 Yiqian Fu
Yiqian Fu G. Danny Esselink
G. Danny Esselink Richard G. F. Visser
Richard G. F. Visser Jaap M. van Tuyl
Jaap M. van Tuyl Paul Arens
Paul Arens- Plant Breeding, Wageningen University and Research Centre (Wageningen UR), Wageningen, Netherlands
For the ornamental crop Gerbera hybrida, breeding at the moment is done using conventional methods. As this has drawbacks in breeding speed and efficiency, especially for complex traits like disease resistance, we set out to develop genomic resources. The leaf and flower bud transcriptomes of four parents, used to generate two gerbera populations, were sequenced using Illumina paired-end sequencing. In total, 36,770 contigs with an average length of 1397 bp were generated and these have been the starting point for SNP identification and annotation. The consensus contig sequences were used to map reads of individual parents, to identify genotype specific SNPs, and to assess the presence of common SNPs between genotypes. Comparison with the non-redundant protein database (nr) showed that 29,146 contigs gave BLAST hits. Of sequences with blast results, 73.3% obtained a clear gene ontology (GO) annotation. EST contigs coding for enzymes were found in Kyoto Encyclopedia of Genes and Genomes maps (KEGG). Through, these annotated data and KEGG molecular interaction network, transcripts associated with the phenylpropanoid metabolism, other secondary metabolite biosynthesis pathways, phytohormone biosynthesis and signal transduction were analyzed in more detail. Identifying genes involved in these processes could provide genetic and genomic resources for studying the mechanism of disease resistance in gerbera.
Introduction
Gerbera hybrida (2n = 2x = 50) is one of the most important ornamental plants and belongs to the Compositae family. Cultivated gerbera, which probably originates from a crossing of two wild species from Africa (G. jamesonii and G. viridifolia; Hansen, 1999), is highly heterozygous. Commercial gerbera cultivars are mainly produced in greenhouses for year round cut flower production (Moyer and Peres, 2008; Simpson, 2009) and ranked fifth in the cut flower sale at the Dutch flower auctions 2014 (https://www.floraholland.com/media/3949227/Kengetallen-2014-Engels.pdf). Gerbera became a model plant to study flower development in composed (Compositea) flowers (Teeri et al., 2006). Furthermore, the high variation in flower color and patterning of ray and disc florets as well as the high levels of secondary metabolites derived from connected pathways make it a putative model crop for biosynthetic research (Teeri et al., 2006). Given the importance of gerbera in floriculture and breeding as well as its potential for fundamental research on flower developmental and regulation of secondary metabolites, there is a demand for genomic resources.
In general, the use of molecular markers in breeding for ornamental crops has been lagging behind other agricultural and horticultural crops (Arens et al., 2012; Smulders et al., 2012). This is partly due to some breeding traits for ornamentals like flower color that are themselves easily visible markers. Also, it is more complicated to develop molecular markers for ornamental crops since they are highly heterozygous with complex genetic background (Debener, 2009). In gerbera, there are only a small amount of SSR and RGA (resistance gene analog) markers (Gong and Deng, 2010, 2012; Seo et al., 2012) available for genetic studies in this species.
With the rapid progress in high-throughput next-generation sequencing (NGS) technologies, new possibilities for creating genomic resources and identifying (SNP) markers have become feasible. Transcriptome RNA sequencing (RNA-seq) provides significant advantages for ornamental crops where genomic resources are still scarce and high levels of heterozygosity are expected. Because gerbera has a relatively large genome size, sequencing transcripts as a genome complexity reduction not only reduces cost and time significantly, but also contributes to establishment of resources by the focus on genes. Furthermore, in species with a very high diversity, many SNPs may not be useable markers because of flanking SNPs. Targeting genic regions which have a lower expected SNP diversity may reduce this and result in more widely applicable markers. At present only two studies have contributed to genomic resources building in gerbera. Using Sanger sequencing, an ESTs database with nearly 17,000 cDNA sequences was already constructed for mining genes involved in gerbera floral development (Laitinen et al., 2005). A transcriptome of the gerbera ray floret sequenced by NGS sequencing was constructed to predict genes involved in gibberellin metabolism and signal transduction (Kuang et al., 2013). Although, these transcriptome analyses have been reported in gerbera, these studies were not focussed on finding SNP markers and focussed strictly on flowers.
SNPs that can be discovered from expressed sequence tags (ESTs) NGS-sequencing are valuable resources for genetic research and accepted as markers for MAS in ornamentals (Shahin et al., 2012; Koning-Boucoiran et al., 2015). RNA-Seq can generate numerous transcripts with sufficient read-depth to guarantee high quality SNP identification (Kim et al., 2014). Development of SNP markers for the highly heterozygous ornamentals is very feasible and 200–1000 SNP markers will be sufficient to construct a genetic map for QTL mapping (Smulders et al., 2012).
