
- 1 Department of Biochemistry, Interdisciplinary Plant Group, Christopher S. Bond Life Sciences Center, University of Missouri, Columbia, MO, USA
- 2 Institute of Biology, State University of Campinas, Campinas, São Paulo, Brazil
- 3 Institute of Biological Chemistry, Washington State University, Pullman, WA, USA
Horsetail (Equisetum hyemale) is a widespread vascular plant species, whose reproduction is mainly dependent on the growth and development of the rhizomes. Due to its key evolutionary position, the identification of factors that could be involved in the existence of the rhizomatous trait may contribute to a better understanding of the role of this underground organ for the successful propagation of this and other plant species. In the present work, we characterized the proteome of E. hyemale rhizomes using a GeLC-MS spectral-counting proteomics strategy. A total of 1,911 and 1,860 non-redundant proteins were identified in the rhizomes apical tip and elongation zone, respectively. Rhizome-characteristic proteins were determined by comparisons of the developing rhizome tissues to developing roots. A total of 87 proteins were found to be up-regulated in both horsetail rhizome tissues in relation to developing roots. Hierarchical clustering indicated a vast dynamic range in the regulation of the 87 characteristic proteins and revealed, based on the regulation profile, the existence of nine major protein groups. Gene ontology analyses suggested an over-representation of the terms involved in macromolecular and protein biosynthetic processes, gene expression, and nucleotide and protein binding functions. Spatial difference analysis between the rhizome apical tip and the elongation zone revealed that only eight proteins were up-regulated in the apical tip including RNA-binding proteins and an acyl carrier protein, as well as a KH domain protein and a T-complex subunit; while only seven proteins were up-regulated in the elongation zone including phosphomannomutase, galactomannan galactosyltransferase, endoglucanase 10 and 25, and mannose-1-phosphate guanyltransferase subunits alpha and beta. This is the first large-scale characterization of the proteome of a plant rhizome. Implications of the findings were discussed in relation to other underground organs and related species.
Introduction
Biological invasions are usually defined as the introduction, establishment, and spread of a species outside their native range, being recognized as a major threat to the economy and environment worldwide (Prentis et al., 2008). With the increase in the environmental pressure for reduction in overall pesticide use, there is a great effort to find sustainable, non-chemical alternatives for weed control (Grundy, 2003). Rhizomes are diageotropic subterranean stems of fundamental importance to plant competitiveness and growth (Jang et al., 2009). They are key elements for propagation and persistence of many weeds (Hu et al., 2003). While rhizomes are an important component of the invasive nature of many noxious aliens; in other species, they are a valuable trait in the establishment, persistence, and massive growth of forage biomass (Jang et al., 2006, 2009). A deeper understanding of the factors that regulate and affect rhizome differentiation and development could impact important sectors of agriculture such as weed management and biofuel production.
Fossils of Equisetum species indicate the first appearance of the genus in the Cretaceous Period, probably being the oldest lineage of extant vascular plants (Gierlinger et al., 2008). Equisetum is a genus of approximately 30 species of non-seed plants, including several widespread and common hybrids (des Marais et al., 2003; Large et al., 2006). Species from this genus are mainly found between 40 and 60° north latitude and generally restricted to seasonally or sometimes perennially wet ground (des Marais et al., 2003). Most Equisetum species, including the extremely weedy E. arvense and the very widespread E. hymale, are recorded as having the potential to become persistent weeds in wetlands (Large et al., 2006). E. hymale also has a long history of use by humans because of the high concentration of silica crystals in its stems, leading to its common name: scouring rush. Ecological success of the dispersion of these species and their occurrence in non-native regions (e.g., tropics) can be attributed to the rhizomatous growth habit and associated vegetative propagation (des Marais et al., 2003). Due to their invasive behavior and the key evolutionary position of the genus within the plant kingdom, the study of the rhizome biology of Equisetum species may allow the identification of unique characteristics of this organ that contributed to the ecological success of modern rhizomatous species and may contribute to the implementation of strategic control programs against rhizome-driven propagation of weeds.
Because proteins play an essential role in biological processes, the characterization and understanding of the proteomic composition of any biological sample can provide important information about complex cellular regulatory networks (Domon and Aebersold, 2006). Furthermore, transcriptomic studies have the central caveat that steady-state protein levels may not follow a similar stoichiometric ratio. Indeed, parallel studies of transcript and protein regulation in plants reveal a correlation of around 0.5–0.6 (Hajduch et al., 2010).
Several different proteomic strategies, designed to comprehensively characterize the proteome of a cell/tissue/organ in different states or living conditions, have been developed in recent years. One of the most facile, but comprehensive and unbiased strategies for protein profiling is SDS-PAGE prefractionation coupled to in-gel digestion and mass spectrometry, also referred to as GeLC-MS. When coupled to techniques such as spectral counting, this approach allows for a relative quantitative assessment of the original proteins. A challenge with any bottom-up proteomic approach is identifying the proper database for interrogating MS/MS data. Homologous databases are preferred although the utility of current RNA sequencing technologies for this purpose is uncertain.
Current advances in sequencing technologies have resulted in an increasing number of plant genome and EST-sequencing projects. However, the number of non-model plant species covered by these projects is still limited and many of these plants with unique biological and physiological characteristics remain “orphan” or poorly studied (Carpentier et al., 2008; Jorrin-Novo et al., 2009; Remmerie et al., 2011). In the present work, we carried out a GeLC-MS spectral counting-based proteome characterization of developing rhizomes of horsetail (E. hyemale) in order to identify proteins that may be involved with different tissues of this organ. E. hyemale rhizome-characteristic proteome was determined by comparisons against root samples; while spatial differences within the rhizome were identified by pairwise analysis between the rhizomes apical tip and the elongation zone.
Experimental Procedures
Plant Material
Equisetum hyemale plants were purchased from a nursery (Mesquite Valley Growers, Tucson, AZ) and maintained in a greenhouse under controlled conditions as described by He et al. (2012). Samples from the rhizome apical tip and elongation zone (Figure 1), as well as root samples, were dissected and immediately frozen in liquid N2. Samples were stored at −80°C until protein extraction.

Figure 1. Rhizome tips from Equisetum hyemale are easily obtained from greenhouse grown plants. (A) Plant removed from pot indicating (arrow) a typical rhizome tip used for analysis. (B) Close up view of excised rhizome that has been washed and prepared for tissue collection. Arrows indicated the regions (tip and elongation zone) that were used in the analysis.
Protein Extraction and Protein Electrophoresis
Frozen samples were ground with a mortar and pestle to produce a fine powder. Aliquots of 200 mg of the powder were resuspended in 1.5 mL of extraction buffer containing 0.1 M Tris (pH 8.0), 10 mM EDTA, 0.9 M sucrose, and 0.1% (w/v) DTT. After incubation on ice for 5 min, 1.5 mL of Tris-buffered phenol (pH 8.0) was added to the extracts and the mixture was vortexed thoroughly for 1 min and incubated on a shaker at 4°C for 30 min. To separate insoluble material, aqueous and organic phases, the samples were centrifuged for 15 min at 5,000 g. The phenolic phase was recovered and transferred into a new tube. For protein precipitation, four volumes of methanol containing 0.1 M ammonium acetate were added and samples were incubated overnight at −20°C. Protein precipitates were collected by centrifugation at 5,000 g for 15 min and the pellets were washed three times with methanol containing 0.1 M ammonium acetate. Finally, proteins were resuspended in 200 μL resuspension buffer [65 mM Tris (pH 6.8), 2% (w/v) SDS] and protein concentration was estimated by the BCA Protein Kit (Thermo Fisher Scientific, Houston, TX) using BSA as standard. Protein extracts were prepared in five biological replicates for rhizome apical tip, rhizome elongation zone, and roots. Prior to gel electrophoresis, sample aliquots containing 100 μg of proteins were mixed with an equal volume of loading buffer containing 125 mM Tris (pH 6.8), 20% (v/v) glycerol, 4% (w/v) SDS, 0.5% (w/v) DTT, and traces of bromophenol blue and incubated for 5 min at 99°C. Gel electrophoresis was performed under denaturing conditions in 12% polyacrylamide gels (11 cm long × 8 cm wide) using 20 mA per gel. Proteins from all the five replicates of each tissue were separated at the same gel and, after protein migration, gels were stained with colloidal Coomassie blue stain under standard conditions.
