
- 1 Bioinformatics Laboratory, Plant Biology Division, Samuel Roberts Noble Foundation, Ardmore, OK, USA
- 2 Genetics and Developmental Biology, Plant and Soil Sciences Division, West Virginia University, Morgantown, WV, USA
Eukaryotic messenger RNA (mRNA) contains not only protein-coding regions but also a plethora of functional cis-elements that influence or coordinate a number of regulatory aspects of gene expression, such as mRNA stability, splicing forms, and translation rates. Understanding the rules that apply to each of these element types (e.g., whether the element is defined by primary or higher-order structure) allows for the discovery of novel mechanisms of gene expression as well as the design of transcripts with controlled expression. Bioinformatics plays a major role in creating databases and finding non-evident patterns governing each type of eukaryotic functional element. Much of what we currently know about mRNA regulatory elements in eukaryotes is derived from microorganism and animal systems, with the particularities of plant systems lagging behind. In this review, we provide a general introduction to the most well-known eukaryotic mRNA regulatory motifs (splicing regulatory elements, internal ribosome entry sites, iron-responsive elements, AU-rich elements, zipcodes, and polyadenylation signals) and describe available bioinformatics resources (databases and analysis tools) to analyze eukaryotic transcripts in search of functional elements, focusing on recent trends in bioinformatics methods and tool development. We also discuss future directions in the development of better computational tools based upon current knowledge of these functional elements. Improved computational tools would advance our understanding of the processes underlying gene regulations. We encourage plant bioinformaticians to turn their attention to this subject to help identify novel mechanisms of gene expression regulation using RNA motifs that have potentially evolved or diverged in plant species.
Background
Messenger RNA (mRNA) is a class of RNA molecules that is transcribed from a DNA template (gene) and carries coding information for protein synthesis. A eukaryotic mRNA molecule is generated by transcription of a gene in the nucleus, at which point it is known as pre-mRNA. The pre-mRNA molecule then undergoes further processing (e.g., 5′ capping, splicing, 3′ polyadenylation) to become mature mRNA, which is transported to the cytoplasm where several translation cycles are processed by the ribosomal machinery. The mRNA molecule is eventually degraded. This process is often referred to as gene expression. The whole process of gene expression is accomplished by dynamic association/dissociation of different regulatory molecules with DNA/mRNA. For instance, the translation of a nucleic acid polymer into an amino acid chain requires several additional molecules, such as cis-acting elements, trans-acting factors, transfer RNAs (tRNAs), and ribosomes. Regulation of gene expression is an important mechanism that increases the versatility and adaptability of an organism by allowing the cell to express specific proteins at a particular time. Gene expression was initially thought to be controlled only at the DNA level by interaction between trans-acting factors and the gene promoter, while transcribed mRNAs were viewed as simple molecules containing only information for protein synthesis. However, later discoveries showed that mRNAs and their precursors also contain various functional elements that can interact with different RNA-binding proteins (RBPs) to modulate gene expression at both the transcriptional and post-transcriptional levels. Gene regulation at the transcriptional level determines whether a gene is transcribed or not and to what extent (Mignone et al., 2002). Post-transcriptional regulation of gene expression acts on transcribed RNA molecules by altering their stability (Bashirullah et al., 2001), translation efficiency (van der Velden and Thomas, 1999), and subcellular localization (Jansen, 2001; Mignone et al., 2002). Functional elements of mRNA can be defined as motifs/segments of the molecule that specifically interact with RBPs or other cis- or trans-acting factors present in the cellular milieu in order to regulate gene expression. Both the primary sequences of these elements and the correct structural conformation are often necessary for function.
Almost all eukaryotes utilize RNA polymerase II (Pol II) to synthesize mRNA transcripts from protein-coding gene sequences. The initial primary transcripts are converted to mature products in the nucleus by undergoing extensively complex processing steps: addition of 7-methyl-guanylate (m7G) cap at 5′-end of the first exon, removal of introns (splicing), cleavage, and addition of poly(A) tail at the 3′-end of the last exon (Reed and Hurt, 2002; Buratowski, 2005). This manipulation results in the formation of a mature mRNA that consists of a 5′ untranslated region (5′-UTR, also called a leader sequence), a coding region, and a 3′ untranslated region (3′-UTR, also called a trailer sequence). The mRNA is then exported to the cytoplasm to be translated into protein. All of these processes are accomplished and regulated with the help of several functional cis-elements, embedded either in the protein-coding or in the untranslated regions (UTRs) of the transcript, by interacting with RBPs.
In order to further our understanding of gene regulation, it is imperative to know the prevalence of different types of functional elements on mRNAs and their characteristic features. It is also important to discern how a functional cis-element in the transcript selectively interacts with an RBP and how mutations in either functional cis-elements or regulatory proteins, such as trans-acting factors, affect gene expression. In this review, we provide an introduction to the major classes of functional elements present in eukaryotic pre-mRNA and mature mRNA. We focus on the available bioinformatics resources (databases and tools) for the analysis of these elements. We also discuss future directions in the development of better computational tools based upon current knowledge of these functional elements. Such tools would advance our understanding of the processes underlying gene regulations. We aim to encourage plant bioinformaticians to turn their attention to this subject and help identify novel mechanisms of gene expression regulation using RNA motifs that have potentially evolved or diverged in plant species. In the following sections, we describe those functional elements that still pose challenges. Understanding the mechanisms of gene regulation in which these elements are involved will require greater attention from investigators.
Functional Elements of an mRNA Molecule
Numerous functional elements and the RBPs with which they interact have already been reported and elements are still being discovered. Figure 1 depicts some important functional elements found on diverse eukaryotic mRNAs. These elements interact with different RBPs expressed in the cell to perform particular functions. Some interactions are highly specific, whereas others acquire specificity only through the binding of auxiliary proteins. The initial step in gene expression is the transcription of pre-mRNA and its subsequent processing to produce mature mRNA. Studies show that several enzymes involved in the maturation of pre-mRNAs are bound to Pol II and, therefore, the transcription and maturation processes are not strictly separated but occur simultaneously. The transcription start point for Pol II commonly lies within the initiator element (Inr) sequence context (Y2CAY5, where Y is any pyrimidine), which is located ∼70–80 bp downstream of the CAAT box, ∼25 bp downstream of the TATA box, and/or ∼24 bp upstream of the DPE element (AGAC) in the gene promoter. As the 5′-end of the new transcript appears during transcription, the γ-phosphate of the triphosphate group of the 5′ nucleotide (usually the adenine within the Inr sequence) is removed by the enzyme RNA triphosphatase, so that the capping enzyme, guanylyl transferase, adds a GMP moiety to the resulting diphosphate group at the 5′-end of the transcript through a non-canonical 5′–5′ linkage reaction. Subsequently, the 7-nitrogen of the capping guanosine is methylated by the enzyme methyl transferase. The result is the formation of the m7GpppN at the 5′-end of the mRNA, which is referred to as the 5′-cap that plays several essential roles such as: preventing degradation of mRNA by exonucleases, assisting in ribosome binding for the initiation of translation, and regulating nuclear export of the mRNA. During transcript termination, the cleavage and polyadenylation specificity factor (CPSF) forms a complex with the polyadenylation signal (PAS) sequence present downstream of the coding sequence (CDS) in the pre-mRNA. Furthermore, binding of the cleavage stimulation factor (CstF) to the complex promotes cleavage of transcript at ∼35 nt downstream of the PAS, and 150–200 adenine nucleotides are rapidly added to the freshly generated 3′-end by the enzyme poly(A) polymerase while poly(A)-binding proteins binds to the poly(A) tail. The resulting poly(A) tail assists in mRNA export from nucleus, provides stability, and facilitates initiation and efficiency of translation.
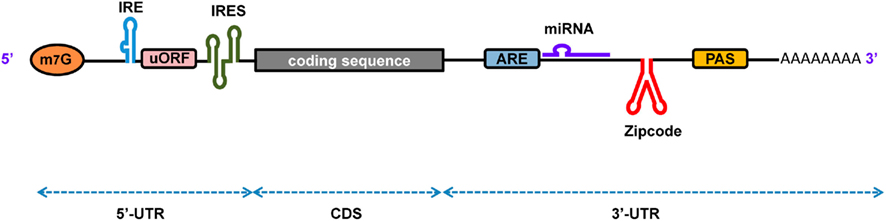
Figure 1. A schematic representation of eukaryotic mRNA with functional elements. UTR, untranslated region; CDS, coding sequence; m7G, 7-methyl-guanosine cap; IRE, iron-responsive element; uORF, upstream open reading frame; IRES, internal ribosome entry site; ARE, AU-rich element; PAS, poly(A) signal.
The removal of introns leads to the formation of a mature mRNA molecule, which assembles with proteins to form ribonucleoprotein (RNP) particles before being exported to the cytoplasm for translation. RNP particles protect the mRNA from degradation and promote the binding of adaptor proteins, which facilitate its transport from the nucleus to the cytoplasm via nuclear pore complexes (NPCs; reviewed in Kohler and Hurt, 2007). Instead of a random dispersion of mRNAs throughout cytoplasm, studies demonstrate that numerous mRNAs are delivered to specific locations in the cytoplasm. The cytosolic localization of most of the mRNAs is achieved by RNA localization elements (zipcodes) located in the 3′-UTR which, together with RBPs, molecular motors, and the cytoskeleton, direct the transport of mRNAs to a specific location in the cytosol for translation (reviewed in Martin and Ephrussi, 2009). In the mRNA, the region that codes for a protein is called the CDS, whereas the remaining UTRs, the 5′-UTR and the 3′-UTR, are responsible for the regulation of translation. During translation initiation, the eIF4F protein complex, which consists of four subunits: eIF4E (the cap-binding protein), eIF4A (the RNA helicase), eIF4G (which interacts with various other proteins including polyadenylate-binding protein), and eIF4B (which activates the RNA helicase activity of eIF4A to unwind secondary structures formed at the 5′-end of the mRNA), binds at the 5′-cap. Concomitantly, the small ribosomal 40S subunit complexes with several protein factors and a GTP-charged initiator tRNA to generate the so-called 43S preinitiation complex, which is then ready to interact with the mRNA and associated proteins, forming the 48S preinitiation complex. This complex scans the mRNA toward its 3′-end to find the start codon (AUG). The complete ribosome (80S) results from the dissociation of several factors and the association of the GTP-charged eIF5A and, most importantly, the large ribosomal 60S subunit. The complete ribosome translates the CDS from the start codon to the stop codon into an amino acid chain. Although most eukaryotic mRNAs contain the 5′ cap, some do not. In mRNA lacking the 5′ cap, the ribosome is guided to the correct start codon with the help of structural motifs located in the 5′-UTR, which are referred to as internal ribosome entry sites (IRES). This process is known as cap-independent initiation, which occurs by directing eIF4G (or the small ribosomal subunit and associated factors directly) to bind to the IRES, allowing the 43S preinitiation complex to bind to the mRNA and to scan downstream of the sequence to identify the start codon.
After several round of translation, the mRNA undergoes degradation. The rate at which a particular mRNA decays is based upon: (1) cis-acting elements present on the mRNA and (2) the enzymatic environment in the cell. The mechanisms for mRNA degradation can be divided into two broad classes. Most mRNA undergoes degradation via the deadenylation-dependent decay pathway, which is initiated by shortening of the 3′-poly(A) tail by the deadenylase enzyme, poly(A) nuclease, followed by decapping and ultimately cleavage of the end nucleotides by exonucleases (Wilusz et al., 2001; Meyer et al., 2004). Alternatively, mRNAs may be degraded by the deadenylation-independent decay pathway, in which mRNA, without deadenylation, undergoes cleavage by endonuclease and exonuclease enzymes via deadenylation-independent decapping, endonucleolytic cleavage, oligo(U) addition (specifically to histone mRNAs), or microRNA (miRNA)-mediated silencing. The trans-factors involved in this degradation recognize specific sequences within the transcript and promote transcript decay. Several mRNAs (8% of mammalian mRNAs) contain AU-rich elements (ARE), which are destabilizing elements located at the 3′-UTR responsible for rapid degradation of transcripts. These elements act by interacting with ARE-binding proteins, which recruit the degradation machinery.