In this study, we aim for the identification of SNP markers from the transcriptomes of four gerbera genotypes based on leaf and flower tissues using NGS sequencing. Through alignment of reads from four genotypes with consensus contigs constructed by de novo assembly, we expect to identify SNPs within and between cultivars and detect reliable SNPs markers that can be used for mapping and other genetic studies. Transcriptomes are analyzed by gene annotation and predicted candidate genes that relate to disease resistance pathways, and to gerbera gray mold in particular, will be shown as examples. Gerbera gray mold is a main problem in gerbera production in greenhouses which is caused by Botrytis cinerea. As a necrotrophic pathogen, a series of plant secondary metabolites from the phenylpropanoid and flavonoid biosynthesis pathway are considered to be involved in plant defense responses (Dixon, 2001; Dixon et al., 2002). Phytohormone jasmonate (JA) and ethylene (ET) also play a role in plant defense against B. cinerea (Thomma et al., 2001). Identifying the gene sequences involved in these pathways will help us to study their gene function in gerbera upon Botrytis infestation. SNPs found will provide genetic tools for gerbera breeding that may help in efficient gerbera improvement.
Materials and Methods
Plant Materials, RNA Isolation and cDNA Library Construction
Three Mini Gerbera breeding lines (“SP1,” “SP2,” “FP1”) and a garden gerbera breeding line (“FP2”) that are also the parents of two gerbera populations were used for cDNA sequencing. The selected 4 parental genotypes show different symptoms on Botrytis susceptibility and the two populations of these parents showed the largest variation on Botrytis susceptibility among 20 populations tested. Young leaves and floral buds of the four parents were collected and stored at −80°C upon RNA isolation.
Total RNA of the leaves and floral buds for the four parents was isolated according to the standard TRIZol reagent protocol (Life Technologies, USA) followed by purification using the RNeasy isolation Kit (Qiagen, Germany). Total RNA of leaves and floral buds was mixed in equal amounts and sent to GATC Biotech (Germany) for sequence library preparation.
Sequencing, Assembly and SNP Detection
The cDNA libraries of all four genotypes were sequenced using 2 × 100 bp paired-end sequencing on an Illumina HiSeq platform (Illumina, USA). Reads were pre-processed using ConDeTri (Content Dependent Read Trimmer) (Smeds and Künstner, 2011) with default settings to trim adapter sequences from the 3′ and 5′ ends from reads and to filter reads with low quality. To improve the quality of assemblies, FLASH (Fast Length Adjustment of Short reads) (Magoč and Salzberg, 2011) was used with default settings to merge overlapping read pairs. For de novo assembly, transcripts of four parents were constructed separately by Trinity (Grabherr et al., 2011) from the merged, single-end and paired-end reads.
Construction of a reference transcriptome was performed in a stepwise procedure. In short, transcriptome of SP1 was assembled de novo and redundancy was removed by reassembling the transcriptome using CAP3 (Huang and Madan, 1999) with default setting and an identity (−p) of 95%. Next, the transcripts of SP2 were added to the CAP3 contigs and singlets of SP1 and assembled again with the same settings. In a similar way the transcripts of FP1 and FP2 were added and contigs were reassembled. The final consensus contig sequences were used as a reference transcriptome for SNP detection.
For SNP detection the raw reads were pre-processed using Prinseq-lite (vs. 0.20.3) which included the trimming of nucleotides having a phred score lower than 25, the trimming of poly A/T tails, the removal of duplicate reads, of low complexity reads (DUST approach), of reads shorter than 50 bp and of reads with more than one ambiguous nucleotide. The remaining reads of each genotype where aligned to the reference transcriptome using Bowtie2 (–very-sensitive setting) and filtered for mapping quality (>2) using SAMtools (Li et al., 2009). The resulting sam files were merged and used for SNP calling using QualitySNPng (Nijveen et al., 2013) with default settings. Retrieved SNP regions were blasted (BLASTn, e-value: 1E-30) to the contigs derived from the EST sequences as a control for possible paralog presence.
GO Annotation, Enzyme Code Annotation and KEGG Annotation
To predict function, assembled unigene contigs were annotated. Gene ontology (GO) annotation in Blast2GO (Conesa et al., 2005) consisted of three steps: blasting, mapping and annotation. The assembled contigs were compared by BLASTX against the NCBI non-redundant protein database (nr) using Blast2GO V.3.0. The expectation value (E-value) threshold was set at 1E-3 for reporting matches and the number of retrieved hits at 20 (default value). After blasting, Gene Ontology (GO) terms associated to the hits were mapped. When a BLAST result is successfully mapped to one or several GO terms, GO annotations were assigned.
Enzyme code (EC) annotation was available only for contig sequences with GO annotations with EC numbers. Additionally, the KEGG mapping was done to display enzymatic functions in the context of the metabolic pathways in which they participate.
Results
Sequencing and SNP Detection
The transcriptome reads of four genotypes were obtained using Illumina 2 × 100 bp paired-end sequencing. For genotype “SP1,” sequencing the cDNA library resulted in a total of 114,519,206 raw paired end reads. After trimming and removing reads with low quality 80,182,250 (70%) paired end reads remained. Merging connected paired-end reads using FLASH software resulted in 46,043,245 single reads and 5,931,379 paired end reads for de novo assembly. The de novo assembly for “SP1” resulted into 113,970 transcripts longer than 200 bp. Results of sequencing assembly data for all four genotypes are shown in Table 1. All raw data has been donated to the SRA (Short Read Archive) and can be found under accession numbers PRJEB12127.
