
- Molecular and Cellular Biology, University of California, Davis, CA, USA
Many introns significantly increase gene expression through a process termed intron-mediated enhancement (IME). Introns exist in the transcribed DNA and the nascent RNA, and could affect expression from either location. To determine which is more relevant to IME, hybrid introns were constructed that contain sequences from stimulating Arabidopsis thaliana introns either in their normal orientation or as the reverse complement. Both ends of each intron are from the non-stimulatory COR15a intron in their normal orientation to allow splicing. The inversions create major alterations to the sequence of the transcribed RNA with relatively minor changes to the DNA structure. Introns containing portions of either the UBQ10 or ATPK1 intron increased expression to a similar degree regardless of orientation. Also, computational predictions of IME improve when both intron strands are considered. These findings are more consistent with models of IME that act at the level of DNA rather than RNA.
Introduction
Many introns are important regulators of gene expression (Fiume et al., 2004; Curi et al., 2005; Rose, 2008; Weise et al., 2008). Removing the introns from a gene often significantly reduces its expression, while the expression of an intronless reporter such as a bacterial gene can be increased substantially by adding an intron (Callis et al., 1987). The difference in expression between an intron-containing and intronless version of the same gene can be 10-fold or more, depending on the intron, and is visible at the level of mRNA accumulation (Rose and Last, 1997; Lu and Cullen, 2003; Nott et al., 2003). The increase in expression caused by introns has been termed intron-mediated enhancement (IME; Mascarenhas et al., 1990) to distinguish it from the effects of more familiar regulatory elements such as enhancers or promoters. IME may reflect a fundamental feature of eukaryotic gene expression because it has been observed in diverse phyla including plants (Vasil et al., 1989), mammals (Palmiter et al., 1991), insects (Zieler and Huynh, 2002), fungi (Lugones et al., 1999), and nematodes (Okkema et al., 1993). Many introns have no discernable effect on expression (Rose, 2002), and even though introns that do stimulate expression could do so in a variety of ways, the characteristics shared by a majority of the introns studied in a number of organisms suggest a common mechanism. The results from experiments in plants described below are consistent with what has been observed in mammals and yeast (Furger et al., 2002; Nott et al., 2003).
The differences between introns and classical transcriptional enhancer elements clearly demonstrate that they must stimulate expression by different mechanisms. While enhancers can elevate expression from upstream or downstream locations that can be considerable distances from the gene (Williamson et al., 2011), most plant introns are only able to affect expression when within transcribed sequences and near the 5′ end of a gene (Callis et al., 1987; Mascarenhas et al., 1990; Clancy et al., 1994; Rose, 2004). A second difference between enhancers and introns is that the boundaries of an enhancer can be defined by deletions (Twell et al., 1991; Itzhaki et al., 1994), indicating the involvement of discrete sequences. In contrast, deletion analysis has been largely unsuccessful in locating the sequences required for IME because the signals are dispersed throughout stimulatory introns and have an additive effect (Rose et al., 2008). A third indication that introns are unlike transcriptional enhancers is that introns generate at most a small increase in the signal in nuclear run-on transcription assays that is insufficient to account for the large change in mRNA abundance they cause (Dean et al., 1989; Rose and Last, 1997; Rose and Beliakoff, 2000). It remains unclear how introns can elevate the steady-state level of mRNA without synthesizing it more quickly, as introns apparently do not increase mRNA stability either (Rethmeier et al., 1997). Introns that contain enhancer elements or promoters have been described (Deyholos and Sieburth, 2000; Vitale et al., 2003; Morello et al., 2006) but these appear less common in plants than they are in Drosophila and mammals (Meredith and Storti, 1993; Hess et al., 2007; Visel et al., 2009).
While the above observations illustrate that introns are operationally different than enhancers, the mechanism of IME remains obscure. The need for introns to be in transcribed sequences near the start of a gene suggests that introns may act during transcription to stimulate elongation, rendering the transcription machinery more likely to traverse the entire gene so that 3′ end processing can generate a stable transcript. Introns could potentially affect the activity of factors known to regulate transcriptional elongation such as the elongator complex (Svejstrup, 2007; Nelissen et al., 2010), FACT (Orphanides et al., 1999), PAF1c (Nagaike et al., 2011), NELF, or DSIF (Wu et al., 2003). The activity of RNA polymerase II could be affected by introns in two ways; either in the form of the DNA that is being transcribed, or as the newly made RNA prior to, or during, splicing. Passage of the transcription machinery over DNA and the emergence of the corresponding RNA sequence occur virtually simultaneously, and the two nucleic acids are initially in close proximity to each other and both contact the polymerase. In addition, many of the enzymes that catalyze the different reactions involved in gene expression interact physically and can influence each other directly and indirectly, creating complicated interconnections that affect the final amount of product made (Pandit et al., 2008; Moore and Proudfoot, 2009; Dahan et al., 2011). Therefore, a direct biochemical analysis of the roles of introns in gene expression will likely prove challenging. However, it is possible to use indirect tests to assess the level at which IME operates.
One such indirect approach pertinent to IME is to ask if introns must be spliced in order to affect expression. Evidence for the involvement of the splicing machinery would support an RNA-based model of IME. Changes to the RNA that occur after transcription may be irrelevant if the stimulation takes place at the DNA level. This is an oversimplification because the physical interactions between the transcription and splicing machineries provide a way for these processes to be coupled and thus affect each other (Alexander et al., 2010; Niu and Yang, 2011). An additional complication is that the intron sequences that are retained in the RNA when splicing is prevented are likely to contain start and stop codons; these can interfere with translation and thereby have secondary effects on mRNA abundance (Harel-Sharvit et al., 2010). An ATG in a retained intron 5′ of the start codon of the reporter gene could interfere with expression due to the inhibitory effects of short upstream open reading frames, cause translation to initiate in the wrong reading frame, or add extraneous amino acids to the reporter enzyme.
