 Amina Ibrahim1 Anna-Lena Schütz1 Jean-Marie Galano2 Cornelia Herrfurth1 Kirstin Feussner1,3 Thierry Durand2 Florian Brodhun1 Ivo Feussner1*
Amina Ibrahim1 Anna-Lena Schütz1 Jean-Marie Galano2 Cornelia Herrfurth1 Kirstin Feussner1,3 Thierry Durand2 Florian Brodhun1 Ivo Feussner1*- 1 Department of Plant Biochemistry, Albrecht von Haller-Institute of Plant Sciences, Georg August University, Göttingen, Germany
- 2 UMR 5247 – CNRS – UM I – UM II, Institut des Biomolécules Max Mousseron, Montpellier, France
- 3 Department of Molecular Microbiology and Genetics, Institute for Microbiology and Genetics, Georg August University, Göttingen, Germany
Galactolipids constitute the major lipid class in plants. In recent years oxygenated derivatives of galactolipids have been detected. They are discussed as signal molecules during leaf damage, since they accumulate in wounded leaves in high levels. Using different analytical methods such as nuclear magnetic resonance, infra-red spectroscopy, and high performance liquid chromatography/mass spectrometry (HPLC/MS) earlier reports focused on the analysis of either oxidized or non-oxidized species and needed high levels of analytes. Here, we report on the analysis of the galactolipid subfraction of the Arabidopsis leaf lipidome by an improved HPLC/MS2-based method that is fast, robust, and comparatively simple in its performance. Due to a combination of phase partitioning, solid phase fractionation, liquid chromatography, and MS2 experiments this method has high detection sensitivity and requires only low amounts of plant material. With this method 167 galactolipid species were detected in leaves of Arabidopsis thaliana. Out of these 79 being newly described species. From all species the head group and acyl side chains were identified via MS2 experiments. Moreover, the structural identification was supported by HPLC/time-of-flight (TOF)-MS and gas chromatography (GC)/MS analysis. The quantification of different galactolipid species that accumulated 30 min after a mechanical wounding in A. thaliana leaves showed that the oxidized acyl side chains in galactolipids are divided into 65% cyclopentenones, 27% methyl-branched ketols, 3.8% hydroperoxides/straight-chain ketols, 2.0% hydroxides, and 2.6% phytoprostanes. In comparison to the free cyclopentenone derivatives, the esterified forms occur in a 149-fold excess supporting the hypothesis that galactolipids might function as storage compounds for cyclopentenones. Additional analysis of the ratio of non-oxidized to oxidized galactolipid species in leaves of wounded plants was performed resulting in a ratio of 2.0 in case of monogalactosyl diacylglycerol (MGD), 8.1 in digalactosyl diacylglycerol (DGD), and 0.6 in the acylated MGD. This indicates that galactolipid oxidation is a major and rapid metabolic process that occurs class specific.
Introduction
Monogalactosyl diacylglycerol (MGD) as well as digalactosyl diacylglycerol (DGD) are ubiquitous non-phosphorous lipid classes in higher plants constituting about 80% of thylakoid membrane lipids and about 60% of all leaf lipids (Douce and Joyard, 1990; Dörmann and Benning, 2002). After the discovery of the first oxidized galactolipid in 2001 (Stelmach et al., 2001), recent studies have demonstrated that wounded leaves of Arabidopsis thaliana accumulate high levels of oxygenated galactolipid derivatives (Buseman et al., 2006). In Arabidopsis these oxidized galactolipids may contain cyclopentenones, i.e., 12-oxo-phytodienoic acid (OPDA) and/or dinor-12-oxo-phytodienoic acid (dnOPDA), esterified either to the glycerol backbone of the lipid at one or at two positions, respectively, and/or to the sugar moiety (Andersson et al., 2006). They are collectively called Arabidopsides (Hisamatsu et al., 2003, 2005). In case of MGD and DGD derivatives, OPDA is either linked to glycerol at both the sn1 and sn2 position as in Arabidopside B (Ara-B) and Arabidopside D (Ara-D), or is attached at the sn1 position while dnOPDA at the sn2 position like in Arabidopside A (Ara-A) and Arabidopside C (Ara-C; Hisamatsu et al., 2003, 2005). In following studies further Arabidopsides were identified containing hexadecatrienoic acid (16:3) linked at the sn2 position and OPDA at the sn1 position (MGD-O; Stelmach et al., 2001) or octadecatrienoic acid (18:3) at the sn1 position and dnOPDA at the sn2 position – the latter one termed Arabidopside F (Ara-F; Nakajyo et al., 2006). Besides these MGD and DGD derivatives also acylated MGD species, like Arabidopside E (Ara-E; Andersson et al., 2006) and Arabidopside G (Ara-G; Kourtchenko et al., 2007) have been identified. Here an extra OPDA molecule is esterified to the galactosyl moiety in addition to the one at the sn1 and sn2 postion (Ara-G; note that in case of Ara-E, dnOPDA is esterified at the sn2 position). Additionally, OPDA linked to monogalactosyl monoacylglycerol MGM was found in Ipomoea tricolor (heavenly blue; Ohashi et al., 2005). Besides the above described galactolipid species, one phospholipid, namely phosphatidylglycerol (PG) has also been demonstrated to contain OPDA esterified at the sn1 position and hexadecenoic acid (16:1) or hexadecanoic acid (16:0) at sn2 position (Buseman et al., 2006).
Knowledge on the biological role of Arabidopsides is still scarce. In several studies it has been suggested that they may act as signal molecules during leaf damage (Stelmach et al., 2001; Buseman et al., 2006; Böttcher and Weiler, 2007). Along this line, it has been demonstrated that different Arabidopsides accumulate during hypersensitive response (Andersson et al., 2006; Kourtchenko et al., 2007). In addition, Arabidopsides may (i) inhibit root growth of cress (Hisamatsu et al., 2003), (ii) promote senescence in oat leaves (Hisamatsu et al., 2006), or (iii) have direct antimicrobial function (Kourtchenko et al., 2007). It has also been discussed that Arabidopsides might act as storage compounds allowing a faster and stronger formation of the well established signal molecule jasmonic acid (JA) and its derivatives (Stelmach et al., 2001; Andersson et al., 2006; Buseman et al., 2006; Kourtchenko et al., 2007).
Several analytical strategies were pursued to elucidate the structure of oxidized galactolipids. In initial experiments galactolipid extracts were treated with lipases in order to release OPDA from lipid molecules allowing the analysis by gas chromatography/mass spectrometry (GC/MS), high performance liquid chromatography/MS (HPLC/MS), and nuclear magnetic resonance (NMR) spectroscopy (Stelmach et al., 2001). In following studies a combination of MS, infra-red (IR) as well as NMR techniques was applied for the analysis of MGD derivatives like Ara-A, Ara-B (Hisamatsu et al., 2003), and Ara-F (Nakajyo et al., 2006), or DGD derivatives such as Ara-C and Ara-D (Hisamatsu et al., 2005). MS and NMR were also used for the elucidation of two oxidized acylated MGD derivatives, Ara-E (Andersson et al., 2006) and Ara-G (Kourtchenko et al., 2007). The latter two metabolites were demonstrated to accumulate preferentially during the hypersensitive response. All these analyses were hampered by the high amount of plant material needed for extraction and the expenditure of time necessary for the analytical analysis. Therefore, Buseman et al. (2006) established an analytical method that based on electrospray ionization (ESI)-tandem MS: it was used for detection and characterization of the already described MGD and DGD derivatives as well as additional oxidized lipid molecules that are distributed among MGD, DGD, and PG classes in wounded A. thaliana leaves.
Besides to the analysis of oxylipin-containing galactolipids, a number of different comprehensive studies have also been carried out on the analysis and identification of non-oxidized galactolipids. These species have been detected in leaves of A. thaliana (Welti et al., 2002; Devaiah et al., 2006; Glauser et al., 2008) and also in several plant species such as Ipomoea batatas (Napolitano et al., 2007) and plant tissues like oat kernels (Moreau et al., 2008). Most of these species were identified and analyzed in parallel using low amounts of plant material either by means of ESI-tandem MS-based methods (Welti et al., 2002; Devaiah et al., 2006; Napolitano et al., 2007; Moreau et al., 2008) or the ultra performance LC (UPLC)/time-of-flight (TOF)-MS technique (Glauser et al., 2008). According to those methods, different numbers of non-oxidized galactolipid species have been identified – with the highest number of identified species reported by Devaiah et al. (2006).
As to date no method was available to analyze a comprehensive galactolipid spectrum that is needed for a functional analysis of Arabidopsides, we aimed with our study to establish a sensitive analytical method that could be used for the analysis of non-oxidized as well as oxidized galactolipids in parallel. Using this method we were able to identify 167 different species of various galactolipid classes in leaves of A. thaliana and 79 of them being newly described species. One hundred forty-six of these species were quantified in A. thaliana leaves 30 min after a mechanical wounding.
Results
Establishment of a HPLC/MS2-Based Method for Identification and Profiling of Galactolipids
In order to analyze the biosynthesis and the role of galactolipid-bound oxylipins in plant cells, several MS-based strategies partially combined with chromatographic separation have been developed and performed during the last years (Andersson et al., 2006; Buseman et al., 2006; Böttcher and Weiler, 2007; Kourtchenko et al., 2007). However, compared to the number of different potential galactolipid species the number of experimentally identified ones was still considerably low. In order to gather a comprehensive picture about the diversity of galactolipids we established a HPLC/MS2-based method that was combined with a solid phase extraction (SPE) step. Using this method we were able to identify and quantify non-oxidized species as well as the oxidized derivatives of the galactolipid classes MGD, DGD (including their acylated forms) as well as SQD.
Extraction Efficiency of Different Media
To quantitatively isolate all galactolipids from the plant material, different solvent systems were tested concerning their extraction efficiency: the extraction was performed either with chloroform/methanol or methyl tert-butyl ether as extraction solvents. Extraction procedures were employed according to three methods which were published by Bligh and Dyer (1959), Heinz and Tulloch (1969), Matyash et al. (2008). The total lipids were extracted from Arabidopsis leaves which were harvested 30 min after wounding. The galactolipids were purified by SPE, analyzed by using HPLC/MS, and the amounts of 12 non-oxidized MGD species, 19 oxidized MGD species, 13 non-oxidized DGD species, 18 oxidized DGD species, 6 acylated MGD species, and 18 oxidized acylated MGD species were determined. As shown in Figure 1 the extraction according to Bligh and Dyer (1959) yielded 16.4 μmol g−1 f.w. whereas with the methods of Heinz and Tulloch (1969) and Matyash et al. (2008) only 9.0 μmol g−1 f.w. could be detected.
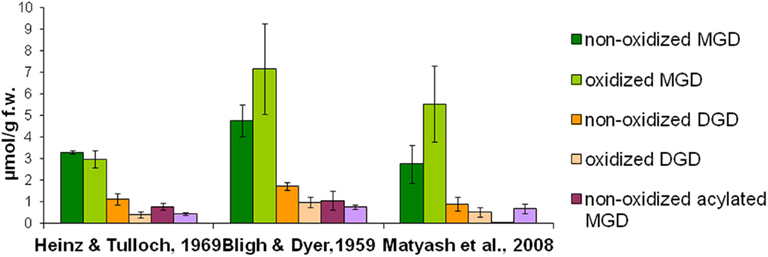
Figure 1. Extraction efficiency of different media. Total lipids were extracted from wounded leaves (30 min harvested after wounding) of A. thaliana according to three extraction methods (Bligh and Dyer, 1959; Heinz and Tulloch, 1969; Matyash et al., 2008). Galactolipids were then purified from these extracts by using SPE and analyzed as well as quantified by HPLC/MS. The amounts of the respective species from the various galactolipid classes were summed. The mean values and SD of three parallel extractions are shown.
Optimization of Analytic HPLC/MS-Parameters
In order to optimize the chromatographic separation of the different galactolipid species different gradient systems consisting of either acetonitrile/water (70:30, v/v) and 2-propanol (binary gradient system) or acetonitrile, water, and 2-propanol (ternary gradient system) were tested. The highest resolution was achieved by using the ternary gradient system (Table 1). In order to improve the ionization efficiency of the galactolipids, 0.1% (v/v) of acetic acid was added. In addition two C18 columns with different dimensions have been tested: 250 mm × 2.1 mm, 5 μm particle size, and 50 mm × 2.1 mm, 1.8 μm particle size. The highest separation efficiency was obtained with the latter one. To optimize quantification, different amounts of plant material were measured. The most reliable and comprehensive peak detection was reached if 100 mg of plant material was extracted (data not shown).
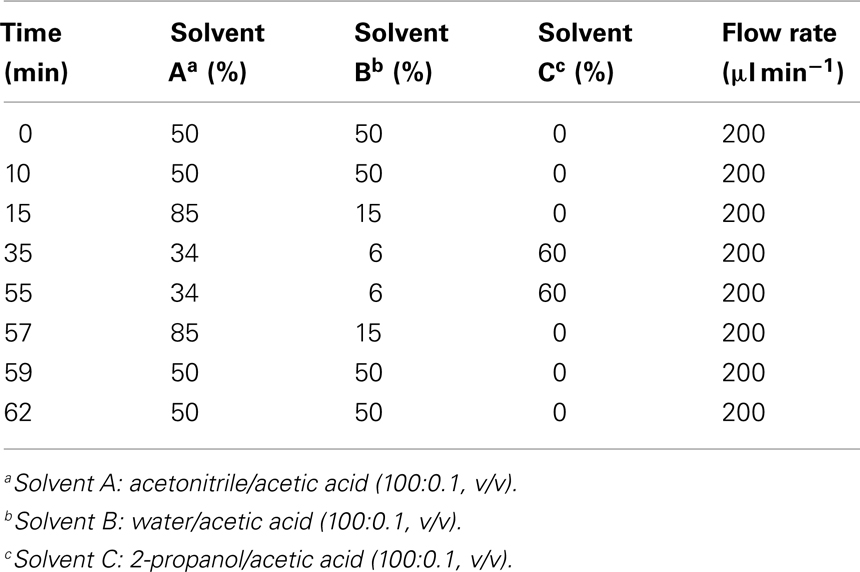
Table 1. Ternary gradient system used for the RP-HPLC/MS2 analysis of different galactolipid species.
These optimized parameters led to a chromatographic separation as shown in Figure 2. For detection of the different galactolipid species positive and negative ESI was used: all species except SQD were detected with highest sensitivity with positive ESI. In addition MS spectra were recorded in a mass range of 450–1,500 amu (details of the respective MS conditions are given in Table 2).
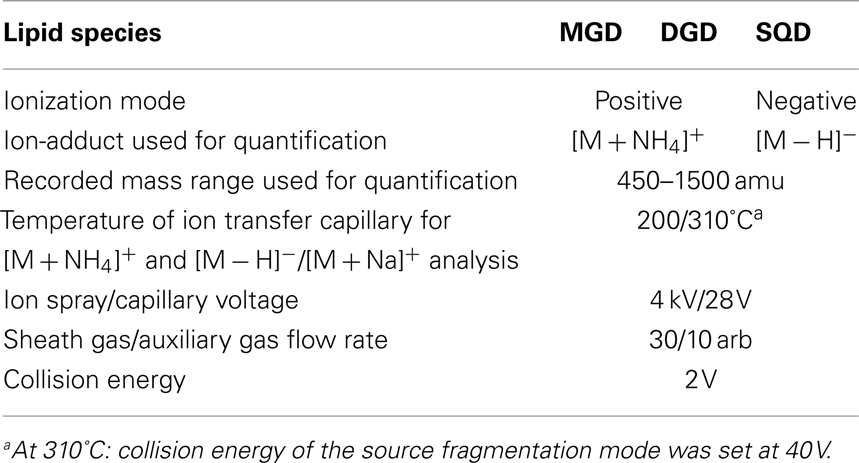
Table 2. Mass spectrometry conditions used for detection and quantification of different lipid species.