In-gel Protein Digestion
Prior to protein digestion, the gel lane for each biological replicate was sliced into 30 equal size segments of approximately 3 mm, diced into approximately 1 mm cubes with a clean scalpel and transferred into a 0.45 μm low-binding hydrophilic PTFE filter plate (MultiScreen Solvinert Plates, Millipore) for in-gel protein digestion. Tryptic digestion was carried out according to Shevchenko et al. (2007). After gel destaining in acetonitrile (ACN): 100 mM ammonium bicarbonate (1:1; AmBic) solution, proteins were reduced and alkylated in 100 mM AmBic solutions containing 10 mM DTT and 50 mM of iodoacetamide, respectively. Protein digestion was performed by the addition of 100 μL of digestion solution (10 mM AmBic and 10% ACN) containing sequencing grade porcine trypsin (Promega, Madison, WI, USA) at 7 ng/μL. After 60 min of cold incubation at 4°C, 100 μL of digestion solution without trypsin was added and samples were digested overnight at 37°C. Upon in-gel digestion, gel pieces were saturated with 400 μL of extraction buffer containing 5% formic acid (FA): ACN (1:2, v/v) and incubated for 30 min at 37°C. Supernatants were collected from the same filtration unit by centrifugal filtration (3,000 g for 30 min), dried down in a vacuum centrifuge and kept at −80°C until LC-MS/MS analyses.
LC-MS/MS Analyses
For each round of LC-MS/MS analysis, extracted peptides were reconstituted in 0.1% (v/v) FA and separated at the flow rate of 150 μL/min into a 10 cm × 150 μm ID in-house packed nanocolumn (C18, 100 Å, 5 μm, Michrom Bioresources) using the following mobile phase gradient: from 5 to 35% of solvent B in 25 min, from 35 to 70% in 25 min, then back to 5% in 5 min. Solvent A was water containing 0.1% FA, solvent B was ACN containing 0.1% FA. After LC separation, peptides were positively ionized at 2.1 kV, at 250°C and injected into the mass spectrometer. Mass spectrometry data were acquired in a ProteomeX-LTQ Workstation (Thermo, San Jose, CA, USA) in data-dependent acquisition (DDA) mode controlled by XCalibur 2.0 software (Thermo Fisher Scientific). The typical DDA cycle consisted of a survey scan within m/z 200–2,000 followed by MS/MS fragmentation of the seven most abundant precursor ions under normalized collision energy of 35%. Fragmented precursor ions were dynamically excluded according to the following: repeat counts: 3, repeat duration: 30 s, exclusion duration: 30 s.
Database Creation and Protein Identification
The database used in the present work was obtained through the isolation of total RNA from E. hyemale rhizome apical tip and elongation zone tissues, followed by the construction of sequencing libraries and Illumina Genome Analyzer or 454 sequencing as described by He et al. (2012).
The final assemblies (unique transcripts) were then translated and the open reading frames (ORF) scanned using the Virtual Ribosome software version 1.1 (Wernersson, 2006). For each nucleotide entry, the longest ORF was reported and used for database searches. For calculations of false discovery rates and further validation of the peptide-spectrum matches (PSMs), randomized (i.e., decoy) sequences were generated and combined with the forward/targeted database. After initial assessment of the Illumina, 454 and the hybrid database (Figure S1 in Supplementary Material), we used the Illumina library containing 70,987 contigs for translation resulting in a decoy concatenated search database containing 139,394 protein entries.
For protein identification, peak lists were initially generated from the raw data using Extract_msn.exe program in Bioworks 3.3.1 SP1 (Thermo) according to the following parameters: MW range: 200–2,000; absolute threshold: 500; precursor ion tolerance: 1,000 ppm; group scan: 1; minimum group count: 1; minimum ion count: 10. Database searches were performed using SEQUEST search engine integrated within the Bioworks 3.3.1 SP1 software package (Thermo). Search parameters were set as follows: oxidation of methionine was allowed as a variable modification and carbamidomethylation of cysteine as a static modification; enzyme: trypsin; number of allowed missed cleavages: 2; mass range: 200–2,000; threshold: 500; minimum ion count: 10; peptide tolerance: 1,000 ppm; fragment ions tolerance: 1 Da. Duplicate peptide matches were reported. After searches, Bioworks SEQUEST output files were converted into SQT files and validation of the PSMs candidates was computationally assessed using the Search Engine Processor tool (Carvalho et al., 2012). For that, the SEQUEST proposed PSMs were divided into nine groups, corresponding to the combination of +1, +2, ≥3 peptide ions and fully, semi, or non-tryptic peptides. The Bayesian discriminant scores were calculated based on the following parameters: normalized XCorr, delta CN, secondary score, number of peaks match, digestion and presence scores, and rank associated to the secondary score. For confident protein identification, spectrum, peptide, and protein cutoffs were adjusted to achieve a false discovery rate of 1% at the protein level for each biological replicate.
Relative Quantification Based on Spectral Counting
To satisfy the principal of parsimony, proteins containing common peptides were grouped and the relative protein quantification was performed based on the number of spectral counts per protein group with care to count only once the spectral counts of the shared peptides within each proposed group. This approach was adopted in order avoid protein identification ambiguity and erroneous quantitative values due to the incorrect distribution of spectral counts among protein isoforms or other proteins with high sequence similarity. Assessments of the quantitative differences were carried out through pairwise analyses. For that, spectral counts were initially normalized according to the Row Sigma Normalization (Carvalho et al., 2008). Detection of differentially regulated proteins was performed using the TFold tests, embedded in the PatternLab for Proteomics suite (Carvalho et al., 2010). The TFold test combines fold-change cutoffs with Student’s t-test and the Benjamini–Hochberg theoretical false-positive estimator to deal with the task of massive hypothesis-testing problem (Benjamini and Hochberg, 1995). We considered as differentially regulated proteins those accessions that presented a fold change higher than 2.5 and a p-value threshold of 0.01 for both t-test and Benjamini–Hochberg estimator. To avoid false interpretation of differential regulation from accessions presenting low spectral counts, we considered only the proteins identified in at least three out of the five biological replicates.
Annotation and Functional Classification
Annotation and classification of the differentially regulated proteins were performed based on matching the protein sequences against the UNIPROTKB/SwissProt database and retrieval of associated GO terms using the Blast2GO tool (Conesa et al., 2005). The sequences of interest were first extracted from the protein database using the MUsite software (Gao et al., 2010). Extracted sequences were then aligned using the BLASTP algorithm against the UNIPROTKB/SwissProt database using the following parameters: report a maximum of five blast hits, 1e−10 for the expected value and minimum high scoring segment pairs (HSPs) length equal to 33. Mapping and annotation steps were also performed using Blast2GO default values (E-value filter of 1e−6, annotation cutoff of 55, and GO weigh equal to 5). After generation of the combined graphs (score alpha 0.6 and sequence filter equal to 1%) for biological process and molecular function, the GO terms distributions were analyzed at the fourth level of depth.
Hierarchical Clustering
To determine similarities of the regulation profile of E. hyemale up-regulated proteins, we performed hierarchical clustering analysis using the software PermutMatrix (Caraux and Pinloche, 2005). The raw spectral counts of the apical tip, elongation zone, and root proteins were subtracted by the average spectral count value of each clustered protein. Then, dissimilarities were calculated based on Euclidean distances and hierarchical clustering was carried out according to the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) method (Sokal and Michener, 1958).
Results and Discussion
Large-Scale Identification of E. hyemale Underground Proteins
Conventional, stringent database searches rely on strict matching of the mass spectrometer-acquired spectra with the theoretical, in silico proposed spectra. Any difference in the sequence of a peptide presented in the database may compromise the scoring significance of the PSM and result in mis-identification of the acquired spectrum. This situation is exacerbated when cross-species protein identification strategies are employed, as the choice of a closely related organism may not be sufficient to significantly cover the proteome of the target species and provide enough information for the biological process under analysis. Thus, a homologous database is more desirable to achieve a significant number of confident PSMs. For spectral counting-based quantitative proteomics, the absence or slight change (e.g., due to sequence polymorphism or sequence error) of a protein sequence in the database may result in mis-identification, which can hamper the correct interpretation of the proteomics data. In order to obtain maximum proteome coverage, we initially evaluated three E. hyemale assembled EST databases from Illumina and 454 sequencing projects as described in He et al. (2012) (Figure S1 in Supplementary Material). SEQUEST searches using the database generated from Illumina sequences resulted in the highest number of spectra, peptides, and protein groups (i.e., proteins sharing the same set of peptides). On the other hand, searches using this database resulted in the lowest number of identified proteins. In a parallel evaluation, searching Illumina database resulted in the lowest number of shared peptides (bottom-left panel in Figure S1 in Supplementary Material), which significantly decreases protein mapping ambiguity and explains the low number of identified proteins while presenting the highest numbers of spectra, peptides, and protein groups. A list of all identified peptides from Illumina searches can be found in Table S1 in Supplementary Material. In order to minimize the protein inference problem, the Illumina database was chosen and E. hyemale quantitative analysis was performed based on the number of spectral counts per protein group (Table S2 in Supplementary Material) and, herein, for the sake of simplicity, protein groups will be only referred to as proteins.