Three studies in which these difficulties were avoided by altering intron sequences to preserve the reading frame of the adjacent exons came to different conclusions about the need for splicing. Point mutations of essential nucleotides at the 5′ splice site reduce the ability of a derivative of the maize Sh1 intron to boost mRNA accumulation from 45- to 2.8-fold (Clancy and Hannah, 2002), indicating that splicing is necessary for IME. Similarly, modifications of the leader intron of the Arabidopsis AtMHX gene that eliminate splicing cause the stimulatory effect of that intron to drop from 270- to 5-fold (Akua et al., 2010). In contrast, the Arabidopsis TRP1 first intron, which normally elevates mRNA accumulation fourfold, still boosts expression 2.6- to 4.5-fold when splicing is prevented in several ways, suggesting that splicing is not required for IME (Rose and Beliakoff, 2000; Rose, 2002). These divergent conclusions may be due to the widely different stimulating abilities of the starting introns; the unspliceable derivatives of each intron increase expression 2.6- to 5-fold, a drastic decline that suggests the need for splicing only for the more strongly enhancing introns. Splicing is clearly not sufficient for IME because several efficiently spliced introns have no effect on expression levels (Rose, 2002).
A second indirect way to differentiate between DNA and RNA-based models of IME is to test the effects of inverting the orientation of the intron. A reversed intron will produce a very different RNA sequence but many features of the double-stranded DNA (such as secondary structure, protein binding sites, ease of strand separation, etc.) will remain unchanged. Several groups have reported that introns fail to increase expression when their orientation is reversed (Callis et al., 1987; Mascarenhas et al., 1990; Clancy et al., 1994; Jeong et al., 2006). However, introns only boost gene expression from within transcribed sequences, but reversed introns in transcribed sequences cannot be spliced due to the loss of essential sequences at both the 5′ and 3′ splice sites (GT…AG becomes CT…AC). Therefore, inverting an entire intron prevents splicing, and the retained sequences can inhibit translation and have secondary effects on mRNA abundance as just described, thereby negating any positive effect of the intron.
Here we report the construction of reversed intron sequences that do not interfere with splicing in Arabidopsis. All intron structures known to be required for splicing, including the conserved sequences at the branch point and 5′ and 3′ splice sites, are located near the ends of introns, as demonstrated by the efficient splicing of introns in which large regions are deleted from the interior (Luehrsen and Walbot, 1994). Previously we showed that hybrid introns composed of sequences from the middle of a stimulatory intron between both ends of a non-stimulatory intron are efficiently spliced and elevate expression (Rose et al., 2008). Therefore we constructed additional hybrids in which the non-stimulating intron ends were preserved in their normal orientation, while sequences from either of two stimulating introns were inserted between them in either their natural orientation or as the reverse complement. For both pairs of hybrid introns, enhancement was similar regardless of the orientation of the stimulating sequences. A computational analysis also supports the idea that the signals involved in IME are found on both strands of intronic DNA. These results show that the magnitude of enhancement caused by an enhancing intron is not a function of the primary sequence of the transcribed strand. Instead the data favor a model in which IME is caused by the presence of signals in the DNA that exert their effect while the intron is being transcribed.
Results
To test the enhancing ability of intron sequences in reverse orientation, a 184-nt region from the middle of UBQ10 intron 1 was inserted as a BamHI–BglII fragment in either orientation between both ends of COR15a intron 1 (Figure 1). The COR15a sequences were maintained in their natural orientation to promote efficient splicing even when the sequences between them were inverted. To confirm that any effect on expression is due to the inserted sequences, a control intron composed of just the COR15a ends also was constructed. These introns will be referred to as CO > UB > CO, CO < UB < CO, and COΔCO, with CO and UB denoting sequences derived from the COR15a and UBQ10 introns, the Δ representing the sequences deleted from the middle of the COR15a intron, and > and < indicating natural and reverse orientations of the UBQ10 sequences respectively. Each intron was placed at the same location in a reporter gene consisting of a translational fusion between the Arabidopsis TRP1 gene (At5g17990) and the Escherichia coli uidA (GUS) gene, which encodes a β-glucuronidase. Genomic DNA blots were used to identify at least two single-copy transgenic Arabidopsis lines for each construct. Transgene expression in homozygous plants was measured by enzyme assay and RNA gel blots and is presented as the amount relative to the intronless control pAR281 (Rose and Beliakoff, 2000). Splicing efficiency was estimated by RT-PCR using primers that flank the intron. The mature mRNAs in all lines will be identical if the introns are correctly spliced.
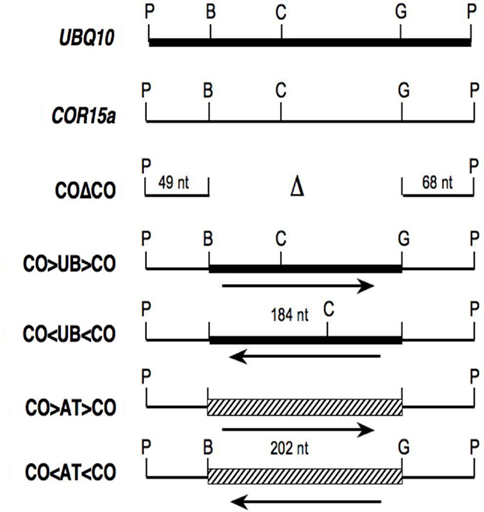
Figure 1. Structure of hybrid introns. Thick lines, thin lines, and the hatched box represent sequences from UBQ10 intron 1, COR15a intron 1, and ATPK1 intron 1 respectively. The letters above vertical lines show the positions of restriction sites for PstI (P), BamHI (B), BclI (C), and BglII (G), while unmarked lines indicate fusions between BamHI and BglII compatible ends.