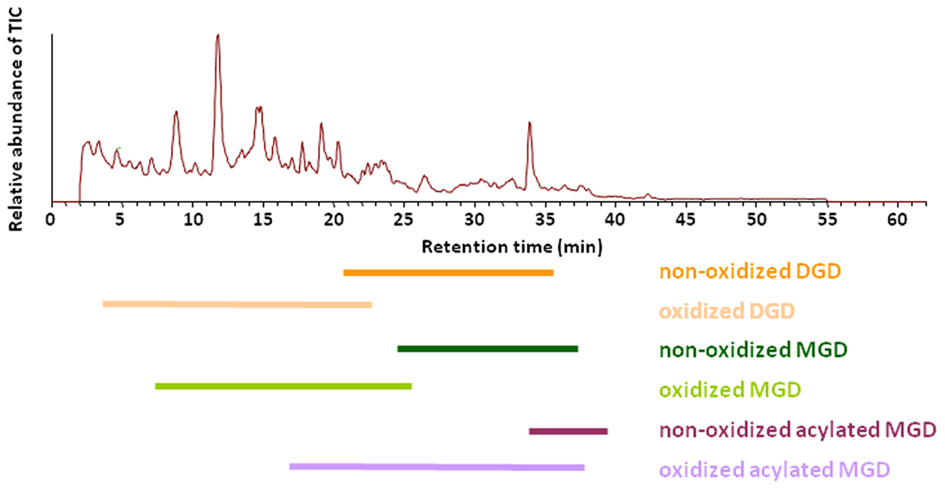
Figure 2. Elution of various galactolipid classes. Total ion chromatogram (TIC) of the RP-HPLC/MS analysis of the galactolipid fraction after purification from the total lipid extract by using SPE. Total lipids were extracted according to Bligh and Dyer (1959) from leaves of A. thaliana 30 min after wounding. Galactolipid species were detected in positive ESI mode using a mass range of 450–1,500 amu.
Structural Elucidation of Different Galactolipid Species Using Ion Trap-MS
At first structure identification MS2 experiments were performed with the ion trap system. Previous reports indicated that it is possible to identify the structure of any galactolipid due to a different fragmentation pattern of its ammonium (Wang et al., 1999) and sodium adduct, respectively (Welti et al., 2003; Devaiah et al., 2006; Napolitano et al., 2007). The fragmentation of the ammonium ion by collision-induced dissociation (CID) leads to a neutral loss of the galactose moiety and an additional water loss whereas the sodium adduct fragments by a neutral loss of the acyl side chains. Moreover, the specific formation of these adducts can be observed by applying different capillary temperatures of either 200 or 310°C (Table 2).
The identification of Ara-E by this strategy is shown as an example in Figure 3. The ammonium adduct (m/z 1066.4) and sodium adduct (m/z 1071.6) of Ara-E were detected in addition to the respective protonated ion (m/z 1049.3) with positive ESI at a capillary temperature of 270°C (Figure 3A). By decreasing the capillary temperature to 200°C the formation of the sodium adduct was almost completely avoided and the ammonium adduct was primarily formed (Figure 3C). The fragmentation of this adduct by CID led to the formation of fragment ions resulting from the neutral loss of the galactose moiety acylated with OPDA (m/z 613.1) as well as of an additional water loss (m/z 595.2, Figure 3E). In contrast to the fragmentation of the ammonium adduct, the sodium adduct occurring primarily at a capillary temperature of 310°C (Figure 3D) fragmented into the ions m/z 779.4 and m/z 807.4 due to the loss of OPDA from the sn1 position and dnOPDA from the sn2 position of the glycerol backbone (Figure 3F). The origin of the fragments from either the sn1 or sn2 position can be deduced from the different signal intensities of the various fragment ions. Fragments resulting from the neutral loss of the acyl chain at sn1 position have in general higher abundances than those resulting from the neutral loss of the acyl chain at the sn2 position (Guella et al., 2003; Napolitano et al., 2007). In addition, the fragmentation of the sodium adduct of several acylated MGD species led to the neutral loss of the acyl residue at the galactosyl moiety. The resulting additional fragments occur with very low abundances (data not shown).
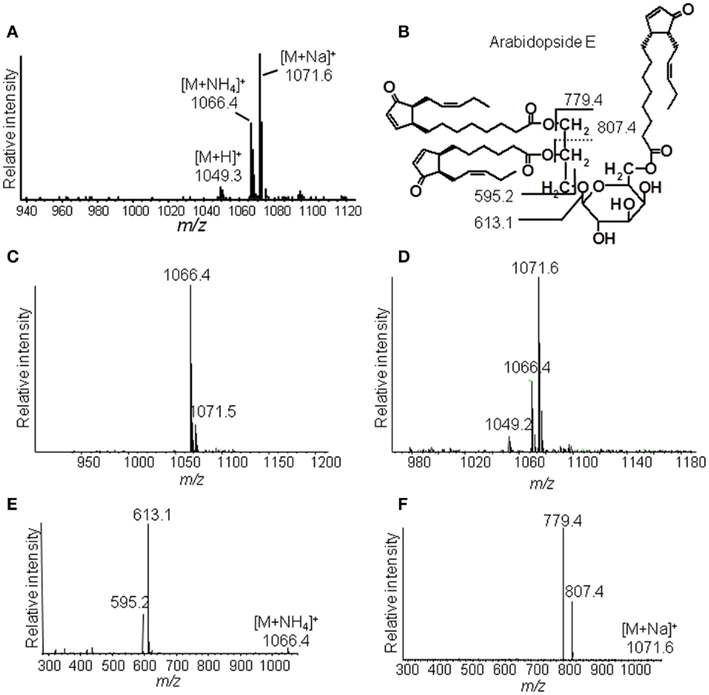
Figure 3. Ion trap mass spectrometric detection. (A) Mass spectrum of Ara-E in positive ESI mode at a capillary temperature of the ion source of 270°C, (B) Structure of Ara-E with fragment ions of the [M + NH4]+ ion or [M + Na]+ ion, (C) Mass spectrum of Ara-E at a capillary temperature of the ion source of 200°C, (D) Mass spectrum of Ara-E at a capillary temperature of the ion source of 310°C, (E) Product ion spectra of [M + NH4]+, (F) Product ion spectra of [M + Na]+. Total lipids were extracted from wounded leaves of A. thaliana according to Bligh and Dyer (1959) and galactolipids were subsequently purified by SPE. For identification of the various molecular species, galactolipids were separated by RP-HPLC and then detected in positive ESI mode using an ion trap mass spectrometer. The galactolipid fraction resulting from 200 to 300 mg of plant material was used. The ammonium and sodium adducts of the various galactolipid species were detected at the respective capillary temperatures of the ion source in a mass range of 450–1,500 amu and at CE of 2 V.
To investigate a comprehensive galactolipid spectrum, all molecular ions with mono-, di-, or acylated galactose moieties were first identified due to the fragment ion spectra at a capillary temperature of 200°C and then further analyzed as described for Ara-E. Most of the detected masses could be identified according to published data on the occurrence of fatty acids and oxylipins in A. thaliana.
Structural Elucidation of Different Galactolipid Species Using TOF-MS
The identity of the galactolipid species that were found with the ion trap MS2 experiments was verified by HPLC–TOF-MS experiments via their accurate molecular masses. In addition, unknown acyl side chains of galactolipids were identified by subtracting the accurate mass of the head group and the other acyl chain, both identified in MS2 experiments, from the accurate masses of the entire molecule. Based on this result it was possible to predict the respective sum formula of the unknown side chain as summarized in Table 3. By using the predicted sum formulas, oxidized acyl side chains could be identified as phytoprostanes, ketols, or hydroperoxides by comparing them with those of known oxylipins from A. thaliana. Notably, for two of the predicted sum formulas (C17H28O4 and C19H32O4) no structure could be assigned. Based on the finding that they contain an odd number of carbon atoms, however, these compounds were tentatively assigned as methyl-branched fatty acid derivatives. This hypothesis was confirmed by GC/MS analysis of the respective compounds (next section).

Table 3. Predicted sum formulas and their predicted structures of some unknown acyl side chains deduced from accurate mass measurements by TOF-MS.
Structural Elucidation of Different Galactolipid Species Using GC/MS
Next GC/MS analysis was performed for identification of the acyl backbone of the galactolipid structures. For this two different fractions were used: the galactolipid fraction resulting from SPE was directly applied to transesterification to obtain the corresponding methyl esters of all acyl side chains. Alternatively, the galactolipid species were further purified and thereby enriched by repetitive HPLC runs before transesterification.
The number and position of the double bonds of the non-oxidized acyl chain derivatives were determined by the analysis of the 4,4-dimethyloxazoline (DMOX) derivatives. However, for some acyl side chains the amounts were too low and it was not possible to determine the position of the double bonds with absolute certainty. This analysis led to the identification of branched-chain fatty acids as acyl residues of galactolipids (Table 4). The respective mass spectra were confirmed by comparison with data of the lipid library1. However, the correct positions of the methyl group at the fatty acid backbone could not always be determined unequivocally by the mass spectra of their methyl esters and DMOX derivatives.
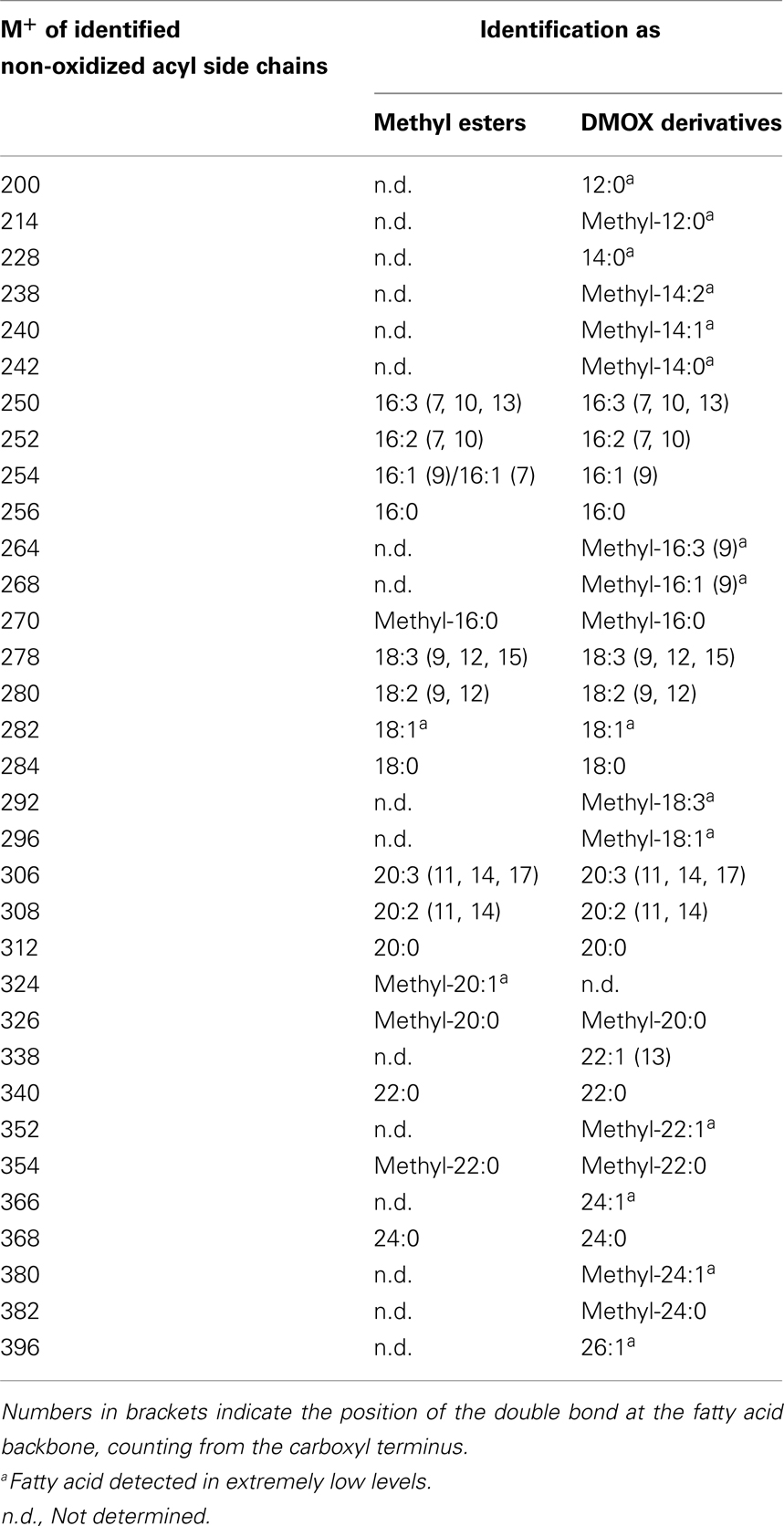
Table 4. Straight- and branched-chain fatty acids identified as methyl esters as well as DMOX derivatives by GC/MS.
For the structural identification of different oxylipins [e.g., OPDA, dnOPDA, hydroxy octadecatrienoic acids (HOTs), and hydroxy octadecadienoic acids (HODs)] that were esterified to the glycerol backbone of galactolipids, their respective trimethylsilyl ether/methyl ester derivatives were analyzed via GC/MS or as methyl esters by HPLC/diode array detection (DAD; data not shown). The identity of those compounds was confirmed by comparison with authentic standards. The structures of the two acyl side chains with the predicted sum formulas of C17H28O4 and C19H32O4 could be resolved from the EI–MS spectra of the corresponding methyl esters as well as trimethylsilyl ester/trimethylsilyl ether derivatives (Figure A1 in Appendix). From these data, the compounds were assigned as ketol derivatives of methyl-branched 16:3 and 18:3 and were therefore being named methyl-ketol-16:2 and methyl-ketol-18:2, respectively.
Qualitative Galactolipid Profile of Wounded A. thaliana Leaves
By combining all data obtained by the different MS experiments the structure of the detected acyl side chains could be assigned. The structures of the non-oxidized and oxidized fatty acid residues are given in Table 5. By combining their structural information with the corresponding information for the head groups a qualitative galactolipid profile of wounded A. thaliana leaves was obtained. Due to different fragment ion pattern, nearly all isobaric species could be identified and their molecular structures resolved. However, in some cases, isobaric species differed in their retention times, but exhibited the same intensities of both ion fragments derived from the sodium adduct rendering it impossible to determine the sn position of the acyl side chains. These species are shown with side chains in brackets in the following and in the tables. Species containing exclusively non-oxidized acyl side chains were grouped together to build the non-oxidized classes. On the other hand, species containing at least one oxidized acyl side chain were arranged into the oxidized classes. This classification led to formation of 10 different galactolipid classes: non-oxidized MGD, oxidized MGD, non-oxidized DGD, oxidized DGD, non-oxidized acylated MGD, oxidized acylated MGD, oxidized acylated DGD, non-oxidized MGM, non-oxidized SQD, and oxidized SQD. The number of species identified are given in Table 6 and compared with the numbers from previous studies. In summary, 22 non-oxidized MGD species (Table A1 in Appendix), 36 oxidized MGD species (Table A2 in Appendix), 31 non-oxidized DGD species (Table A3 in Appendix), 26 oxidized DGD species (Table A4 in Appendix), 13 non-oxidized acylated MGD species (Table A5 in Appendix), 27 oxidized acylated MGD species (Table A6 in Appendix), one oxidized acylated DGD species (Table A7 in Appendix), two non-oxidized lyso-MGD species (Table A8 in Appendix) seven non-oxidized SQD species (Table A9 in Appendix) as well as two oxidized SQD species (Table A10 in Appendix) were identified.