The rhizomatous trait is widely distributed across the plant kingdom; however, few studies have been carried out to characterize the proteome of this key subterranean organ. One of the first dedicated studies was carried out by Lum et al. (2002) aiming to detect specific and common two-dimensional electrophoresis (2-DE) protein spots that could be used as markers for different ginseng species and rhizome parts. This approach resulted in the identification of two proteins (ribonucleases I and II) from the nine protein spots commonly identified in all 2-DE gels. Migliore et al. (2007) also studied the 2-DE protein spot distribution patterns of Posidonia oceanica rhizome samples naturally grown in different areas and proposed the use of a combined approach of phenol assay and 2-DE protein analysis as a “diagnostic” tool to monitor the health state of this species in contaminated areas. More recently, He et al. (2012), using a proteome-wide quantitative profiling, identified 1,280 proteins in rhizomes of Phragmites australis producing an extensive survey of proteins related to rhizome development. Using the semi-throughput approach described here, we confidently identified, at a maximum of 1% false discovery rate, 2,377 total, non-redundant proteins in the rhizome and root samples of E. hyemale (Figure 2; Table S2 in Supplementary Material). From the analyzed tissues, the highest number of proteins was identified in the rhizome apical tip (1,911) followed by the rhizome elongation zone (1,860). Root samples comprised a set of proteins equal to 1,374. This extensive protein identification list provided us with a solid framework for a spectral counting-based comparative analysis against the developing root proteins (used as reference) in order to identify up-regulated, enriched proteins in E. hyemale rhizomes. To detect these rhizome-enriched proteins (proteins up-regulated in both rhizome apical tip and elongation zone in relation to the roots), we carried out two pairwise analyses: rhizome apical tip versus roots and rhizome elongation zone versus roots. These proteins were termed rhizome-characteristic proteins. Surprisingly, only 87 non-redundant proteins, or approximately 3.9% of total proteins identified, were up-regulated in rhizomes (Figure 2; Table S3 in Supplementary Material). These data reveal a highly similar proteome profile between these two underground organs, even though the tissues are quite distinct morphologically. This suggests that the proteins responsible for tissue and organ patterning and differentiation in Equisetum roots and rhizomes are likely to be limited in number, perhaps within that list of 87, or of low abundance to avoid detection in this investigation.
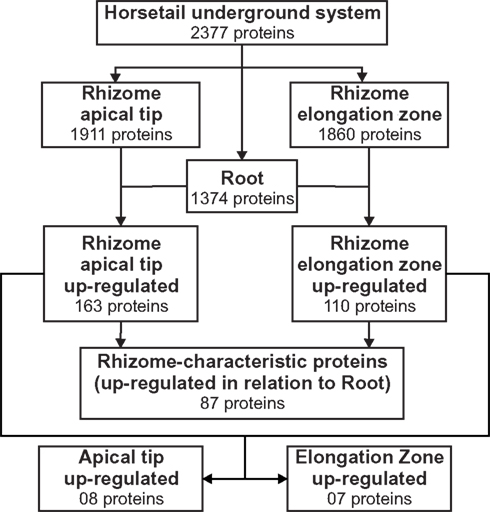
Figure 2. Downstream analysis for the identification of Equisetum hyemale rhizome-characteristic proteins and identification of differential regulation between the apical tip and the elongation zone proteins. Proteins identified in the apical tip and elongation zone were compared with those detected in roots. Proteins found to be up-regulated in both apical tip and elongation zone were combined to create a non-redundant list of the up-regulated, characteristic proteins of E. hyemale rhizomes. Spatial differences were determined by comparing the expression level of the apical tip and elongation zone proteins after exclusion of those detected in the same or lower expression level than in the developing roots.
Functional Classification of the 87 Proteins Reveals Active Gene Expression and Protein Metabolism in the Rhizomes
Gene ontology (GO) terms distribution analysis was carried for the 87 rhizome-characteristic proteins in relation to the total proteome of E. hyemale rhizomes (2,238 proteins). The biological process GO term distribution indicated that the terms cellular macromolecule metabolic process (mapped by 11% of the characteristic proteins), protein metabolic process (9%), cellular biosynthetic process (8%), gene expression (7%), and macromolecule biosynthetic process (6%) were over-represented in the rhizome-characteristic proteome in relation to the total E. hyemale rhizome-proteome (Figure 3). The molecular function term distribution indicated an over-representation of the terms RNA-binding (mapped by 12% of the characteristic proteins), ribonucleotide binding (12%), purine nucleotide binding (12%), purine nucleoside binding (11%), hydrolase activity (6%), translation factor activity (5%), identical and unfolded protein binding (4%), and carbon–carbon lyase activity (3%; Figure 3). Our GO analyses revealed an over-representation of the terms associated with macromolecular and protein biosynthetic processes, gene expression, and nucleotide binding functions, indicating an active metabolism preferentially shifted to cellular proliferation activities. Because these tissues were the apical meristematic region and the closely associated stem elongation zone, abundance of proteins associated with such processes is not surprising.
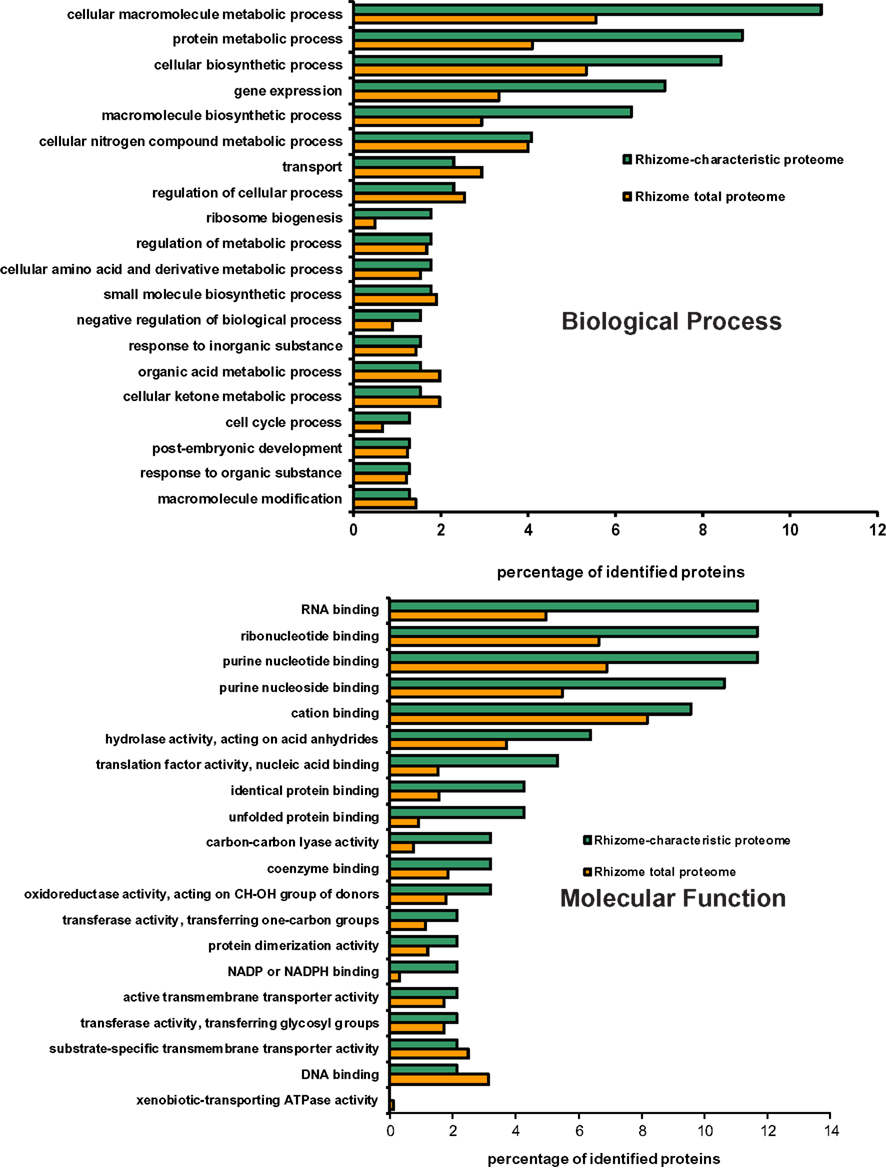
Figure 3. Distribution of GO biological process and molecular function terms mapped from Equisetum hyemale rhizome proteins. Proteins identified in rhizome samples were aligned against the UNIPROTKB/SwissProt entries and the retrieved accessions mapped to the biological process and molecular function GO terms. The set of rhizome-characteristic proteins was compared to the total rhizome-proteome at the fourth level.