As shown in Figure 2 and Table 1, the CO > UB > CO intron stimulates TRP1:GUS mRNA accumulation 7.5 ± 0.8-fold relative to the intronless control. The UBQ10 sequences that are present in the CO > UB > CO intron are composed of two regions that were each previously shown to stimulate mRNA accumulation approximately fourfold (Rose et al., 2008), illustrating the additive nature of their effects. The increased expression must be due to the UBQ10 sequences in the hybrid intron because the COΔCO intron has virtually no effect on mRNA accumulation (Table 1; Figure 2A), consistent with previous findings that the entire COR15a intron increases mRNA accumulation less than twofold (Rose et al., 2008). The effect of the CO < UB < CO intron on mRNA accumulation (7.2 ± 1.2-fold) was indistinguishable from that of the CO > UB > CO intron, indicating that the UBQ10 sequences increased expression to the same degree regardless of orientation. Both hybrid introns were efficiently spliced, as RT-PCR product from unspliced TRP1:GUS mRNA in these lines was not detected (Figure 2C).
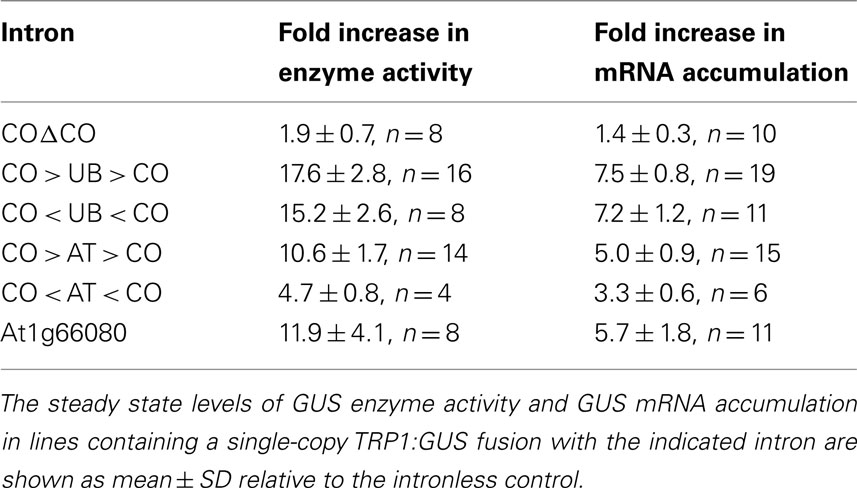
Table 1. The effect of different introns on TRP1:GUS expression.
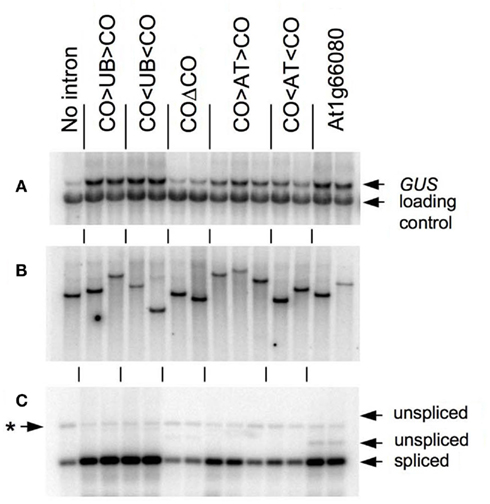
Figure 2. Analysis of TRP1:GUS transgenic lines. (A) RNA gel blot of total RNA from lines transgenic for TRP1:GUS fusions containing the indicated intron, probed with GUS and a loading control. (B) Genomic DNA blot digested with PstI and probed with GUS. (C) RT-PCR analysis of intron splicing with arrows indicating the predicted positions of products derived from spliced and unspliced mRNA as determined by amplification of genomic DNA with the same primers. The asterisk marks a non-specific amplification product. The blots in (A–C) are vertically aligned so that lanes in the same position in each of the three panels are from the same independent transgenic line.
One possible explanation for the ability of the CO < UB < CO intron to stimulate expression is that the UBQ10 intron fragment used may fortuitously contain sequences that contain IME signals on the opposite strand. To test the possibility that the UBQ10 sequences are unique in their ability to stimulate expression when reversed, a 202-nt fragment of the ATPK1 first intron was inserted between the COR15a intron ends to make the hybrid introns CO > AT > CO and CO < AT < CO. The increase in mRNA accumulation caused by the CO < AT < CO hybrid intron was slightly less than that caused by the CO > AT > CO intron (3.3 ± 0.6- and 5.0 ± 0.9-fold, respectively, Figure 2A and Table 1), although both stimulated expression by more than did the COΔCO control intron. The reduced enhancement relative to the CO > UB > CO and CO < UB < CO introns was not due to splicing problems as both the CO > AT > CO and CO < AT < CO introns were efficiently spliced (Figure 2C). Therefore, the ability to stimulate expression in either orientation was not limited to the UBQ10 intron sequences tested.
While the sequences involved in IME remain unknown, there is a strong correlation between the ability of an intron to stimulate expression and its score from the IMEter algorithm, and this can be used to predict which introns will stimulate expression (Rose et al., 2008). IMEter scores express the degree to which the oligomer composition of an individual sequence resembles that of all promoter-proximal introns. A revised version (IMEter 2.0) weights sequences based on their distance from the start of the intron and counts only those oligomers that make a positive contribution to IMEter score (Parra et al., 2011).
The observation that intron sequences can increase expression in either orientation suggests that introns that have a high IMEter score on their non-transcribed strand might also stimulate expression. To test this, the IMEter scores for both strands of 30,330 Arabidopsis introns were calculated. Approximately two-thirds (20,411) of the transcribed intron sequences had a negative score, and approximately 5% of these (1050) represented instances where the corresponding sequence from the reverse strand had a positive IMEter score. We chose the first intron from the At1g66080 gene for further study because it has the highest IMEter score on its reverse strand of all 20,411 introns with negative scores on the transcribed strand. This intron would not be expected to affect expression on the basis of its low IMEter score (−3) in the normal orientation, but its reverse complement has a very high IMEter score (35) that would predict enhancement (IMEter 2.0 scores are 2.9 and 30 respectively). This entire intron was inserted in its natural orientation into the TRP1:GUS reporter gene, whose expression and splicing efficiency was measured in single-copy transgenic plants as above. This intron increased mRNA accumulation 5.7 ± 1.8-fold relative to the intronless control (Figure 2). A small amount of RT-PCR product derived from unspliced mRNA was detected that constitutes approximately 5% of all RT-PCR products (Figure 2C), indicating that splicing is nearly complete. The stimulation caused by this intron was much more consistent with the IMEter score of the reverse complement than that of the natural intron. This increase in expression was substantially higher than that from any of the other six introns previously tested that have negative IMEter scores on both strands, none of which increases mRNA accumulation more than twofold (Rose et al., 2008). This suggests that high IMEter scores on either strand can be used to identify introns that are capable of stimulating expression.