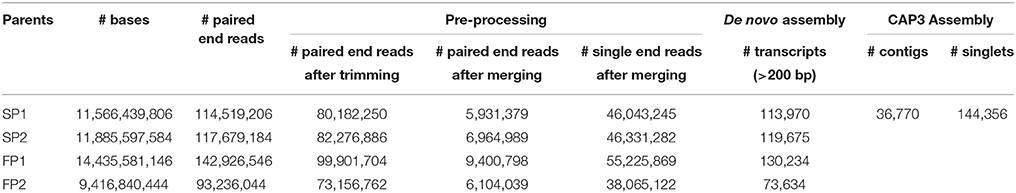
Table 1. Summary of the sequence assembly data.
Transcripts of parent “SP1” were first used to construct a reference transcriptome after which transcripts of the other genotypes were one by one added to reach an overall consensus assembly (Figure 1). The final consensus transcriptome yielded 36,770 consensus contigs and 144,356 singletons. The average length of consensus contigs was 1397 bp, and the N50 was equal to 1889 bp (The minimum length of 201 bp, median length of 1130 bp and maximum length of 15746 bp). This consensus transcriptome (named “Cap3Contigs_All”) was the starting point for SNP identification and annotation. All paired-end and single-end reads of the four genotypes were mapped to the 36,770 “Cap3Contigs_All” consensus contigs for SNP detection. In total 398,917 SNPs polymorphic within or between the four genotypes were detected. Genetic diversity of the consensus sequences on average is 7.8 SNPs per kb. The average SNP density within the four genotypes varied from 3.7 to 4.8 SNPs per kb of sequence. They all harbor quite a lot parent specific SNPs and population specific SNPs polymorphic in only that specific parent or population (Table 2).
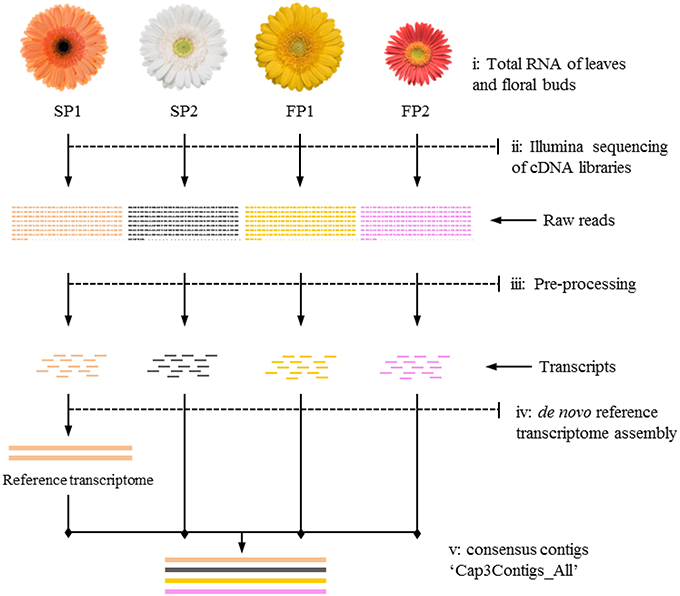
Figure 1. Workflow of transcriptome sequencing for four parents. (i) Leaves and floral buds of the four gerbera genotypes used to isolate RNA; (ii) Mixed cDNA libraries of four genotypes sequenced on an Illumina platform; (iii) Raw reads used for pre-processing to trim adapter sequences and to filter reads with low quality; (iv) Transcripts of SP1 assembled as first step toward reference transcriptome construction; (v) Transcripts of other genotypes mapped to reference to yield consensus contigs named “Cap3Contigs_All” for SNP detection and annotation.
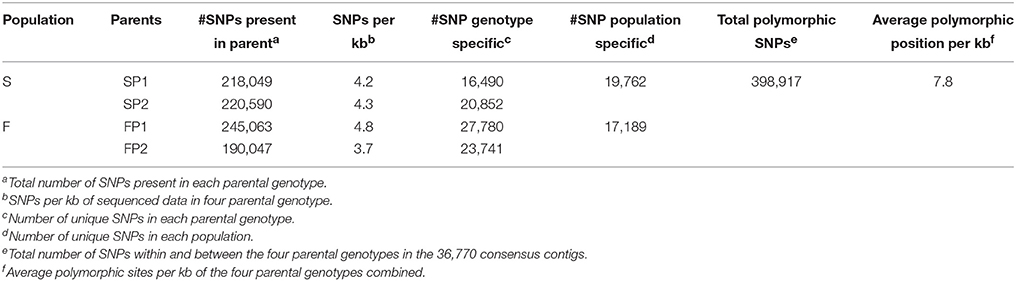
Table 2. SNPs on parents and consensus sequences.