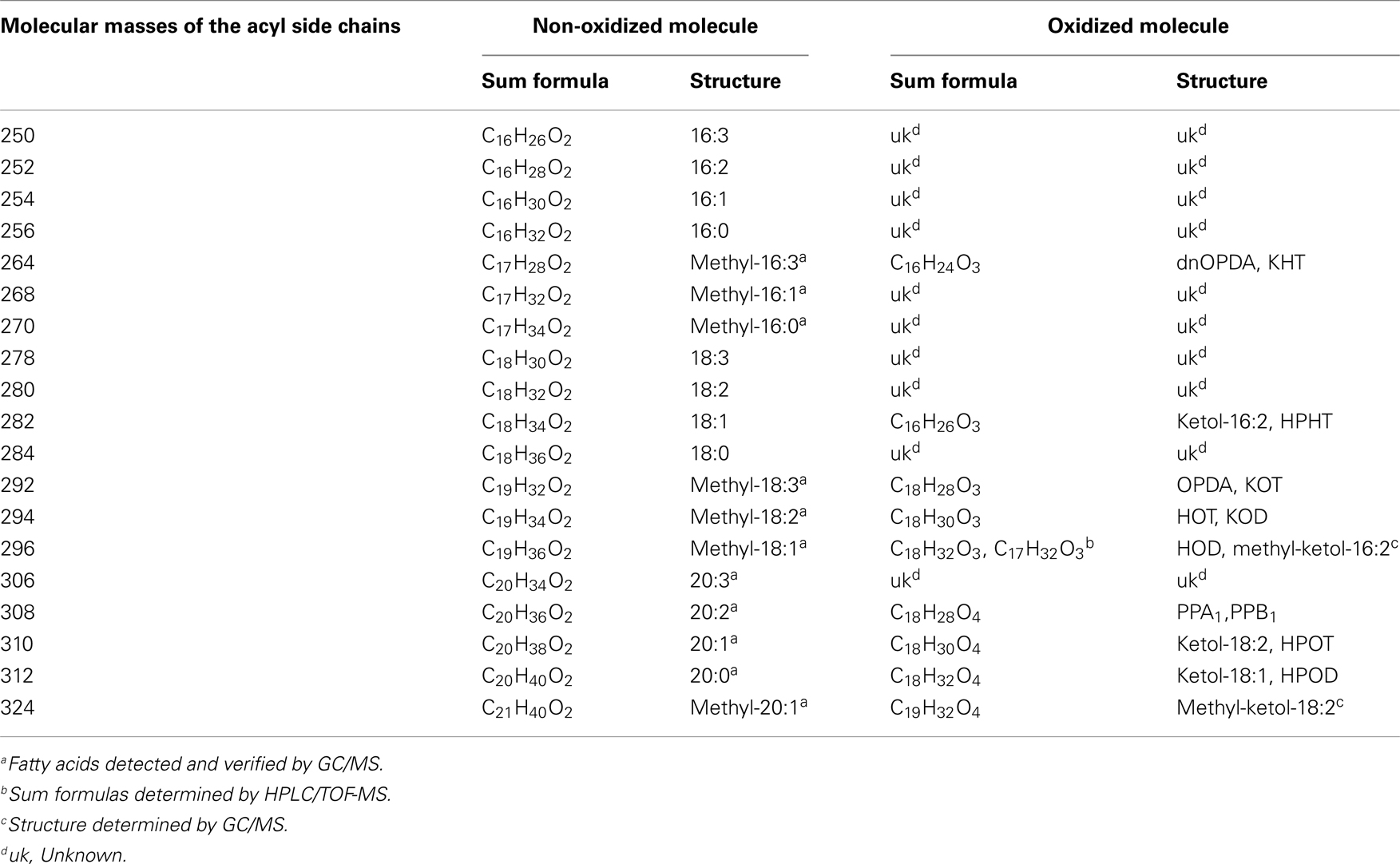
Table 5. Molecular masses of the detected acyl side chains and the appropriate sum formulas and structures of the respective non-oxidized or oxidized molecular versions.
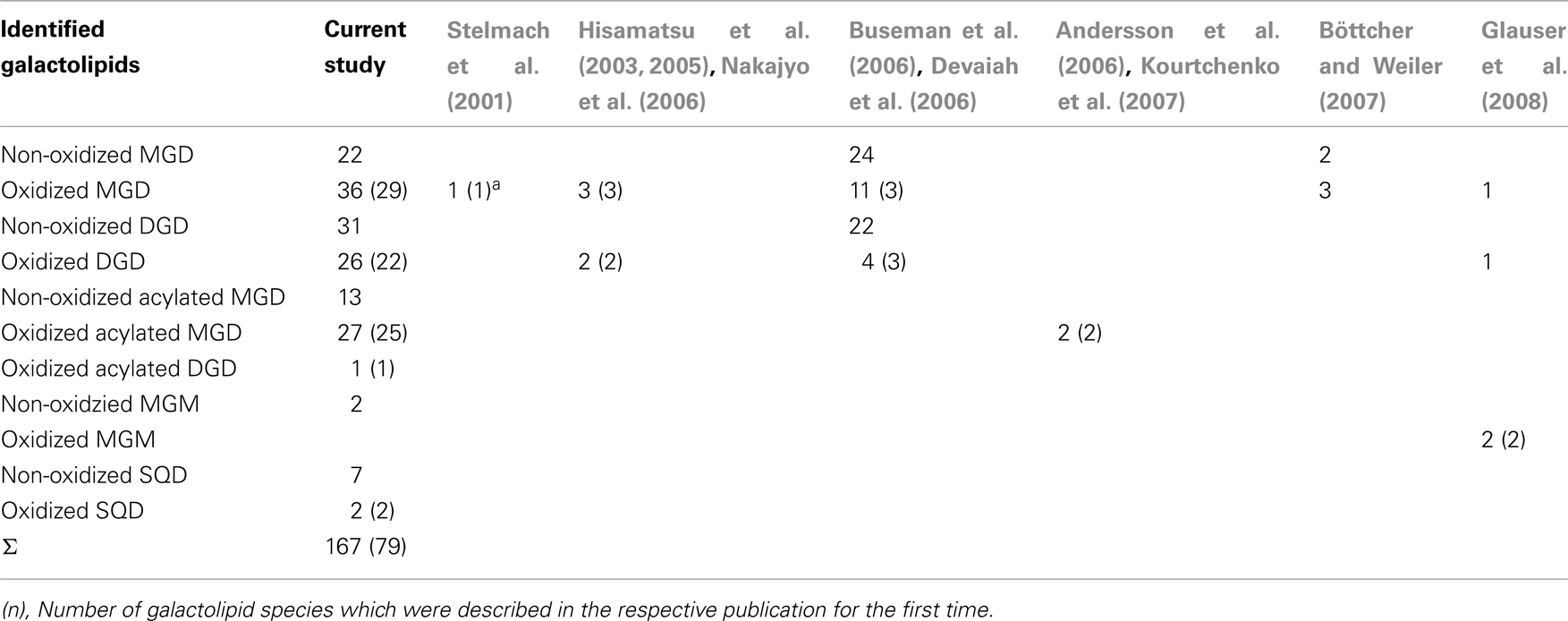
Table 6. Number of identified galactolipid species detected in wounded leaves of A. thaliana.
Quantitative Galactolipid Profile of Wounded A. thaliana Leaves
In summary 167 species could be distinguished by the HPLC/MS2 experiment. However, some galactolipid species could not be quantified since their signals were either too small or they were not fully separated under the used experimental conditions. Therefore, only 146 of the 167 species were quantified by the HPLC/MS-based method.
In the non-oxidized MGD class, the most abundant species is the plastid-derived prokaryotic species 34:6-MGD (x:y-MGD, where x:y represents the sum of both acyl side chains with x carbons and y double bonds bound to MGD) with an amount of 737.5 ± 166.3 nmol g−1 f.w. followed by the endoplasmic reticulum-derived eukaryotic species 36:6-MGD (511.5 ± 88.1 nmol g−1 f.w.; Table A1 in Appendix). Other prokaryotic species like 34:5-MGD and 34:4-MGD had amounts of 215.1 ± 25.4 and 221.1 ± 40.5 nmol g−1 f.w. Another abundant eukaryotic species is 36:5-MGD with an amount of 174.3 ± 36.6 nmol g−1 f.w. Seven further non-oxidized MGD species were determined at levels lower than 100 nmol g−1 f.w. The total amount of the quantified non-oxidized MGD species was 2.15 ± 0.19 μmol g−1 f.w. The corresponding oxidized MGD species are shown in Table A2 in Appendix. The prokaryotic species methyl-ketol-18:2/16:3-MGD [X/Y-MGD, where fatty acid (derivative) X is esterified at the sn1 position and Y at the sn2 position of the glycerol backbone of MGD] and Ara-A derived from 34:6-MGD were the most abundant oxidized species with amounts of 257.1 ± 80.6 and 194.0 ± 184.7 mmol g−1 f.w., respectively. The next most intensive species were (HPOT/ketol-18:2)/16:3-MGD, 18:3/methyl-ketol-16:2-MGD, OPDA/methyl-ketol-16:2-MGD, and OPDA/16:3-MGD//18:3/dnOPDA-MGD (MGD-O//Ara-F) containing partially methyl-ketol-16:2 and methyl-ketol-18:2 as acyl side chains. The levels of these species were between 70 and 100 nmol g−1 f.w. Within this MGD class, all further species including Ara-B derived from 36:6-MGD were determined with levels lower than 50 nmol g−1 f.w. The total amount of the oxidized MGD species was 1.07 ± 0.54 μmol g−1 f.w.
The non-oxidized DGD class was mainly composed of the eukaryotic species 36:6-DGD (416.9 ± 104.2 nmol g−1 f.w.) and 34:3-DGD composed of 16:0 at sn1 and 18:3 at sn2 (351.9 ± 107.0 nmol g−1 f.w.) as well as the prokaryotic species 34:6-DGD (294.5 ± 64.9 nmol g−1 f.w.; Table A3 in Appendix). The species 34:5-DGD, 34:2-DGD, 34:1-DGD, and 36:3-DGD had amounts between 70 and 170 nmol g−1 f.w. All other species were detected with amounts lower than 60 nmol g−1 f.w. The total amount of the non-oxidized DGD species was 1.69 ± 0.41 μmol g−1 f.w.
In accordance with the non-oxidized DGD profile, Ara-D derived from the eukaryotic 36:6-DGD was together with methyl-ketol-18:2/18:3-DGD and OPDA/OPDA-DGD (Ara-D) the most abundant oxidized species in the oxidized DGD class with amounts of 40.7 ± 29.9 and 67.0 ± 34.9 nmol g−1 f.w., respectively (Table A4 in Appendix). Two other abundant oxidized species were methyl-ketol-18:2/methyl-ketol-18:2-DGD and 16:0/OPDA-DGD//OPDA/16:0-DGD with levels above 10 nmol g−1 f.w. The other oxidized DGD species including OPDA/dnOPDA-DGD (Ara-C) were found to have levels lower than 8 nmol g−1 f.w. In total, the oxidized DGD species had an amount of 0.21 ± 0.09 μmol g−1 f.w.
Within the non-oxidized acylated MGD class, three species, i.e., the eukaryotic species 54:9-MGD (90.0 ± 41.0 nmol g−1 f.w.), 52:9-MGD representing a mixture of a prokaryotic and a eukaryotic species (69.8 ± 41.9 nmol g−1 f.w.), and the prokaryotic species 50:9-MGD (23.6 ± 13.8 nmol g−1 f.w.; Table A5 in Appendix), were determined with decreasing amounts. Four additional species were quantified with levels lower than 20 nmol g−1 f.w. The total amount of the non-oxidized acylated MGD species was 0.18 μmol ± 0.07 g−1 f.w.
The most abundant oxidized acylated MGD species was the prokaryotic OPDA/dnOPDA–MGD–OPDA [X/Y-MGD-Z, where fatty acid (derivative) X is esterified at the sn1 position, Y at the sn2 position of the glycerol backbone and Z at the galactose moiety of MGD; Ara-E] with an amount of 181.4 ± 76.1 nmol g−1 f.w. (Table A6 in Appendix). The second most abundant species was OPDA/OPDA-MGD-OPDA (Ara-G) with an amount of 67.7 ± 26.8 nmol g−1 f.w. The other species were determined with levels lower than 20 nmol g−1 f.w. leading to a total amount of the oxidized acylated MGD class of 0.33 ± 0.12 μmol g−1 f.w.
For the class of oxidized acylated DGD and non-oxidized MGM only three different species were detected. The level of these metabolites were comparatively low with levels of 9.4 ± 5.0 nmol g−1 f.w. for OPDA/OPDA–DGD–OPDA (Table A7 in Appendix) and 0.5 ± 0.4 nmol g−1 f.w. (18:1-MGM) to 28.8 ± 26.4 nmol g−1 f.w. (18:3-MGM; Table A8 in Appendix).
While the non-oxidized SQD species gave amounts of 0.3 ± 0.5 nmol g−1 f.w. (for 16:1–16:0-SQD) to 502.9 ± 629.4 nmol g−1 f.w. (for 18:3/16:0-SQD; Table A9 in Appendix) the amount of the oxidized species was too low for quantification (Table A10 in Appendix).
In addition to the galactolipids, the non-esterified oxylipins jasmonic acid (JA), OPDA, and dnOPDA were analyzed. Due to the different requirements of these free oxylipins in the chromatographic and ionization conditions, these metabolites were analyzed via HPLC/quadrupole-linear ion trap-MS. Using this method the free oxylipins were measured with amounts of 0.73 ± 0.14 nmol g−1 f.w. for JA, 4.7 ± 2.2 nmol g−1 f.w. of dnOPDA, and 2.7 ± 1.1 nmol g−1 f.w. of OPDA giving a total amount of 8.16 ± 3.14 nmol g−1 f.w. The sum of all OPDA- and dnOPDA-containing galactolipid species is 1.10 ± 0.45 μmol g−1 f.w. and is therefore about 150-fold higher than the total amount of non-esterified OPDA and dnOPDA.
Validation of the Established HPLC/MS2-Based Method
In order to evaluate the quality and reliability of the established method, the sensitivity, the quantification range and precision of this method were examined according to the Guidance for Industry-Bioanalytical method validation2 (2001).
The sensitivity of the method was determined with the galactolipid species 17:0/17:0-MGD to be 2 pmol. The lower limit of quantification was 20 pmol. Next the quantification range for the six main non-oxidized galactolipids as well as the six main Arabidopsides was examined. The linear range achieved for 34:6-MGD was 2.0–20 nmol (r2 = 0.98), for 36:6-MGD 1.8–17 nmol (r2 = 0.98), for 34:6-DGD 0.6–6.5 nmol (r2 = 0.99), for 36:6-DGD 0.5–10 nmol (r2 = 0.99), for 52:9-MGD 0.03–2.6 nmol (r2 > 0.99), and for 54:9-MGD 0.03–3.8 nmol (r2 > 0.99), respectively. In case of the six main Arabidopsides, the linear range achieved for Ara-A was 1.4–64 nmol (r2 = 0.99), for Ara-B 0.12–15 nmol (r2 = 0.99), for Ara-C 0.04–1.8 nmol (r2 = 0.99), for Ara-D 0.07–16 nmol (r2 = 0.99), for Ara-E 0.37–21.5 nmol (r2 = 0.98), and for Ara-G 0.12–8.5 nmol (r2 = 0.99), respectively.