To determine if considering the sequence of both strands can improve the performance of the IMEter, 1080 permutations of the IMEter and IMEter 2.0 were tested with different settings for the variable parameters of oligomer size, the distance values for assigning introns as promoter-proximal or distal, and whether or not the sequence of the reverse complement was included in the calculations. Performance was evaluated as the R2 value of the best fit line comparing IMEter scores with the measured effect on TRP1:GUS mRNA accumulation of all 15 wild-type Arabidopsis introns for which data are available. All but one of the 25 parameter sets that give the highest R2 values utilize the sequence of the reverse complement, as do 48 of the top 50 and 88 of the top 100 parameter sets. There are cases where using intron sequences from the plus strand alone yielded better performance than using both strands. For example, of the 45 possible combinations of thresholds tested, 9 gave better IMEter performance using plus strand only when the K-mer size was 4 or 5, although using both strands gave better performance in all cases when the K-mer size is 3. An example of the difference in performance is presented in Table 2. As was seen with other parameter sets, the benefit of using both strands as well as overall performance declined at larger K-mer sizes. The correlation between IMEter scores and the known effects on expression of numerous introns further supports the idea that the signals involved in IME can be found on both strands of intronic DNA.
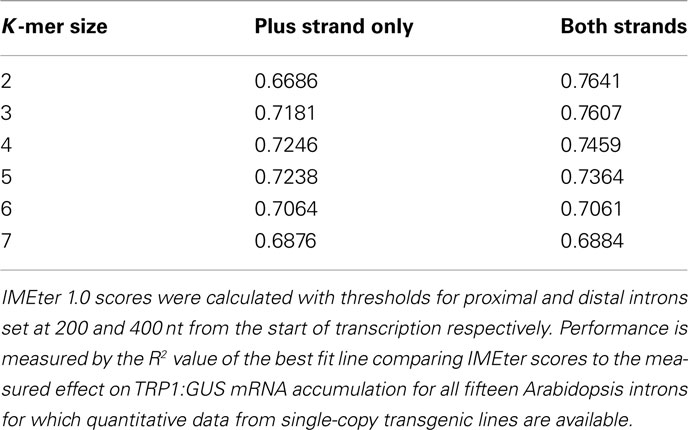
Table 2. A sample comparison of IMEter performance with and without minus strand sequence.
Even though the primary sequence of the transcribed strand changes substantially when the orientation of an intron is reversed, it is possible that important features of RNA sequence or secondary structure with relevance to IME could be preserved. For example, IME could be caused by factors that bind to palindromic sequences or stem loop structures in the RNA that would also be present when the reverse complement of an intron is transcribed. Potential secondary structures and palindromic sequences were sought in individual introns whose effects on expression have been quantified. In additional genome-wide analyses, introns with high IMEter scores and thus large predicted effects on expression were compared to low-scoring and presumably non-stimulating introns.
Palindromes within individual introns were visualized by creating dot plots in which every mark indicates the locations of matches of 5 nt in length between the sequence of an intron and its reverse complement. The lack of long diagonal lines in Figure 3 indicates that there are no extensive regions of palindromic sequence in either stimulatory introns such as the UBQ10 or ATPK1 introns or the non-stimulating COR15a intron. To explore possible connections between palindrome frequency and effect on expression, a Perl script was written that compares the frequency of short palindromes and IMEter scores of all the introns in the Arabidopsis genome; palindromes were required to have pairs of identical sequences of 4–10 nt, separated by 0–5 nt of any sequence. The 20% of introns in the genome with the highest IMEter scores had on average 18.7 palindromes per kilobase, slightly lower than the 20.7 palindromes per kilobase found in the 20% of introns with the lowest IMEter scores. Thus, the sequences that contribute to high IMEter scores and possibly cause IME are unlikely to be simple palindromes.

Figure 3. Dot plots of introns. The sequence of each intron (x axis) compared to its reverse complement (y axis), with dots indicating the position of each match of 5 nt in length.
To test the possibility that stem loop structures could be responsible for the different effects on expression of stimulatory and non-stimulatory introns, the potential of the UBQ10, COR15a, ATPK1, and the hybrid introns to form stable secondary structures was predicted using the RNA fold application of the Vienna package (Gruber et al., 2008). The overall free energy of the predicted secondary structure was similar for all introns of a comparable length and did not correlate with their effect on expression. Reversing the UBQ10 or ATPK1 sequences in the hybrid introns dramatically changed their predicted secondary structures (Figure 4). Only one predicted stem loop in the UBQ10 intron is conserved in the CO > UB > CO intron, but this structure is absent from the CO < UB < CO intron predicted structure. Conversely, the only region of predicted base-pairing that is conserved in either pair of hybrids is one small stem loop in the CO > UB > CO and CO < UB < CO introns that is predicted to be unpaired in the native UBQ10 intron. None of the predicted secondary structures can account for the observed abilities of these introns to stimulate expression.