GO, EC, KEGG Annotation
The 36,770 consensus contigs “Cap3Contigs_All” were used for blasting against the NCBI non-redundant protein database (nr) to look for the most similar proteins for each contig (Table 3). A total number of 29,146 contigs gave BLAST hits (79.2%). Out of the best-hit for every contig with BLAST result (See Table S1), the E-value of 9398 contigs (~32%) is below an value of 1e−180, and 2481 contigs (8.5%) with the E-value greater than 1e−20, others are in between. Sequences similarity distribution chart (see Figure S1A) shows that most (91.2%) of the BLAST hits have sequence similarity values higher than 60% and half of them (50.4%) higher than 80% with our gerbera consensus contigs. Most blast hits were found from grape (Vitis vinifera), soybean (Glycine max), poplar (Populus trichocarpa), potato (Solanum tuberosum), tomato (S. lycopersicum) and cacao tree (Theobroma cacao) (Figure S1B). Most of these species also feature in the top hits distribution with grape as main contributor (Figure S1C).

Table 3. Transcriptomes annotation process results.
The GO terms were obtained during the mapping step. Out of sequences with blast results, 73.3% (21,357 contigs) could be GO annotated. The sequences with GO annotation were described in terms of biological processes, cellular components and molecular functions. Top 20 GO terms of the three separate aspects were listed in Table S2.
Enzyme annotations were also done in contigs with GO annotations. A total of 8761 contigs eventually showed EC numbers and the enzyme code distribution is shown in Table 4. KEGG mapping displayed enzymatic functions in the context of the metabolic pathways in which they participate. The EC annotated contigs are involved in a total of 144 different metabolic pathways, including all kinds of carbohydrate metabolic pathways, amino acid metabolic pathways, nitrogen metabolic pathways, as well as a series of secondary metabolic biosynthesis pathways. The top 30 pathways in overall sequence coverage and the details of all 144 pathways with contig identity and enzyme code can be found in Tables S3, S4, respectively.

Table 4. Gerbera transcripts enzyme code distribution.
Transcripts Related to Phenylpropanoid Biosynthesis and Flavonoid Biosynthesis Pathway
Based on the EC annotated sequences, enzymes involved in phenylpropanoid and flavonoid biosynthesis pathway that are considered to be involved in flower color and disease resistance were retrieved and highlighted in different colors in the pathway-maps from KEGG (see Figures S2, S3). There are 137 contigs that translate to 14 enzymes in the phenylpropanoid biosynthesis pathway. Eleven enzymes represented by 71 contigs were found for the flavonoid biosynthesis pathway. These two pathway-maps loaded from KEGG (see Figures S2, S3) included all possible enzymes and metabolites in a broad perspective, but we can see clearly from the simplified phenylpropanoid and flavonoid biosynthetic pathway (Figure 2) that the key enzymes in the pathways are well represented. The three key regulatory enzymes in the phenylpropanoid biosynthesis are phenylalanine ammonia lyase (PAL, EC:4.3.1.24), cinnamate-4-hydroxylase (C4H, EC:1.14.13.11), 4-coumarate-CoA ligase (4CL, EC:6.2.1.12), which were represented by 11, 2, and 12 homologous contigs respectively. The sequence similarities of the best-hit for these 25 contigs are above 90% and 17 of the best-hits come from other species within the Compositae like, Helianthus tuberosus, Lactuca sativa, Artemisia sieberi, Cynara cardunculus, etc indication that these pathways are well conserved within the family. After the formation of p-coumaroyl-CoA, the next step is into the central flavonoid pathway. Chalcone synthase (CHS, EC:2.3.1.74) and chalcone isomerase (chalcone-flavanone isomerase, CHI, EC:5.5.1.6.) are the first two enzymes in the flavonoid pathway leading to subsequent metabolite synthesis. Eight out of the 10 contigs which are predicted as CHS gene are highly identical to the gerbera CHS genes in public databases (Z38096.1, Z38097.2, Z38098.1, AM906210.1, AM906211.1, X91339.1) with a very low (or zero) E-value and similarity close to 100%. No contig were found specific for stilbene synthase (STS, EC:2.3.1.95) which is the key enzyme for stilbene synthesis. Enzymes in this pathway and the number of contigs homologous to these enzymes are show in Figure 2.
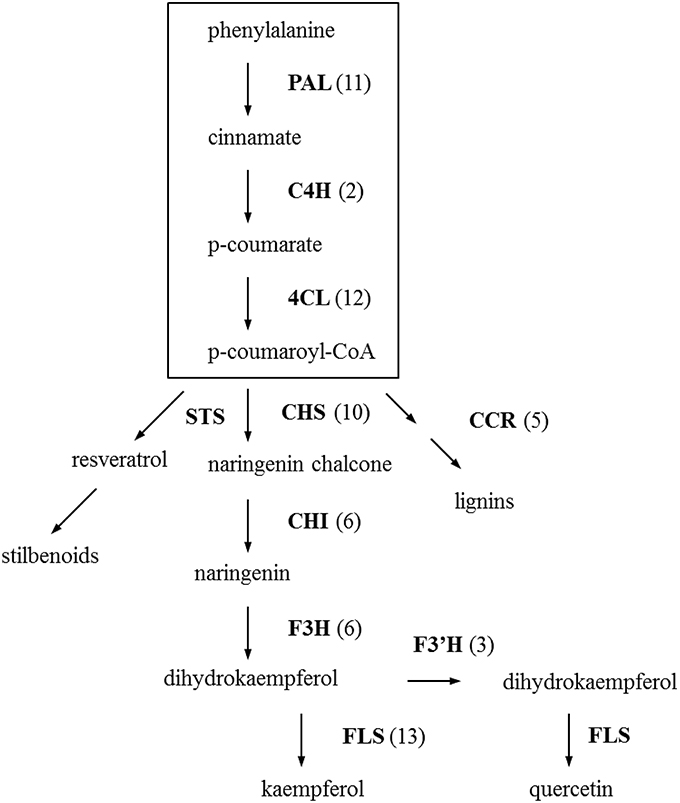
Figure 2. Distribution of Gerbera transcripts in the simplified phenylpropanoid and flavonoid biosynthesis pathway (Dixon et al., 2002; Ainasoja, 2008; Ali et al., 2011; Ferreyra et al., 2012; Boubakri et al., 2013). The processes in the box indicate the general phenylpropanoid pathway and the rest is flavonoid pathway. Each enzyme name is followed with the number of contigs homologous to the gene family encoding this enzyme between brackets. PAL, phenylalanine ammonia-lyase; C4H, cinnamate-4-hydroxylase; 4CL, 4-coumarate-CoA ligase; CHS, chalcone synthase; STS, stilbenes synthase; CCR, cinnamoyl-CoA reductase; CHI, chalcone isomerase/chalcone-flavanone isomerase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; FLS, flavonol synthase.