In order to maintain iron homeostasis in the cell, some mRNAs form several stem–loop structures in the 5′- or 3′-UTR that act as stabilizing motifs called iron-responsive elements (IREs). Under conditions of iron starvation, the iron regulatory protein (IRP) binds to the IRE and induces an allosteric change in order to control the expression of target mRNAs either by promoting mRNA degradation or repressing translation. Similarly, many prokaryotic mRNAs use riboswitches as functional element to sense the presence of specific molecules which can help to switch gene expression on or off. Bacteria and viruses possess small genomes and utilize functional elements to expand the number of possible protein products. To enhance the information content of an mRNA, motifs called ribosome frameshift signals (RFS) are used, which shift the ribosome to an alternative coding frame and yield two different protein products from the same mRNA or a single protein from two overlapping ORFs.
A decade ago, it was discovered that eukaryotic cells evolved RNA interference mechanisms (RNAi) to control gene expression at the post-transcriptional level. This process produces small RNAs of ∼21–24 nt called miRNA, which are derived from hairpin structure embedded in a long miRNA primary transcript (pri-miRNA). The pri-miRNA undergoes two subsequent cleavages by members of the RNase III superfamily, Drosha, in a complex with the essential cofactor Pasha, and Dicer (in animals) or DCL1 (in plants), which generate the miRNA:miRNA* duplex. Based on sequence and structural features, only one strand of the duplex, the miRNA, incorporates into the RNA-induced silencing complex (RISC), while the passenger strand, the miRNA*, is thought to be degraded (Schwarz et al., 2003; Ahmed et al., 2009a). The activated RISC, containing the miRNA, binds to the target mRNA (which possesses some base complementarity to the miRNA), resulting in either cleavage of the mRNA or arrest of translation. Interestingly, miRNA binding sites are mostly found in the 3′-UTR of animal mRNAs, whereas in plants, both CDS and UTR regions are targeted (Dai and Zhao, 2011; Dai et al., 2011). Other small non-coding RNA classes exist with diverse mechanisms involved to control gene expression via RNA degradation, translation arrest, DNA methylation and heterochromatin formation. Here, we focus on intrinsic RNAi mechanisms of gene expression control. Trans-acting small interfering RNAs (tasiRNAs) are ∼21-nt long, found in plants, and generated by a pathway that involves miRNA-mediated cleavage of a precursor mRNA sequence. They are often associated with the silencing of hormone-related developmental genes. (Dai and Zhao, 2008). Among the other small RNA species, it is worth mentioning Piwi-interacting RNAs (piRNAs), which are found in animal germ cells. These small RNAs are produced by a different pathway, are longer (26–31 nt) and more complex (with a lower level of sequence conservation) than miRNAs, and their function is related to the epigenetic and post-transcriptional silencing of retrotransposable elements. Repeat-associated siRNAs (rasiRNAs) are a subclass of piRNA that act in germline cells to control heterochromatin architecture and silence transposable and repetitive elements.
Splicing Regulatory Elements
Eukaryotic pre-mRNA contains small segments of coding regions known as exons (∼50- to 250-bp long), usually interrupted by large segments of non-coding regions known as introns (hundreds to thousands of basis point long). In order to generate the functional mature mRNA, it is necessary to splice exons at the intron–exon boundary, removing the introns, and ligating the exons. For splicing to occur at the correct position, the intron has well-conserved cis-elements known as the 5′ splice site (GU), the branch site (UACUAAC), a polypyrimidine (Y) tract, and the 3′ splice site (AG; Figure 2). All of these elements are known as the core splicing signals on which the spliceosome complex [which contains five small nuclear RNPs (snRNPs) and various auxiliary proteins] is assembled in order to catalyze proper intron excision and exon ligation.
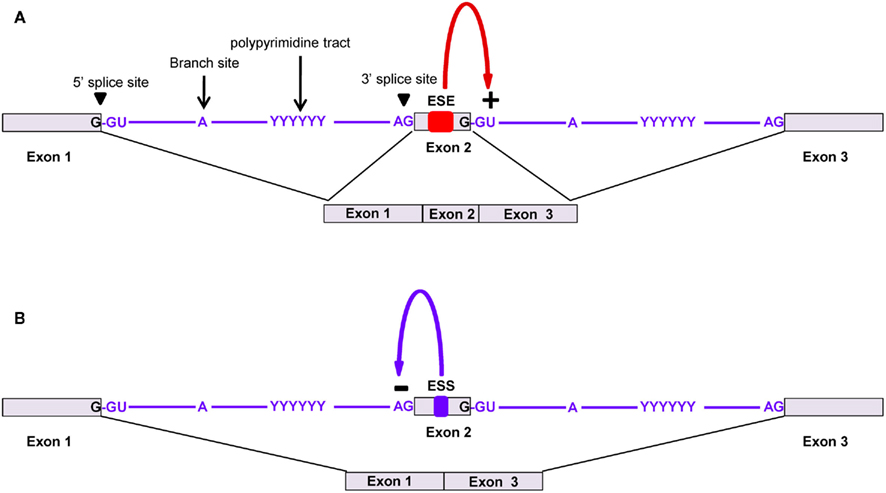
Figure 2. Alternative splicing of pre-mRNA using splicing regulatory elements. Core splicing signals help to recruit the spliceosome complex. The assembly of five ribonucleoprotein subunits (U1, U2, U4, U5, and U6 snRNPs) with several other proteins make up the spliceosome, which catalyzes the splicing and ligation of exons. (A) Exonic splicing enhancers (ESEs) promote the binding of the spliceosome complex, which leads to the splicing and removal of introns for exon inclusion. (B) Exonic splicing silencers (ESSs) bind to trans-acting factors that prevent the binding of the spliceosome complex or other protein factors, which leads to exon exclusion.
Minor spliceosome machinery exists in multicellular eukaryotes that performs a less canonical splicing between AU-AC sites rather than between the GU-AG sites targeted by the major spliceosome machinery. Moreover, there are some pre-mRNAs that undergo alternative splicing (AS) in which exons are alternatively integrated into the mature mRNA depending on the cell type and physiological conditions. Such pre-mRNA presents weaker patterns of core splicing sites, which require some auxiliary elements known as splicing regulatory elements (SREs) for correct identification of splicing sites. The process of AS allows the generation of multiple forms of mRNA from a single pre-mRNA species, leading to protein diversification. This process not only helps to add or remove protein domains, but it can also shift the reading frame to translate a different protein. When it occurs in the 5′ or 3′-UTR of pre-mRNA, regulatory elements can be added or removed to modulate the fate of the mRNA.
The SREs that stimulate or repress splicing site selection are called splicing enhancers or splicing silencers, respectively. Enhancers promote the inclusion of exons in the mature mRNA by assisting in the assembly of the spliceosome complex. Splicing silencers promote exon exclusion by binding to repressor proteins to block either the assembly of the spliceosome complex or the interaction between proteins responsible for intron excision (Ule et al., 2006). Furthermore, enhancers and silencers may be present in exonic or intronic regions. Thus, based on function and location, SREs can be classified as exonic splicing enhancers (ESEs), intronic splicing enhancers (ISEs), exonic splicing silencers (ESSs), or intronic splicing silencers (ISSs). For proper function, SREs are bound by specific classes of RBPs, such as serine/arginine-rich proteins (SR) and heterogeneous nuclear RNPs (hnRNP; Black, 2003). Thus, tissue-specific expression of particular RBPs determines the inclusion or exclusion of alternative exons. Several examples demonstrating the importance of SREs for correct splice site identification and the mechanism of alternative splicing have been well documented in recent reviews (Smith and Valcarcel, 2000; Ladd and Cooper, 2002; House and Lynch, 2008; Wang and Burge, 2008).
It has been reported that some SREs can act as either enhancers or silencers of splicing, depending on their locations relative to alternative exons. For instance, YCAY clusters, the binding sites of the Nova protein, act as silencers when located in an exon or in the preceding intron while they act as splicing enhancers when found in the downstream intron (Sugnet et al., 2006; Ule et al., 2006). Mutation or loss of SREs leads to incorrect splicing that generates toxic proteins, which have been implicated in severe pathologies. To date, the patterns that determine alternative splicing have not been fully elucidated; therefore, it is necessary to further study specific alternative splicing choices and gene products under different cell conditions.
Several computational methods have been developed to predict splicing sites in eukaryotic genes by considering sequence features around the core splicing sites consensus of GT…AG), such as SplicePredictor (Usuka and Brendel, 2000), GeneSplicer (Pertea et al., 2001), SpliceMachine (Degroeve et al., 2005), and others (Pavy et al., 1999; Lim et al., 2011). In spite of the high prevalence of alternative splicing in animals and plants (Wang et al., 2008; Koscielny et al., 2009), an important factor missing in most of these genome annotation tools is the prediction of SREs. SREs play an important role in determining the boundaries of introns when core splicing signals are weak. A recent study has shown that integration of predicted ESEs increases the splice site prediction accuracy of GeneSplicer and SpliceMachine (Pertea et al., 2007). In the study, 50-nt regions on either side of internal exons in Arabidopsis thaliana were analyzed by searching for conserved patterns. They identified 84 hexameric ESE candidates, of which 35 were experimentally reported as ESEs (Pertea et al., 2007).
Expression of tissue/cell-specific splicing factors may provide mechanistic insights into AS. However, patterns that determine alternative splicing are still poorly understood, especially in plants (Kazan, 2003). AS databases are valuable resources for the study of SREs, and several of them are publicly available (Table 1). Generally, AS can be identified by aligning ESTs/mRNA to the genome assembly using tools such as BLAST or BLAT (Kim and Lee, 2008). Determining how alternative splicing is regulated under different physiological conditions remains a challenging problem. Data mining on how transcripts are expressed at specific stages or in specific tissues can help detect novel and specific SREs. Moreover, instead of analyzing terminal sequences at exons–intron borders, the whole transcript sequence should be analyzed. There are several tools available for sequence pattern recognition, such as MEME (Bailey and Elkan, 1994), GIMSAN (Ng and Keich, 2008), and Gibbs sampling (Stormo, 2010). Furthermore, new insights are emerging on the role of secondary structures in pre-mRNA during AS that are still unexplored (Jin et al., 2011). The compiled AS sequence data could be made more reliable by comparing it with the amino acid sequences of known protein isoforms. Subsequently, the discovery of important patterns in introns and exons can be utilized to develop more accurate computational methods for gene annotation.