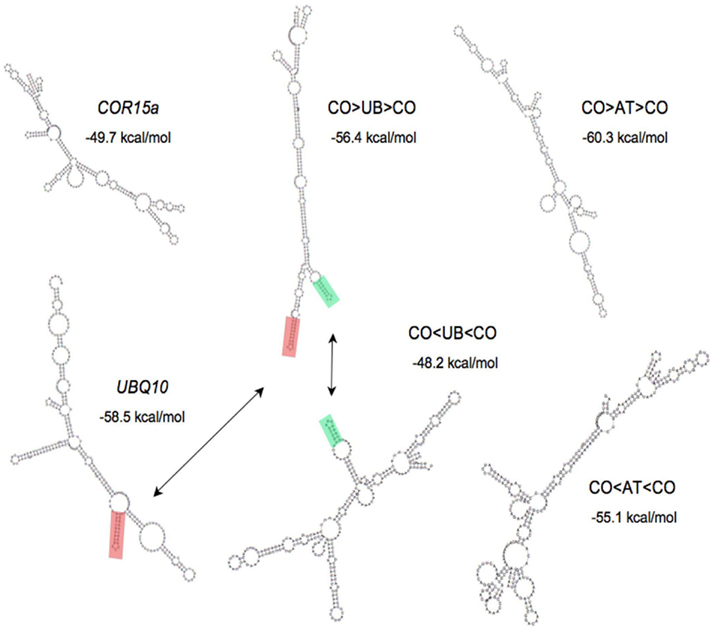
Figure 4. Predicted secondary structure of introns. The minimum free energy structure of introns as determined by the Vienna package is shown. Stem loop structures conserved between introns are highlighted in red and green.
Discussion
The ability of sequences from two separate introns to stimulate expression remained similar when those sequences were reversed within the context of a spliced intron. Even though reversibility is a hallmark of transcriptional enhancer elements, the need for introns to be within transcribed sequences near the start of transcription and the dispersed nature of the intron sequences capable of stimulating expression indicate that the intron sequences tested do not contain conventional enhancer elements. Therefore, IME must increase gene expression by a mechanism distinct from that of enhancers.
The ends of the COR15a intron supported efficient splicing of the hybrid introns even when the UBQ10 or ATPK1 intron sequences between these ends were inverted and therefore became more A-rich than typical Arabidopsis introns, whose U-richness may be important for intron recognition (Goodall and Filipowicz, 1989). The intron composed of just the COR15a ends also was efficiently spliced but had little effect on expression, confirming that splicing is not sufficient for IME and that the stimulating effect of the hybrids was entirely due to the UBQ10 or ATPK1 sequences they contained. The observation that the ability to increase expression caused by both fragments was preserved when their orientation was reversed indicates that this characteristic is not unique to a specific intron but more likely is a general property of IME. The ability of the IMEter to predict the enhancement caused by the At1g66080 intron, and the increase in performance of the IMEter when the reverse complements of introns are considered, provide additional support for the idea that the signals recognized by the IMEter and presumably involved in the mechanisms of IME are not limited to the transcribed strand. It will be interesting to test the At1g66080 intron in reverse orientation as this would add high-scoring sequences to the transcribed strand that may or may not influence expression levels.
The possibility that IME is catalyzed by palindromic sequences or stem loops that might be similar in either orientation was explored but no plausible candidate structures were found. There is experimental evidence that many of the secondary structures predicted using the Vienna package actually exist in RNA (Kertesz et al., 2010), although the accuracy of the predictions of this and similar programs remains imperfect. None of the RNA secondary structures predicted by the Vienna package are conserved in the UBQ10, CO > UB > CO, and CO < UB < CO introns, nor in the ATPK1, CO > AT > CO, and CO < AT < CO introns. One reason that an intron sequence and its reverse complement have such different predicted secondary structures is that inverting the sequence changes the nucleotides involved in G:U base pairs into As and Cs that are unable to pair with each other. There could be IME-relevant secondary or tertiary nucleic acid structures that are difficult to identify from sequence alone because of their small size or because they involve multiple dispersed degenerate sequences.
Any model of IME must account for the reversibility of stimulating sequences, and for the observations that the stimulatory sequences are distributed throughout the intron and are additive in their effect on expression, at least for the UBQ10 intron. The reversibility suggests that IME operates at the DNA level. That is, the structure of intronic DNA or factors associated with the DNA affect the activity of (and possibly modify) the transcription machinery as it transcribes the intron, leading to greater production of mature mRNA. One explanation for the dispersed nature and low sequence specificity of stimulating regions is that IME could depend on the physical properties of the intron, rather than factors that bind to it. Possible ways in which the DNA in stimulatory introns could be physically different from the DNA in exons or non-stimulatory introns include the ease with which the DNA strands are separated, its propensity to form or resolve R-loops between newly transcribed intronic RNA and the complementary DNA strand (Skourti-Stathaki et al., 2011), or its ability to dissipate the torsional strain generated by transcription of the helical DNA substrate (Roca, 2011). Any of these characteristics could influence the movement of the polymerase along a gene. U-rich sequences, which should have lower melting temperatures than more GC-rich exons, have been implicated in IME (Clancy and Hannah, 2002; Rose, 2002), although the correlation between U-richness and IME in tested introns is weak. The degree to which intronic DNA is methylated is another feature of DNA structure with potential implications for gene expression (Laurent et al., 2010).
Intron-mediated enhancement could be caused by factors that associate with intronic DNA with a low degree of sequence specificity. The simplest model would involve an as yet unspecified protein that binds to weakly conserved sequences in multiple locations throughout the intron. Such a protein would have to be able to stimulate the transcription machinery in either orientation, or bind to short or palindromic sequences, to explain the reversibility. This hypothetical IME-inducing protein could also be a histone or some other chromatin component. Nucleosome density is lower on introns than exons in Arabidopsis and mammals (Schwartz et al., 2009; Tilgner et al., 2009; Chodavarapu et al., 2010), and the lack of nucleosomes could aid passage of the transcription machinery over introns. Trimethylation of histone H3 lysine 4, a modification associated with active transcription, is only found within one kilobase downstream of the transcription start site of Arabidopsis genes (Roudier et al., 2009; Luo and Lam, 2010). The similarity between this distribution and the intron position requirements for IME suggest a possible connection between introns and gene expression through histone modification. An influence on chromatin structure may explain why some introns stimulate expression to a much higher degree as part of a stably integrated transgene than they do in transient expression assays (Rollfinke et al., 1998; Plesse et al., 2001).
Models in which IME operates at the RNA level are more difficult to reconcile with the results presented here but cannot be ruled out entirely. While secondary structure predictions and palindrome finding programs failed to identify obvious RNA features shared between stimulating introns and their reverse complements, the sequence requirements for IME remain undefined but cannot be stringent. Key RNA structures could conceivably be present regardless of the strand transcribed, and these may affect the polymerase either directly or through interacting factors.