The precision of the method was determined in relation to the six main non-oxidized galactolipids as well as the six main Arabidopsides (Table 7). For these 12 compounds, the coefficient of variation (CV) of the analytical precision was calculated from data resulting from analysis of identical biological material which was extracted in triplicates and from each extract three aliquots were injected. The mean values (MV) and SD of the absolute peak areas of each nine measurements were calculated and CV was estimated by the following equation: CV = SD/MV*100. The CV values referring to the different galactolipid species were between 4.0 and 35.4. To evaluate these values the biological variation of the plant material was determined as well. For this, plant material originating from three biological replicates was extracted in parallel and for each extract a single measurement was performed. The CV of these nine measurements was also calculated as described above for the evaluation of the analytical precision. As shown in Table 7 the biological variation of the used plant material is higher than the analytical precision (with one exception for Ara-B). Therefore, three parallel extractions of the same plant material shall be always performed and subsequently measured once for the intended use.
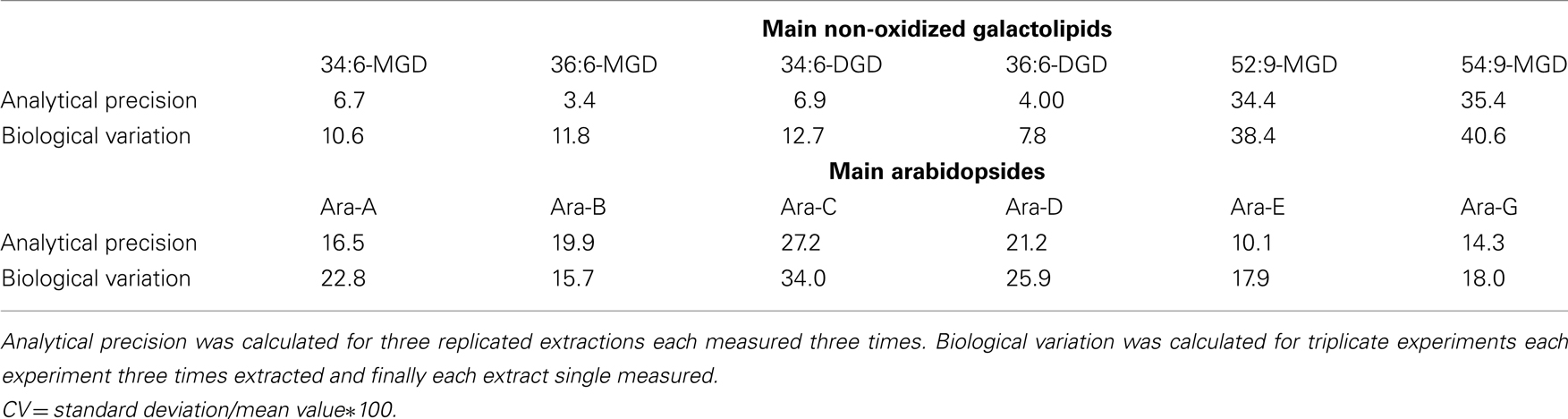
Table 7. Calculated coefficients of variation (CV) of the analytical precision and biological variation using the established HPLC/MS2-based method.
Discussion
Galactolipids are the most abundant lipids on earth (Dörmann and Benning, 2002). Interestingly, in A. thaliana the majority of them occurs oxidized at the acyl side chains already after short times of wounding and during hypersensitive response (Stelmach et al., 2001; Andersson et al., 2006; Buseman et al., 2006; Grun et al., 2007; Kourtchenko et al., 2007). Here we describe a comprehensive analysis of galactolipids by a HPLC/MS2-based method that qualitatively and quantitatively determine the non-oxidized species and oxidized derivatives of the galactolipid classes MGD, DGD (including the acylated forms), and SQD.
Establishment of a Method for Comprehensive Galactolipid Analysis
Recently, the analysis of galactolipids and particularly of the oxidized species has been intensified due to the discovery of the Arabidopsides in the genius Arabidopsis (Böttcher and Weiler, 2007). For the first structure identification spectroscopic analyses, i.e., IR and NMR spectroscopy were typically used. These methods need high amounts of ideally purified substances and are time consuming (Stelmach et al., 2001; Hisamatsu et al., 2003, 2005; Andersson et al., 2006; Kourtchenko et al., 2007). The spectrum of identified galactolipid species was later augmented by using a MS-based method coupled with chromatographic separation (Glauser et al., 2008). Notably, the most comprehensive analyses, which either mainly focused on non-oxidized galactolipid species or on oxidized species, were performed so far with ESI-MS2-based approaches (Welti et al., 2002; Buseman et al., 2006; Devaiah et al., 2006). These methods, however, may have the disadvantage that due to the missing chromatographic separation or insufficient resolution or sensitivity of the mass spectrometer, galactolipid species with the same molecular mass can only be differentiated by MS2 experiments and therefore, the sn-specific molecular composition of those species cannot be deduced from the signal abundance of the respective fragment ions. In addition, direct ESI-MS is highly prone to ionization suppression and/or enhancement which can affect the quantification process of low abundant galactolipid species. A task of this study was to establish a fast, sensitive, and robust method for analysis of the comprehensive galactolipid spectrum composed of the non-oxidized species as well as the oxidized species. As analytical system, ESI-MS2 detection was chosen in conjunction with HPLC separation. In combination with SPE as an additional purification step 167 non-oxidized and oxidized species could be identified. To our knowledge this study presents the most comprehensive analysis of galactolipids so far (Table 6).
The analysis was focused on the determination of MGD, DGD, acylated MGD, acylated DGD, MGM species as well as SQD species. While the SQD species were analyzed with negative ESI all other galactolipid species were measured with positive ESI. Since MGD and DGD represent about 80% of the plants lipid membrane in which MGD is the most abundant one with about 50% (Douce and Joyard, 1990; Dörmann and Benning, 2002), the positive ESI parameters were focused on the analysis of the main galactolipid classes.
Structural characterization of the main galactolipids has been achieved in this study by using MS2 experiments based on the identification of the neutral loss fragments derived from either the ammonium adducts or the sodium adducts. The preferred formation of the different adducts could be forced by optimizing the temperature of the ion transfer capillary of the ion source either at 200 or at 310°C. Since the two different adducts lead to the formation of specific fragment ions either derived from the neutral loss of the hexose moiety or from the neutral loss of the acyl side chains (Napolitano et al., 2007; Pacetti et al., 2007), the structure of the main galactolipid species could be characterized in the positive ESI mode by two measurements at different temperatures of the ion transfer capillary. The acyl side chains can also be identified as acyl anions derived from the deprotonated ion [M − H]– and the acetate adduct [M + OAc]− (Buseman et al., 2006; Devaiah et al., 2006).
The regiochemical distribution of the acyl side chains could be deduced from the intensity ratio of the fragment ions corresponding to [M + Na-sn1]+ and [M + Na-sn2]+. Since the loss of the acyl side chain from the sn1 position is the preferred fragmentation process compared to the one form the sn2 position (Guella et al., 2003), the ratio of the fragment ion intensities derived from the sodium adducts was used to establish the position of the acyl side chains at the glycerol backbone. The chromatographic separation supported the structural identification, because at least in case of the oxidized galactolipids regioisomers could be chromatographically resolved. The structures of the acyl side chains were determined based on their molecular masses which were used to predict the appropriate sum formula. These data were, however, not capable to distinguish between compounds with identical m/z values. Additionally, the position of double bonds as well as of hydro(pero)xy and keto groups could not be elucidated based on the MS2 fragment ions. In contrast, Kim et al. (2001) were able to determine the positions of the double bonds in the acyl side chains of MGD and DGD species in wheat flour using the complex MS2 ion spectra of the sodium adducts desorbed by fast atom bombardment (Kim et al., 2001). These experiments, however, were carried out with purified galactolipids and could not be applied for the compound identification in lipid extracts. In the current study, it was possible according to the used solvent gradient of the chromatographic separation to predict whether the respective galactolipid species contained non-oxidized or oxidized acyl side chains, because the oxidized species eluted earlier than the corresponding non-oxidized species. Unequivocal identification of most acyl side chains was achieved by analyzing the respective DMOX and trimethylsilyl ether/methyl ester derivatives via GC/MS.
For two compounds with m/z values of 296 and 324 the structures could not be directly deduced based from MS2 spectra since they have been so far not been identified as acyl side chains of galactolipids and as oxylipins. However, the elucidation of these structures was possible by TOF-MS and GC/MS analyses: the accurate masses and the EI mass spectra of the respective methyl esters and trimethylsilyl ester/trimethylsilyl ether derivatives strongly suggest that these compounds are ketol derivatives derived from methyl-branched fatty acids (Figure A1 in Appendix). They could result from activities of lipoxygenases (LOXs) and an allene oxide synthase (AOS) of A. thaliana against methyl-branched fatty acids leading to the formation of allene oxides which are non-enzymatically further hydrolyzed into ketols.
The quantification of the various galactolipid species was carried out using only 17:0/17:0-MGD (a non-natural galactolipid that is not found in A. thaliana) as internal standard. In some studies, however, 18:0/18:0- and 18:0/16:0-MGD as well as 18:0/18:0- and 18:0/16:0-DGD derived from the natural galactolipids by hydrogenation have been used as internal standards (Welti et al., 2002; Buseman et al., 2006; Devaiah et al., 2006), since those species were found in the galactolipid extracts only in low levels. It should be emphasized at this point, that the use of only one single internal standard as in the present study could affect the quantification process, because the ionization efficiency of the distinct galactolipid species could be different within the used gradient system of the chromatographic separation. The gradient system and ionization conditions were therefore optimized and different extraction methods were tested in order to obtain the highest quality of the quantification results. Furthermore, the precision of the quantification process has been validated in the corresponding signal intensity range in which all galactolipid species were detected in the HPLC/MS2-based method. Due to the fact that the fatty acid composition of a lipid can influence its ionization behavior, it should be stressed, however, that the amount of lipids analyzed in this study can differ from that actually accumulating in the leaves.
Galactolipid Profiles in Wounded A. thaliana Leaves
A number of studies demonstrated that non-esterified oxylipins such as JA, OPDA, and dnOPDA as well as the oxylipin-containing galactolipids accumulate in A. thaliana leaves after wounding (Farmer and Ryan, 1992; Blechert et al., 1997; Stelmach et al., 2001; Stintzi et al., 2001; Buseman et al., 2006). Therefore we used our method to quantify the identified galactolipid species by analyzing leaves 30 min after wounding. Since knowledge on the biosynthesis and the function of the oxidized galactolipid species is rather scarce till now, the levels of the non-esterified dnOPDA, OPDA, and JA, which are the potential precursors or products of the galactolipid-bound forms, were determined in parallel. Due to the different requirements of the galactolipids and the non-esterified oxylipins in the chromatographic and ionization conditions both compound classes could not be determined by a single analytical method, but were quantified by two different HPLC/MS2-based methods.
The ratio of the total amount of non-esterified OPDA and dnOPDA and the total amount of the oxylipin-containing galactolipids in leaves after wounding could provide hints whether the galactolipid species have functions as storage forms of the cyclopentenones, which could be liberated as signaling compounds after leaf damage (Mosblech et al., 2009). In this case the oxidized galactolipid species may be synthesized by direct oxidation of the acyl side chains of the galactolipids. Alternatively, it has been suggested that non-esterified dnOPDA, OPDA, and JA may be esterified into galactolipids subsequently after their biosynthesis (Stelmach et al., 2001). The quantitative galactolipid profiles revealed that the total amount of the OPDA- and dnOPDA-containing galactolipids were around 150-fold higher than those of non-esterified OPDA and dnOPDA. This finding goes in line with that of a previous study by Andersson et al. (2006), who demonstrated that the hypersensitive response resulted in a 200-fold induction of esterified OPDA and an accumulation of esterified OPDA that is 10 times higher compared to the free fatty acid derivative. Due to this huge difference in the amounts of the non-oxidized and oxidized galactolipids one may assume that non-esterified OPDA and dnOPDA molecules are esterified to the glycerol backbones directly after their biosynthesis producing the respective oxidized galactolipid species in plastids. Consequently, only small amounts remain as free OPDA and dnOPDA or enter the peroxisome where they are converted by a series of reactions leading to the synthesis of even lower levels of JA. Alternatively, JA biosynthesis could proceed with the release of defined amounts of esterified OPDA and dnOPDA from the respective oxidized galactolipid species in the plastids, which are then converted into JA in the peroxisome. Up to now, the proportion of both metabolic pathways on the overall JA biosynthesis cannot be elucidated. In addition, the biosynthesis of the free oxylipins as well as of the lipid-bound oxylipins can be directly linked to the availability of the respective substrates. Taking into account that more than 99% of the polyunsaturated fatty acids α-linolenic acid and roughanic acid occur esterified within lipids in plant cells (Conconi et al., 1996), one may assume that the synthesis of the huge amounts of oxylipin-containing galactolipids occurs using lipid-bound substrates, even because free fatty acids are only present in low cellular levels. They are liberated from lipids by the catalytic activity of lipases and can serve as substrate for sequential oxidation and isomerization reactions via the oxylipin pathway leading to low oxylipin levels (Andreou et al., 2009).
The total amount of non-oxidized galactolipid species was 4.82 ± 1.37 μmol g−1 f.w. and that of the oxidized galactolipid species was 1.81 ± 0.71 μmol g−1 f.w. This finding demonstrates that 30 min after wounding the non-oxidized galactolipids were 2.7-fold higher than the possible oxidized species. Interestingly, in the major abundant galactolipid class MGD, this ratio was only 2.0 whereas it was even 8.1 in case of the DGD species and only 0.6 in the acylated MGD class. This may indicate that the oxidation degree of the galactolipids may be class-specifically regulated after wounding or different substrate specificities for the enzymes of the oxylipin pathway against the different lipid molecules may exist. Moreover, these different proportions may hint at specific functions of the respective species. One conclusion that one may derive from this observation is that the primary substrate of oxylipin forming enzymes under these conditions are MGD species. From these oxidized MGD molecules oxylipins may be transferred by acyl editing reactions onto acylated MGD- and DGD species.
Most of the so far described oxylipin-containing galactolipids contain either OPDA- and/or dnOPDA as acyl side chains (Stelmach et al., 2001; Hisamatsu et al., 2003, 2005; Andersson et al., 2006; Kourtchenko et al., 2007). In addition eight species have been described as ketol-containing galactolipids (Buseman et al., 2006). In the current study, several additional oxylipin species were identified to be esterified into galactolipids. Besides the most abundant cyclopentenones OPDA and dnOPDA, which comprised of 65 mol%, of the total oxidized acyl side chains, two novel methyl-branched ketols were identified as the second most abundant oxylipins with 27 mol%. Acyl side chains corresponding to either hydroperoxides or common oxylipin pathway-derived ketols were found to represent 3.8 mol% of the total oxidized acyl side chains. The distinct composition of these galactolipid species could not be further deduced since the respective acyl side chains have an identical molecular mass. Hydroxides and phytoprostanes were determined with 2.0 and 2.6 mol%, respectively. These data indicate that the majority of the oxidized galactolipids is derived from enzymatic reactions, since cyclopentenones in these amounts may be exclusively formed by the coordinated activities of 13-LOX, 13-AOS, and 13-allene oxide cyclase. Indeed, the analysis of the enantiomeric composition of the esterified hydroxides and hydroperoxides revealed that the main 13-regioisomers occurred over 90% as S-enantiomers confirming the enzymatic origin of these galactolipid acyl side chains (data not shown). In contrast, phytoprostanes were the only acyl side chains determined within the galactolipid fraction of exclusively non-enzymatic origin. In summary, these data revealed that the oxidized galactolipid species accumulating after wounding in leaves of A. thaliana are the result of enzymatic reactions assuming that the novel methyl-branched ketols are also derived from an enzymatic activity.