Protein Regulation Profile Indicates Vast Dynamic Range and Suggests Differential Regulation between Apical Tip and Elongation Zone
Hierarchical cluster analysis of the rhizome-characteristic proteins suggested a considerable dynamic range of the E. hyemale rhizome-proteome, resulted from abundance differences up to 563:1 based upon spectral counts. Histidine decarboxylase (UNIPROT/SwissProt accession P54772) and the 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase were the most abundant proteins (O50008); while the 26S protease regulatory subunit 7 (Q9FXT9), ubiquilin-1 (Q8R317), putative H/ACA ribonucleoprotein complex (Q8VZT0), arogenate dehydratase (Q9SGD6), 26S proteasome non-ATPase regulatory subunit 4 (P55034), and glucose-6-phosphate 1 (Q43839) were the least abundant detected proteins within E. hyemale rhizome-characteristic proteome (Figure 4 and Table S3 in Supplementary Material). Due to this vast dynamic range, nine main clusters were defined based on the regulation profile of the 87 characteristic proteins within the rhizomes samples (Figure 4). Cluster A consisted of proteins with the highest levels in both tissues. Clusters C and H accounted for 11 proteins presenting similar regulation in both apical tip and elongation zone. Clusters B, D, and E accounted for 6, 7, and 13 proteins, respectively, with regulation profile higher in the apical tip in relation to the elongation zone. In contrast, clusters G (five proteins) and I (six proteins) were characterized by proteins presenting higher level in the elongation zone in relation to the apical tip. Finally, major cluster F comprised all proteins that presented minor differences between the elongation zone and apical tip regions.

Figure 4. Regulation profile of Equisetum hyemale rhizome-characteristic proteins. Hierarchical clustering was carried out for the 87 rhizome-characteristic proteins. A larger image can be found Figure S2 in Supplementary Material.
Regarding the most abundant, rhizome-characteristic proteins, Wang et al. (2000) reported the induction of the gene histidine decarboxylase (EC 4.1.1.22) after nitrate supplementation in Arabidopsis seedlings. Picton et al. (1993) identified a histidine decarboxylase-like mRNA up-regulated during tomato ripening. However, the role of this abundant enzyme in the underground tissues of Equisetum is still unknown. The cytosolic enzyme 5-methyltetrahydropteroyltriglutamate, also known as cobalamin-independent methionine synthase (EC 2.1.1.1.14), was also detected in high levels in theses organs. Zeh et al. (2002), studying RNA expression in potato plants, suggested that the gene that encodes this enzyme is a low-copy gene with differential expression across the various evaluated organs. Although methionine synthase transcripts were detected in roots, the highest levels were found in flowers. In a proteomic study of A. stolonifera, a Poaceae family member, levels of this enzyme varied significantly according to the salinity stress imposed on different organs (Xu et al., 2010). In the same manner, proteome analysis of soybean roots revealed higher regulation of methionine synthase under flood stress (Komatsu et al., 2010). As horsetail is a wetland plant, constitutive high levels of this enzyme indicate a possible metabolic strategy used to cope with flood and salinity stress.
Another protein related to stress tolerance and is among the 87 characteristic proteins is the enzyme aldehyde dehydrogenase (EC 1.2.1.3). Recent reports indicate that the over-regulation of this enzyme confers stress tolerance in different plant species (Huang et al., 2008; Brocker et al., 2010; Missihoun et al., 2011; Stiti et al., 2011). In E. hyemale, this identification was over-represented in the elongation zone, a region that is commonly affected by water stress (Zhu et al., 2007; Spollen et al., 2008; Yamaguchi et al., 2010). In addition, elongation of the cells in this region is a necessary process to allow growth and development of the whole rhizome organ. As cell expansion in plants is limited by the cell wall matrix, the rigid structure elements that constraint cell growth should be degraded and resynthesized. An aldehyde dehydrogenase has been proposed to act in sinapoyl-malate formation in Arabidopsis (Nair et al., 2004) and ferulate ester formation in grasses (Barriere et al., 2007). Thus, this enzyme may play a role in plant cell wall reorganization allowing cell expansion in this region and, consequently, plant growth. It is intriguing to consider that such processes may have evolved as long ago as during the origin of Equisetum.
A significant fraction of the rhizome-characteristic proteins was comprised of ribosomal proteins: 17 accessions in total. These proteins were mainly identified as type 40 and 60S ribosomal proteins, presenting higher levels in the apical tip in relation to the elongation zone. This scenario is clear for cluster B, as shown in Figure 4, as all proteins that compose this group are 40 or 60S ribosomal protein types. Recently, mutations of the genes that encode ribosomal proteins have proven to be deleterious to the differentiation of meristematic tissues (Horiguchi et al., 2003; Minnebruggen et al., 2010; Szakonyi and Byrne, 2011). In our work, regulation profile analysis of the identified 40S ribosomal protein S6 (RPS6) resulted in the classification of this protein in cluster D (Figure 4), which is characterized by proteins with higher levels in the apical tip in relation to the elongation zone (Table 1, accession number EhRi_039920). This protein may thus play an important role in the differentiation of the Equisetum apical tip.
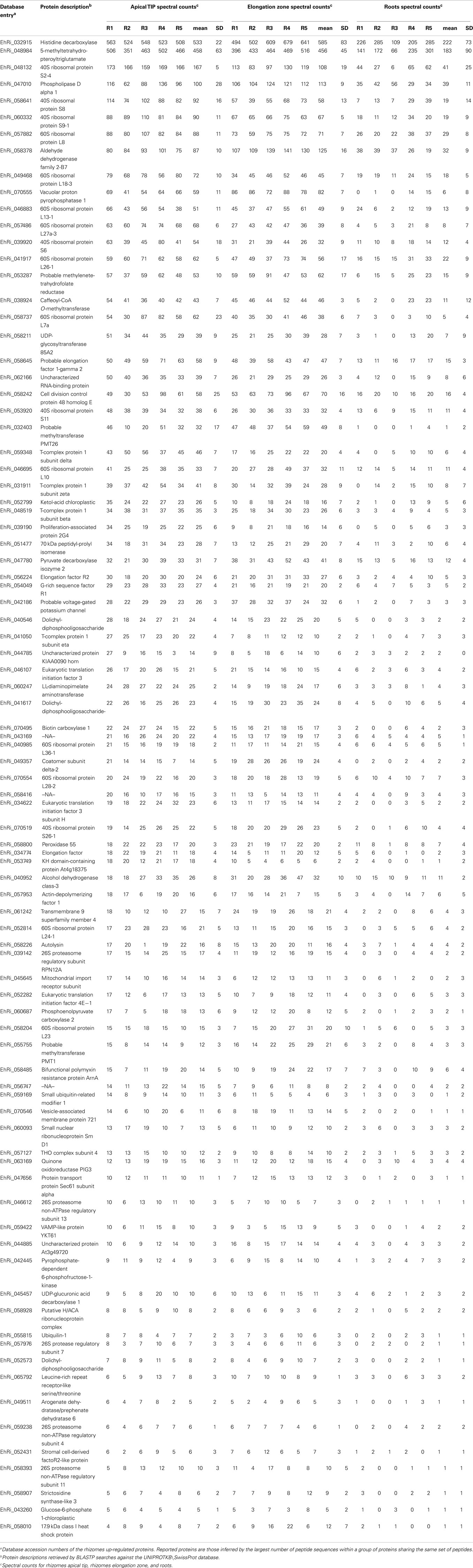
Table 1. List of the 87 characteristic proteins identified in the Equisetum hyemale rhizomes.
Spatial Differences between the Apical Tip and Elongation Zone Reflects the Different Roles of These Regions during the Growth and Development of the Rhizomes
Proteome spatial differences within E. hyemale rhizomes were further studied using a two-step sequential pairwise analysis (Figure 2). In the first comparison, the proteins identified in the rhizome apical tip and elongation zone were compared to those identified in root samples. Then, the up-regulated proteins in the rhizome apical tip and elongation zone were pairwise compared for a final list of the up-regulated proteins in a particular rhizome region (Table 2). This strategy was adopted in order to detect the proteins that were highly expressed in only one of the rhizome tissues in relation to the roots, and thus not included in the previously described set of the 87 rhizomes characteristic proteins. A total of 15 proteins showed differential regulation between the studied tissues (Figures 5 and 6).
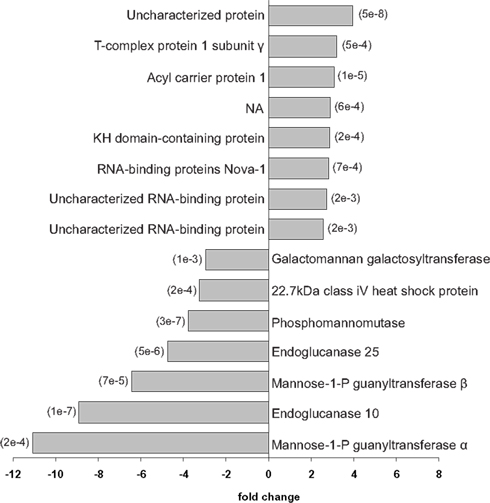
Figure 5. Differentially regulated proteins between Equisetum hyemale rhizome apical tip elongation zone. Gray bars indicate the fold changes in the regulation of the 15 differentially regulated proteins. The p-values for each pairwise comparison (apical tip versus elongation zone) are presented in parenthesis at the end of the gray bars. Apical tip and elongation zone up-regulated proteins are represented as positive and negative fold changes, respectively.