The finding that stimulating intron sequences are equally effective in both orientations helps to delineate the mechanism of IME by eliminating models based on unique or asymmetric RNA sequences. An intriguing hint that the splicing machinery might not be involved in IME is that IMEter scores are almost as high in 5′-untranslated regions as they are in promoter-proximal introns (Parra et al., 2011). This suggests that the IME mechanism might not be limited to introns, and that expression can be stimulated by the appropriate signals encountered by RNA polymerase II at any location within the first kilobase after transcription initiates. Determining the exact nature of those signals and how they work should substantially advance our understanding of eukaryotic gene expression.
Materials and Methods
Construction of Hybrid Introns
The modifications to introduce BamHI and BglII restriction sites (which produce compatible cohesive ends) into the stimulating UBQ10 (At4g05320) intron 1 and the non-stimulating COR15a (At2g42540) intron 1 are described in Rose et al. (2008). The ends of each hybrid intron shown in Figure 1 are composed of 49 nt from the 5′ end and 68 nt from the 3′ end of the COR15a intron in their natural orientation. The 189-nt BamHI–BglII region from the middle of the COR15a intron was either deleted (in the COΔCO intron) or replaced with either the 184-nt BamHI–BglII fragment from the UBQ10 intron or a 202-nt BamHI–BglII fragment from the middle of the stimulating ATPK1 (At3g08730) intron. The ATPK1 region was generated by PCR amplification using the primers ATPK1F (5′-CCAATAGATCTGAATTATCGAAATTGC) and ATPK1R (5′-GTCCAGGGATCCTTTTACTAATTGAG), which contain a natural BglII site and change a single nucleotide (bold) to introduce a BamHI site (underlined). Both orientations of each insert were isolated, verified by sequencing, and each intron was inserted as a PstI fragment into a TRP1:GUS reporter gene fusion in the binary vector pEND4K as described (Rose, 2002).
Measuring Expression
Each construct was introduced into Arabidopsis thaliana ecotype Columbia by Agrobacterium-mediated transformation using the floral dip method (Clough and Bent, 1998). At least 36 lines of each were screened by segregation analysis and genomic DNA blots with three different restriction enzymes to determine transgene copy number. All single-copy lines obtained (between 2 and 7 for each fusion) were used in at least two separate expression experiments performed with 3-week-old homozygous plants in the T3 generation as described (Rose et al., 2008). The steady state level of TRP1:GUS mRNA in each line was measured in RNA gel blots by Storm PhosphorImager quantification (Molecular dynamics, Sunnyvale, CA, USA), and is expressed relative to that in a line containing an intronless TRP1:GUS fusion line after correcting for slight differences in loading with an internal control as described (Rose, 2004). As previously observed, the expression in single-copy lines containing the same construct was very consistent, with less variation between independent lines in the same experiment than in a single line in different biological replicates. Therefore, every lane (n ≥ 6) in the blots from lines with the same transgene was given equal weight in calculating the degree to which a particular intron increased TRP1:GUS mRNA accumulation. Splicing efficiency was estimated by reverse transcribing total RNA with random hexamer primers, PCR amplifying with primers that flank the intron, and using PhosphorImager to measure band intensities of the resulting products in gel blots as described (Rose, 2002).
Sequence Analysis
Dot plots were prepared using the website http://www.vivo.colostate.edu/molkit/dnadot/. RNA secondary structure predictions were performed using the Vienna Package (http://rna.tbi.univie.ac.at/). The Perl script written to search for palindromes and stem loop structures is available upon request.
The IMEter Algorithm
The IMEter algorithm (Rose et al., 2008) can be summarized as follows. For each possible subsequence of length K in a test intron, its observed frequency in introns less than a threshold distance from the transcription start site is divided by its expected occurrence in sequence with the average nucleotide composition of all introns in the genome. The logarithms of the observed/expected ratios are summed, and the process is repeated using the observed frequencies in introns that are greater than a threshold distance (equal to or larger than the first threshold) from the start of transcription. The difference between these two sums is the IMEter score, which is positive for introns that resemble promoter-proximal introns and whose magnitude reflects the degree of similarity. A revised version (IMEter 2.0) applies a weighting factor based on the distance of each oligomer from the 5′ end of the test intron, and ignores observed/expected ratios that have negative logarithms, generating scores with a lower boundary of 0 (Parra et al., 2011). To include the opposite strand in IMEter calculations, each K-mer, as well as its reverse complement, are counted at every position in the intron. The effectiveness of considering the opposite strand in the IMEter was evaluated in a total of 1,080 parameter sets, comprising seven thresholds for promoter-proximal introns (50, 75, 100, 125, 150, 175, 200, 300, or 400 nt), six thresholds for distal introns (100, 200, 300, 400, 500, or 1000 nt), and seven K-mer sizes (2–8 nt) using the IMEter or IMEter 2.0. Several combinations of large oligomers and small thresholds for promoter-proximal introns could not be used because the number of introns that occur very early in the set of Arabidopsis genes is too small to calculate meaningful frequencies of large K-mers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health grant number HG00064 to Ian Korf. We thank Jeff Lu for technical assistance.
References
Akua, T., Berezin, I., and Shaul, O. (2010). The leader intron of AtMHX can elicit, in the absence of splicing, low-level intron-mediated enhancement that depends on the internal intron sequence. BMC Plant Biol. 10, 93. doi:10.1186/1471-2229-10-93
Alexander, R. D., Innocente, S. A., Barrass, J. D., and Beggs, J. D. (2010). Splicing-dependent RNA polymerase pausing in yeast. Mol. Cell 40, 582–593.
Callis, J., Fromm, M., and Walbot, V. (1987). Introns increase gene expression in cultured maize cells. Genes Dev. 1, 1183–1200.