Non-enzymatic peroxidation of membrane lipids and in particularly of the most abundant polyunsaturated fatty acids roughanic acid, linoleic acid, and α-linolenic acid is another process in lipid metabolism and results in the formation of lipid-bound phytoprostanes or hydroxy fatty acids (Müller, 2004). However, we detected only four minor abundant oxidized galactolipid species being lipid-bound phytoprostanes. This finding was unexpected, since during the hypersensitive response of A. thaliana non-enzymatically formed oxylipins accumulated to much higher amounts (Grun et al., 2007).
Concerning the non-oxidized galactolipid species it is notable that in addition to the C16 and C18 fatty acids, several C20 fatty acids as well as methyl-branched fatty acids were identified as acyl residues of the galactolipids. These fatty acid structures were mainly identified within the DGD class, esterified either at the sn1 or the sn2 position. Even though the levels of these species were very low in comparison to the other non-oxidized DGD species their biosynthesis seems to be specific for DGD.
In summary, we have explored the high diversity of galactolipids in A. thaliana and identified 167 different species. Our results indicate that galactolipid oxidation upon wounding occurs class specific and that esterifed cyclopentenone derivatives are more abundant compared to the free forms. Our data will be valuable for analyzing comprehensive galactolipid profiles in different biological contexts and might help to understand their function and biosynthetic pathways.
Materials and Methods
Material
Solvents for lipid extraction and galactolipid purification were purchased either (chloroform or acetone) from Carl Roth (Karlsruhe, Germany) or (2-propanol, acetonitrile (HPLC/MS grade) and methanol (HPLC/MS and HPLC grade) from Acros (Geel, Belgium). Acetic acid was from Sigma-Aldrich (Steinheim, Germany). Deionized water was filtered with cartridge to 0.055 μs/cm (arium® 611, Sartorius, Göttingen, Germany). Di-heptadecanoate-monogalactosyldiacylglycerol (17:0/17:0-MGD) standard was provided by Prof. E. Heinz (Hamburg, Germany) and used for galactolipid quantification. (10-2H,11-2H2, 12-2H3)-JA (D6-JA) and (17-2H2, 18-2H3)-OPDA (D5-OPDA) standards were provided by Dr. O. Miersch (Halle/Saale, Germany) for quantification of non-esterified JA, OPDA, and dnOPDA. Phytoprostanes were synthesized using our published procedures (El Fangour et al., 2004, 2005; Pinot et al., 2008).
Plant Material and Growth Conditions
In this study the Columbia-0 (Col-0) ecotype of Arabidopsis was used as wild type plants. Plants were grown for 7–8 weeks under light period of 8 h at 22°C with 90 μmol/m2 s light intensity and a dark period of 16 h at 18°C. Wounding of rosette leaves was mechanically performed by using forceps. Wounded plants were further incubated for 30 min at 90 μmol/m2 s light intensity until the harvest. The harvested leaves were frozen directly in liquid nitrogen and stored at −80°C until use. Prior to extraction, leaf material was ground into fine powder under liquid nitrogen using a beat mill (Retsch, Haan, Germany) and kept again at −80°C until use.
Sample Preparation, Extraction Processes
Galactolipids and their oxidized derivatives were extracted according to (Bligh and Dyer, 1959; Heinz and Tulloch, 1969; Matyash et al., 2008) with some minor modifications as described below in order to compare the extraction efficiency of the respective method. In standard experiments 0.2–2 g of plant material was used for extraction. As internal standards, 25 μg of 17:0/17:0-MGD for the quantification of galactolipids and 25 ng of each D5-OPDA and D6-JA for the quantification of non-esterified JA, OPDA, and dnOPDA were added.
Chloroform/Methanol Extraction (According to Bligh and Dyer, 1959)
Briefly, total lipids were extracted from 0.2 g of wounded leaves of A. thaliana by the addition of 1.5 ml of chloroform/methanol (1:2, v/v) and incubation at 6°C for 4 h. The above mentioned internal standards were added. The extract was centrifuged at 450 × g at 4°C for 10 min and the supernatant was stored under argon atmosphere at −20°C. The pellet was resolved in 1.5 ml of chloroform/methanol (2:1, v/v), incubated for 20 h and centrifuged at 450 × g at 4°C for 10 min. The resulting supernatant of both extraction steps were combined and 0.7 ml of 0.45% (w/v) sodium chloride was added. After manual shaking the mixture was centrifuged at 450 × g at 4°C for 5 min and sodium sulfate was added to the organic phase in order to remove residual water from the sample. The total lipid extract was dried under streaming nitrogen and the remaining lipids were re-dissolved in 0.5 ml methanol and stored under argon atmosphere at −20°C. About 0.1 ml of each individual sample was used for the quantification of non-esterified JA, OPDA, and dnOPDA whereas the remaining sample was used for the identification and quantification of galactolipid species and their oxidized derivatives. In experiments where 2.0 g of plant material were used, the volume of the extraction solvents adjusted adequately.
Chloroform/Methanol Extraction (According to Heinz and Tulloch, 1969)
Total lipids were extracted in a procedure similar to the one of Bligh and Dyer (1959) described above with the following modifications: the composition of chloroform/methanol in both extraction steps was 1:1 (v/v) instead of 1:2 (v/v) and 2:1 (v/v) in the first and in the second step, respectively.
Methyl tert-butyl Ether Extraction (According to Matyash et al., 2008)
Wounded leaves of A. thaliana were incubated with 1.5 ml methanol containing the above mentioned internal standards under constant shaking. Five microliter of methyl tert-butyl ether were added and the resulting mixture was shaken for 1 h in the dark. About 1.25 ml of water was added and after manual shaking and incubation at room temperature for 10 min, the extract was centrifuged at 450 × g at 4°C for 15 min. The upper phase was stored under argon atmosphere at −20°C and the lower phase was re-extracted with 1.4 ml of methanol/water (3:2.5, v/v) and 2.5 ml of methyl tert-butyl ether. After manually shaking the mixture was incubated at room temperature for 10 min and centrifuged at 450 × g at 4°C for 15 min. The resulting upper phase was combined with the one from the first reaction step and dried under streaming nitrogen. The remaining lipids were re-dissolved in 0.5 ml of methanol and stored under argon atmosphere at −20°C. For the quantification of non-esterified JA, OPDA, and dnOPDA, 0.1 ml of each individual sample was used. The remaining sample was used for the quantification of galactolipid species and their oxidized derivatives.
Separation of Lipid Classes by SPE
The lipid extract was dried under streaming nitrogen and remaining lipids were re-dissolved in 1 ml of chloroform. The solution was applied on SPE column (Strata SI-1 Silica, 500 mg/6 ml; Phenomenex, Aschaffenburg, Germany) which was pre-equilibrated with 1 ml of chloroform. Neutral lipids were eluted from the column with 14 ml of chloroform, while galactolipids were eluted with 15 ml of acetone/2-propanol (9:1, v/v). The galactolipid fraction was dried under streaming nitrogen and subsequently re-dissolved in 0.5 ml of methanol and stored at −20°C until use.
Identification and Quantification of Galactolipid Species via HPLC/MS2
Galactolipid species were identified by using a HPLC Surveyor system (Thermo Finnigan, San Jose, CA, USA) equipped with an EC 50/2 Nucleodur C18 column, gravity 1.8 μm (50 mm × 2.1 mm, 1.8 μm particle size; Macherey and Nagel, Düren, Germany) and with a PDA coupled with an LCQ Advantage mass spectrometer (Thermo Finnigan, San Jose, CA, USA). A ternary gradient system consisting of acetonitrile as solvent A, water as solvent B, and 2-propanol as solvent C was used. The gradient program started with an isocratic run at 50% of each solvent A and B for 10 min, followed by a linear gradient reaching 85% of solvent A and 15% of solvent B within 5 min, then continued with a linear gradient reaching 34% of solvent A, 6% of solvent B, and 60% of solvent C within 20 min and followed by an isocratic run for 20 min. The flow rate was 0.2 ml/min.
For analysis the dried sample was re-dissolved in 16 μl of methanol. By using the partial loop injection mode, 4 μl of the sample were injected. The tray temperature was at 10°C and column temperature was at 25°C. The Surveyor PDA was performed to collect spectral absorbance from 200 nm up to 340 nm. The MS analysis was performed in positive (MGD and DGD species) and negative (SQD species) ESI mode and the resulting ions were collected in a mass range of 450–1,500 amu in full scan mode. The ion spray voltage was 4 kV, capillary voltage was 28 V, the flow rate of the sheath gas was at 30 arbitrary units (arb) and the auxiliary gas flow rate at 10 arb. The temperature of the ion transfer capillary was either at 200°C to detect the ammonium adducts [M + NH4]+ or at 310°C in addition to the setting-up of the collision energy (CE) of the source fragmentation mode at 40 V in order to analyze the sodium adducts [M + Na]+. For ion identification the data-dependent scan mode was used or alternatively specific MS/MS experiments were performed. The CE was 2 V. For quantification of the galactolipid species, the HPLC/MS analysis was performed without PDA detection and in full scan MS mode without using data-dependent MS/MS scan mode. The capillary temperature was at 200°C and for quantification of MGD and DGD species the ammonium adducts were used. SQD species were analyzed and quantified as deprotonated molecular ion ([M − H]−) in the negative ESI mode using the same MS conditions described above (Table 2). The molar values of the galactolipid species were quantified by using a defined amount of 17:0/17:0-MGD as internal extraction standard. The data were smoothed and peak areas were integrated using Xcalibur™ software version 1.4 (Thermo Finnigan, San Jose, CA, USA).
Identification of the Acyl Side Chains Using HPLC/TOF-MS and GC/MS
Unknown fatty acids and the respective derivatives esterified either to the glycerol backbone or the galactosyl moiety of MGD and DGD were identified by using a combination of HPLC/TOF-MS and GC/MS analyses. For this purpose on the one hand the accurate mass of a galactolipid species was determined by using an orthogonal TOF mass spectrometer (LCT Premier™; Waters Corporation, Milford, USA) that was coupled to the same RP-HPLC system as described above. The same solvent system and gradient elution profile were used. The TOF-MS analysis was performed in positive ESI mode using the W optic system of the mass analyzer and with a mass resolution larger than 104. Data were acquired by MassLynx™ software (Waters Corporation, Milford, USA) in centroided format over a mass range of 400–1,200 amu with scan duration of 0.5 s and interscan delay of 0.1 s. The capillary voltage was maintained at 2.7 kV and the cone voltage at 30 V. The desolvation and source temperature were set at 250 and 80°C, respectively. Nitrogen was used as cone gas and desolvation gas at a flow rate of 30 and 600 l h−1, respectively. For accurate mass measurement of <5 ppm root mean squared, the TOF mass spectrometer was calibrated with 0.01% (v/v) phosphoric acid in acetonitrile/water (50:50, v/v) and the dynamic range enhancement mode was used for data recording. All analyses were monitored by using leucine–enkephalin ([M + H]+ 556.2771 as well as the 2 × 13C isotopomer [M + H]+ 558.2828 (Sigma-Aldrich, Steinheim, Germany) as lock spray reference compound at a concentration of 1 μg ml−1 in acetonitrile/water (50:50, v/v) and at a flow rate of 20 μl min−1 delivered by a 515 HPLC pump (Waters Corporation, Milford, USA).
In order to identify and characterize esterified fatty acids as well as esterified oxylipins in more detail on the other hand GC/MS analysis was employed. For this purpose fatty acids were converted to the corresponding methyl esters by a transesterification reaction that was performed according to op den Camp et al. (2003). Briefly, the galactolipid fraction from SPE was dried under streaming nitrogen and 330 μl of toluene/methanol (2:1, v/v) and 170 μl of sodium methoxide solution (0.5 M in methanol; Sigma-Aldrich, Steinheim, Germany) were added and incubated for 20 min at room temperature. Then 500 μl of saturated sodium chloride solution and 50 μl of 32% (v/v) hydrochloride acid were added. The resulting methyl esters were extracted three times with 1 ml of hexane each. The combined organic phases were dried under streaming nitrogen; remaining methyl esters were re-dissolved in 10 μl of acetonitrile and stored under argon atmosphere at - 20°C until use.
In case of hydroxy fatty acid derivatives, the sample was additionally treated with 1–2 μl N,O-bis(trimethylsilyl)trifluoroacetamide (Sigma-Aldrich, Steinheim, Germany) directly before analysis.
For identification of the double bond positions of esterified fatty acids, DMOX derivatives of the esterified fatty acids were injected to GC/MS in addition to the corresponding fatty acid methyl esters. DMOX derivatization of fatty acids that are esterified to galactolipid backbones was performed according to Fay and Richli (1991). The galactolipid fraction from SPE was dried under streaming nitrogen and 200 μl of 2-amino-2-methyl-1-propanol solution (Sigma-Aldrich, Steinheim, Germany) were added. The reaction mixture was enriched with argon to eliminate autooxidation and incubated for 18 h at 180°C. After cooling at room temperature DMOX derivatives were extracted with 1 ml of dichloromethane, 5 ml of hexane, and 2 ml of water. The organic phase was dried under streaming nitrogen and the remaining DMOX derivatives were re-dissolved in 30 μl chloroform. DMOX derivatives were purified by thin layer chromatography (silica gel 60, 20 cm × 20 cm; Merck, Darmstadt, Germany) using a petrol ether/diethyl ether (2:1, v/v) system. DMOX derivatives were extracted from the silica gel by adding 0.4 ml of water, 2 ml of methanol, 2 ml of chloroform, and 2 ml of saturated sodium chloride solution. Residual water was removed from the organic phase by the addition of sodium sulfate. The resulting DMOX derivatives were dried under streaming nitrogen, re-dissolved in 10 μl acetonitrile and stored at −20°C until use.
Gas chromatography/MS was carried out using an Agilent 5973 network mass selective detector connected to an Agilent 6890 GC equipped with a capillary HP-5 column (30 m × 0.25 mm, 0.25 μm coating thickness; J&W Scientific, Agilent, Waldbronn, Germany). Helium was used as carrier gas at a flow of 1 ml min−1. The temperature gradient was as follows: 150°C for 1 min, 150–200°C at 4 K min−1, 200–250°C at 5 K min−1, 250–320°C at 20 K min−1, and 320°C hold for 15 min. Mass range was set of 50–650 amu. Electron energy of 70 eV, an ion source temperature of 230°C and a temperature of 320°C for the transfer liner were used.
