1002/0471143030.cb0322s30
Tian, R., and Abel, E. D. (2001). Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 103, 2961–2966. doi: 10.1161/01.CIR.103.24.2961
Troncoso, R., Ibarra, C., Vicencio, J. M., Jaimovich, E., and Lavandero, S. (2014). New insights into IGF-1 signaling in the heart. Trends Endocrinol. Metab. 25, 128–137. doi: 10.1016/j.tem.2013.12.002
Ullrich, A., Bell, J. R., Chen, E. Y., Herrera, R., Petruzzelli, L. M., Dull, T. J., et al. (1985). Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761. doi: 10.1038/313756a0
Vered, A., Battler, A., Segal, P., Liberman, D., Yerushalmi, Y., Berezin, M., et al. (1984). Exercise-induced left ventricular dysfunction in young men with asymptomatic diabetes mellitus (diabetic cardiomyopathy). Am. J. Cardiol. 54, 633–637. doi: 10.1016/0002-9149(84)90263-7
Vicencio, J. M., Yellon, D. M., Sivaraman, V., Das, D., Boi-Doku, C., Arjun, S., et al. (2015). Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Coll. Cardiol. 65, 1525–1536. doi: 10.1016/j.jacc.2015.02.026
Voulgari, C., Papadogiannis, D., and Tentolouris, N. (2010). Diabetic cardiomyopathy: from the pathophysiology of the cardiac myocytes to current diagnosis and management strategies. Vasc. Health Risk Manag. 6, 883–903. doi: 10.2147/VHRM.S11681
Wang, X., Huang, W., Liu, G., Cai, W., Millard, R. W., Wang, Y., et al. (2014). Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 74, 139–150. doi: 10.1016/j.yjmcc.2014.05.001
Wright, J. J., Kim, J., Buchanan, J., Boudina, S., Sena, S., Bakirtzi, K., et al. (2009). Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc. Res. 82, 351–360. doi: 10.1093/cvr/cvp017
Wyatt, A. W., Steinert, J. R., Wheeler-Jones, C. P., Morgan, A. J., Sugden, D., Pearson, J. D., et al. (2002). Early activation of the p42/p44MAPK pathway mediates adenosine-induced nitric oxide production in human endothelial cells: a novel calcium-insensitive mechanism. FASEB J. 16, 1584–1594. doi: 10.1096/fj.01-0125com
Yang, G. K., Fredholm, B. B., Kieffer, T. J., and Kwok, Y. N. (2012). Improved blood glucose disposal and altered insulin secretion patterns in adenosine A(1) receptor knockout mice. Am. J. Physiol. Endocrinol. Metab. 303, E180–E190. doi: 10.1152/ajpendo.00050.2012
Yellon, D. M., and Davidson, S. M. (2014). Exosomes: nanoparticles involved in cardioprotection? Circ. Res. 114, 325–332. doi: 10.1161/circresaha.113.300636
Zhao, S. M., Wang, Y. L., Guo, C. Y., Chen, J. L., and Wu, Y. Q. (2014). Progressive decay of Ca2+ homeostasis in the development of diabetic cardiomyopathy. Cardiovasc. Diabetol. 13:75. doi: 10.1186/1475-2840-13-75
Zhao, Z., Francis, C. E., and Ravid, K. (1997). An A3-subtype adenosine receptor is highly expressed in rat vascular smooth muscle cells: its role in attenuating adenosine-induced increase in cAMP. Microvasc. Res. 54, 243–252. doi: 10.1006/mvre.1997.2044
Zheng, D., Ma, J., Yu, Y., Li, M., Ni, R., Wang, G., et al. (2015). Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 58, 1949–1958. doi: 10.1007/s00125-015-3622-8
Zhu, Y., Pereira, R. O., O'Neill, B. T., Riehle, C., Ilkun, O., Wende, A. R., et al. (2013a). Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol. Endocrinol. 27, 172–184. doi: 10.1210/me.2012-1210
Zhu, Y., Pires, K. M., Whitehead, K. J., Olsen, C. D., Wayment, B., Zhang, Y. C., et al. (2013b). Mechanistic target of rapamycin (Mtor) is essential for murine embryonic heart development and growth. PLoS ONE 8:e54221. doi: 10.1371/journal.pone.0054221
Keywords: insulin, diabetic cardiomyopathy, miRNAs, exosomes, adenosine, adenosine receptors
Citation: Westermeier F, Riquelme JA, Pavez M, Garrido V, Díaz A, Verdejo HE, Castro PF, García L and Lavandero S (2016) New Molecular Insights of Insulin in Diabetic Cardiomyopathy. Front. Physiol. 7:125. doi: 10.3389/fphys.2016.00125
Received: 15 December 2015; Accepted: 22 March 2016;
Published: 12 April 2016.
Edited by:
Michael A. Hill, University of Missouri, USAReviewed by:
James Sowers, University of Missouri, USAAaron J. Trask, Nationwide Children's Hospital, USA
Copyright © 2016 Westermeier, Riquelme, Pavez, Garrido, Díaz, Verdejo, Castro, García and Lavandero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio Lavandero, slavander@uchile.cl; sergio.lavandero@utsouthwestern.edu