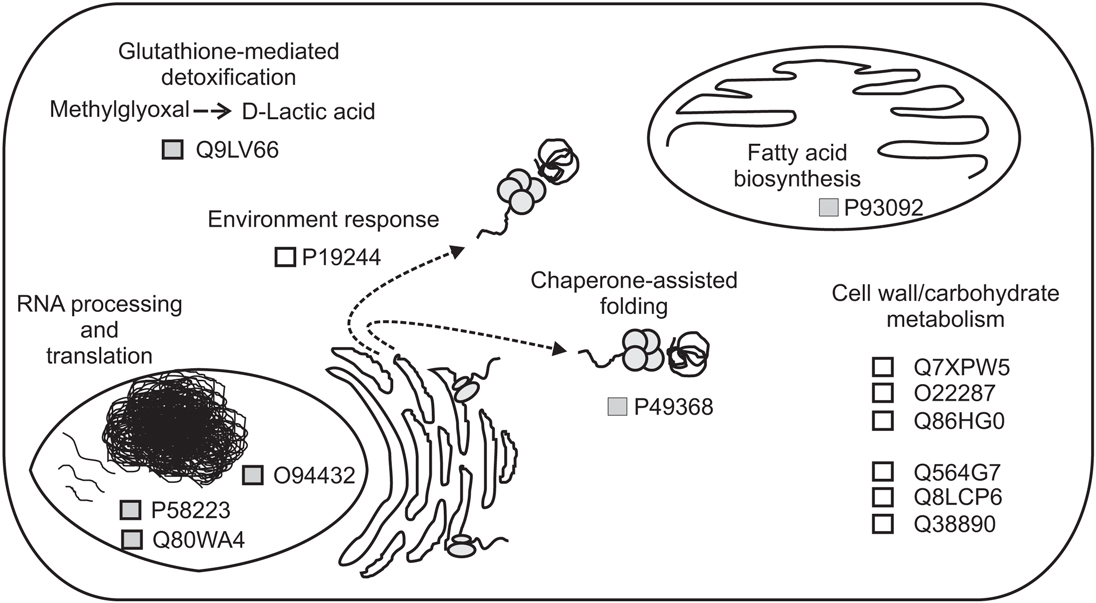
Figure 6. Schematic representation of the proposed functions for proteins that are differentially regulated between Equisetum hyemale apical tip and elongation zone. Proteins are represented by the retrieved UniProtKB/SwissProt accession numbers.
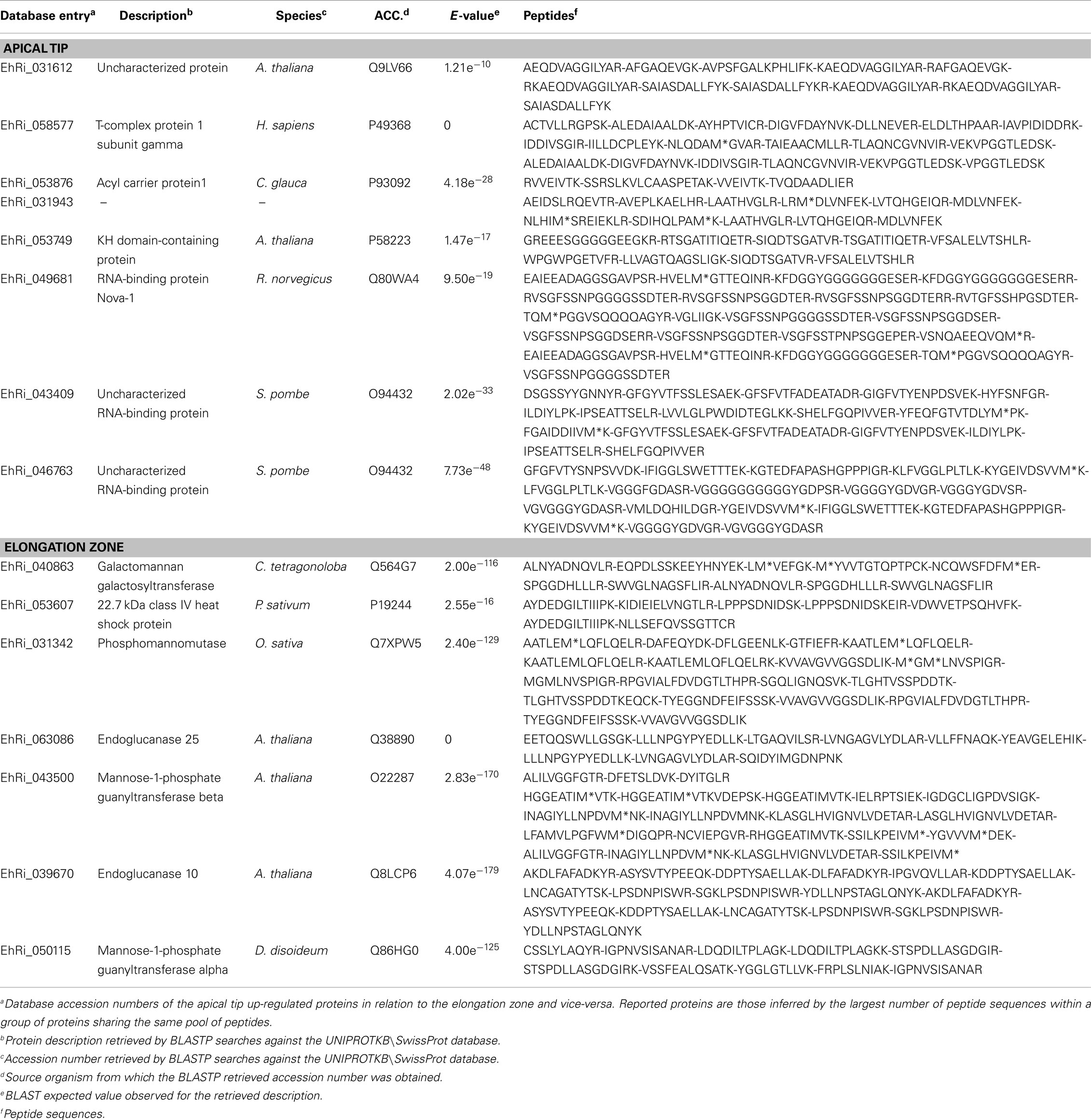
Table 2. List of differentially regulated proteins within the Equisetum hyemale rhizomes.
In the apical tip samples, four up-regulated proteins are described as RNA-binding proteins (UniProtKB/SwissProt accessions P58223, Q80WA4 and 2 proteins with accession number equal to O94432). These multifunctional proteins are involved in numerous RNA-mediated processes, including the regulation of gene expression. High expression of RNA-binding proteins in actively proliferative regions was already described for Arabidopsis (Suzuki et al., 2000; Fusaro et al., 2007). Due to its inherent functions, it is not a surprise to identify high levels of RNA-binding proteins in highly active tissues, such as in the rhizome apical tip. Another protein involved in the gene information path and up-regulated in the apical tip was the protein described as a T-complex protein 1 subunit gamma (P49368). The cDNA encoding TCP-1 in Arabidopsis was first cloned by Mori et al. (1992). TCP-1 is a subunit of the chaperone containing TCP-1 (Iijima et al., 1998), a protein that acts in the cytosol during the post-translation protein folding process of several proteins, including actin and tubulin (Lewis et al., 1992; Yaffe et al., 1992). Although no microtubule-related protein was found to be spatial regulated within the rhizomes, actin-depolymerizing factor 1 appeared at similar levels in both apical tip and elongation zone as a rhizome-characteristic protein (Figure 4).
Acyl carrier proteins (ACP, accession P93092) are small, acidic proteins that carry the nascent acyl chains during the synthesis of 16- and 18-carbon acyl groups (Bonaventure and Ohlrogge, 2002), playing an essential role during plant lipid biosynthesis (Ohlrogge and Kuo, 1985). The higher level of this enzyme in the rhizome apical tip in relation to the elongation zone and roots are in accordance with the usually highest regulation in the meristematic zones of vegetative tissues in Arabidopsis (Baerson and Lamppa, 1993) and cells with high rates of division (Bonaventure and Ohlrogge, 2002), suggesting a high demand of fatty acids in tissues where multiplication and differentiation processes occur at high rates.