Quantification of Non-Esterified Oxylipins Using HPLC/QLIT-MS Analysis
For the quantification of non-esterified oxylipins JA, OPDA, and dnOPDA in the total lipid extracts HPLC–QLIT-MS were utilized. About 1/5 of the total lipid extract was dried under streaming nitrogen and re-dissolved in 100 μl acetonitrile/water/acetic acid (20:80:0.1, v/v/v). The analysis was carried out using an Agilent 1100 HPLC system (Agilent, Waldbronn, Germany) equipped with an EC 50/2 Nucleodur C18, gravity 1.8 μm column (50 × 2, 1.8 μm particle size; Macherey and Nagel, Düren, Germany) and coupled to an Applied Biosystems 3200 hybrid triple quadrupole/linear ion trap mass spectrometer (MDS Sciex, ON, Canada). Nanoelectrospray ionization (nanoESI) analysis was achieved using a chip ion source (TriVersa NanoMate; Advion BioSciences, Ithaca, NY, USA). A binary gradient system was used: solvent A, acetonitrile/water/acetic acid (20:80:0.1, v/v/v) and solvent B, acetonitrile/acetic acid (100:0.1, v/v) with the following gradient program: 90% solvent A and 10% solvent B for 2 min, followed by linear increase of solvent B up to 90% within 6 min and an isocratic run at 10% solvent A and 90% solvent B for 2 min. The flow rate was 0.3 ml min−1. For stable nanoESI, 100 μl min−1 of 2-propanol/acetonotrile/water/formic acid (70:20:10.0.1, v/v/v/v) were added just after the column via a mixing tee valve. By using another post column splitter 990 nl min−1 of the eluent were directed to the nanoESI chip. Ionization voltage was set to −1.7 kV. The non-esterified oxylipins were ionized in a negative mode and determined in multiple reactions monitoring mode. The mass transitions were as follows: 215/59 [declustering potential (DP) −45 V, entrance potential (EP) −9.5 V, CE −22 V] for D6-JA, 209/59 (DP −45 V, EP −9.5 V, CE −22 V) for JA, 296/170 (DP −70 V, EP −8.5 V, CE −28 V) for D5-OPDA, 291/165 (DP −70 V, EP −8.5 V, CE −28 V) for OPDA, and 263/59 (DP −70 V, EP −8.5 V, CE −28 V) for dnOPDA. The mass analyzers were adjusted to a resolution of 0.7 amu full width at half-height. The ion source temperature was 40°C and the curtain gas was set at 10 arb. The data were smoothed and peak areas were integrated using Applied Biosystems Analyst software. The quantification was carried out by comparison to two internal standards D6-JA and D5-OPDA using a calibration curve (unlabeled/deuterium-labeled) vs. molar amounts of unlabeled (0.3–1,000 pmol).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the perfect technical assistance of Sabine Freitag and Pia Meyer. We also thank Dr. Otto Miersch (IPB, Halle) and Prof. Dr. Ernst Heinz (Hamburg University) for providing us with lipid standards. Funding of AI by Damascus University is also gratefully acknowledged.
Abbreviations
AOS, allene oxide synthase; arb, arbitrary units; Col-0, Columbia-0; CP, chiral phase; DAD, diode array detector; DGD, digalactosyl diacylglycerol; DMOX, 4,4-dimethyloxazolines; dnOPDA, (7R,8Z,11S,13Z)-10-oxo-8,13-dinor-phytodienoic acid; DP, declustering potential; EP, entrance potential; EI, electron impact; ESI, electrospray ionization; GC, gas chromatography; HPHT, hydro(pero)xy hexadecatrienoic acid; HPLC, high performance liquid chromatography; H(P)OD, hydro(pero)xy octadecadienoic acid; H(P)OT, hydro(pero)xy octadecatrienoic acid; JA, jasmonic acid; KHT, keto hexadecatrienoic acid; KOT, keto octadecatrienoic acid; α-LeA, α-linolenic acid; LOX, lipoxygenase; MGD, monogalactosyl diacylglycerol; MS, mass spectrometry; OPDA, (9R,10Z,13S,15Z)-12-oxo-10,15-phytodienoic acid; PPA1, phytoprostane A1; PPB1, phytoprostane B1; QLIT, quadrupole-linear ion trap; RP, reversed phase; RT, retention time; SPE, solid phase extraction; SQD, sulfoquinovosyl diacylglycerol; UPLC/TOF-MS, ultra performance liquid chromatography/time-of-flight mass spectrometry.
Footnotes
References
Andersson, M. X., Hamberg, M., Kourtchenko, O., Brunnstrom, A., McPhail, K. L., Gerwick, W. H., Göbel, C., Feussner, I., and Ellerström, M. (2006). Oxylipin profiling of the hypersensitive response in Arabidopsis thaliana: formation of a novel oxo-phytodienoic acid-containing galactolipid, Arabidopside E. J. Biol. Chem. 281, 31528–31537.
Andreou, A., Brodhun, F., and Feussner, I. (2009). Biosynthesis of oxylipins in non-mammals. Prog. Lipid Res. 48, 148–170.
Blechert, S., Bockelmann, C., Brümmer, O., Füß lein, M., Gundlach, H., Haider, G., Hölder, S., Kutchan, T. M., Weiler, E. W., and Zenk, M. H. (1997). Structural separation of biological activities of jasmonates and related compounds. J. Chem. Soc. Perkin Trans. 1, 3549–3559.
Bligh, E. G., and Dyer, W. J. (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917.
Böttcher, C., and Weiler, E. W. (2007). cyclo-Oxylipin-galactolipids in plants: occurrence and dynamics. Planta 226, 629–637.
Buseman, C. M., Tamura, P., Sparks, A. A., Baughman, E. J., Maatta, S., Zhao, J., Roth, M. R., Esch, S. W., Shah, J., Williams, T. D., and Welti, R. (2006). Wounding stimulates the accumulation of glycerolipids containing oxophytodienoic acid and dinor-oxophytodienoic acid in Arabidopsis leaves. Plant Physiol. 142, 28–39.
Conconi, A., Miquel, M., Browse, J. A., and Ryan, C. A. (1996). Intracellular levels of free linolenic and linoleic acids increase in tomato leaves in response to wounding. Plant Physiol. 111, 797–803.
Devaiah, S. P., Roth, M. R., Baughman, E., Li, M., Tamura, P., Jeannotte, R., Welti, R., and Wang, X. (2006). Quantitative profiling of polar glycerolipid species from organs of wild-type Arabidopsis and a PHOSPHOLIPASE D[alpha]1 knockout mutant. Phytochemistry 67, 1907–1924.
Dörmann, P., and Benning, C. (2002). Galactolipids rule in seed plants. Trends Plant Sci. 7, 112–118.
Douce, R., and Joyard, J. (1990). Biochemistry and function of the plastid envelope. Annu. Rev. Cell Biol. 6, 173–216.
El Fangour, S., Guy, A., Despres, V. R., Vidal, J.-P., Rossi, J.-C., and Durand, T. (2004). Total synthesis of the eight diastereomers of the syn-anti-syn phytoprostanes F1 types I and II. J. Org. Chem. 69, 2498–2503.
El Fangour, S., Guy, A., Vidal, J.-P., Rossi, J.-C., and Durand, T. (2005). A flexible synthesis of the phytoprostanes B1 type I and II. J. Org. Chem. 70, 989–997.
Farmer, E. E., and Ryan, C. A. (1992). Octadecanoid precursors of jasmonic acid activate the synthesis of wound-inducible proteinase inhibitors. Plant Cell 4, 129–134.
Fay, L., and Richli, U. (1991). Location of double bonds in polyunsaturated fatty acids by gas chromatography-mass spectrometry after 4,4-dimethyloxazoline derivatization. J. Chromatogr. A 541, 89–98.
Glauser, G., Grata, E., Rudaz, S., and Wolfender, J. -L. (2008). High-resolution profiling of oxylipin-containing galactolipids in Arabidopsis extracts by ultra-performance liquid chromatography/time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 22, 3154–3160.
Grun, C., Berger, S., Matthes, D., and Müller, M. J. (2007). Early accumulation of non-enzymatically synthesised oxylipins in Arabidopsis thaliana after infection with Pseudomonas syringae. Funct. Plant Biol. 34, 65–71.
Guella, G., Frassanito, R., and Mancini, I. (2003). A new solution for an old problem: the regiochemical distribution of the acyl chains in galactolipids can be established by electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 1982–1994.
Heinz, E., and Tulloch, A. P. (1969). Reinvestigation of the structure of acyl galactosyl diglyceride from spinach leaves. Hoppe-Seyler’s Z. Physiol. Chem. 350, 493–498.
Hisamatsu, Y., Goto, N., Hasegawa, K., and Shigemori, H. (2003). Arabidopsides A and B, two new oxylipins from Arabidopsis thaliana. Tetrahedron Lett. 44, 5553–5556.
Hisamatsu, Y., Goto, N., Hasegawa, K., and Shigemori, H. (2006). Senescence-promoting effect of arabidopside A. Z. Naturforsch. 61c, 363–366.
Hisamatsu, Y., Goto, N., Sekiguchi, M., Hasegawa, K., and Shigemori, H. (2005). Oxylipins arabidopsides C and D from Arabidopsis thaliana. J. Nat. Prod. 68, 600–603.
Kim, Y. H., Gil, J. H., Hong, J., and Yoo, J. S. (2001). Tandem mass spectrometric analysis of fatty acyl groups of galactolipid molecular species from wheat flour. Microchem. J. 68, 143–155.
Kourtchenko, O., Andersson, M. X., Hamberg, M., Brunnstrom, A., Göbel, C., McPhail, K. L., Gerwick, W. H., Feussner, I., and Ellerstrom, M. (2007). Oxo-phytodienoic acid-containing galactolipids in Arabidopsis: jasmonate signaling dependence. Plant Physiol. 145, 1658–1669.
Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A., and Schwudke, D. (2008). Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146.
Moreau, R., Doehlert, D., Welti, R., Isaac, G., Roth, M., Tamura, P., and Nuñez, A. (2008). The Identification of mono-, di-, tri-, and tetragalactosyl-diacylglycerols and their natural estolides in oat kernels. Lipids 43, 533–548.
Mosblech, A., Feussner, I., and Heilmann, I. (2009). Oxylipins: structurally diverse metabolites from fatty acid oxidation. Plant Physiol. Biochem. 47, 511–517.
Müller, M. J. (2004). Archetype signals in plants: the phytoprostanes. Curr. Opin. Plant Biol. 7, 441–448.
Nakajyo, H., Hisamatsu, Y., Sekiguchi, M., Goto, N., Hasegawa, K., and Shigemori, H. (2006). Arabidopside F, a new oxylipin from Arabidopsis thaliana. Heterocycles 69, 295–301.
Napolitano, A., Carbone, V., Saggese, P., Takagaki, K., and Pizza, C. (2007). Novel galactolipids from the leaves of Ipomoea batatas L.: characterization by liquid chromatography coupled with electrospray ionization-quadrupole time-of-flight tandem mass spectrometry. J. Agric. Food Chem. 55, 10289–10297.
Ohashi, T., Ito, Y., Okada, M., and Sakagami, Y. (2005). Isolation and stomatal opening activity of two oxylipins from Ipomoea tricolor. Bioorg. Med. Chem. Lett. 15, 263–265.
op den Camp, R. G. L., Przybyla, D., Ochsenbein, C., Laloi, C., Kim, C., Danon, A., Wagner, D., Hideg, E., Göbel, C., Feussner, I., Nater, M., and Apel, K. (2003). Rapid induction of distinct stress responses after the release of singlet oxygen in Arabidopsis. Plant Cell 15, 2320–2332.
Pacetti, D., Boselli, E., Lucci, P., and Frega, N. G. (2007). Simultaneous analysis of glycolipids and phospholids molecular species in avocado (Persea americana Mill) fruit. J. Chromatogr. A 1150, 241–251.
Pinot, E., Guy, A., Fournial, A., Balas, L., Rossi, J.-C., and Durand, T. (2008). Total synthesis of the four enantiomerically pure diasteroisomers of the phytoprostanes E1 type II and of the 15-E2t-isoprostanes. J. Org. Chem. 73, 3063–3069.
Stelmach, B. A., Müller, A., Hennig, P., Gebhardt, S., Schubert-Zsilavecz, M., and Weiler, E. W. (2001). A novel class of oxylipins, sn1-O-(12-Oxophytodienoyl)-sn2-O-(hexadecatrienoyl)-monogalactosyl diglyceride, from Arabidopsis thaliana. J. Biol. Chem. 276, 12832–12838.
Stintzi, A., Weber, H., Reymond, P., Browse, J., and Farmer, E. E. (2001). Plant defense in the absence of jasmonic acid: the role of cyclopentenones. Proc. Natl. Acad. Sci. U.S.A. 98, 12837–12842.
Wang, W., Liu, Z., Ma, L., Hao, C., Liu, S., Voinov, V. G., and Kalinovskaya, N. I. (1999). Electrospray ionization multiple-stage tandem mass spectrometric analysis of diglycosyldiacylglycerol glycolipids from the bacteria Bacillus pumilus. Rapid Commun. Mass Spectrom. 13, 1189–1196.
Welti, R., Li, W., Li, M., Sang, Y., Biesiada, H., Zhou, H. E., Rajashekar, C. B., Williams, T. D., and Wang, X. (2002). Profiling membrane lipids in plant stress responses. Role of phospholipase D alpha in freezing-induced lipid changes in Arabidopsis. J. Biol. Chem. 277, 31994–32002.
Welti, R., Wang, X., and Williams, T. D. (2003). Electrospray ionization tandem mass spectrometry scan modes for plant chloroplast lipids. Anal. Biochem. 314, 149–152.
Appendix
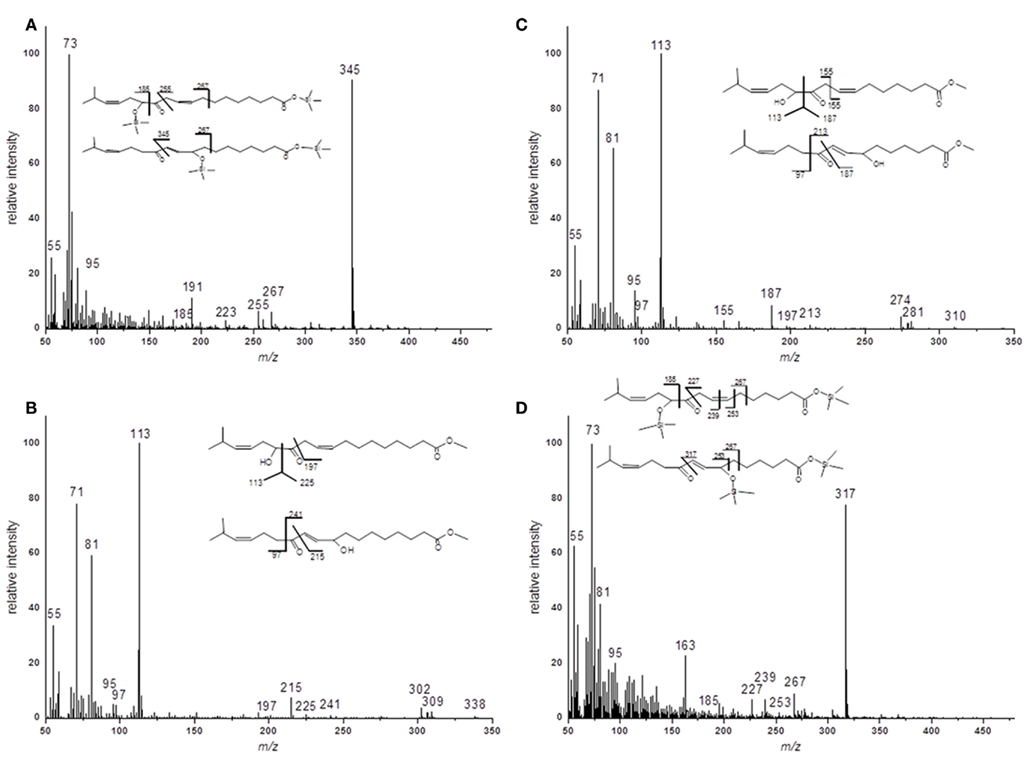
Figure A1. EI–MS spectra of methyl ester and trimethylsilyl ester/trimethylsilyl ether derivatives of methyl-branched ketols. (A) Racemic methyl ester of 17-methyl-α-/γ-ketol-18:2 (B) Trimethylsilyl ester/trimethylsilyl ether of 17-methyl-α-/γ-ketol-18:2 (C) Racemic methyl ester of 17-methyl-α-/γ-ketol-16:2 (D) Trimethylsilyl ester/trimethylsilyl ether of 17-methyl-α-/γ-ketol-16:2.