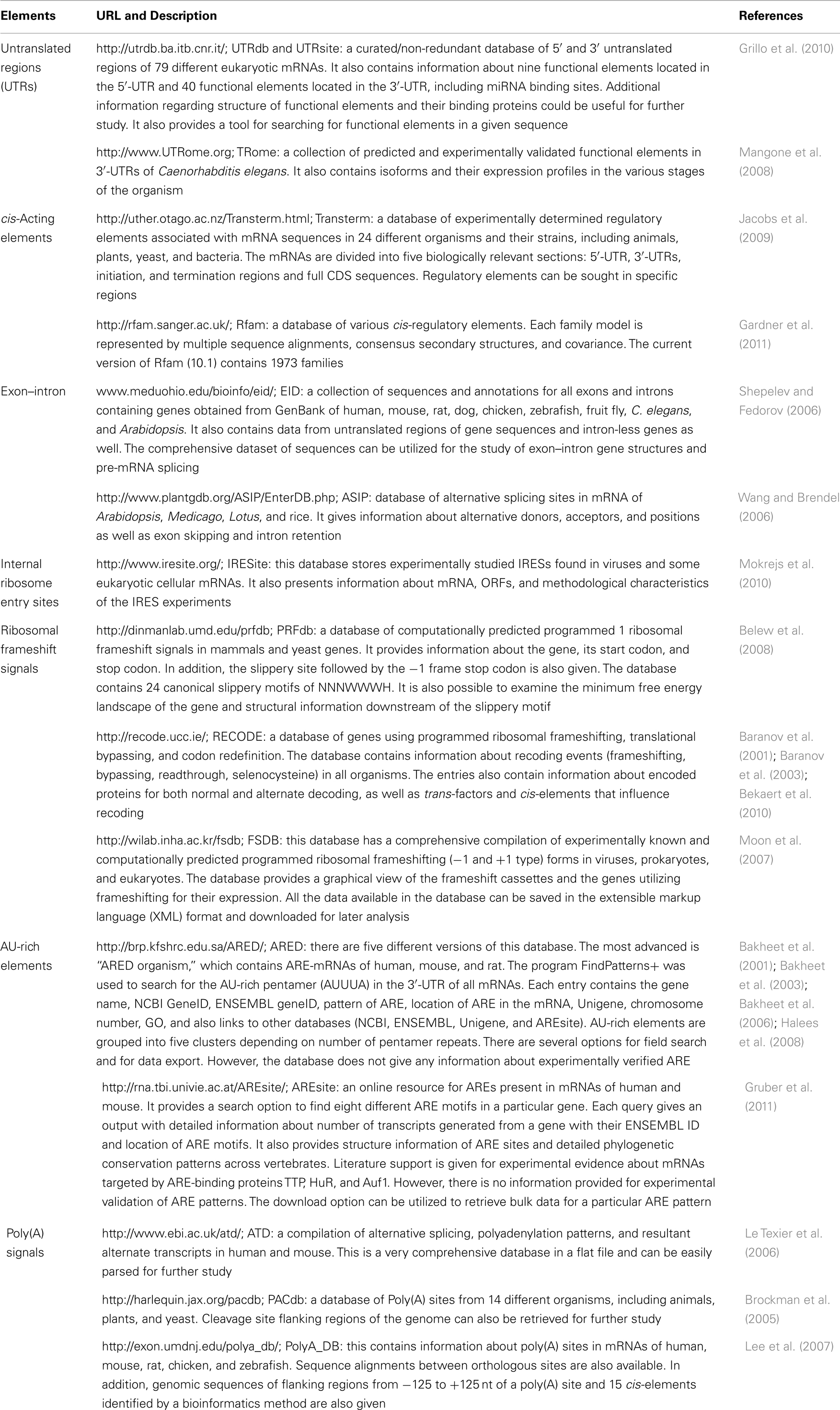
Table 1. Databases of different functional elements of mRNA and their components.
Internal Ribosomal Entry Sites
These cis-elements enable the cap-independent recruitment of the 40S ribosomal subunits to the 5′-UTR of the transcript, thus bypassing the requirement of some translation initiation factors. Many viruses, such as the hepatitis C virus and HIV, contain transcripts with strong IRESs to manipulate the host cell to translate viral protein-coding RNAs. The bacterial Shine–Dalgarno element (AGGAGG) is a well-characterized motif located ∼10 bp upstream of the initiation codon of the regularly uncapped transcripts. It induces high translation rates with minimal recruitment of initiation factors due to its base-pairing property with the small ribosomal subunit. In addition to its canonical localization in the promoter region, it can also be positioned downstream of the first start codon, thus acting as an alternate IRES to permit polycistronic transcripts that encode alternative proteins in prokaryotes. It has been demonstrated that eukaryotic transcripts also use this mechanism to induce high translation rates for proteins like p27/Kip1 (a cyclin-dependent kinase inhibitor that controls the cell cycle; Zheng and Miskimins, 2011) and the insulin-like growth factor receptor, IGF1R, (Meng et al., 2010) under certain physiological conditions. IRES-regulated (i.e., cap-independent) translation has been shown to be involved not only in the etiology of viral diseases, but also other diseases, such as several cancer types and Alzheimer’s (Beaudoin et al., 2008; Allam and Ali, 2010). Certainly, a better understanding of the secondary and tertiary structures of this type of eukaryotic mRNA element (Spriggs et al., 2009) will shed more light on the eukaryotic cap-independent recruitment mechanisms of the translational machinery, which will contribute to the discovery of novel eukaryotic IRES-containing transcripts. Bioinformatics will unquestionably play a crucial role in this development, starting with the development of IRES databases (Mokrejs et al., 2010), which may lead to the construction of a model to understand and discover genes under the control of IRESs that induce cap-independent translation.
Ribosome Frameshift Signals
These signals direct elongating ribosomes to shift the reading frame a single nucleotide (forward or reverse) on the coding mRNA, which can lead to premature stop codons (which can result in mRNA destabilization via the nonsense-mediated decay pathway) or the production of an alternative protein from the same transcript. This phenomenon is well-characterized in viruses, prokaryotes, and organelle systems. Programmed −1 RFSs are present in eukaryotic transcripts as heptameric “slippery” sites (e.g., AAAAAAT) followed by a spacer and a secondary structure (pseudoknot) that together induce the ribosome to reverse one nucleotide (Figure 3). The pseudoknot structure has also been predicted to halt translation (perhaps under certain situations when the secondary structure is more stable, such as cold temperatures in non-homeothermal organisms or high intracellular levels of salt), inducing mRNA destabilization via the no-go decay (NGD) pathway. Databases of computationally predicted frameshift signals in eukaryotes and prediction tools are available (Table 1 and 2). Phylogenetic analyses indicate that these RNA cis-elements evolve rapidly, and sequence analysis with yeast identified −1 RFSs with high confidence in ∼10% of the genes in eukaryotic genomes (Belew et al., 2011). The potential impact of RFSs in post-transcriptional control of gene expression or production of alternative protein forms from the same transcript in higher eukaryotes, including plants and humans, still awaits full investigation.

Table 2. Tools for predicting different functional elements in mRNA.
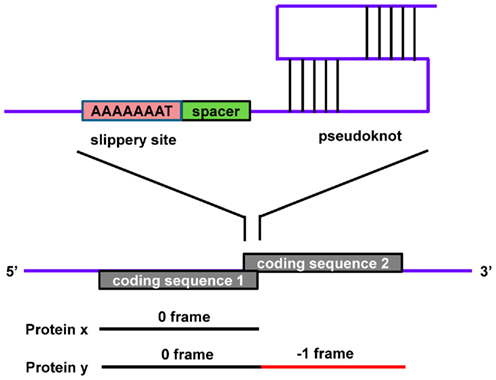
Figure 3. A typical −1 ribosomal frameshift signal contains a slippery site, a spacer, and a pseudoknot. Protein x is produced in the 0 frame from CDS 1 while a −1 frameshift bypasses the stop codon, resulting in the fusion of CDS 1 and CDS 2, which produces a longer protein, y.
Riboswitches
These are catalytic domains of mRNA molecules that undergo conformational changes when bound to small molecules or metal ions, leading to changes in transcription, translation, or splicing, independently of protein regulators (Mandal et al., 2003; Winkler and Breaker, 2003). The aptamer region of the riboswitch, usually in the 5′-UTR, forms a secondary structure where the ligand is recognized, which induces an allosteric change of the riboswitch configuration that causes a reaction, such as cleavage of the mRNA or early termination of transcription to prevent gene expression. In this way, riboswitches adopt two different conformations. The repressed conformation, with a bound ligand, causes premature termination of transcription or inhibition of translation initiation. The de-repressed conformation, without binding bound ligand, allows translation and transcription to proceed normally. The repressed conformation contains base pairs that either cover the translation initiation site or form a terminator of transcription. Such conformations are eliminated during the de-repressing stage. Riboswitches are involved in the regulation of several metabolic pathways, including the biosynthesis of vitamins, purines, and amino acids. The advantage of riboswitches over other elements is that they directly bind to ligands without the help of additional proteins. In this way, riboswitches can sense the concentration of ligands in the cellular environment, and if found in sufficient amount, inhibit gene expression. Riboswitches are also conserved across diverse organisms and are thus considered one of the oldest regulatory elements (Vitreschak et al., 2004).
The potential applications of riboswitches in biotechnology and medical fields are associated with their ability to control gene expression based on the presence of specific molecules (such as metabolites or ingested drugs; Mulhbacher et al., 2010a,b; Verhounig et al., 2010). Although these cis-elements are mostly characterized in prokaryotes, with an estimated 2% of bacterial genes regulated by riboswitches (Mandal et al., 2003), there is also evidence of a riboswitch in eukaryotic systems: the thiamine pyrophosphate (TPP) riboswitch that binds to vitamin B1 (Cressina et al., 2011), leading to changes in mRNA stability via alteration of the polyadenylation pattern. There are six different classes of riboswitches that have been reported: RFN, THI, B12, G-box, S-box, and LYS-elements (reviewed in Vitreschak et al., 2004). These differ in structural conformation and ligand binding, but all possess base stems, hairpins, and loops. For instance, the RFN-element is present upstream of several mRNAs involved in riboflavin biosynthesis (e.g., the rib operon and the ypaA gene) in a variety of bacterial genomes. The RFN-element binds to FMN when it is present in high concentrations, which causes premature termination of transcription and inhibition of translation initiation (Mironov et al., 2002; Winkler et al., 2002). In plants, there are suggestions that the expression of hormone receptors may be under the control of riboswitches with auxins and cytokinins acting as ligands (Meli et al., 2002; Grojean and Downes, 2010). This hypothesis is compelling, although it remains experimentally untested. The human cleavage factor Im (CFIm), a key component of the pre-mRNA 3′ processing complex involved in poly(A) site recognition, interacts with RNA only in the presence of the signaling molecule diadenosine tetraphosphate (Ap4A), indicating a possible role for ligands in mRNA 3′ processing in eukaryotes (Yang et al., 2010). Computational methods to discover riboswitches are an obvious choice. In addition, given the evolutionary conservation of aptamer sequences belonging to the same riboswitch class, a recently developed algorithm based on profile hidden Markov models (pHMMs) can be used to identify known classes of riboswitches, especially in prokaryotes (Singh et al., 2009). Despite their prokaryotic origins, mitochondria, and plastids do not hold known riboswitch genes. However, a synthetic theophylline riboswitch introduced into tobacco chloroplasts was recently demonstrated to function in planta (Verhounig et al., 2010), opening avenues to control gene expression in cell organelles. Given the paucity of known eukaryotic riboswitch systems and the complexity of predicting the interaction between the secondary structure of mRNAs and small molecules, methods to discover novel riboswitch classes using computational programs are still a far-reaching goal. However, attempts to achieve this goal coupled with experimental confirmation might generate successful results. A more tangible goal, however, might be the use of programs to design organellar (plastidial, potentially also mitochondrial) riboswitches using well-established systems.
Iron-Responsive Elements
These highly conserved stem–loop structures are mainly responsible for maintaining iron homeostasis in vertebrates (Muckenthaler et al., 2008). IREs are located in the 5′ or 3′-UTR of various mRNAs that encode proteins involved in iron metabolism, such as ferritin, transferrin receptor, erythroid 5-aminolevulinic-acid synthase, mitochondrial aconitase, ferroportin, and the divalent metal transporter 1. IREs undergo conformational changes upon interacting with IRPs, and the fate of the mRNA is modulated depending on the UTR location of the IRE. These elements are grouped into nine classes based on the mRNA in which they are present (Piccinelli and Samuelsson, 2007). They possess a 26- to 30-nt long hairpin structure with a conserved central loop of CAGWGN and an unpaired C residue (UGC in ferritin) in the stem region (Figure 4). NMR-spectroscopy has shown base pair formation between C1 and G5 of the CAGWGN hexaloop, which pushes the AGW segment into the solvent, facilitating IRP binding with the hairpin (Addess et al., 1997). Substitution of C-G for AU prevents IRP binding, which also indicates its crucial role in the RNA-protein interaction. Directed mutagenesis studies have shown that the residues between the hexaloop and the C-bulge or loop bulge, also contribute to protein binding. Several diseases have been found to be related to impaired interactions between IREs and IRPs (Girelli et al., 1995; Kato et al., 2001; LaVaute et al., 2001).