Finally, the protein Q9LV66, an uncharacterized protein with high sequence similarity with glyoxalases (glutathione-mediated detoxification enzymes), was also identified as up-regulated in E. hyemale apical tip. Altered glyoxalase regulation has been implicated with several human diseases and disorders (Landgraf et al., 2007; Hambsch, 2011; Rabbani and Thornalley, 2011; Urscher et al., 2011). In plants, the expression of this enzyme is influenced by environmental conditions, being up-regulated during stress or conferring abiotic tolerance in several plant species (Espartero et al., 1995; Singla-Pareek et al., 2003, 2006; Lee et al., 2009; Zhou et al., 2009; Xue et al., 2011). Up-regulation of this stress-related enzyme in E. hyemale apical tip is in contrast with the pattern observed for the enzyme aldehyde dehydrogenase, which was detected in higher levels in the elongation zone, suggesting differential stress-induced machinery in close, but different, tissues.
Up-regulated proteins in the elongation zone were mostly related to carbohydrate and cell wall metabolism (Figure 6). Equisetum species are known to have the 1,3;1,4-beta-D-glucan as a major hemicellulose in plant cell walls (Fry et al., 2008; Sørensen et al., 2008). Our work showed that several glucan metabolic enzymes were identified in both apical tip and elongation zone (Table S2 in Supplementary Material). However, from the seven up-regulated proteins in the elongation zone, three were involved with mannose metabolism: Q7XPW5 (phosphomannomutase), O22287 (mannose-1-phosphate guanyltransferase beta), and Q86HG0 (mannose-1-phosphate guanyltransferase alpha). The enzyme phosphomannomutase catalyzes the conversion of mannose 6-phosphate and mannose-1-phosphate; while the mannose-1-phosphate guanyltransferase catalyzes the conversion of mannose-1-phosphate and GDP-mannose (Kruszewska et al., 1998; Qian et al., 2007; Hoeberichts et al., 2008; Badejo et al., 2009). Regulation of both enzymes leads to the formation of GDP-mannose, a central molecule that can be allocated in three different paths: protein glycosylation (Kruszewska et al., 1998), ascorbic acid biosynthesis (Qian et al., 2007; Hoeberichts et al., 2008; Badejo et al., 2009), and mannan biosynthesis (Gilbert et al., 2009). Identification of the galactomannan galactosyltransferase (Q564G7), an enzyme that catalyzes the polymerization of galactomannan (Reid et al., 2003; Edwards et al., 2004), suggests metabolism toward the biosynthesis of the polysaccharide mannan. Recently, Silva et al. (2011) demonstrated that the composition of the plant cell wall in ferns is different from higher plants, as they contain higher levels of mannan and lower levels of pectin. The identification of the enzymes involved in mannose metabolism in the elongation zone suggests that horsetails may have cell walls that are similar to ferns and indicate an active plant cell wall metabolism in this rhizome region.
Besides the enzymes involved in the biosynthesis of the cell wall, two enzymes involved in cellulose hydrolysis were also up-regulated in the rhizome elongation zone, the endoglucanase 10 (Q8LCP6) and the endoglucanase 25 (Q38890). The elongation zone as the preferential localization site of these enzymes is in accordance with their inherent activity as wall loosing agents (Yuan et al., 2001), promoting cell wall relaxation and, thus, rhizome growth.
Concluding Remarks and Implications
We employed large-scale proteome analysis of the underground system of the fern E. hyemale. The strategy used here allowed the identification of a vast number of proteins from the studied organs and characterization of the rhizome-proteome in this species. These proteins can be used as reference for comparisons with other plant species that propagate via rhizomes. Due to its key phylogenetic position, identification of rhizome-characteristic proteins in E. hyemale paves the way for a better understanding of rhizomatousness in other, distantly related species and how evolution shaped this trait. Besides the spatial characterization described in the present work, a temporal characterization of proteins and transcripts needs to be further studied in order to confirm the identity of the region-specific proteins suggested here. In addition, due to active carbohydrate metabolism in the rhizome elongation zone, analysis of protein glycosylation and identification of glycoproteins need further investigation as this may contribute to the elucidation of the signaling processes involved in the differentiation of this region. It is also important to note that tissue specific isoforms may be present in both apical tip and elongation zone tissues and that, due to the protein grouping approach used herein, we did not consider them in the present work. A complete list of all the peptides and proteins identified in this work for the species E. hyemale can be downloaded from http://www.plantrhizome.org/peptides/. All SePro filtered MS/MS spectra and the corresponding Sequest scores for each PSM may be found in the Table S4 (apical tip), Table S5 (elongation zone), and Table S6 (roots) in Supplementary Material.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Materials for this article can be found online at http://www.frontiersin.org/Plant_Proteomics/10.3389/
Figure S1. Database search evaluation for Equisetum hyemale samples. Protein sequences obtained from the translation of transcript assemblies generated from lllumina, 454 and both sequencing projects (He et al., 2012) were used for Sequest-driven searches. Candidate matches were filtered using the program SEPro (Carvalho et al., 2010). The number of spectra, peptides, proteins, and protein groups identified for each observed false discovery rate (FDR) was calculated. The number of peptides shared by two or more proteins was also computed for each evaluated database (left bottom panel).
Figure S2. Expression profile of Equisetum hyemale rhizome-characteristic proteins, hierarchical clustering was carried out for the 87 rhizome-characteristic proteins.
Table S1. List of identified peptides in Equisetum hyemale rhizomes and roots.
Table S2. List of all groups and associated proteins identified in Equisetum hyemale rhizomes and root samples. Protein grouping was carried out based on the creation of a minimal list of proteins that map to a set of peptides. For the quantitative analyses, the number of spectra acquired for each non-redundant peptide within a protein group was summed and reported for the entire group. For the GO analyses, only one protein per group (the one with the highest number of matched peptides) was selected to avoid overestimation of terms due to the presence of conserved peptides and proteins.
Table S3. List of up-regulated proteins in the Equisetum hyemale rhizomes in relation to root samples.
Table S4. List of SePro filtered MS/MS spectra containing the SEQUEST scores and proposed peptide sequences.
Table S5. List of SePro filtered MS/MS spectra containing the SEQUEST scores and proposed peptide sequences.
Table S6. List of SePro filtered MS/MS spectra containing the SEQUEST scores and proposed peptide sequences.
Acknowledgments
The authors acknowledge Cari Soderlund for providing the assembled horsetail database for querying. This project was supported by NSF grant IOS-1044821.
References
Badejo, A. A., Eltelib, H. A., Fukunaga, K., Fujikawa, Y., and Esaka, M. (2009). Increase in ascorbate content of transgenic tobacco plants overexpressing the acerola (Malpighia glabra) phosphomannomutase gene. Plant Cell Physiol. 50, 423–428.
Baerson, S. R., and Lamppa, G. K. (1993). Developmental regulation of an acyl carrier protein gene promoter in vegetative and reproductive tissues. Plant Mol. Biol. 22, 255–267.
Barriere, Y., Riboulet, C., Mechin, V., Maltese, S., Pichon, M., Cardinal, A., Lapierre, C., Lubberstedt, T., and Martinant, J.-P. (2007). Genetics and genomics of lignification in grass cell walls based on maize as model species. Genes Genomes Genomics 1, 133–156.
Benjamini, Y., and Hochberg, T. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Bonaventure, G., and Ohlrogge, J. B. (2002). Differential regulation of mRNA levels of acyl carrier protein isoforms in Arabidopsis. Plant Physiol. 128, 223–235.
Brocker, C., Lassen, N., estey, T., Pappa, A., Cantore, M., Orlova, V. V., Chavakis, T., Kavanagh, K. L., Oppermann, U., and Vasiliou, V. (2010). Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J. Biol. Chem. 285, 18452–18463.
Caraux, G., and Pinloche, S. (2005). PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21, 1280–1281.
Carpentier, S. C., Panis, B., Vertommen, A., Swennen, R., Sergeant, K., Renaut, J., Laukens, K., Witters, E., Samyn, B., and Devreese, B. (2008). Proteome analysis of non-model plants: a challenging but powerful approach. Mass Spectrom. Rev. 27, 354–377.
Carvalho, P. C., Fischer, J. S. G., Chen, E. I., Yates, J. R. III, and Barbosa, V. C. (2008). PatternLab for proteomics: a tool for differential shotgun proteomics. BMC Bioinformatics 9, 316–329. doi:10.1186/1471-2105-9-316
Carvalho, P. C., Fischer, J. S. G., Tao, X., Cociorva, D., Balbuena, T. S., Valente, R., Perales, J., Yates, J. R. III, and Barbosa, V. C. (2012). Search engine processor: filtering and organizing peptide spectrum matches. Proteomics 12, 944–949.
Carvalho, P. C., Yates, J. R. III, and Barbosa, V. C. (2010). Analyzing shotgun proteomic data with PatternLab for proteomics. Curr. Protoc. Bioinformatics 30, 13.13.1–13.13.15.
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676.
des Marais, D. L., Smith, A. R., Britton, D. M., and Pryer, K. M. (2003). Phylogenetic relationships and evolution of extant horsetails, Equisetum, based on chloroplast DNA sequence data (rbcL and trnL-F). Int. J. Plant Sci. 164, 737–751.