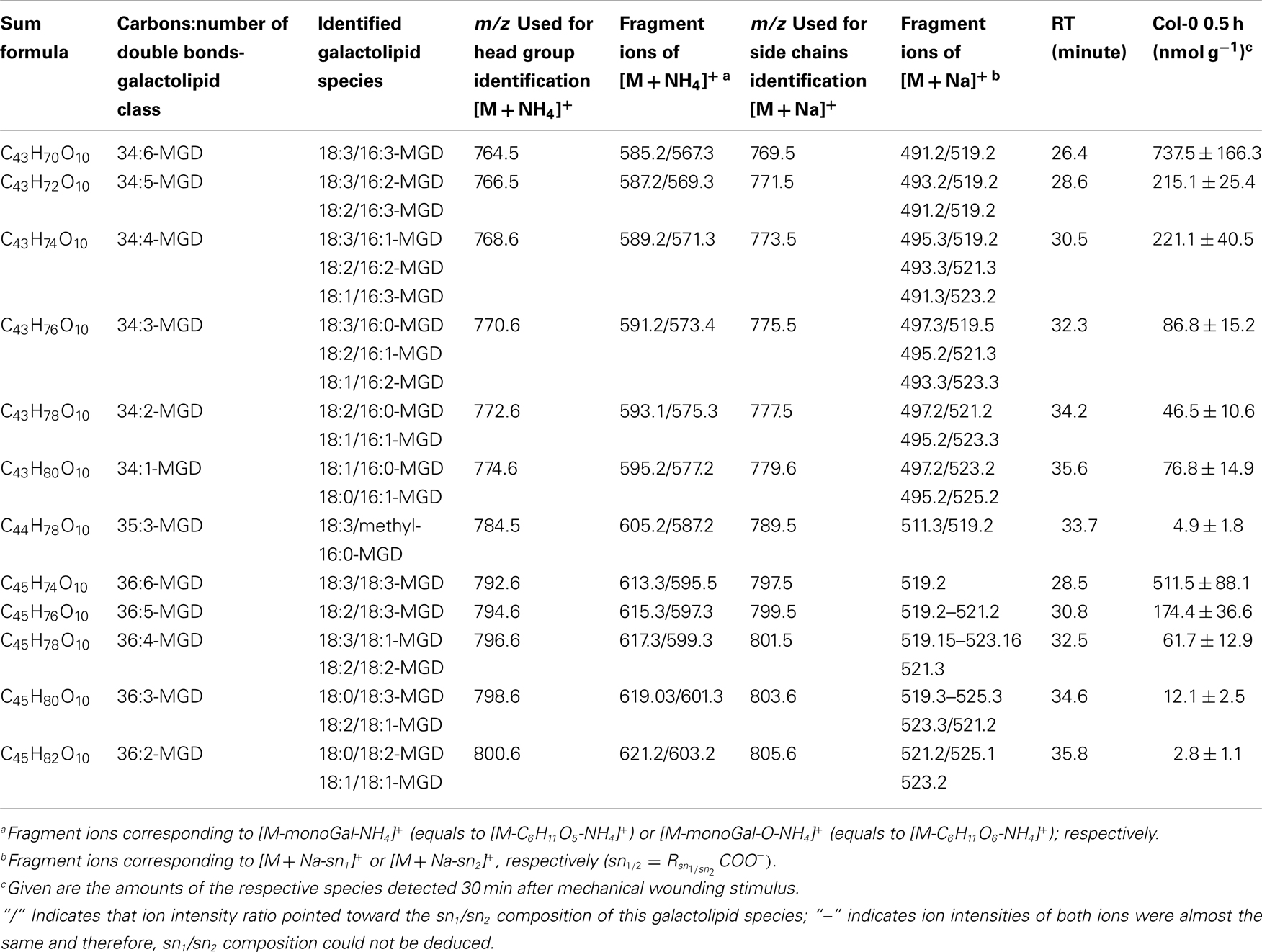
Table A1. Non-oxidized MGD species identified in wounded A. thaliana leaves.
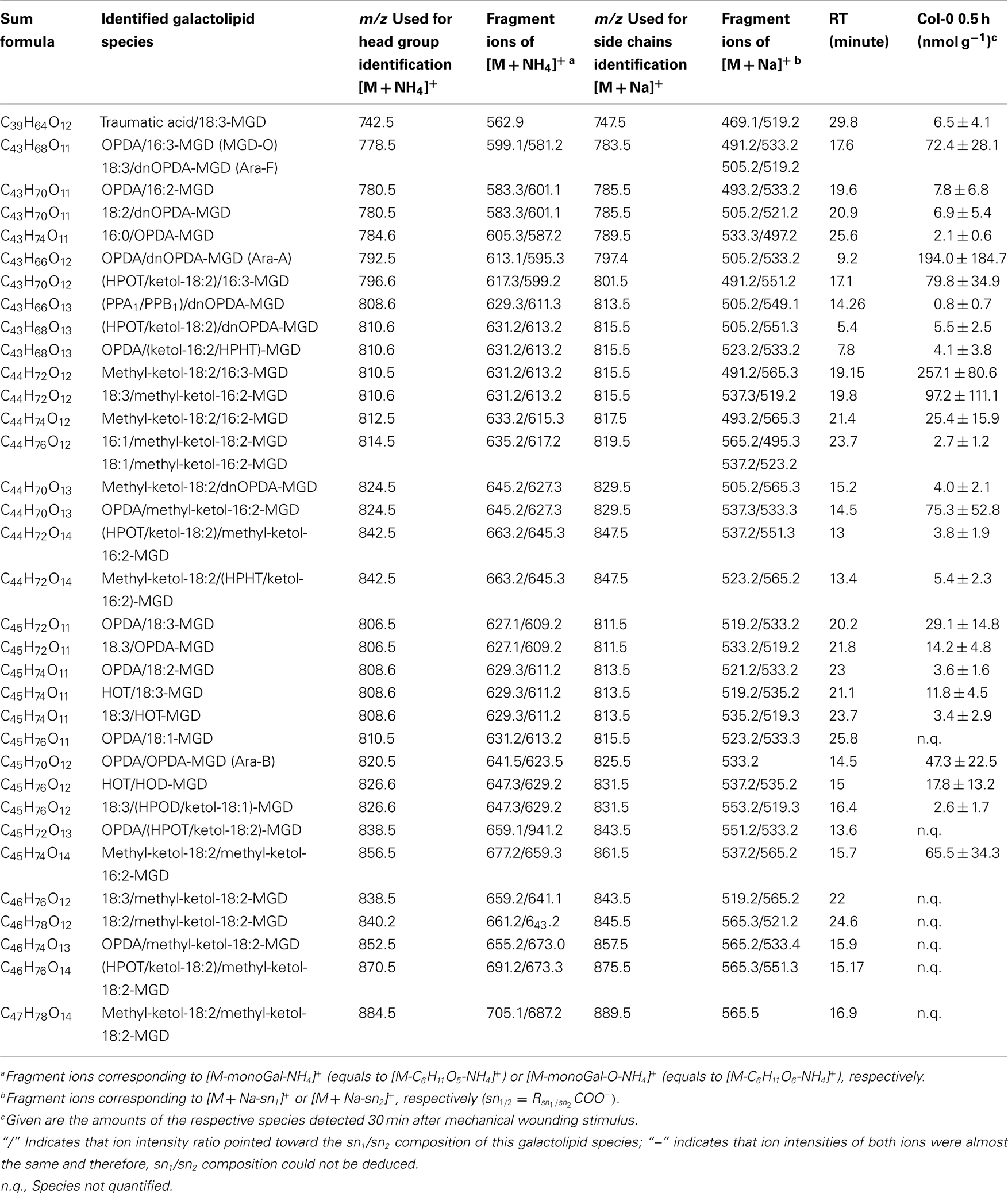
Table A2. Oxidized MGD species identified in wounded A. thaliana leaves.
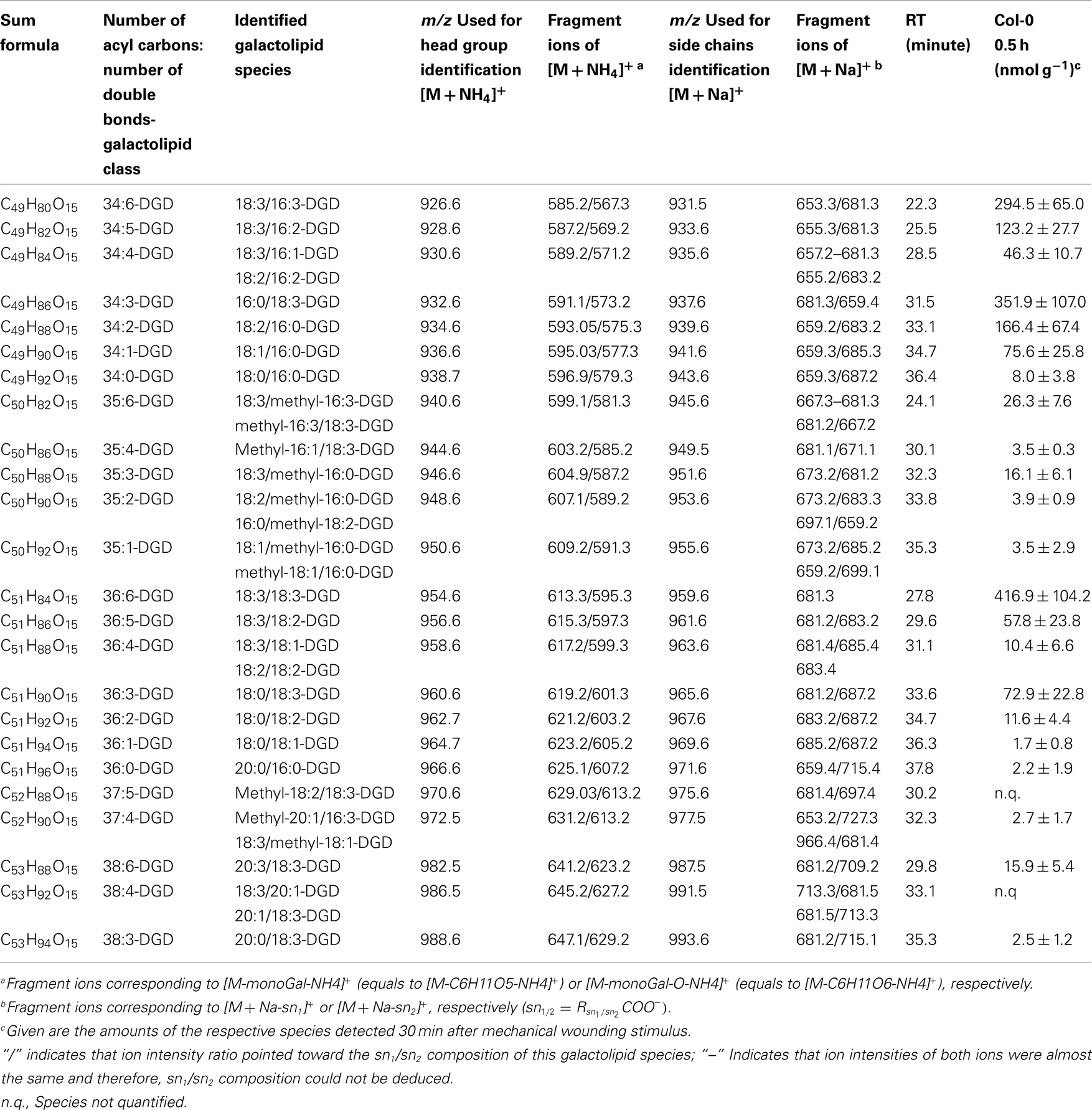
Table A3. Non-oxidized DGD species identified in wounded A. thaliana leaves.
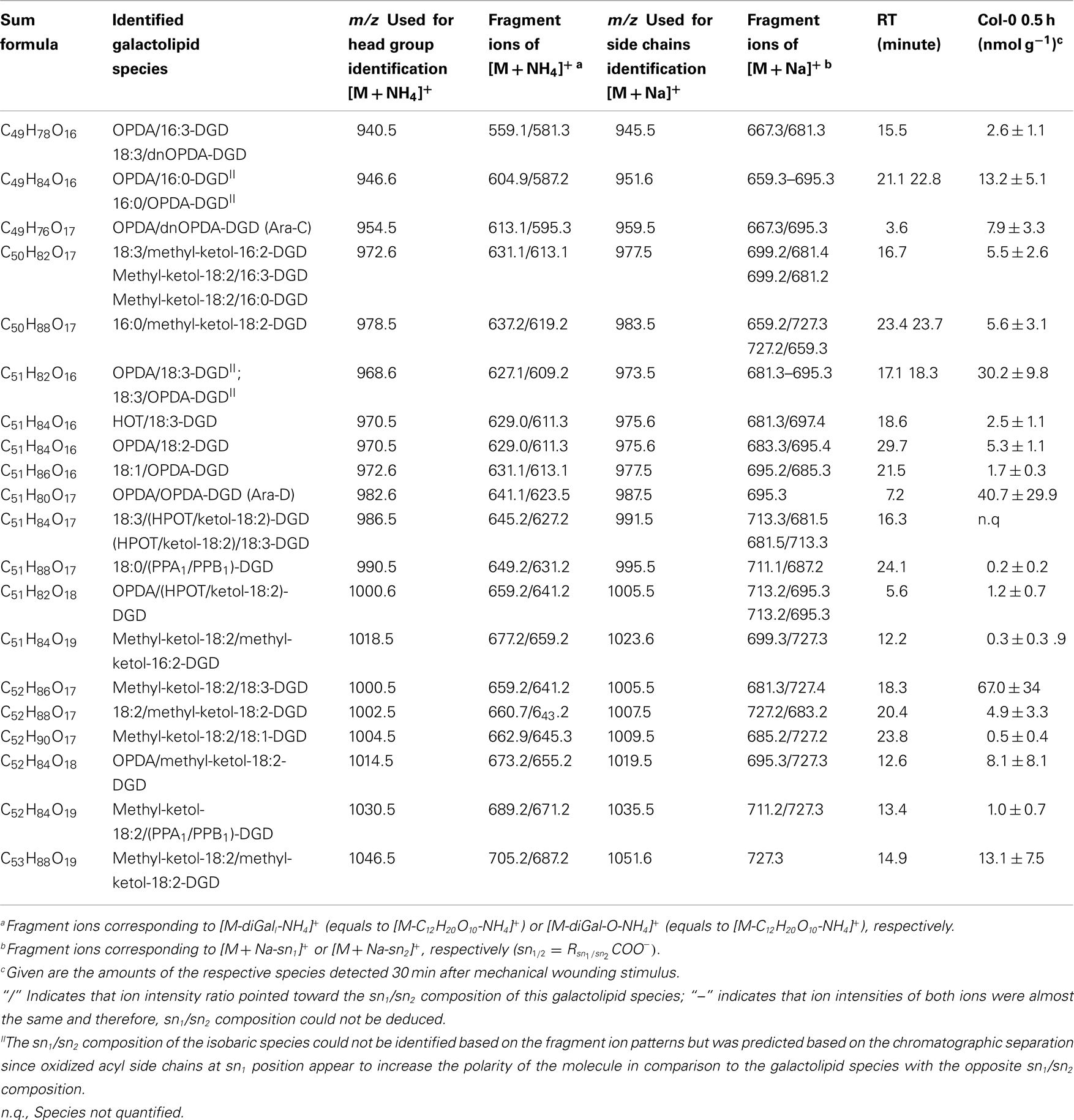
Table A4. Oxidized DGD species identified in wounded A. thaliana leaves.