Transcripts Related to Phytohormone Biosynthesis and Signaling
The initial precursor for ethylene synthesis is the amino acid methionine. The three key regulatory enzymes in the pathway are S-adenosyl-l-methionine synthase (SAMS, EC:2.5.1.6), ACC synthase (ACS, EC:4.4.1.14) and ACC oxidase (AOC, EC:1.14.17.4). Our gerbera EST database contains multiple contigs encoding these three enzymes (see Figure 3). Jasmonate biosynthesis start from linolenic acid. Lipoxygenase (LOX, EC:1.13.11.12), allene oxide synthase (AOS, EC:4.2.1.92), allene oxide cyclase (AOC, EC:5.3.99.6) and 12-oxo-phytodienoic acid reductase (OPDR, EC:1.3.1.42) participant in the synthesis. Numbers of contigs encoding these enzymes are show in Figure 4. We also found the multiple contigs connected with these two plant hormone signaling pathway which were shown on Figure 5, although some of them still remained without coverage.

Figure 3. Distribution of Gerbera transcripts in the ethylene biosynthetic pathway (Wang et al., 2002). Each enzyme name is followed with the number of contigs homologous to the gene family encoding this enzyme between brackets. SAMS, S-adenosyl methionine synthase; ACS, 1-aminocyclopropane-1-carboxylic acid (ACC) synthase; ACO, ACC oxidase.
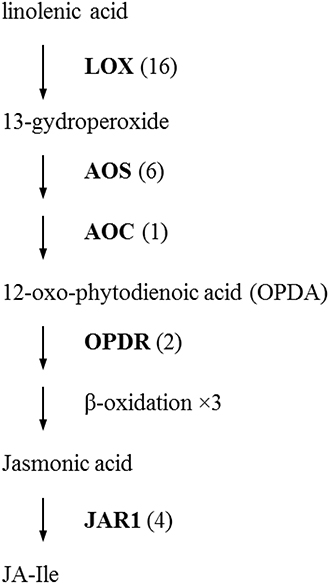
Figure 4. Distribution of Gerbera transcripts in the Jasmonic acid biosynthetic pathway (Howe, 2001; Wasternack, 2007). Each enzyme name is followed with the number of contigs homologous to the gene family encoding this enzyme between brackets. LOX, lipoxygenase; AOS, allene oxide synthase; AOC, allene oxide cyclase; OPDR, 12-oxo-phytodienoic acid reductase; JAR1, JA amino acid conjugate synthase; JA-Ile, jasmonate-isoleucine.
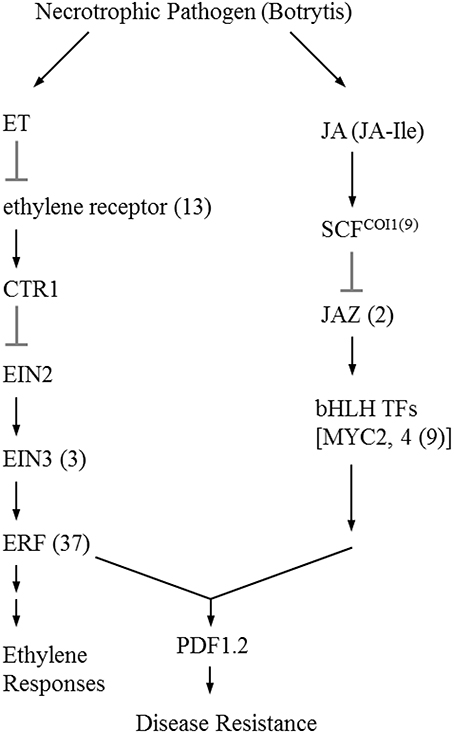
Figure 5. Simplified signaling ethylene and jasmonic acid signal transduction during Botrytis infection (Wang et al., 2002; Katsir et al., 2008a). Each enzyme name is followed with the number of contigs homologous to the gene family encoding this enzyme between brackets. ET, ethylene; CTR1, constitutive triple response 1; EIN2, ethylene insensitive 2; EIN3, Ethylene insensitive 3; ERF; ethylene response factor; JA, jasmonate; JA-Ile, jasmonate-isoleucine; SCF, Skp/Cullin/F-box; COI1, coronatine-insensitive 1; JAZ, jasmonate ZIM domain; bHLH TFs, basic-helix-loop-helix transcription factors; PDF1.2, plant defensin 1.2.