Edwards, M. E., Choo, T.-S., Dickson, C. A., Scott, C., Gidley, M., and Reid, J. S. G. (2004). The seeds of Lotus japonicus lines transformed with sense, antisense and sense/antisense galactomannan galactosyltransferase constructs have structurally altered galactomannans in their endosperm cell walls. Plant Physiol. 134, 1153–1162.
Espartero, J., Sanchez-Aquayo, I., and Pardo, J. M. (1995). Molecular characterization of glyoxalase-I from a higher plant; upregulation by stress. Plant Mol. Biol. 29, 1223–1233.
Fry, S. C., Nesselrode, B. H., Miller, J. G., and Mewburn, B. R. (2008). Mixed-linkage (1 → 3,1 → 4)-beta-D-glucan is a major hemicellulose of Equisetum (horsetail) cell walls. New Phytol. 179, 104–115.
Fusaro, A. F., Bocca, S. N., Ramos, R. L. B., Barroco, R. M., Magioli, C., Jorge, V. C., Coutinho, T. C., Rangel-Lima, C. M., Rycke, R. D., Inze, D., Engler, G., and Sachetto-Martins, G. (2007). AtGRP2, a cold-induced nucleo-cytoplasmic RNA-binding protein, has a role in flower and seed development. Planta 225, 1339–1351.
Gao, J., Thelen, J. J., Dunker, A. K., and Xu, D. (2010). Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol. Cell. Proteomics 9, 2586–2600.
Gierlinger, N., Sapei, L., and Paris, O. (2008). Insights into the chemical composition of Equisetum hyemale by high resolution Raman imaging. Planta 227, 969–980.
Gilbert, L., Alhagdow, M., Nunes-Nesi, A., Quemener, B., Guillon, F., Bouchet, B., Faurobert, M., Gouble, B., Page, D., Garcia, V., Petit, J., Stevens, R., Causse, M., Fernie, A. R., Lahaye, M., Rothan, C., and Baldet, P. (2009). GDP-D-mannose 3,5-epimerase (GME) plays a key role at the intersection of ascorbate and non-cellulosic cell-wall biosynthesis in tomato. Plant J. 60, 499–508.
Grundy, A. C. (2003). Predicting weed emergence: a review of approaches and future challenges. Weed Res. 43, 1–11.
Hajduch, M., Hearne, L. B., Miernyk, J. A., Casteel, J. E., Joshi, T., Agrawal, G. K., Song, Z., Zhou, M., Xu, D., and Thelen, J. J. (2010). Systems analysis of seed filling in Arabidopsis: using general linear modeling to assess concordance of transcript and protein expression. Plant Physiol. 152, 2078–2087.
Hambsch, B. (2011). Altered glyoxalase 1 expression in psychiatric disorders: cause or consequence? Semin. Cell Dev. Biol. 22, 302–308.
He, R., Kim, M.-J., Nelson, W., Balbuena, T. S., Kim, R., Kramer, R., Crow, J. A., May, G. D., Thelen, J. J., Soderlund, C. A., and Gang, D. R. (2012). Next generation sequencing based transcriptomic and proteomic analysis of the common reed, Phragmites australis (Poaceae), reveals genes involved in invasiveness and rhizome specificity. Am. J. Bot. 99, 232–247.
Hoeberichts, F., Vaeck, E., Kiddle, G., Coppens, E., Cotte, B. V. D., Adamantidis, A., Ormenese, S., Foyer, C. H., Zabeau, M., Inze, D., Perilleux, C., Breusegem, F. V., and Vuylsteke, M. (2008). A temperature-sensitive mutation in the Arabidopsis thaliana phosphomannomutase gene disrupts protein glycosylation and triggers cell death. J. Biol. Chem. 283, 5708–5718.
Horiguchi, G., Kodama, H., and Iba, K. (2003). Mutations in a gene for plastid ribosomal protein S6-like protein reveal a novel developmental process required for the correct organization of lateral root meristem in Arabidopsis. Plant J. 33, 521–529.
Hu, F. Y., Tao, D. Y., Sacks, E., Fu, B. F., Xu, P., Li, J., Yang, Y., McNally, K., Khush, G. S., Paterson, A. H., and Li, Z. K. (2003). Convergent evolution of perenniality in rice and sorghum. Proc. Natl. Acad. Sci. U.S.A. 100, 4050–4054.
Huang, W., Ma, X., Wang, Q., Gao, Y., Xue, Y., Niu, X., Yu, G., and Liu, Y. (2008). Significant improvement of stress tolerance in tobacco plants by overexpressing a stress-responsive aldehyde dehydrogenase gene from maize (Zea mays). Plant Mol. Biol. 68, 451–463.
Iijima, M., Shimizu, H., Tanaka, Y., and Urushihara, H. (1998). A Dictyostelium discoideum homologue to Tcp-1 is essential for growth and development. Gene 213, 101–106.
Jang, C. S., Kamps, T. L., Skinnner, D. N., Schulze, S. R., Vencill, W. K., and Paterson, A. H. (2006). Functional classification, genomic organization putatively cis-acting regulatory elements, and relationship to quantitative trait loci of Sorghum genes with rhizome-enriched expression. Plant Physiol. 142, 1148–1159.
Jang, C. S., Kamps, T. L., Tang, H., Bowers, J. E., Lemke, C., and Paterson, A. H. (2009). Evolutionary fate of rhizome-specific genes in a non-rhizomatous Sorghum genotype. Heredity 102, 266–273.
Jorrin-Novo, J. V., Maldonado, A. M., Echevarria-Zomeno, S., Valledor, L., Castillejo, M. A., Curto, M., Valero, J., Sghaier, B., Donoso, G., and Redondo, I. (2009). Plant proteomics update (2007–2008). Second-generation proteomic techniques, an appropriate experimental design, and data analysis to fulfill MIAPE standards, increase plant proteome coverage and expand biological knowledge. J. Proteomics 72, 285–314.
Komatsu, S., Kobayashi, Y., Nishizawa, K., Nanjo, Y., and Furukawa, K. (2010). Comparative proteomics analysis of differentially expressed proteins in soybean cell wall during flooding stress. Amino Acids 39, 1435–1449.
Kruszewska, J. S., Saloheimo, M., Penttila, M., and Palamarczyk, G. (1998). Isolation of a Trichoderma reesei cDNA encoding GTP: α-D-mannose-e-phosphate guanyltransferase involved in early steps of protein glycosylation. Curr. Genet. 33, 445–450.
Landgraf, R., Kesler, M. S., Bunck, M., Murgatroyd, C., Spengler, D., Zimbelmann, M., Nusbaumer, M., Czibere, L., Turck, C. W., Singewald, N., Rujescu, D., and Frank, E. (2007). Candidate genes of anxiety-related behavior in HAB/LAB rats and mice: focus on vasopressin and glyoxalase-I. Neurosci. Biobehav. Rev. 31, 89–102.
Large, M. F., Blanchon, D. J., and Angus, M. L. (2006). Devitalisation of imported horsetail (Equisetum hyemale). N. Z. J. Crop Hortic. Sci. 34, 151–153.
Lee, D.-G, Ahsan, N., Lee, S.-H., Lee, J.-J., Bahk, J. D., Kang, K. Y., and Lee, B.-H. (2009). Chilling stress-induced proteomic changes in rice roots. J. Plant Physiol. 166, 1–11.
Lewis, V. A., Hynes, G. M., Zheng, D., Saibil, H., and Willison, K. (1992). T-complex polypeptide-1 is a subunit of a heteromeric particle in the eukaryotic cytosol. Nature 358, 249–252.
Lum, J. H.-K., Fung, K.-L., Cheung, P.-Y., Wong, M.-S, Lee, C.-H., Kwok, F. S.-L., Leung, M. C.-P., Hui, P.-K., and Lo, S. C.-L. (2002). Proteome of oriental ginseng Panax ginseng C. A. Meyer and the potential to use it as an identification tool. Proteomics 2, 1123–1130.
Migliore, L., Rotini, A., Randazzo, D., Albanese, N. N., and Giallongo, A. (2007). Phenols content and 2-D electrophoresis protein pattern: a promising tool to monitor Posidonia meadows health state. BMC Ecol. 7, 6. doi:10.1186/1472-6785-7-6
Minnebruggen, A. V., Neyt, P., Groeve, S. D., Coussens, G., Ponce, M. R., Micol, J. L., and Lijsebettens, M. V. (2010). The ang3 mutation identified the ribosomal protein gene RPL5B with a role in cell expansion during organ growth. Physiol. Plant. 138, 91–101.
Missihoun, T. D., Schmitz, J., Klug, R., Kirch, H.-H., and Bartels, D. (2011). Betaine aldehyde dehydrogenase genes from Arabidopsis with different sub-cellular localization affect stress responses. Planta 233, 369–382.