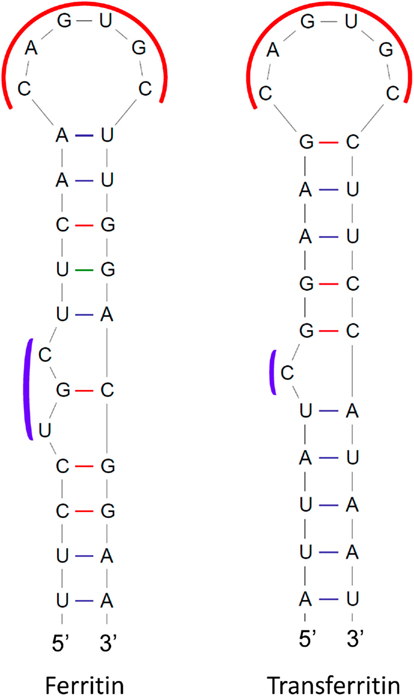
Figure 4. Structures of iron-responsive elements (IREs) in mRNAs encoding human ferritin and transferrin receptor proteins. Conserved regions are shown in red and blue curves.
Binding of IRPs to IREs in the 5′-UTR reduces rates of translation, while IPR binding to IREs in the 3′-UTR has been reported to increase mRNA stability. For instance, ferritin is an iron storage protein and its mRNA has an IRE in the 5′-UTR. When concentrations of iron become low, IRPs bind to the ferritin mRNA, resulting in repression of translation. However, at high iron concentrations, the IRP has lower affinity for the IRE and the ferritin mRNA is translated. The mRNA of the transferrin receptor contains five copies of IREs, which are all located in the 3′-UTR. Under conditions of low iron concentration, the IRP binds to the IREs, resulting in stabilization of the transferrin receptor mRNA. In contrast, under conditions of high iron concentration, the IRP has low affinity for the IREs, and the transferrin receptor mRNA is degraded. Many oxygen metabolism proteins, heat shock housekeeping proteins, proto-oncogenes, chlorophyll-binding proteins, and ribonucleotide reductases also have IREs in their transcripts. It is noteworthy that animal and plant ferritin CDSs are highly conserved, but plant ferritin mRNAs lack IREs (Kimata and Theil, 1994).
Recently, an informatics approach has been utilized to understand the sequence and structural characteristics of IREs and their phylogenetic conservation among eukaryotes (Piccinelli and Samuelsson, 2007). In the study, 49 known IREs were used to generate HMM profiles for each class of IREs. Moreover, they also used Rfam models of IREs, consisting of sequence, structure, and covariance features. Using these models, they scanned metazoan genomes and mRNAs, resulting in the discovery of 107 novel IREs. The study also indicated that the ferritin IRE is the most primitive and ancestral of all IREs found in other mRNAs. Very recently, an experimental study identified multiple sequence and structural determinants in IREs found in the 5′-UTR that are responsible for efficient binding with IRP1 (Goforth et al., 2010). This work provided useful data to generate bioinformatics models for IREs and to identify IRE-regulated mRNAs. Several tools are available for RNA secondary structure formation and pattern searches, such as PatScan. However, each tool has its own limitations. Thus, instead of using a single tool, various suitable tools should be utilized for model generation and pattern searching. Furthermore, the occurrences of single nucleotide polymorphisms (SNPs) in IREs have been largely underexplored. Thus, this is an area worth examining to enhance our understanding of the genetic basis of complex diseases in humans, and plant adaptation.
AU-Rich Elements
These are adenine- and uridine-rich segments embedded in the 3′-UTR of mammalian mRNAs that are responsible for rapid mRNA turnover (Caput et al., 1986). The control of mRNA stability is essential for adjusting mRNA levels and for limiting protein expression in order to prevent detrimental effects caused by overexpression (Bakheet et al., 2001). AREs are commonly found repeats of a core sequence of the AUUUA pentamer and are classified according to two distinct methods. According to Chen’s method, AREs are grouped into three different classes based on the number and distribution of repeats of the core sequence (Chen and Shyu, 1995). Class I is defined by a single AUUUA motif dispersed along a U-rich region, such as in c-myc and p21 transcripts. Class II is defined by an overlapping pattern of two or more motifs, such as WWAUUUAUUUAWW, like those in TNF-α and interferon-α transcripts. Class III AREs are poorly defined AREs in U-rich regions without the AUUUA motif, such as in the c-jun transcript. On the other hand, according to Bakheet’s method, AREs are classified into five groups based on the number of continuous repeats of the core sequence of AUUUA. Groups 1, 2, 3, and 4 possess 5, 4, 3, and 2 continuous repeats of AUUUA, respectively, whereas group 5 AREs possess only one core pattern in the transcript (Bakheet et al., 2001). These AREs are functional after interacting with AU-binding proteins (AUBPs), which further recruit different enzymes responsible for mRNA degradation in a process described as ARE-mediated decay (AMD; Figure 5; Barreau et al., 2005). There are several different AUBPs, such as AUF1, TTP, and HuR. Most of these destabilize, but some stabilize the mRNA and also modulate translation efficiency (reviewed in Barreau et al., 2005). For instance, binding of the protein TTP to AREs decreases mRNA stability, while binding of HuR to AREs increases mRNA stability. It has also been reported that different ARE-binding proteins bind to the same mRNA molecule and their relative levels may determine mRNA stability. However, the AMD mechanism is still poorly understood (von Roretz and Gallouzi, 2008).
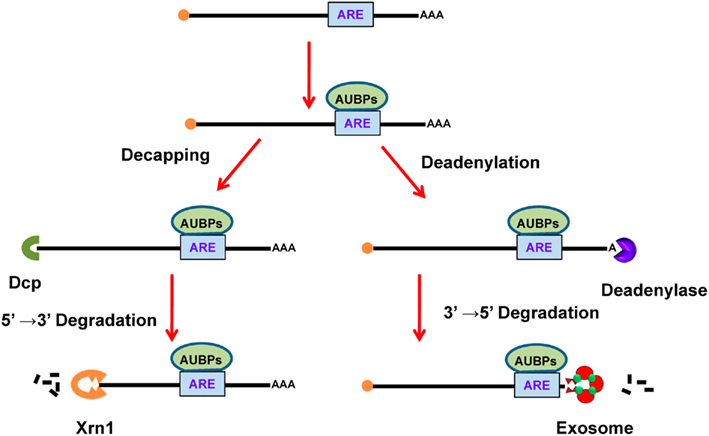
Figure 5. ARE-mediated decay of mRNA. In the cytoplasm, AUBPs (like TTP) bind to ARE (AUUUA) in the mRNA. The binding of AUBPs recruits either Dcp, which promotes decapping of mRNA, or deadenylase, which removes the poly(A) tail. After exposure of terminal mRNA, exonucleases act to degrade the mRNA in either the 5′–3′ direction using Xrn1 or in the 3′–5′ direction using the exosome. ARE, AU-rich element; AUBPs, AU-binding proteins.
In order to understand the significance of AREs, a computational analysis was carried out to identify the prevalence of AREs in human mRNAs (Bakheet et al., 2001). Fifty-seven experimentally verified 3′-UTR ARE-containing transcript sequences were extracted and revealed the conserved ARE motif “UAUUUAWW” via the MEME tool (Bailey and Elkan, 1994). Considering the flanking region around the motif, the authors identified a 13-bp conserved string (WWWUAUUUAUWWW). Subsequently, they searched for this 13-bp pattern in 3′-UTRs of 13057 human mRNAs using the program FindPatterns and reported 897 (∼8%) sequences that possess AREs. It was also revealed that ARE-containing mRNAs encode functionally diverse proteins. However, Gene Ontology (http://www.geneontology.org/)-based analysis showed that most of them play important roles in regulatory processes, such as cell communication, nucleic acid metabolism, cell proliferation, signal transduction, and transcription (Bakheet et al., 2006). A more recent study showed that the frequency of AREs is high among the most unstable mRNAs (half-life <2 h), while its presence decreases among mRNAs with increasing half-life (>8 h; Lam et al., 2001). In a very recent work, eight different consensus ARE motifs starting from AUUUA to WWWWAUUUAWWWW were scanned in human 3′-UTRs (Gruber et al., 2011). The work also considered the probability of AREs being unpaired in the sequence as well as conservation across different species. It was estimated that ∼13% of protein-coding genes in the human genome contain AREs (Gruber et al., 2011). However, conservation analysis of AREs in mRNA orthologs in human, mouse, and rat showed that a significant number of genes (25%) differ with respect to ARE patterns, indicating that species quickly evolved different AREs according to their own gene regulation requirements (Halees et al., 2008).
The association between turnover of defective mRNA and diseases underscores the significance of a deep understanding of the AMD mechanism (Espel, 2005; Khabar, 2005; Eberhardt et al., 2007). Yet, a compilation of mRNAs and their AREs along with half-life is still missing in available databases. This might open a new avenue to further establish the relationship between ARE types and positions and the transcript half-life. Furthermore, inclusion of more experimentally verified AREs in the training data should help uncover novel ARE motifs. Computational analysis has suggested that sequence features of the flanking region around poly(A) signals helped to more accurately identify poly(A) signals in the genome (Ahmed et al., 2009b). Therefore, the analysis of ARE-flanking regions to discover motifs promises to assist in the identification of weak and non-conserved AREs. Furthermore, integrating sequence and structural features of ARE-flanking regions should be adopted to develop a robust machine learning-based model for genome annotation.
Zipcodes
These are important elements generally present in the 3′-UTR of some eukaryotic mRNAs but also reported in the 5′-UTR and CDS region. Zipcodes are responsible for the delivery of mRNAs to subcellular compartments, thus this element is also known as the “localization element.” Restricting protein synthesis to a specialized compartment within polarized (e.g., Drosophila oocytes) and asymmetric (e.g., neurons) cells is vital for normal function because it prevents the diffusion of the mRNA throughout cytoplasm, provides protection against degradation, and translates the protein at the site at which it functions. Studies have shown that zipcodes are highly variable in length, sequence, and structure, which makes it difficult to define their exact characteristics (Chartrand et al., 1999). Zipcodes can be a tandem repeat of “ACACCC,” as found across a 54-nt region in β-actin mRNA that is responsible for its localization to lamellipodia of chicken embryo fibroblasts (Kislauskis et al., 1994). Alternatively, it can be a complex secondary structure, such as the five stem–loops found in bicoid mRNA spanning over a 625-nt segment in the 3′-UTR that is essential for its localization to the anterior pole of Drosophila oocytes (Macdonald and Kerr, 1997; Weil et al., 2006). Moreover, the existence of multiple localization elements of the same or different types in an mRNA makes it challenging to define its localization properties. In the case of Ash1 mRNA in budding yeast, several localization elements were reported, including E1, E2a, and E2b in the CDS region, and E3 in both the CDS region and the 3′-UTR, all working synergistically to deliver mRNA to the bud tip (Chartrand et al., 1999, 2002). During the processing of pre-mRNA in the nucleus, RBPs bind to zipcodes, and form a RNP complex. Several similar RNPs are assembled into a large granule that transports mRNAs to their final destination via motor proteins along the cytoskeleton (Figure 6; Knowles et al., 1996). Staufen is a well-characterized RBP that interacts with zipcodes within bicoid mRNAs and runs along myosin fibers to travel to the anterior pole of Drosophila oocytes (Macdonald and Kerr, 1997; Weil et al., 2006).
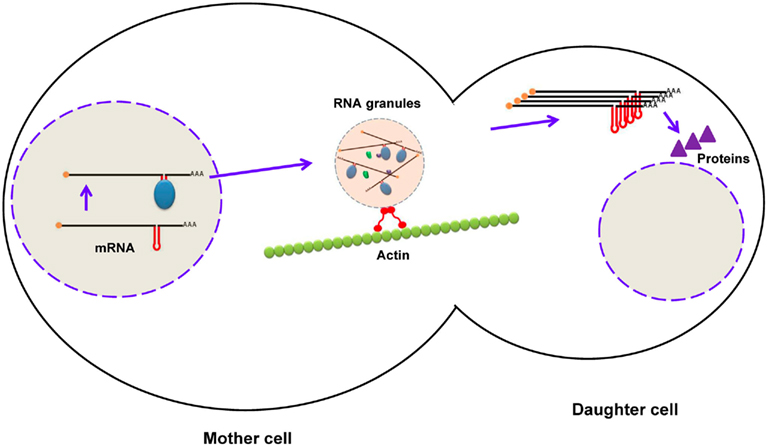
Figure 6. Localization of mRNA in budding yeast using zipcodes. During maturation of mRNA (ASH1), RNA-binding proteins bind to zipcodes and form ribonucleoprotein complexes. After export to the cytoplasm, some proteins are added to form RNA granules, which are transported to the daughter cell along the actin cytoskeleton. After mRNA reaches the distal pole, it is translated into Ash1p (a transcription factor), which enters the nucleus of the daughter cell.