Discussion
In this study, we developed transcriptomes (based on leaf and flower tissues) of gerbera using RNA sequencing in four gerbera genotypes. After de novo assembly, we generated 36,770 consensus contigs with an average length of 1397 bp and a N50 length of 1889 bp. Average length and N50 length are markedly larger than a transcriptome of flower development (845 bp and 1321 bp respectively; Kuang et al., 2013). This is likely because four gerbera genotypes were used instead of one which increases chances for higher read coverage and wider sequence overlap.
SNPs present within genotypes and between genotypes were detected from the alignment of reads of all parents with the consensus contigs. In all those consensus contigs with a total length of 51,360,054 bp, 398,917 polymorphic loci (SNPs present either in parent or between parents) were identified with an average SNP density of 7.8 SNPs per 1kb whereas within genotype SNP density ranged from 3.7 to 4.8 SNPs per 1 kb. These numbers are comparable to for instance rose 4–6 SNP/kb (Koning-Boucoiran et al., 2015) and sunflower 6.1 SNP/kb (Bachlava et al., 2012). In eleven safflower (Carthamus tinctorius) individuals exons and introns sequences of 7 genes showed a SNP density of 10.5 SNP/kb (Chapman and Burke, 2007) whereas SNP densities varied from 2.6 to 26.9 SNP/kb in 10 intron regions from eight safflower accessions (Chapman et al., 2007). The frequency of polymorphisms in safflower seems higher than that in overall consensus gene sequences of gerbera but this could be biased because of the small set of genes studied in safflower. Furthermore, polymorphism rates in intron are higher than in exons since introns are under less strict selection pressure (Tamura et al., 2013).
The highest numbers of homologs were found with Vitis vinifera (grapevine). Interestingly, grapevine is a crop that also is known for its high number of secondary metabolites and its interaction with B. cinerea. Many studies have been performed on this crop-pathogen from multiple aspects (Jeandet et al., 1991; Hain et al., 1993; Goetz et al., 1999; Coutos-Thévenot et al., 2001; Bézier et al., 2002; Poinssot et al., 2003; Trotel-Aziz et al., 2006; Deytieux-Belleau et al., 2009; Timperio et al., 2012) that could be instructive for the interaction of the pathogen with gerbera as well.
There is general recognition that various natural secondary metabolites play an important role in plant defense (Dixon, 2001; Dixon et al., 2002; Howlett, 2006; van Baarlen et al., 2007). Plants combating the necrotrophic pathogen Botrytis especially utilize this tool (Oliver and Ipcho, 2004; Glazebrook, 2005). The precursors for these compounds, which are involved in physical and chemical barriers, such as lignin and phytoalexins, are derived from the phenylpropanoid pathway. Enzymes involved in the biosynthesis of the general phenylpropanoid pathway have been well studied (see Dixon et al., 2002). For instance, enzyme activities increased lignification in wheat upon B. cinerea infection (Maule and Ride, 1976, 1983). In our study, we identified homologs for the three key genes (PAL, C4H, and 4CL) in the core of the phenylpropanoid pathway in gerbera, and found multiple transcripts encoding the three enzymes. For 17 out of the 25 best-hits homologs come from Compositae species.
The flavonoid pathway is closely linked with the phenylpropanoid pathway and the precursor is a phenylpropanoid-derived compound. Flavonoid biosynthesis will yield in different flower color pigments (Winkel-Shirley, 2001) but may also produce a range of plant defense compounds (Treutter, 2005). For instance, chalcone synthase (CHS) belongs to the type III polyketide synthase (PKS) superfamily (Austin and Noel, 2003; Abe and Morita, 2010) and is the key enzyme toward the flavonoid biosynthesis. Members in the type III PKS superfamily, including stilbene synthases (STS) and 2-pyrone synthase (2-PS) in gerbera, share high amino acid similarities and are highly correlated with Botrytis resistance. Grapevine synthesizes stilbenes upon Botrytis infection (Jeandet et al., 1991; Goetz et al., 1999). Tobacco transformed with a stilbene synthase gene from grapevine showed increased resistance to B. cinerea (Hain et al., 1993). The 2-pyrone synthase (2-PS) codes for polyketide synthase which synthesizes a putative precursor for two phytoalexins in gerbera. Knocking out this gene resulted in increased susceptibility to B. cinerea (Koskela et al., 2011). Deng et al. (2014) confirmed that CHS enzymes in gerbera are encoded by a family of three genes. We found at least 10 transcripts annotated to the chalcone and stilbene synthase family protein. Eight of them showed high similarity at nucleotide level with gerbera CHS or CHS-like genes in public databases, whereas the other two showed only low amino acid similarity (40%) to known gerbera sequences. The latter two give the best hits to chalcone and stilbene synthases from T. cacao (XP_007041944.1, 90%) and a putative chalcone synthase from Artemisia annua (ACY74337.1, 93%). Based on the phylogenetic tree of the amino acid sequences of chalcone- and stilbene-like synthases (Figure S4 and Table S5), these two transcripts (GhCHS1 and GhCHS2 in Figure S4) belong to a clearly separate group of putative chalcone and stilbene synthases.