Mori, M., Murata, K., Kubota, H., Yamamoto, A., Matsushiro, A., and Morita, T. (1992). Cloning of a cDNA encoding the Tcp-1 (t complex polypeptide 1) homologue of Arabidopsis thaliana. Gene 381–382.
Nair, R. B., Bastress, K. L., Ruegger, M. O., Denault, J. W., and Chapple, C. (2004). The Arabidopsis thaliana REDUCED EPIDERMAL FLUORESCENCE 1 gene encodes an aldehyde dehydrogenase involved in ferulic acid and sinapic acid biosynthesis. Plant Cell 16, 544–554.
Ohlrogge, J. B., and Kuo, T. M. (1985). Plants have different isoforms for acyl carrier protein that are expressed differently in different tissues. J. Biol. Chem. 260, 8032–8037.
Picton, S., Gray, J. E., Payton, S., Barton, S. L., Lowe, A., and Grierson, D. (1993). A histidine decarboxylase-like mRNA is involved in tomato fruit ripening. Plant Mol. Biol. 23, 627–631.
Prentis, P. J., Wilson, J. R. U., Dormontt, E. E., Richardson, D. M., and Lowe, A. J. (2008). Adaptive evolution in invasive species. Trends Plant Sci. 13, 288–293.
Qian, W., Yu, C., Qin, H., Liu, X., Zhang, A., Johansen, I. E., and Wang, D. (2007). Molecular and functional analysis of phosphomannomutase (PMM) from higher plants and genetic evidence for the involvement of PMM in ascorbic acid biosynthesis in Arabidopsis and Nicotiana benthaminana. Plant J. 49, 399–413.
Rabbani, N., and Thornalley, P. J. (2011). Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol. 22, 309–317.
Reid, J. S. G., Edwards, M. E., Dickson, C. A., Scott, C., and Gidley, M. J. (2003). Tobacco transgenic lines that express fenugreek galactomannan galactosyltransferase constitutively have structurally altered galactomannans in their seed endosperm cell walls. Plant Physiol. 131, 1487–1495.
Remmerie, N., Vijlder, T., Laukens, K., Dang, T. H., Lemiere, F., Mertens, I., Valkenborg, D., Blust, R., and Witters, E. (2011). Next generation functional proteomics in non-model plants: a survey on techniques and applications for the analysis of protein complexes and post-translational modifications. Phytochemistry 72, 1192–1218.
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V., and Mann, M. (2007). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860.
Silva, G. B., Ionashiro, M., Carrara, T. B., Crivellari, A. C., Tine, M. A. S., Prado, J., Carpita, N. C., and Buckeridge, M. S. (2011). Cell wall polysaccharides from fern leaves: evidence for a mannan-rich type III cell wall in Adiantum raddianum. Phytochemistry 72, 2352–2360.
Singla-Pareek, S. L., Reddy, M. K., and Sopory, S. K. (2003). Genetic engineering of the glyoxalase pathway in tobacco leads to enhanced salinity tolerance. Proc. Natl. Acad. Sci. U.S.A. 100, 14672–14677.
Singla-Pareek, S. L., Yadav, S. K., Pareek, A., Reddy, M. K., and Sopory, S. K. (2006). Transgenic tobacco overexpressing glyoxalase pathway enzymes grow and set viable seeds in zinc-spiked soils. Plant Physiol. 140, 613–623.
Sokal, R. R., and Michener, C. D. (1958). A statistical method for evaluating systematic relationships. Univ. Kans. Sci. Bull. 38, 1409–1438.
Sørensen, I., Pettolino, F. A., Wilson, S. M., Doblin, M. S., Johansen, B., Bacic, A., and Willats, W. G. (2008). Mixed-linkage (1 → 3),(1 → 4)-beta-D-glucan is not unique to the Poales and is an abundant component of Equisetum arvense cell walls. Plant J. 54, 510–521.
Spollen, W. G., Tao, W., Valliyodan, B., Chen, K., Hejlek, L. G., Kim, J.-J., LeNoble, M. E., Zhu, J., Bohnert, H. J., Henderson, D., Schachtman, D. P., Davis, G. E., Springer, G. K., Sharp, R. E., and Nguyen, H. T. (2008). Spatial distribution of transcript changes in the maize primary root elongation zone at low water potential. BMC Plant Biol. 8, 32. doi:10.1186/1471-2229-8-32
Stiti, N., Adewale, I. O., Petersen, J., Bartels, D., and Kirch, H.-H. (2011). Engineering the nucleotide coenzyme specificity and sulfhydryl redox sensitivity of two stress-responsive aldehyde dehydrogenase isoenzymes of Arabidopsis thaliana. Biochem. J. 434, 459–471.
Suzuki, M., Kato, A., and Komeda, Y. (2000). Na RNA-binding protein, AtRBP1, is expressed in actively proliferative regions in Arabidopsis thaliana. Plant Cell Physiol. 41, 282–288.
Szakonyi, D., and Byrne, M. E. (2011). Involvment of ribosomal protein RPL27a in meristem activity and organ development. Plant Signal. Behav. 6, 712–714.
Urscher, M., Alisch, R., and Deponte, M. (2011). The glyoxalase system of malaria parasites-implications for cell biology and general glyoxalase research. Semin. Cell. Dev. Biol. 22, 262–270.
Wang, R., Guegler, K., LaBrie, S. T., and Crawford, N. M. (2000). Genomic analysis of a nutrient response in Arabidopsis reveals diverse expression patterns and novel metabolic and potential regulatory genes induced by nitrate. Plant Cell 12, 1491–1509.
Wernersson, R. (2006). Virtual ribosome – a comprehensive translation tool with support for sequence feature integration. Nucleic Acids Res. 34, W385–W388.
Xu, C., Sibicky, T., and Huang, B. (2010). Protein profile analysis of salt-responsive proteins in leaves and roots in two cultivars of creeping bentgrass differing in salinity tolerance. Plant Cell Rep. 29, 595–615.
Xue, G.-P., Way, H. M., Richardson, T., Drenth, J., Joyce, P. A., and McIntyre, C. L. (2011). Overexpression of TaNAC69 leads to enhanced transcript levels of stress up-regulated genes and dehydration tolerance in bread wheat. Mol. Plant 4, 697–712.
Yaffe, M. B., Farr, G. W., Miklos, D., Horwich, A. L., Sternlicht, M. L., and Sternlich, H. (1992). TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature 358, 245–248.
Yamaguchi, M., Valliyodan, B., Zhang, J., LeNoble, M. E., Yu, O., Rogers, E. E., Nguyen, H. T., and Sharp, R. E. (2010). Regulation of growth response to water stress in the soybean primary root. I. Proteomic analysis reveals region-specific regulation of phenylpropanoid metabolism and control of free iron in the elongation zone. Plant Cell Environ. 33, 223–243.
Yuan, S., Wu, Y., and Cosgrove, D. (2001). A fungal endoglucanase with plant cell wall extension activity. Plant Physiol. 127, 324–333.
Zeh, M., Leggewie, G., Hoefgen, R., and Hesse, H. (2002). Cloning and characterization of a cDNA encoding a cobalamin-independent methionine synthase from potato (Solanum tuberosum L.). Plant Mol. Biol. 48, 255–265.
Zhou, S., Sauve, R., and Thannhauser, T. W. (2009). Proteome changes induced by aluminuim stress in tomato roots. J. Exp. Bot. 60, 1849–1857.
Zhu, J., Alvarez, S., Marsh, E. L., LeNoble, M. E., Cho, I.-J., Sivaguru, M., Chen, S., Nguyen, H. T., Wu, Y., Schachtman, D. P., and Sharp, R. E. (2007). Cell wall proteome in the maize primary root elongation zone. II. Region-specific changes in water soluble and lightly ionically bound proteins under water deficit. Plant Physiol. 145, 1533–1548.
Keywords: rough horsetail, ferns, label-free proteomics, spectral counting
Citation: Balbuena TS, He R, Salvato F, Gang DR and Thelen JJ (2012) Large-scale proteome comparative analysis of developing rhizomes of the ancient vascular plant Equisetum hyemale. Front. Plant Sci. 3:131. doi: 10.3389/fpls.2012.00131
Received: 27 March 2012; Accepted: 01 June 2012;
Published online: 26 June 2012.
Edited by:
Alex Jones, The Sainsbury Laboratory, UKReviewed by:
Sebastien Carpentier, KU Leuven, BelgiumLaurence Veronique Bindschedler, University of Reading, UK
Copyright: © 2012 Balbuena, He, Salvato, Gang and Thelen. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Tiago Santana Balbuena, Instituto de Biologia-Bloco J, Universidade Estadual de Campinas, Rua Monteiro Lobato 970, CEP 13.083-970 Campinas, São Paulo, Brazil. e-mail: tsbalbuena@yahoo.com.br