Two commonly used experimental techniques for deciphering zipcodes and their characteristics are in situ hybridization and tagging mRNA with a fluorescently tagged RBP (Weil et al., 2010). Employing fluorescent in situ hybridization has demonstrated that 71% of 3370 mRNAs show subcellular localization in the Drosophila embryo (Lecuyer et al., 2007). However, these experimental methods are tedious, time-consuming, and impractical for deciphering zipcodes at a genome-wide level. Several studies have shown that, by using bioinformatics methods, subcellular localization of proteins can be successfully addressed (Garg et al., 2005; Garg and Raghava, 2008; Kaundal and Raghava, 2009; Kaundal et al., 2010). However, studies addressing zipcodes by exploiting bioinformatics approaches are very limited, despite promising results (Cohen et al., 2005; Hamilton et al., 2009; Hamilton and Davis, 2011). Very recently, a computational method, RNA2DSearch, was developed for discovering localization signals in transposable elements (Hamilton et al., 2009). Two localization signals were studied that are present in gurken and I factor retrotransposon mRNA, GLS, and ILS, respectively. These signals target the transcript to the dorso-anterior area of Drosophila oocytes. The study found that despite sending both transcripts to the same subcellular position, the signals only show 35% sequence similarity. However, the signals are very similar at the secondary structure level. In order to discover new transposons with similar characteristics, they compiled transposable element and folding data with RNALfold. Furthermore, the secondary structures were compared with those of GLS and ILS by using RNAdistance and RNAforester, resulting in 48 potential candidates. Among these, 22 were tested, and only two transposons, G2 and Jockey, were experimentally confirmed to localize to the predicted position. This finding indicates that although bioinformatics might be a good alternative approach to discover new candidate zipcodes, at least at this point wet experiments are still needed to prove in silico results. Additionally, there is room for developing better prediction tools for zipcodes with high accuracy using machine learning techniques.
Defective sequences in localization signals of mRNA in neuronal cells have been reported as the etiology of several diseases, demonstrating the importance of subcellular localization of mRNAs (Jin and Warren, 2003; Mutsuddi et al., 2004). On the other hand, it is still not clear how many mRNAs contain zipcodes in animals and plants. Several studies have shown the existence of multiple pathways for mRNA localization in plants and, among these, some have zipcodes that are still largely unexplored (Bouget et al., 1996; Vermerris et al., 2001; Okita and Choi, 2002; Crofts et al., 2004). The field of mRNA subcellular localization is still at a very early stage and requires more attention regarding the prevalence and characteristic features of zipcodes in both the plant and animal kingdoms. Developing a database of experimentally verified zipcodes with their sequence and structural characteristics with localization properties would be a very good initiative. Furthermore, sequence and structural patterns could be extracted for each zipcode using a computational approach (Hamilton and Davis, 2007, 2011; Lan et al., 2010) and could be integrated to develop algorithms for zipcode prediction and genome annotation. Inclusion of RBP features associated with zipcodes may further refine the confidence of zipcode identification. This will not only help to understand gene regulation at the subcellular level but it will also be useful in engineering an mRNA to be delivered to a specific destination within the cell, which may have important biotechnological and medical applications.
Poly(A) Signals
Polyadenylation signals are very important functional elements in all eukaryotic mRNAs at the 3′-end that are responsible for transcript maturation. A PAS is composed of a hexamer with a canonical signal (AAUAAA). This motif recruits several protein factors and determines the position of cleavage, which occurs ∼35 nt downstream of the PAS at the 3′-end of the nascent pre-mRNA. This is also the position of the addition of a poly(A) tail of ∼200 adenine residues by the poly(A) polymerase (Danckwardt et al., 2007). The importance of PAS’s has been revealed by several studies showing that a mutation in PAS’s alters the cleavage site and generates aberrant transcripts that lead to several diseases (Orkin et al., 1985; Rund et al., 1992). A computational analysis extracted 13 variants of PAS’s, covering 93% of the human transcriptome (Tian et al., 2005). Among them, AAUAAA and AUUAAA are highly prevalent in both animals and plants, but alternative PAS’s (UAUAAA, AGUAAA, AAGAAA, AAUAUA, AAUACA, CAUAAA, GAUAAA, AAUGAA, UUUAAA, ACUAAA, and AAUAGA) also regulate a few mRNAs.
Studies have shown that the PAS sequence is surrounded by other cis-elements and works cooperatively in 3′-end processing (Hu et al., 2005; Salisbury et al., 2006). In order to gain more insight, several investigations have been carried out to detect hidden cis-elements around poly(A) sites in animals and plants using computational methods (Beaudoing et al., 2000; Hu et al., 2005; Loke et al., 2005; Dong et al., 2007; Shen et al., 2008). These analyses involved the use of sequence data from authentic polyadenylation sites by retrieving EST sequences with oligo(A) stretches and comparing these to their genomic DNA sequences to ensure that the oligo(A) stretches were added post-transcriptionally, keeping in mind that the first adenine of the poly(A) tail is present in the gene. After this filter, the genomic region containing the poly(A) site was extracted (≥100 nt upstream and ≥100 nt downstream). Computational methods were applied to extract statistically significant cis-elements (PAS’s or other hexamers) and the distribution patterns of nucleotides around the poly(A) site were calculated. In mammals, it was found that the PAS signal, AAUAAA, is highly conserved and located about 10–30 nt upstream of the cleavage site (Figure 7A; Zhao et al., 1999; Hu et al., 2005; Tian et al., 2005; Salisbury et al., 2006). Another study identified 15 cis-elements in four regions surrounding human poly(A) sites using a hexamer enrichment method with the PROBE tool (Hu et al., 2005). Furthermore, several other statistically significant mononucleotide and dinucleotide patterns were observed adjacent to PAS elements (Ahmed et al., 2009b). In contrast, plants lack highly conserved PAS sequences in near upstream elements (NUEs; Figure 7B; Graber et al., 1999; Loke et al., 2005). Other elements were also observed, such as a far upstream element (FUE) that enhances processing efficiency, and a cleavage element (CE) that consists of a cleavage site flanked by U-rich regions at both sides (Zhao et al., 1999; Loke et al., 2005; Shen et al., 2008). NUEs and cleavage site signals are more conserved in plants. One study identified 12 hexameric patterns of these three elements (NUE, FUE, and CE) in rice (Shen et al., 2008). It has been observed that in spite of the similarity in pattern distribution of poly(A) sites in closely related organisms, some differences exist (Tian et al., 2005). Significantly, the AAUAAA string was found in only 7 and 10% of NUEs in the rice and Arabidopsis transcriptomes, respectively (Loke et al., 2005; Shen et al., 2008). Transcripts may also undergo alternative polyadenylation resulting in distinct 3′-UTRs, which leads to the expression of protein isoforms. In the physiological conditions of the cell, expression of trans-factors and the presence of cis-elements on pre-mRNA are primarily responsible for alternative polyadenylation. Studies have shown that alternative polyadenylation is highly prevalent in plants and animals, including humans (Meyers et al., 2004; Tian et al., 2005; Shen et al., 2008, 2011; Shepard et al., 2011; Wu et al., 2011). Interestingly, proximal poly(A) sites tend to produce mRNAs with shorter 3′-UTRs, whereas distal poly(A) sites generate mRNAs with longer 3′-UTRs (Sandberg et al., 2008; Ji and Tian, 2009). Thus, in addition to protein isoforms, alternative polyadenylation may also have an impact on miRNA regulation due to changes in 3′-UTR target sites. An investigation showed that oncogenes in cancer cell lines undergo more frequent alternative polyadenylation to produce shorter 3′-UTRs, resulting in higher protein expression due to loss of miRNA target sites in the mRNA (Mayr and Bartel, 2009). Several studies analyzed and experimentally identified the global pattern of alternative polyadenylation in various cells using microarrays (Flavell et al., 2008; Sandberg et al., 2008) and most recently by using PAS-Seq, a high-throughput next-generation sequencing method (Shepard et al., 2011).
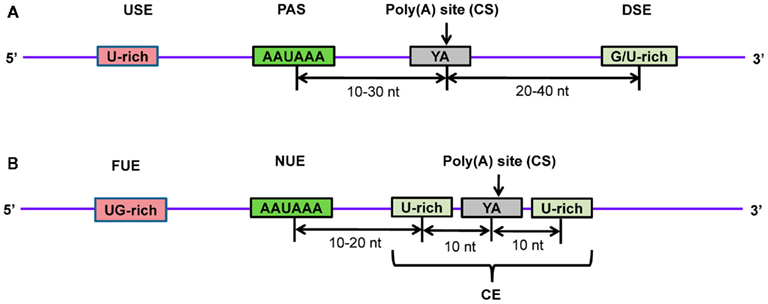
Figure 7. Schematic diagram of poly(A) signals in the 3’-end of pre-mRNA in (A) human and (B) Arabidopsis. Several proteins recognize and bind to poly(A) signals and other cis-elements to facilitate proper cleavage at the CS and subsequent polyadenylation. Transcripts are cleaved at the poly(A) site and poly(A) polymerase carries out polymerization at the newly formed 3′-end to generate the mature mRNA. USE, upstream element; PAS, poly(A) signal; CS, cleavage site; DSE, downstream element; FUE, far upstream element; NUE, near upstream element; CE, cleavage element.
The characteristics of PASs and their adjacent patterns facilitate the development of several computational methods for predicting poly(A) signals for gene annotation in animals, plants, and yeast. A support vector machine (SVM)-based method, polya_svm, was developed for predicting poly(A) sites in human mRNA by using position-specific scoring matrices of 15 cis-regulatory signals (Cheng et al., 2006). Recently, a more accurate method, PolyApred, was developed for analyzing poly(A) signals in the human genome (Ahmed et al., 2009b). It exploits nucleotide frequencies in four sub-regions upstream and downstream of PAS’s and employs SVMs for modeling and prediction. PolyApred achieved a precision of 75.8–95.7% with a sensitivity of 57% evaluated on independent data that were not used during model development. SVM methods are a set of related, supervised learning methods used for classification and regression problems, such as the prediction of functional sites in DNA sequences (Vapnik, 1995). It is based on statistical and optimization theories that handle complex high-dimensional features. The main concept underlying the SVM method is the generation of a hyperplane that separates the positive and negative examples within the multidimensional data space while maximizing the differences between them. Other methods developed for predicting PAS elements in plants are PASS for prediction in Arabidopsis (Ji et al., 2007), and PASS-Rice for rice (Shen et al., 2008). Both of these models are based on generalized hidden Markov models (GHMM), which recognize nucleotide signals in one direction from left to right. Performances of these models were tested on various datasets with a reported sensitivity of ∼90%, and specificity ranging from 60 to 100%.
Despite recent advances in high-throughput DNA sequencing and data collection, the mechanisms underlying 3′-end maturation of mRNA during different physiological conditions are still poorly understood. Furthermore, several challenges remain to be addressed to develop more reliable algorithms for PAS prediction such as: (1) improvement in prediction accuracy, (2) development of models that can predict the transition of functional to non-functional PAS elements or vice-versa, upon nucleotide mutation in functional elements, and (3) the development of models for predicting alternative polyadenylation sites and relative amounts of mRNA isoforms.
These challenges could be addressed by collecting the latest experimentally annotated transcriptome data from several species and extracting new features with advanced bioinformatics approaches. The genomes of many organisms have been sequenced recently, opening an avenue to explore characteristic features of PAS’s (Sato et al., 2007; Xu et al., 2011). This will not only enhance our understanding about phylogenetically conserved sequence motifs and species-specific motifs around PAS sites but it will also help to develop a robust model for improved gene annotation. For the prediction of PAS’s and cleavage sites, studies carried out so far have been mainly focused on sequence patterns in pre-mRNAs (Liu et al., 2005; Cheng et al., 2006; Ji et al., 2007; Shen et al., 2008; Ahmed et al., 2009b). However, new insights into the role of secondary RNA structures at PAS’s and their influence on cleavage site selection in some genes are emerging (Loke et al., 2005). There is a great need to explore more deeply the formation of these secondary structure patterns and their role in 3′-end maturation at the genome-wide level. It is clear from several studies that integrating secondary structure features of mRNA during siRNA design generates more functionally potent siRNA (Tafer et al., 2008; Ahmed and Raghava, 2011). Therefore, new algorithms integrating both sequence and structural features may be helpful for predicting more accurate PAS’s.