In this study, we also exampled the possibilities of the presented transcriptome on plant hormone ethylene and jasmonate biosynthesis and signaling networks that are considered to play an important role in plant resistance in general and Botrytis in specific. For instance, ethylene production plays an important role in plant resistance against B. cinerea (Broekaert et al., 2006). The rate-limiting step of ethylene synthesis is the conversion of SAMe to ACC by ACC synthase (ACS; Kende, 1993). A multigene family codes for ACC synthase in plants. Nine contigs were found in our data related with ACS genes. Similarly, the Arabidopsis and tomato genomes contain nine ACS genes (Harpaz-Saad et al., 2012). Two knockout mutants of type I ACS isoforms in Arabidopsis, acs2 and acs6, reduced B. cinerea-induced ethylene biosynthesis (Han et al., 2010). ACS-silenced apple fruit was more susceptible to B. cinerea than untransformed apple (Akagi et al., 2011). The activity of lipoxygenase (LOX), a key JA biosynthetic enzyme, is also highly related to Botrytis resistance (Azami-Sardooei et al., 2010). Our gerbera EST database contained multiple transcripts encoding key enzymes in ethylene and jasmonate synthesis pathways. An efficient defense response to Botrytis also need genes in signaling transduction pathways, such as EIN2 in ethylene signaling (Thomma et al., 1999), COI1 and JAZ in jasmonate signaling (Cerrudo et al., 2012). COI1 protein was shown to mediate JAZ degradation to release its bound downstream TFs (e.g., MYC2) for defense gene expression (Katsir et al., 2008b; Kazan and Manners, 2013). Jasmonoyl–isoleucine (JA-Ile), which is the only bioactive jasmonates derivative by a JA conjugate synthase (JAR1) confirmed so far, directly promotes their interaction (Katsir et al., 2008a; Wasternack, 2007). In Arabidopsis, the coi1 and other mutations that block functional JA signaling, showed increased susceptibility to Botrytis and decreased induction of the plant antimicrobial metabolite camalexin after infection (Rowe et al., 2010). Some of these components in the signal transduction pathway still remained without coverage which may be related to the RNA-seq source that is from unchallenged material as the main focus was building generic genomic resources and SNP detection.
Through analysis of the large gerbera EST database that was generated from next-generation sequencing, we identified a series of SNP markers for further linkage mapping and also identified transcripts that might be involved in interesting pathways for both fundamental as well as applied studies as was exampled for Botrytis resistance. We expect these genes can provide genetic resources for studying the mechanism of disease resistance and developing markers for gerbera breeding in the future.
Author Contributions
Conception of the study: YF, PA, JV and RV; Practical design of the study: PA, YF and GE; Development of methodology: YF, GE and PA; Assembly and SNP detection: GE; Annotation of transcripts: YF; Writing of the manuscript: YF, GE, and PA. Manuscript modification and discussions: PA, RV and JV. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful for the support from the Foundation Technological Top Institute Green Genetics (3CFL030RP) and from gerbera breeding companies Florist Holland BV and Schreurs Holland BV.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00247
References
Abe, I., and Morita, H. (2010). Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat. Prod. Rep. 27, 809–838. doi: 10.1039/b909988n
Ainasoja, M. (2008). Secondary Metabolites in Gerbera hybrida. [PhD Thesis], University of Helsinki, Helsinki.
Akagi, A., Dandekar, A. M., and Stotz, H. U. (2011). Resistance of Malus domestica Fruit to Botrytis cinerea depends on Endogenous Ethylene Biosynthesis. Phytopathology 101, 1311–1321. doi: 10.1094/PHYTO-03-11-0087
Ali, M. B., Howard, S., Chen, S., Wang, Y., Yu, O., Kovacs, L. G., et al. (2011). Berry skin development in Norton grape: distinct patterns of transcriptional regulation and flavonoid biosynthesis. BMC Plant Biol. 11:7. doi: 10.1186/1471-2229-11-7
Arens, P., Bijman, P., Tang, N., Shahin, A., and van Tuyl, J. M. (2012). “Mapping of disease resistance in ornamentals: a long haul,” in Proceedings of the 24th International Eucarpia Symposium Section Ornamentals “Ornamental breeding worldwide,” Vol. 953, ed T. Orlikowska (Warsaw), 231–238.