Chodavarapu, R. K., Feng, S., Bernatavichute, Y. V., Chen, P. Y., Stroud, H., Yu, Y., Hetzel, J. A., Kuo, F., Kim, J., Cokus, S. J., Casero, D., Bernal, M., Huijser, P., Clark, A. T., Kramer, U., Merchant, S. S., Zhang, X., Jacobsen, S. E., and Pellegrini, M. (2010). Relationship between nucleosome positioning and DNA methylation. Nature 466, 388–392.
Clancy, M., and Hannah, L. C. (2002). Splicing of the maize Sh1 first intron is essential for enhancement of gene expression, and a T-rich motif increases expression without affecting splicing. Plant Physiol. 130, 918–929.
Clancy, M., Vasil, V., Hannah, L. C., and Vasil, I. K. (1994). Maize Shrunken-1 intron and exon regions increase gene expression in maize protoplasts. Plant Sci. 98, 151–161.
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743.
Curi, G. C., Chan, R. L., and Gonzalez, D. H. (2005). The leader intron of Arabidopsis thaliana genes encoding cytochrome c oxidase subunit 5c promotes high-level expression by increasing transcript abundance and translation efficiency. J. Exp. Bot. 56, 2563–2571.
Dahan, O., Gingold, H., and Pilpel, Y. (2011). Regulatory mechanisms and networks couple the different phases of gene expression. Trends Genet. 27, 316–322.
Dean, C., Favreau, M., Bond-Nutter, D., Bedbrook, J., and Dunsmuir, P. (1989). Sequences downstream of translation start regulate quantitative expression of two petunia rbcS genes. Plant Cell 1, 201–208.
Deyholos, M. K., and Sieburth, L. E. (2000). Separable whorl-specific expression and negative regulation by enhancer elements within the AGAMOUS second intron. Plant Cell 12, 1799–1810.
Fiume, E., Christou, P., Giani, S., and Breviario, D. (2004). Introns are key regulatory elements of rice tubulin expression. Planta 218, 693–703.
Furger, A., O’sullivan, J. M., Binnie, A., Lee, B. A., and Proudfoot, N. J. (2002). Promoter proximal splice sites enhance transcription. Genes Dev. 16, 2792–2799.
Goodall, G. J., and Filipowicz, W. (1989). The AU-rich sequences present in the introns of plant nuclear pre-mRNAs are required for splicing. Cell 58, 473–483.
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neubock, R., and Hofacker, I. L. (2008). The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74.
Harel-Sharvit, L., Eldad, N., Haimovich, G., Barkai, O., Duek, L., and Choder, M. (2010). RNA polymerase II subunits link transcription and mRNA decay to translation. Cell 143, 552–563.
Hess, N. K., Singer, P. A., Trinh, K., Nikkhoy, M., and Bernstein, S. I. (2007). Transcriptional regulation of the Drosophila melanogaster muscle myosin heavy-chain gene. Gene Expr. Patterns 7, 413–422.
Itzhaki, H., Maxson, J. M., and Woodson, W. R. (1994). An ethylene-responsive enhancer element is involved in the senescence-related expression of the carnation glutathione-S-transferase (GST1) gene. Proc. Natl. Acad. Sci. U.S.A. 91, 8925–8929.
Jeong, Y. M., Mun, J. H., Lee, I., Woo, J. C., Hong, C. B., and Kim, S. G. (2006). Distinct roles of the first introns on the expression of Arabidopsis profilin gene family members. Plant Physiol. 140, 196–209.
Kertesz, M., Wan, Y., Mazor, E., Rinn, J. L., Nutter, R. C., Chang, H. Y., and Segal, E. (2010). Genome-wide measurement of RNA secondary structure in yeast. Nature 467, 103–107.
Laurent, L., Wong, E., Li, G., Huynh, T., Tsirigos, A., Ong, C. T., Low, H. M., Kin Sung, K. W., Rigoutsos, I., Loring, J., and Wei, C. L. (2010). Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331.
Lu, S., and Cullen, B. R. (2003). Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA 9, 618–630.
Luehrsen, K. R., and Walbot, V. (1994). Addition of A- and U-rich sequence increases the splicing efficiency of a deleted form of a maize intron. Plant Mol. Biol. 24, 449–463.
Lugones, L. G., Scholtmeijer, K., Klootwijk, R., and Wessels, J. G. (1999). Introns are necessary for mRNA accumulation in Schizophyllum commune. Mol. Microbiol. 32, 681–689.
Luo, C., and Lam, E. (2010). ANCORP: a high-resolution approach that generates distinct chromatin state models from multiple genome-wide datasets. Plant J. 63, 339–351.
Mascarenhas, D., Mettler, I. J., Pierce, D. A., and Lowe, H. W. (1990). Intron-mediated enhancement of heterologous gene expression in maize. Plant Mol. Biol. 15, 913–920.
Meredith, J., and Storti, R. V. (1993). Developmental regulation of the Drosophila tropomyosin II gene in different muscles is controlled by muscle-type-specific intron enhancer elements and distal and proximal promoter control elements. Dev. Biol. 159, 500–512.
Moore, M. J., and Proudfoot, N. J. (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136, 688–700.
Morello, L., Bardini, M., Cricri, M., Sala, F., and Breviario, D. (2006). Functional analysis of DNA sequences controlling the expression of the rice OsCDPK2 gene. Planta 223, 479–491.
Nagaike, T., Logan, C., Hotta, I., Rozenblatt-Rosen, O., Meyerson, M., and Manley, J. L. (2011). Transcriptional activators enhance polyadenylation of mRNA precursors. Mol. Cell 41, 409–418.
Nelissen, H., De Groeve, S., Fleury, D., Neyt, P., Bruno, L., Bitonti, M. B., Vandenbussche, F., Van Der Straeten, D., Yamaguchi, T., Tsukaya, H., Witters, E., De Jaeger, G., Houben, A., and Van Lijsebettens, M. (2010). Plant Elongator regulates auxin-related genes during RNA polymerase II transcription elongation. Proc. Natl. Acad. Sci. U.S.A. 107, 1678–1683.
Niu, D. K., and Yang, Y. F. (2011). Why eukaryotic cells use introns to enhance gene expression: splicing reduces transcription-associated mutagenesis by inhibiting topoisomerase I cutting activity. Biol. Direct 6, 24.