Conclusion, Current Challenges, and Perspectives
Technical advances in high-throughput sequencing methods are producing vast amounts of genomics, transcriptomics, and proteomics data. Furthermore, the continuous improvement of computational resources enables the better management and analysis of these data. To gain a deeper understanding of patterns of gene expression and regulation, it is necessary to decipher every component involved in the protein-coding messenger molecule, including its synthesis (transcription), usage (translation), and turnover (decay). It is well-known that mRNA and its precursor possess several cis-elements that interact with trans-acting factors at various steps during gene expression, which raises a number of interesting questions regarding the regulation of gene expression. However, discovering and characterizing mRNA functional elements, especially in recently sequenced or non-model organisms, still poses great challenges for bioinformaticians as well as experimental biologists.
Bioinformatics plays a vital role in addressing some of these challenges, especially regarding the discovery of evolutionarily conserved elements and the correlation of these elements with gene expression mechanisms. Several functional elements and the RBPs with which they interact have been reported, but only a few are functionally characterized. Table 1 shows a list of publicly available databases of different functional mRNA elements. However, it is important to integrate additional information into these databases in order to make them more applicable. For example, information about half-life of mRNAs and their associated AREs may enable the correlation of half-life of specific transcripts with their AREs. A recent study on mRNA and protein turnover may prove to be very helpful (Schwanhausser et al., 2011). Additionally, most of these databases focus on animal species, largely neglecting plant and yeast systems. Comparative analysis of the genomes of different species has been demonstrated as a better approach to discover regulatory elements (Xie et al., 2005). Furthermore, the sequence polymorphisms have also been reported to assist in the identification of functional elements in humans. Indeed, by analyzing genetic variation in intronic regions across different human populations, SREs were found to be very well preserved across populations, which allowed using genetic diversity features to develop an algorithm for predicting splicing enhancers in intronic regions (Lomelin et al., 2010). It would also be interesting to extract sequence and structure patterns in subsets of gene expression data at specific stages or in specific tissues, in order to understand the motifs involved in gene regulation.
In order to discover functional elements in new sequences, a number of freely accessible computational tools have been developed (Table 2). However, it remains crucial to improve the specificity and sensitivity of these tools. In model development, one of the main challenges is to reduce the high rates of false positive and false negative predictions. This could be addressed by using non-redundant sequence data and defining precise sequence and structural features of functional elements. There are several tools available for deciphering sequence motifs (Bailey, 2008) and structural patterns in functional elements (Hamilton and Davis, 2007, 2011). The sequence and structure patterns could also be implemented with machine learning techniques for the development of highly accurate methods for the prediction of functional elements. Since mutations and sequence variations in regulatory elements have been associated with the etiology of several diseases (Cazzola and Skoda, 2000; Chen et al., 2006), the analysis of SNPs within functional elements should also enhance our understanding of the role of these motifs in disease.
Furthermore, the selection of the most suitable bioinformatics analysis tools to uncover regulatory elements of mRNAs should be based on the specific datasets used and the questions to be addressed. Despite recent advances in our understanding of transcriptional and post-transcriptional regulation of gene expression dictated by cis-regulatory elements and trans-regulatory factors, much is still unknown, especially in non-model organisms and non-metazoan systems. Continuously advancing bioinformatics techniques certainly will play a major role in the discovery of regulatory sequence patterns present in mRNAs and extend our understanding of the evolution of these patterns across species.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants to Patrick X. Zhao from the National Science Foundation (NSF ABI-0960897), the Oklahoma Center for the Advancement of Science and Technology (OCAST PSB11-004), and the Samuel Roberts Noble Foundation.
References
Addess, K. J., Basilion, J. P., Klausner, R. D., Rouault, T. A., and Pardi, A. (1997). Structure and dynamics of the iron responsive element RNA: implications for binding of the RNA by iron regulatory binding proteins. J. Mol. Biol. 274, 72–83.
Ahmed, F., Ansari, H. R., and Raghava, G. P. (2009a). Prediction of guide strand of microRNAs from its sequence and secondary structure. BMC Bioinform. 10, 105. doi:10.1186/1471-2105-10-105
Ahmed, F., Kumar, M., and Raghava, G. P. (2009b). Prediction of polyadenylation signals in human DNA sequences using nucleotide frequencies. In silico Biol. 9, 135–148.
Ahmed, F., and Raghava, G. P. (2011). Designing of highly effective complementary and mismatch siRNAs for silencing a gene. PLoS ONE 6, e23443. doi:10.1371/journal.pone.0023443
Allam, H., and Ali, N. (2010). Initiation factor eIF2-independent mode of c-Src mRNA translation occurs via an internal ribosome entry site. J. Biol. Chem. 285, 5713–5725.
Bailey, T. L., and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36.
Bakheet, T., Frevel, M., Williams, B. R., Greer, W., and Khabar, K. S. (2001). ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 29, 246–254.
Bakheet, T., Williams, B. R., and Khabar, K. S. (2006). ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 34, D111–D114.
Bakheet, T., Williams, B. R., and Khabar, K. S. (2003). ARED 2.0: an update of AU-rich element mRNA database. Nucleic Acids Res. 31, 421–423.
Baranov, P. V., Gurvich, O. L., Fayet, O., Prère, M. F., Miller, W. A., Gesteland, R. F., Atkins, J. F., and Giddings, M. C. (2001). RECODE: a database of frameshifting, bypassing and codon redefinition utilized for gene expression. Nucleic Acids Res. 29, 264–267.
Baranov, P. V., Gurvich, O. L., Hammer, A. W., Gesteland, R. F., and Atkins, J. F. (2003). Recode 2003. Nucleic Acids Res. 31, 87–89.
Barreau, C., Paillard, L., and Osborne, H. B. (2005). AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33, 7138–7150.
Bashirullah, A., Cooperstock, R. L., and Lipshitz, H. D. (2001). Spatial and temporal control of RNA stability. Proc. Natl. Acad. Sci. U.S.A. 98, 7025–7028.
Beaudoin, M. E., Poirel, V. J., and Krushel, L. A. (2008). Regulating amyloid precursor protein synthesis through an internal ribosomal entry site. Nucleic Acids Res. 36, 6835–6847.
Beaudoing, E., Freier, S., Wyatt, J. R., Claverie, J. M., and Gautheret, D. (2000). Patterns of variant polyadenylation signal usage in human genes. Genome Res. 10, 1001–1010.
Bekaert, M., Firth, A. E., Zhang, Y., Gladyshev, V. N., Atkins, J. F., and Baranov, P. V. (2010). Recode-2: new design, new search tools, and many more genes. Nucleic Acids Res. 38, D69–D74.
Belew, A. T., Advani, V. M., and Dinman, J. D. (2011). Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res. 39, 2799–2808.
Belew, A. T., Hepler, N. L., Jacobs, J. L., and Dinman, J. D. (2008). PRFdb: a database of computationally predicted eukaryotic programmed −1 ribosomal frameshift signals. BMC Genomics 9, 339. doi:10.1186/1471-2164-9-339
Bengert, P., and Dandekar, T. (2003). A software tool-box for analysis of regulatory RNA elements. Nucleic Acids Res. 31, 3441–3445.
Bengert, P., and Dandekar, T. (2004). Riboswitch finder – a tool for identification of riboswitch RNAs. Nucleic Acids Res. 32, W154–W159.
Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336.
Bouget, F. Y., Gerttula, S., Shaw, S. L., and Quatrano, R. S. (1996). Localization of actin mRNA during the establishment of cell polarity and early cell divisions in fucus embryos. Plant Cell 8, 189–201.
Brockman, J. M., Singh, P., Liu, D., Quinlan, S., Salisbury, J., and Graber, H. (2005). PACdb: polyA cleavage site and 3′-UTR database. Bioinformatics 21, 3691–3693.
Buratowski, S. (2005). Connections between mRNA 3′ end processing and transcription termination. Curr. Opin. Cell Biol. 17, 257–261.
Campillos, M., Cases, I., Hentze, M. W., and Sanchez, M. (2010). SIREs: searching for iron-responsive elements. Nucleic Acids Res. 38, W360–W367.
Caput, D., Beutler, B., Hartog, K., Thayer, R., Brown-Shimer, S., and Cerami, A. (1986). Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc. Natl. Acad. Sci. U.S.A. 83, 1670–1674.
Cazzola, M., and Skoda, R. C. (2000). Translational pathophysiology: a novel molecular mechanism of human disease. Blood 95, 3280–3288.
Chang, T. H., Huang, H. D., Wu, L. C., Yeh, C. T., Liu, B. J., and Horng, J. T. (2009). Computational identification of riboswitches based on RNA conserved functional sequences and conformations. RNA 15, 1426–1430.
Chartrand, P., Meng, X. H., Huttelmaier, S., Donato, D., and Singer, R. H. (2002). Asymmetric sorting of ash1p in yeast results from inhibition of translation by localization elements in the mRNA. Mol. Cell 10, 1319–1330.
Chartrand, P., Meng, X. H., Singer, R. H., and Long, R. M. (1999). Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr. Biol. 9, 333–336.
Chen, C. Y., and Shyu, A. B. (1995). AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20, 465–470.
Chen, J. M., Ferec, C., and Cooper, D. N. (2006). A systematic analysis of disease-associated variants in the 3′ regulatory regions of human protein-coding genes II: the importance of mRNA secondary structure in assessing the functionality of 3′ UTR variants. Hum. Genet. 120, 301–333.
Cheng, Y., Miura, R. M., and Tian, B. (2006). Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics 22, 2320–2325.
Cohen, R. S., Zhang, S., and Dollar, G. L. (2005). The positional, structural, and sequence requirements of the Drosophila TLS RNA localization element. RNA 11, 1017–1029.
Cressina, E., Chen, L., Moulin, M., Leeper, F. J., Abell, C., and Smith, A. G. (2011). Identification of novel ligands for thiamine pyrophosphate (TPP) riboswitches. Biochem. Soc. Trans. 39, 652–657.
Crofts, A. J., Washida, H., Okita, T. W., Ogawa, M., Kumamaru, T., and Satoh, H. (2004). Targeting of proteins to endoplasmic reticulum-derived compartments in plants. The importance of RNA localization. Plant Physiol. 136, 3414–3419.
Dai, X., and Zhao, P. X. (2008). pssRNAMiner: a plant short small RNA regulatory cascade analysis server. Nucleic Acids Res. 36, W114–W118.
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159.
Dai, X., Zhuang, Z., and Zhao, P. X. (2011). Computational analysis of miRNA targets in plants: current status and challenges. Brief. Bioinform. 12, 115–121.
Danckwardt, S., Kaufmann, I., Gentzel, M., Foerstner, K. U., Gantzert, A. S., Gehring, N. H., Neu-Yilik, G., Bork, P., Keller, W., Wilm, M., Hentze, M. W., and Kulozik, A. E. (2007). Splicing factors stimulate polyadenylation via USEs at non-canonical 3′ end formation signals. EMBO J. 26, 2658–2669.
Degroeve, S., Saeys, Y., De Baets, B., Rouzé, P., and Van de Peer, Y. (2005). SpliceMachine: predicting splice sites from high-dimensional local context representations. Bioinformatics 21, 1332–1338.
Dong, H., Deng, Y., Chen, J., Wang, S., Peng, S., Dai, C., Fang, Y., Shao, J., Lou, Y., and Li, D. (2007). An exploration of 3′-end processing signals and their tissue distribution in Oryza sativa. Gene 389, 107–113.
Eberhardt, W., Doller, A., el Akool, S., and Pfeilschifter, J. (2007). Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 114, 56–73.