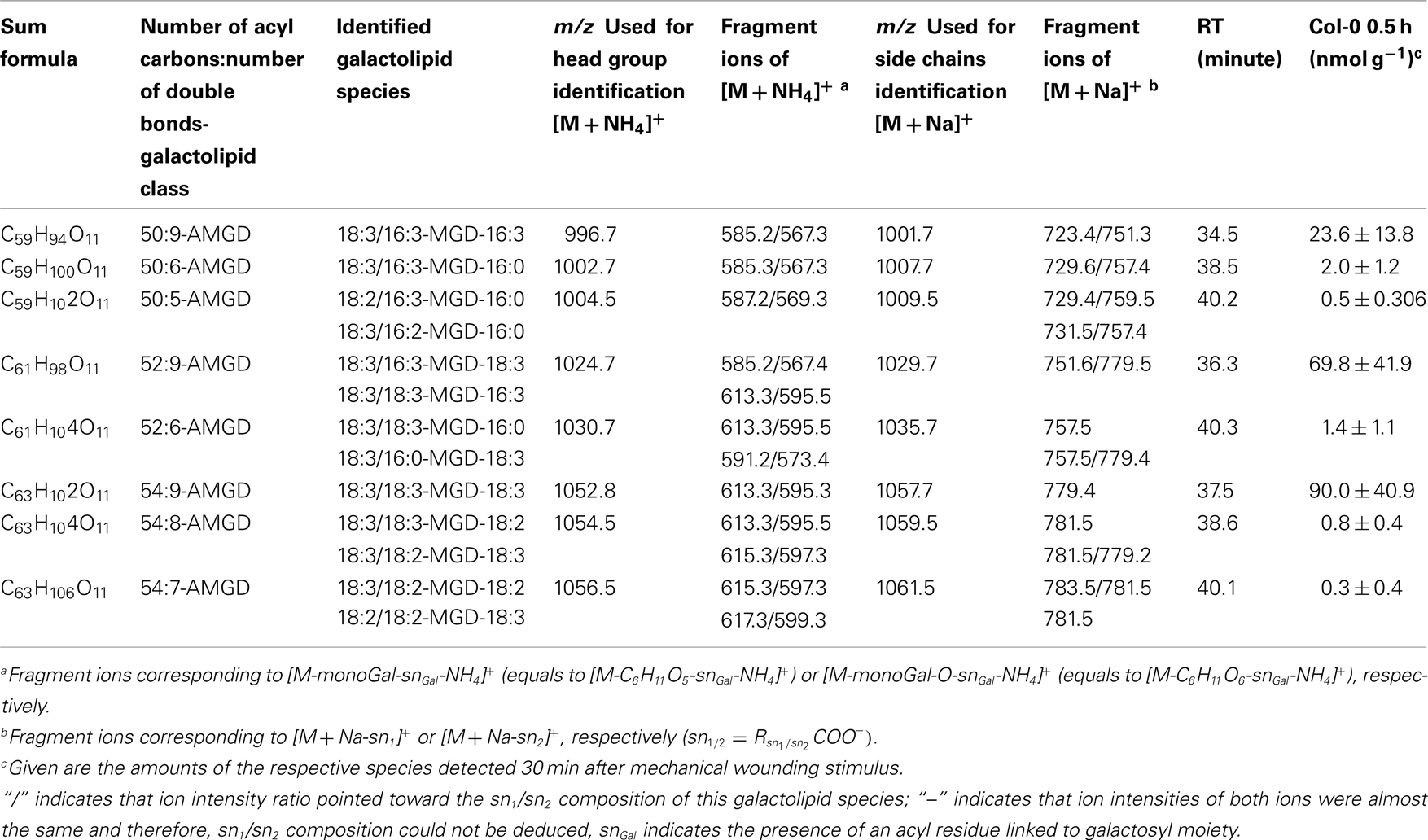
Table A5. Non-oxidized acylated MGD species identified in wounded A. thaliana leaves.
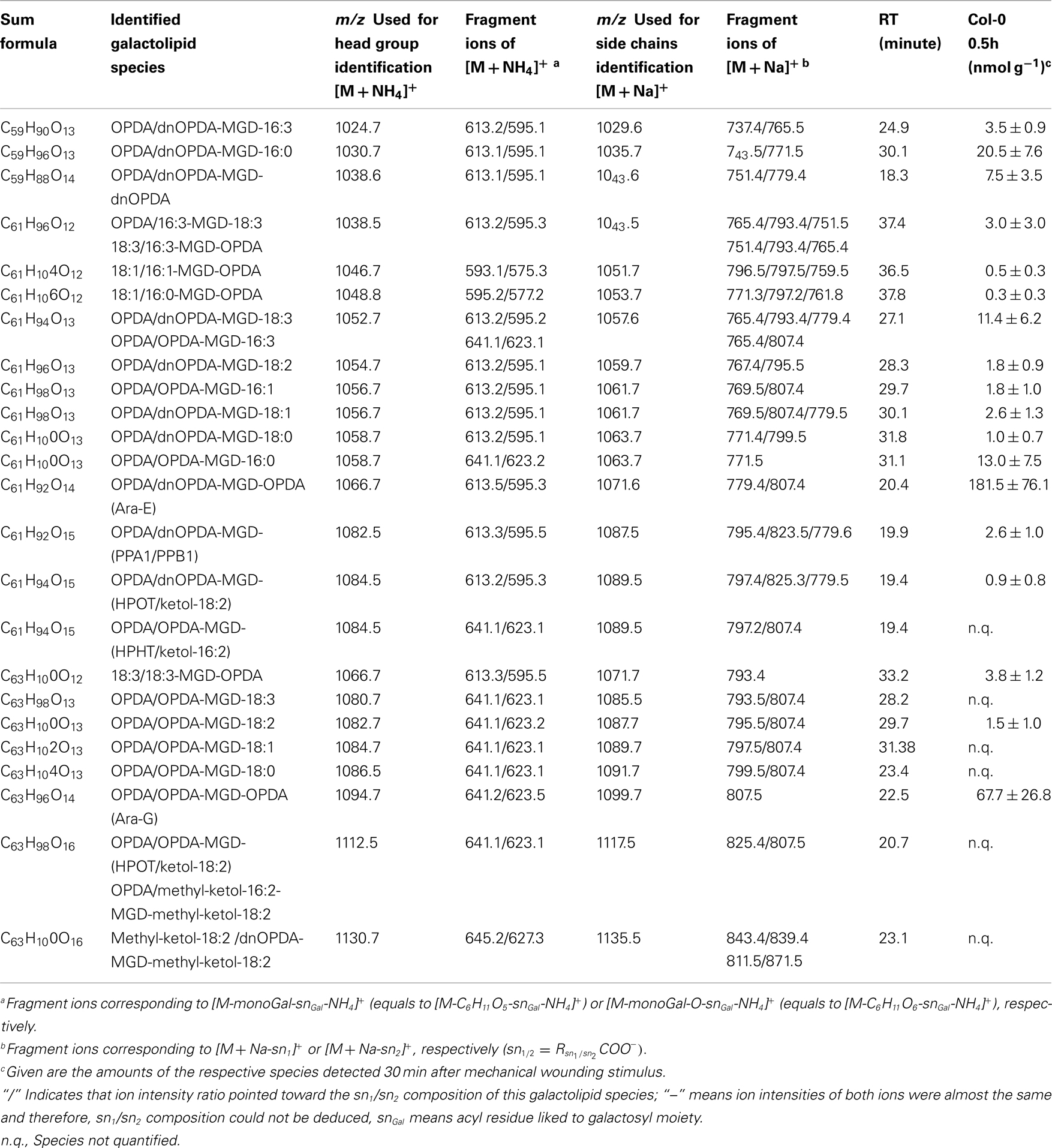
Table A6. Oxidized acylated MGD species identified in wounded A. thaliana leaves.

Table A7. Oxidized acylated DGD species identified in wounded A. thaliana leaves.
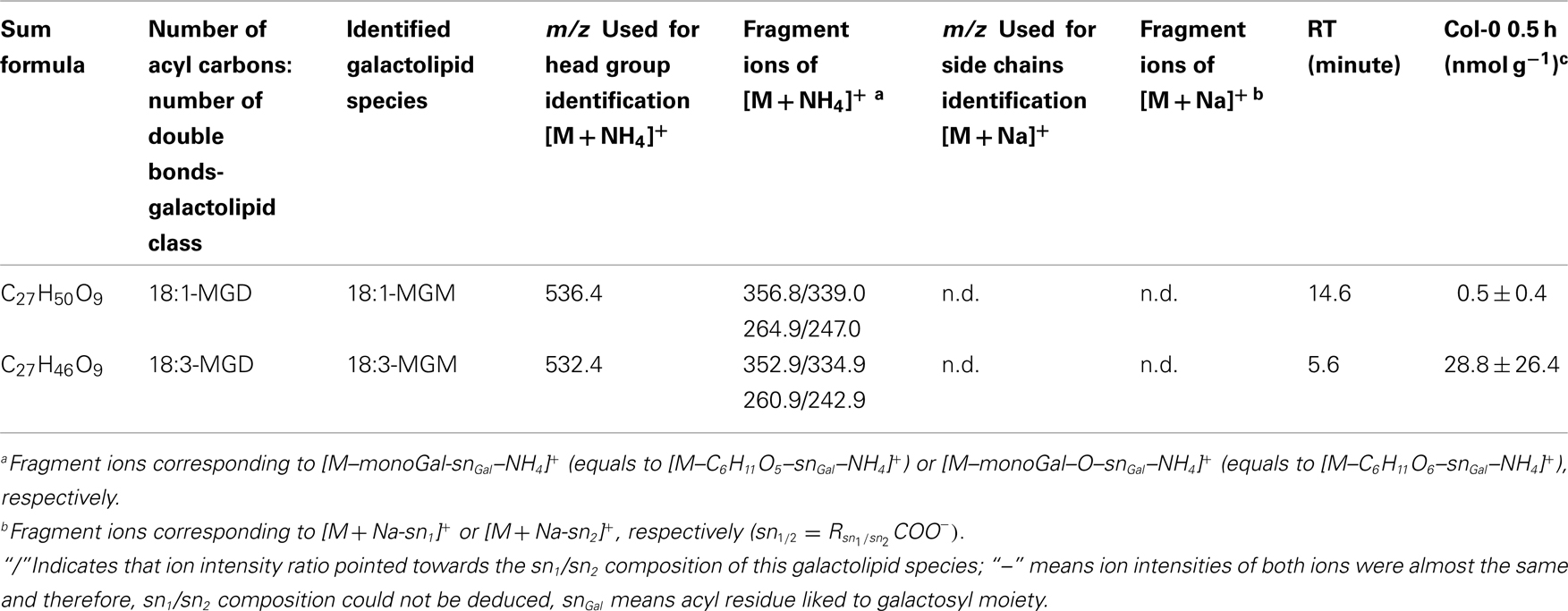
Table A8. Non-oxidized MGM species identified in wounded A. thaliana leaves.
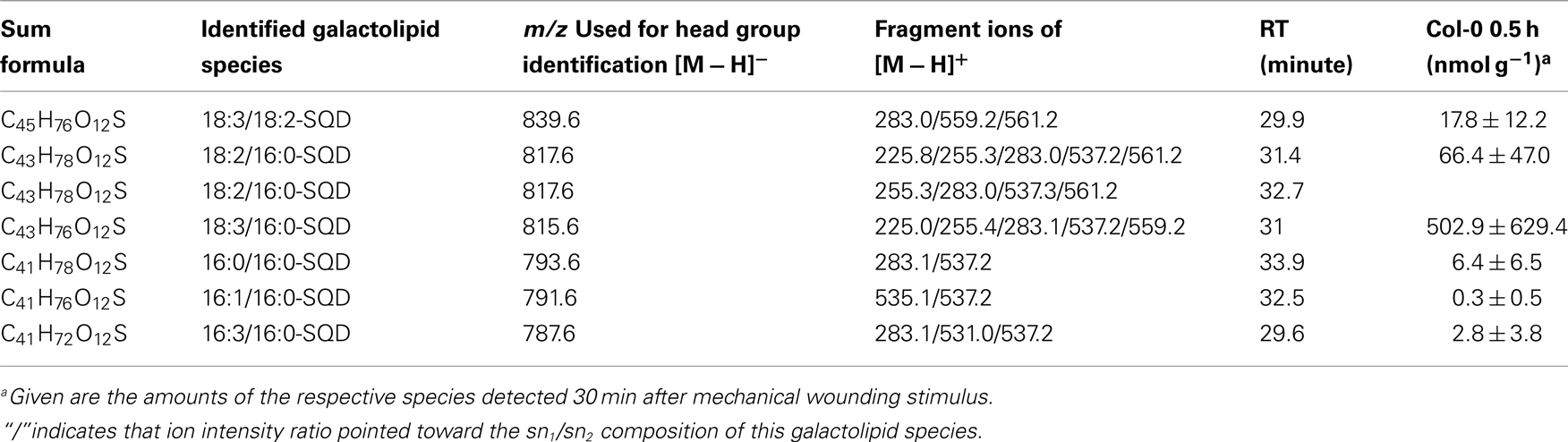
Table A9. Non-oxidized SQD species identified in wounded A. thaliana leaves.

Table A10. Oxidized SQD species identified in wounded A. thaliana leaves.
Keywords: arabidopsides, esterified oxylipins, HPLC/MS, lipid peroxidation
Citation: Ibrahim A, Schütz A-L, Galano J-M, Herrfurth C, Feussner K, Durand T, Brodhun F and Feussner I (2011) The alphabet of galactolipids in Arabidopsis thaliana. Front. Plant Sci. 2:95. doi: 10.3389/fpls.2011.00095
Received: 21 October 2011; Accepted: 24 November 2011;
Published online: 12 December 2011.
Edited by:
Kent D. Chapman, University of North Texas, USAReviewed by:
Ruth Welti, Kansas State University, USAGustavo Bonaventure, Max Planck Institute for Chemical Ecology, Germany
Copyright: © 2011 Ibrahim, Schütz, Galano, Herrfurth, Feussner, Durand, Brodhun and Feussner. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ivo Feussner, Department of Plant Biochemistry, Albrecht von Haller-Institute of Plant Sciences, Georg August University, Justus-von-Liebig-Weg 11, D-37075 Göttingen, Germany. e-mail: ifeussn@uni-goettingen.de