Nott, A., Meislin, S. H., and Moore, M. J. (2003). A quantitative analysis of intron effects on mammalian gene expression. RNA 9, 607–617.
Okkema, P. G., Harrison, S. W., Plunger, V., Aryana, A., and Fire, A. (1993). Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics 135, 385–404.
Orphanides, G., Wu, W. H., Lane, W. S., Hampsey, M., and Reinberg, D. (1999). The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400, 284–288.
Palmiter, R. D., Sandgren, E. P., Avarbock, M. R., Allen, D. D., and Brinster, R. L. (1991). Heterologous introns can enhance expression of transgenes in mice. Proc. Natl. Acad. Sci. U.S.A. 88, 478–482.
Pandit, S., Wang, D., and Fu, X. D. (2008). Functional integration of transcriptional and RNA processing machineries. Curr. Opin. Cell Biol. 20, 260–265.
Parra, G., Bradnam, K., Rose, A. B., and Korf, I. (2011). Comparative and functional analysis of intron-mediated enhancement signals reveals conserved features among plants. Nucleic Acids Res. 39, 5328–5337.
Plesse, B., Criqui, M. C., Durr, A., Parmentier, Y., Fleck, J., and Genschik, P. (2001). Effects of the polyubiquitin gene Ubi. U4 leader intron and first ubiquitin monomer on reporter gene expression in Nicotiana tabacum. Plant Mol. Biol. 45, 655–667.
Rethmeier, N., Seurinck, J., Van Montagu, M., and Cornelissen, M. (1997). Intron-mediated enhancement of transgene expression in maize is a nuclear, gene-dependent process. Plant J. 12, 895–899.
Rollfinke, I. K., Silber, M. V., and Pfitzner, U. M. (1998). Characterization and expression of a heptaubiquitin gene from tomato. Gene 211, 267–276.
Rose, A. B. (2002). Requirements for intron-mediated enhancement of gene expression in Arabidopsis. RNA 8, 1444–1453.
Rose, A. B. (2004). The effect of intron location on intron-mediated enhancement of gene expression in Arabidopsis. Plant J. 40, 744–751.
Rose, A. B. (2008). “Intron-mediated regulation of gene expression,” in Nuclear Pre-mRNA Processing in Plants, eds A. S. N. Reddy and M. Golovkin (Berlin: Springer-Verlag), 277–290.
Rose, A. B., and Beliakoff, J. A. (2000). Intron-mediated enhancement of gene expression independent of unique intron sequences and splicing. Plant Physiol. 122, 535–542.
Rose, A. B., Elfersi, T., Parra, G., and Korf, I. (2008). Promoter-proximal introns in Arabidopsis thaliana are enriched in dispersed signals that elevate gene expression. Plant Cell 20, 543–551.
Rose, A. B., and Last, R. L. (1997). Introns act post-transcriptionally to increase expression of the Arabidopsis thaliana tryptophan pathway gene PAT1. Plant J. 11, 455–464.
Roudier, F., Teixeira, F. K., and Colot, V. (2009). Chromatin indexing in Arabidopsis: an epigenomic tale of tails and more. Trends Genet. 25, 511–517.
Schwartz, S., Meshorer, E., and Ast, G. (2009). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 16, 990–995.
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 42, 794–805.
Svejstrup, J. Q. (2007). Elongator complex: how many roles does it play? Curr. Opin. Cell Biol. 19, 331–336.
Tilgner, H., Nikolaou, C., Althammer, S., Sammeth, M., Beato, M., Valcarcel, J., and Guigo, R. (2009). Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 16, 996–1001.
Twell, D., Yamaguchi, J., Wing, R. A., Ushiba, J., and Mccormick, S. (1991). Promoter analysis of genes that are coordinately expressed during pollen development reveals pollen-specific enhancer sequences and shared regulatory elements. Genes Dev. 5, 496–507.
Vasil, V., Clancy, M., Ferl, R. J., Vasil, I. K., and Hannah, L. C. (1989). Increased gene expression by the first intron of maize Shrunken-1 locus in grass species. Plant Physiol. 91, 1575–1579.
Visel, A., Rubin, E. M., and Pennacchio, L. A. (2009). Genomic views of distant-acting enhancers. Nature 461, 199–205.
Vitale, A., Wu, R. J., Cheng, Z., and Meagher, R. B. (2003). Multiple conserved 5′ elements are required for high-level pollen expression of the Arabidopsis reproductive actin ACT1. Plant Mol. Biol. 52, 1135–1151.
Weise, A., Lalonde, S., Kuhn, C., Frommer, W. B., and Ward, J. M. (2008). Introns control expression of sucrose transporter LeSUT1 in trichomes, companion cells and in guard cells. Plant Mol. Biol. 68, 251–262.
Williamson, I., Hill, R. E., and Bickmore, W. A. (2011). Enhancers: from developmental genetics to the genetics of common human disease. Dev. Cell 21, 17–19.
Wu, C. H., Yamaguchi, Y., Benjamin, L. R., Horvat-Gordon, M., Washinsky, J., Enerly, E., Larsson, J., Lambertsson, A., Handa, H., and Gilmour, D. (2003). NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev. 17, 1402–1414.
Keywords: gene expression, intron-mediated enhancement, Arabidopsis
Citation: Rose AB, Emami S, Bradnam K and Korf I (2011) Evidence for a DNA-based mechanism of intron-mediated enhancement. Front. Plant Sci. 2:98. doi: 10.3389/fpls.2011.00098
Received: 08 September 2011;
Accepted: 30 November 2011;
Published online: 13 December 2011.
Edited by:
Anireddy S. N. Reddy, Colorado State University, USAReviewed by:
William Brad Barbazuk, University of Florida, USASteve M. Mount, University of Maryland, USA
Copyright: © 2011 Rose, Emami, Bradnam and Korf. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alan B. Rose, Molecular and Cellular Biology, University of California, 1 Shields Avenue, Davis, CA 95616, USA. e-mail: abrose@ucdavis.edu