Espel, E. (2005). The role of the AU-rich elements of mRNAs in controlling translation. Semin. Cell Dev. Biol. 16, 59–67.
Flavell, S. W., Kim, T. K., Gray, J. M., Harmin, D. A., Hemberg, M., Hong, E. J., Markenscoff-Papadimitriou, E., Bear, D. M., and Greenberg, M. E. (2008). Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022–1038.
Gardner, P. P., Daub, J., Tate, J., Moore, B. L., Osuch, I. H., Griffiths-Jones, S., Finn, R. D., Nawrocki, E. P., Kolbe, D. L., Eddy, S. R., and Bateman, A. (2011). Rfam: wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39, D141–D145.
Garg, A., Bhasin, M., and Raghava, G. P. (2005). Support vector machine-based method for subcellular localization of human proteins using amino acid compositions, their order, and similarity search. J. Biol. Chem. 280, 14427–14432.
Garg, A., and Raghava, G. P. (2008). ESLpred2: improved method for predicting subcellular localization of eukaryotic proteins. BMC Bioinform. 9, 503. doi:10.1186/1471-2105-9-503
Girelli, D., Corrocher, R., Bisceglia, L., Olivieri, O., De Franceschi, L., Zelante, L., and Gasparini, P. (1995). Molecular basis for the recently described hereditary hyperferritinemia-cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the “Verona mutation”). Blood 86, 4050–4053.
Goforth, J. B., Anderson, S. A., Nizzi, C. P., and Eisenstein, R. S. (2010). Multiple determinants within iron-responsive elements dictate iron regulatory protein binding and regulatory hierarchy. RNA 16, 154–169.
Graber, J. H., Cantor, C. R., Mohr, S. C., and Smith, T. F. (1999). In silico detection of control signals: mRNA 3′-end-processing sequences in diverse species. Proc. Natl. Acad. Sci. U.S.A. 6, 14055–14060.
Grillo, G., Turi, A., Licciulli, F., Mignone, F., Liuni, S., Banfi, S., Gennarino, V. A., Horner, D. S., Pavesi, G., Picardi, E., and Pesole, G. (2010). UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 38, D75–D80.
Grojean, J., and Downes, B. (2010). Riboswitches as hormone receptors: hypothetical cytokinin-binding riboswitches in Arabidopsis thaliana. Biol. Direct 5, 60.
Gruber, A. R., Fallmann, J., Kratochvill, F., Kovarik, P., and Hofacker, I. L. (2011). AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 39, D66–D69.
Halees, A. S., El-Badrawi, R., and Khabar, K. S. (2008). ARED Organism: expansion of ARED reveals AU-rich element cluster variations between human and mouse. Nucleic Acids Res. 36, D137–D140.
Hamilton, R. S., and Davis, I. (2007). RNA localization signals: deciphering the message with bioinformatics. Semin. Cell Dev. Biol. 18, 178–185.
Hamilton, R. S., and Davis, I. (2011). Identifying and searching for conserved RNA localisation signals. Methods Mol. Biol. 714, 447–466.
Hamilton, R. S., Hartswood, E., Vendra, G., Jones, C., Van De Bor, V., Finnegan, D., and Davis, I. (2009). A bioinformatics search pipeline, RNA2DSearch, identifies RNA localization elements in Drosophila retrotransposons. RNA 15, 200–207.
House, A. E., and Lynch, K. W. (2008). Regulation of alternative splicing: more than just the ABCs. J. Biol. Chem. 283, 1217–1221.
Hu, J., Lutz, C. S., Wilusz, J., and Tian, B. (2005). Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 11, 1485–1493.
Huang, H. Y., Chien, C. H., Jen, K. H., and Huang, H. D. (2006). RegRNA: an integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 34, W429–W434.
Jacobs, G. H., Chen, A., Stevens, S. G., Stockwell, P. A., Black, M. A., Tate, W. P., and Brown, C. M. (2009). Transterm: a database to aid the analysis of regulatory sequences in mRNAs. Nucleic Acids Res. 37, D72–D76.
Ji, G., Zheng, J., Shen, Y., Wu, X., Jiang, R., Lin, Y., Loke, J. C., Davis, K. M., Reese, G. J., and Li, Q. Q. (2007). Predictive modeling of plant messenger RNA polyadenylation sites. BMC Bioinform. 8, 43. doi:10.1186/1471-2105-8-43
Ji, Z., and Tian, B. (2009). Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 4, e8419. doi:10.1371/journal.pone.0008419
Jin, P., and Warren, S. T. (2003). New insights into fragile X syndrome: from molecules to neurobehaviors. Trends Biochem. Sci. 28, 152–158.
Jin, Y., Yang, Y., and Zhang, P. (2011). New insights into RNA secondary structure in the alternative splicing of pre-mRNAs. RNA Biol. 8, 450–457.
Kato, J., Fujikawa, K., Kanda, M., Fukuda, N., Sasaki, K., Takayama, T., Kobune, M., Takada, K., Takimoto, R., Hamada, H., Ikeda, T., and Niitsu, Y. (2001). A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet. 69, 191–197.
Kaundal, R., and Raghava, G. P. (2009). RSLpred: an integrative system for predicting subcellular localization of rice proteins combining compositional and evolutionary information. Proteomics 9, 2324–2342.
Kaundal, R., Saini, R., and Zhao, P. X. (2010). Combining machine learning and homology-based approaches to accurately predict subcellular localization in Arabidopsis. Plant Physiol. 154, 36–54.
Kazan, K. (2003). Alternative splicing and proteome diversity in plants: the tip of the iceberg has just emerged. Trends Plant Sci. 8, 468–471.
Khabar, K. S. (2005). The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J. Interferon Cytokine Res. 25, 1–10.
Kim, N., and Lee, C. (2008). Bioinformatics detection of alternative splicing. Methods Mol. Biol. 452, 179–197.
Kimata, Y., and Theil, E. C. (1994). Posttranscriptional regulation of ferritin during nodule development in soybean. Plant Physiol. 104, 263–270.
Kislauskis, E. H., Zhu, X., and Singer, R. H. (1994). Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J. Cell Biol. 127, 441–451.
Knowles, R. B., Sabry, J. H., Martone, M. E., Deerinck, T. J., Ellisman, M. H., Bassell, G. J., and Kosik, K. S. (1996). Translocation of RNA granules in living neurons. J. Neurosci. 16, 7812–7820.
Kohler, A., and Hurt, E. (2007). Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 8, 761–773.
Koscielny, G., Le Texier, V., Gopalakrishnan, C., Kumanduri, V., Riethoven, J. J., Nardone, F., Stanley, E., Fallsehr, C., Hofmann, O., Kull, M., Harrington, E., Boué, S., Eyras, E., Plass, M., Lopez, F., Ritchie, W., Moucadel, V., Ara, T., Pospisil, H., Herrmann, A. G., Reich, J., Guigó, R., Bork, P., Doeberitz, M. K., Vilo, J., Hide, W., Apweiler, R., Thanaraj, T. A., and Gautheret, D. (2009). ASTD: the alternative splicing and transcript diversity database. Genomics 93, 213–220.
Ladd, A. N., and Cooper, T. A. (2002). Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 3, reviews0008.
Lam, L. T., Pickeral, O. K., Peng, A. C., Rosenwald, A., Hurt, E. M., Giltnane, J. M., Averett, L. M., Zhao, H., Davis, R. E., Sathyamoorthy, M., Wahl, L. M., Harris, E. D., Mikovits, J. A., Monks, A. P., Hollingshead, M. G., Sausville, E. A., and Staudt, L. M. (2001). Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2, RESEARCH0041.
Lan, L., Lin, S., Zhang, S., and Cohen, R. S. (2010). Evidence for a transport-trap mode of Drosophila melanogaster gurken mRNA localization. PLoS ONE 5, e15448. doi:10.1371/journal.pone.0015448
LaVaute, T., Smith, S., Cooperman, S., Iwai, K., Land, W., Meyron-Holtz, E., Drake, S. K., Miller, G., Abu-Asab, M., Tsokos, M., Switzer, R. III, Grinberg, A., Love, P., Tresser, N., and Rouault, T. A. (2001). Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 27, 209–214.
Le Texier, V., Riethoven, J. J., Kumanduri, V., Gopalakrishnan, C., Lopez, F., Gautheret, D., and Thanaraj, T. A. (2006). AltTrans: transcript pattern variants annotated for both alternative splicing and alternative polyadenylation. BMC Bioinform. 7, 169. doi:10.1186/1471-2105-7-169
Lecuyer, E., Yoshida, H., Parthasarathy, N., Alm, C., Babak, T., Cerovina, T., Hughes, T. R., Tomancak, P., and Krause, H. M. (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174–187.
Lee, J. Y., Yeh, I., Park, J. Y., and Tian, B. (2007). PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res. 35, D165–D168.
Lim, K. H., Ferraris, L., Filloux, M. E., Raphael, B. J., and Fairbrother, W. G. (2011). Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl. Acad. Sci. U.S.A. 108, 11093–11098.
Liu, H., Han, H., Li, J., and Wong, L. (2005). DNAFSMiner: a web-based software toolbox to recognize two types of functional sites in DNA sequences. Bioinformatics 21, 671–673.
Loke, J. C., Stahlberg, E. A., Strenski, D. G., Haas, B. J., Wood, P. C., and Li, Q. Q. (2005). Compilation of mRNA polyadenylation signals in Arabidopsis revealed a new signal element and potential secondary structures. Plant Physiol. 138, 1457–1468.
Lomelin, D., Jorgenson, E., and Risch, N. (2010). Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 20, 311–319.
Macdonald, P. M., and Kerr, K. (1997). Redundant RNA recognition events in bicoid mRNA localization. RNA 3, 1413–1420.
Mandal, M., Boese, B., Barrick, J. E., Winkler, W. C., and Breaker, R. R. (2003). Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell 113, 577–586.
Mangone, M., Macmenamin, P., Zegar, C., Piano, F., and Gunsalus, K. C. (2008). UTRome.org: a platform for 3′UTR biology in C. elegans. Nucleic Acids Res. 36, D57–D62.
Martin, K. C., and Ephrussi, A. (2009). mRNA localization: gene expression in the spatial dimension. Cell 136, 719–730.
Mayr, C., and Bartel, D. P. (2009). Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684.
Meli, M., Vergne, J., Décout, J. L., and Maurel, M. C. (2002). Adenine-aptamer complexes: a bipartite RNA site that binds the adenine nucleic base. J. Biol. Chem. 277, 2104–2111.
Meng, Z., Jackson, N. L., Shcherbakov, O. D., Choi, H., and Blume, S. W. (2010). The human IGF1R IRES likely operates through a Shine-Dalgarno-like interaction with the G961 loop (E-site) of the 18S rRNA and is kinetically modulated by a naturally polymorphic polyU loop. J. Cell. Biochem. 110, 531–544.
Meyer, S., Temme, C., and Wahle, E. (2004). Messenger RNA turnover in eukaryotes: pathways and enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 197–216.
Meyers, B. C., Vu, T. H., Tej, S. S., Ghazal, H., Matvienko, M., Agrawal, V., Ning, J., and Haudenschild, C. D. (2004). Analysis of the transcriptional complexity of Arabidopsis thaliana by massively parallel signature sequencing. Nat. Biotechnol. 22, 1006–1011.
Mignone, F., Gissi, C., Liuni, S., and Pesole, G. (2002). Untranslated regions of mRNAs. Genome Biol. 3, reviews0004.
Mironov, A. S., Gusarov, I., Rafikov, R., Lopez, L. E., Shatalin, K., Kreneva, R. A., Perumov, D. A., and Nudler, E. (2002). Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 111, 747–756.
Mokrejs, M., Masek, T., Vopálensky, V., Hlubucek, P., Delbos, P., and Pospísek, M. (2010). IRESite – a tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 38, D131–D136.
Moon, S., Byun, Y., and Han, K. (2007). FSDB: a frameshift signal database. Comput. Biol. Chem. 31, 298–302.
Moon, S., Byun, Y., Kim, H. J., Jeong, S., and Han, K. (2004). Predicting genes expressed via −1 and +1 frameshifts. Nucleic Acids Res. 32, 4884–4892.
Muckenthaler, M. U., Galy, B., and Hentze, M. W. (2008). Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 28, 197–213.
Mulhbacher, J., Brouillette, E., Allard, M., Fortier, L. C., Malouin, F., and Lafontaine, D. A. (2010a). Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 6, e1000865. doi:10.1371/journal.ppat.1000865
Mulhbacher, J., St-Pierre, P., and Lafontaine, D. A. (2010b). Therapeutic applications of ribozymes and riboswitches. Curr. Opin. Pharmacol. 10, 551–556.
Mutsuddi, M., Marshall, C. M., Benzow, K. A., Koob, M. D., and Rebay, I. (2004). The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr. Biol. 14, 302–308.
Ng, P., and Keich, U. (2008). GIMSAN: a Gibbs motif finder with significance analysis. Bioinformatics 24, 2256–2257.
Okita, T. W., and Choi, S. B. (2002). mRNA localization in plants: targeting to the cell’s cortical region and beyond. Curr. Opin. Plant Biol. 5, 553–559.
Orkin, S. H., Cheng, T. C., Antonarakis, S. E., and Kazazian, H. H. Jr. (1985). Thalassemia due to a mutation in the cleavage-polyadenylation signal of the human beta-globin gene. EMBO J. 4, 453–456.
Pavy, N., Rombauts, S., Déhais, P., Mathé, C., Ramana, D. V., Leroy, P., and Rouzé, P. (1999). Evaluation of gene prediction software using a genomic data set: application to Arabidopsis thaliana sequences. Bioinformatics 15, 887–899.
Pertea, M., Lin, X., and Salzberg, S. L. (2001). GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 29, 1185–1190.
Pertea, M., Mount, S. M., and Salzberg, S. L. (2007). A computational survey of candidate exonic splicing enhancer motifs in the model plant Arabidopsis thaliana. BMC Bioinform. 8, 159. doi:10.1186/1471-2105-8-159
Piccinelli, P., and Samuelsson, T. (2007). Evolution of the iron-responsive element. RNA 13, 952–966.
Reed, R., and Hurt, E. (2002). A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108, 523–531.
Rund, D., Dowling, C., Najjar, K., Rachmilewitz, E. A., Kazazian, H. H. Jr., and Oppenheim, A. (1992). Two mutations in the beta-globin polyadenylylation signal reveal extended transcripts and new RNA polyadenylylation sites. Proc. Natl. Acad. Sci. U.S.A. 89, 4324–4328.
Salisbury, J., Hutchison, K. W., and Graber, J. H. (2006). A multispecies comparison of the metazoan 3′-processing downstream elements and the CstF-64 RNA recognition motif. BMC Genomics 7, 55. doi:10.1186/1471-2164-7-55
Sandberg, R., Neilson, J. R., Sarma, A., Sharp, P. A., and Burge, C. B. (2008). Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320, 1643–1647.
Sato, S., Nakamura, Y., Asamizu, E., Isobe, S., and Tabata, S. (2007). Genome sequencing and genome resources in model legumes. Plant Physiol. 144, 588–593.
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., Chen, W., and Selbach, M. (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342.
Schwarz, D. S., Hutvagner, G., Du, T., Xu, Z., Aronin, N., and Zamore, P. D. (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199–208.
Shen, Y., Ji, G., Haas, B. J., Wu, X., Zheng, J., Reese, G. J., and Li, Q. Q. (2008). Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 36, 3150–3161.
Shen, Y., Venu, R. C., Nobuta, K., Wu, X., Notibala, V., Demirci, C., Meyers, B. C., Wang, G. L., Ji, G., and Li, Q. Q. (2011). Transcriptome dynamics through alternative polyadenylation in developmental and environmental responses in plants revealed by deep sequencing. Genome Res. 21, 1478–1486.
Shepard, P. J., Choi, E. A., Lu, J., Flanagan, L. A., Hertel, K. J., and Shi, Y. (2011). Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17, 761–772.
Shepelev, V., and Fedorov, A. (2006). Advances in the exon-intron database (EID). Brief. Bioinform. 7, 178–185.
Singh, P., Bandyopadhyay, P., Bhattacharya, S., Krishnamachari, A., and Sengupta, S. (2009). Riboswitch detection using profile hidden Markov models. BMC Bioinform. 10, 325. doi:10.1186/1471-2105-10-325
Smith, C. W., and Valcarcel, J. (2000). Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 25, 381–388.
Spriggs, K. A., Cobbold, L. C., Jopling, C. L., Cooper, R. E., Wilson, L. A., Stoneley, M., Coldwell, M. J., Poncet, D., Shen, Y. C., Morley, S. J., Bushell, M., and Willis, AE. (2009). Canonical initiation factor requirements of the Myc family of internal ribosome entry segments. Mol. Cell. Biol. 29, 1565–1574.
Stormo, G. D. (2010). Motif discovery using expectation maximization and Gibbs’ sampling. Methods Mol. Biol. 674, 85–95.
Sugnet, C. W., Srinivasan, K., Clark, T. A., O’Brien, G., Cline, M. S., Wang, H., Williams, A., Kulp, D., Blume, J. E., Haussler, D., and Ares, M. Jr. (2006). Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput. Biol. 2, e4. doi:10.1371/journal.pcbi.0020004
Tafer, H., Ameres, S. L., Obernosterer, G., Gebeshuber, C. A., Schroeder, R., Martinez, J., and Hofacker, I. L. (2008). The impact of target site accessibility on the design of effective siRNAs. Nat. Biotechnol. 26, 578–583.
Theis, C., Reeder, J., and Giegerich, R. (2008). KnotInFrame: prediction of −1 ribosomal frameshift events. Nucleic Acids Res. 36, 6013–6020.
Tian, B., Hu, J., Zhang, H., and Lutz, C. S. (2005). A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 33, 201–212.
Ule, J., Stefani, G., Mele, A., Ruggiu, M., Wang, X., Taneri, B., Gaasterland, T., Blencowe, B. J., and Darnell, R. B. (2006). An RNA map predicting Nova-dependent splicing regulation. Nature 444, 580–586.
Usuka, J., and Brendel, V. (2000). Gene structure prediction by spliced alignment of genomic DNA with protein sequences: increased accuracy by differential splice site scoring. J. Mol. Biol. 297, 1075–1085.
van der Velden, A. W., and Thomas, A. A. (1999). The role of the 5′ untranslated region of an mRNA in translation regulation during development. Int. J. Biochem. Cell Biol. 31, 87–106.
Verhounig, A., Karcher, D., and Bock, R. (2010). Inducible gene expression from the plastid genome by a synthetic riboswitch. Proc. Natl. Acad. Sci. U.S.A. 107, 6204–6209.
Vermerris, W., Vreugdenhil, D., and Visser, R. G. F. (2001). mRNA localization in in vitro grown microtubers of potatoas a tool to study starch metabolism. Plant Physiol. Biochem. 39, 161–166.
Vitreschak, A. G., Rodionov, D. A., Mironov, A. A., and Gelfand, M. S. (2004). Riboswitches: the oldest mechanism for the regulation of gene expression? Trends Genet. 20, 44–50.
von Roretz, C., and Gallouzi, I. E. (2008). Decoding ARE-mediated decay: is microRNA part of the equation? J. Cell Biol. 181, 189–194.
Wang, B. B., and Brendel, V. (2006). Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. U.S.A. 103, 7175–7180.
Wang, B. B., O’Toole, M., Brendel, V., and Young, N. D. (2008). Cross-species EST alignments reveal novel and conserved alternative splicing events in legumes. BMC Plant Biol. 8, 17. doi:10.1186/1471-2229-8-17
Wang, Z., and Burge, C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813.
Weil, T. T., Forrest, K. M., and Gavis, E. R. (2006). Localization of bicoid mRNA in late oocytes is maintained by continual active transport. Dev. Cell 11, 251–262.
Weil, T. T., Parton, R. M., and Davis, I. (2010). Making the message clear: visualizing mRNA localization. Trends Cell Biol. 20, 380–390.
Wilusz, C. J., Wormington, M., and Peltz, S. W. (2001). The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2, 237–246.
Winkler, W. C., and Breaker, R. R. (2003). Genetic control by metabolite-binding riboswitches. Chembiochem 4, 1024–1032.
Winkler, W. C., Cohen-Chalamish, S., and Breaker, R. R. (2002). An mRNA structure that controls gene expression by binding FMN. Proc. Natl. Acad. Sci. U.S.A. 99, 15908–15913.
Wu, T. Y., Hsieh, C. C., Hong, J. J., Chen, C. Y., and Tsai, Y. S. (2009). IRSS: a web-based tool for automatic layout and analysis of IRES secondary structure prediction and searching system in silico. BMC Bioinform. 10, 160. doi:10.1186/1471-2105-10-160
Wu, X., Liu, M., Downie, B., Liang, C., Ji, G., Li, Q. Q., and Hunt, A. G. (2011). Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. U.S.A. 108, 12533–12538.
Xie, X., Lu, J., Kulbokas, E. J., Golub, T. R., Mootha, V., Lindblad-Toh, K., Lander, E. S., and Kellis, M. (2005). Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434, 338–345.
Xu, X., Pan, S., Cheng, S., Zhang, B., Mu, D., Ni, P., Zhang, G., Yang, S., Li, R., Wang, J., Orjeda, G., Guzman, F., Torres, M., Lozano, R., Ponce, O., Martinez, D., De la Cruz, G., Chakrabarti, S. K., Patil, V. U., Skryabin, K. G., Kuznetsov, B. B., Ravin, N. V., Kolganova, T. V., Beletsky, A. V., Mardanov, A. V., Di Genova, A., Bolser, D. M., Martin, D. M., Li, G., Yang, Y., Kuang, H., Hu, Q., Xiong, X., Bishop, G. J., Sagredo, B., Mejía, N., Zagorski, W., Gromadka, R., Gawor, J., Szczesny, P., Huang, S., Zhang, Z., Liang, C., He, J., Li, Y., He, Y., Xu, J., Zhang, Y., Xie, B., Du, Y., Qu, D., Bonierbale, M., Ghislain, M., Herrera, Mdel, R., Giuliano, G., Pietrella, M., Perrotta, G., Facella, P., O’Brien, K., Feingold, S. E., Barreiro, L. E., Massa, G. A., Diambra, L., Whitty, B. R., Vaillancourt, B., Lin, H., Massa, A. N., Geoffroy, M., Lundback, S., DellaPenna, D., Buell, C. R., Sharma, S. K., Marshall, D. F., Waugh, R., Bryan, G. J., Destefanis, M., Nagy, I., Milbourne, D., Thomson, S. J., Fiers, M., Jacobs, J. M., Nielsen, K. L., Sønderkær, M., Iovene, M., Torres, G. A., Jiang, J., Veilleux, R. E., Bachem, C. W., de Boer, J., Borm, T., Kloosterman, B., van Eck, H., Datema, E., Hekkert, B. L., Goverse, A., van Ham, R. C., Visser, R. G., and Potato Genome Sequencing Consortium. (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195.
Yang, Q., Gilmartin, G. M., and Doublié, S. (2010). Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3′ processing. Proc. Natl. Acad. Sci. U.S.A. 107, 10062–10067.
Zhao, J., Hyman, L., and Moore, C. (1999). Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev. 63, 405–445.
Keywords: bioinformatics, cis-elements, splicing regulatory elements, internal ribosome entry sites, iron-responsive elements, AU-rich elements, zipcodes, polyadenylation signals
Citation: Ahmed F, Benedito VA and Zhao PX (2011) Mining functional elements in messenger RNAs: overview, challenges, and perspectives. Front. Plant Sci. 2:84. doi: 10.3389/fpls.2011.00084
Received: 31 August 2011;
Accepted: 03 November 2011;
Published online: 30 November 2011.
Edited by:
Patrick J. Krysan, University of Wisconsin-Madison, USACopyright: © 2011 Ahmed, Benedito and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Patrick Xuechun Zhao, Bioinformatics Laboratory, Plant Biology Division, Samuel Roberts Noble Foundation, 2510 Sam Noble Parkway, Ardmore, OK 73401, USA e-mail: pzhao@noble.org