Austin, M. B., and Noel, A. J. P. (2003). The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110. doi: 10.1039/b100917f
Azami-Sardooei, Z., França, S. C., De Vleesschauwer, D., and Höfte, M. (2010). Riboflavin induces resistance against Botrytis cinerea in bean, but not in tomato, by priming for a hydrogen peroxide-fueled resistance response. Physiol. Mol. Plant Pathol. 75, 23–29. doi: 10.1016/j.pmpp.2010.08.001
Bachlava, E., Taylor, C. A., Tang, S., Bowers, J. E., Mandel, J. R., Burke, J. M., et al. (2012). SNP discovery and development of a high-density genotyping array for sunflower. PLoS ONE 7:e29814. doi: 10.1371/journal.pone.0029814
Bézier, A., Lambert, B., and Baillieul, F. (2002). Study of defense-related gene expression in grapevine leaves and berries infected with Botrytis cinerea. Euro. J. Plant Pathol. 108, 111–120. doi: 10.1023/A:1015061108045
Boubakri, H., Poutaraud, A., Wahab, M. A., Clayeux, C., Baltenweck-Guyot, R., Steyer, D., et al. (2013). Thiamine modulates metabolism of the phenylpropanoid pathway leading to enhanced resistance to Plasmopara viticola in grapevine. BMC Plant Biol. 13:31. doi: 10.1186/1471-2229-13-31
Broekaert, W. F., Delaure, S. L., De Bolle, M. F. C., and Cammue, B. P. A. (2006). The role of ethylene in host-pathoven interactions Annual Review of Phytopathology 44, 393–416. doi: 10.1146/annurev.phyto.44.070505.143440
Cerrudo, I., Keller, M. M., Cargnel, M. D., Demkura, P. V., de Wit, M., Patitucci, M. S., et al. (2012). Low Red/Far-Red ratios reduce arabidopsis resistance to Botrytis cinerea and jasmonate responses via a COI1-JAZ10-Dependent, Salicylic Acid-Independent Mechanism. Plant Physiol. 158, 2042–2052. doi: 10.1104/pp.112.193359
Chapman, M. A., and Burke, J. M. (2007). DNA sequence diversity and the origin of cultivated safflower (Carthamus tinctorius L., Asteraceae). BMC Plant Biol. 7:60. doi: 10.1186/1471-2229-7-60
Chapman, M. A., Chang, J., Weisman, D., Kesseli, R. V., and Burke, J. M. (2007). Universal markers for comparative mapping and phylogenetic analysis in the Asteraceae (Compositae). Theor. Appl. Genet. 115, 747–755. doi: 10.1007/s00122-007-0605-2
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Coutos-Thévenot, P., Poinssot, B., Bonomelli, A., Yean, H., Breda, C., Buffard, D., et al. (2001). In vitro tolerance to Botrytis cinerea of grapevine 41B rootstock in transgenic plants expressing the stilbene synthase Vst1 gene under the control of a pathogen-inducible PR 10 promoter. J. Exp. Bot. 52, 901–910. doi: 10.1093/jexbot/52.358.901
Debener, T. (2009). “Current strategies and future prospects of resistance breeding in ornamentals,” in 23rd International Eucarpia Symposium, Section Ornamentals: Colourful Breeding and Genetics, Vol. 836, eds J. M. van Tuyl and D. P. de Vries (Leiden: Acta Horticulturae), 125–130.
Deng, X., Bashandy, H., Ainasoja, M., Kontturi, J., Pietiäinen, M., Laitinen, R. A. E., et al. (2014). Functional diversification of duplicated chalcone synthase genes in anthocyanin biosynthesis of Gerbera hybrida. New Phytol. 201, 1469–1483. doi: 10.1111/nph.12610
Deytieux-Belleau, C., Geny, L., Roudet, J., Mayet, V., Donèche, B., and Fermaud, M. (2009). Grape berry skin features related to ontogenic resistance to Botrytis cinerea. Eur. J. Plant Pathol. 125, 551–563. doi: 10.1007/s10658-009-9503-6
Dixon, R. A. (2001). Natural products and plant disease resistance. Nature 411, 843–847. doi: 10.1038/35081178
Dixon, R. A., Achnine, L., Kota, P., Liu, C. J., Reddy, M. S. S., and Wang, L. (2002). The phenylpropanoid pathway and plant defence - a genomics perspective. Mol. Plant Pathol. 3, 371–390. doi: 10.1046/j.1364-3703.2002.00131.x
Ferreyra, M. L. F., Rius, S. P., and Casati, P. (2012). Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3:222. doi: 10.3389/fpls.2012.00222
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227. doi: 10.1146/annurev.phyto.43.040204.135923
Goetz, G., Fkyerat, A., Métais, N., Kunz, M., Tabacchi, R., Pezet, R., et al. (1999). Resistance factors to grey mould in grape berries: identification of some phenolics inhibitors of Botrytis cinerea stilbene oxidase. Phytochemistry 52, 759–767. doi: 10.1016/S0031-9422(99)00351-9
Gong, L., and Deng, Z. (2010). EST-SSR markers for gerbera (Gerbera hybrida). Mol. Breed. 26, 125–132. doi: 10.1007/s11032-009-9380-x
Gong, L., and Deng, Z. (2012). Selection and application of SSR markers for variety discrimination, genetic similarity and relation analysis in gerbera (Gerbera hybrida). Sci. Hortic. 138, 120–127. doi: 10.1016/j.scienta.2012.02.020
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Hain, R., Reif, H. J., Krause, E., Langebartels, R., Kindl, H., Vornam, B., et al. (1993). Disease resistance results from foreign phytoalexin expression in a novel plant. Nature 361, 153–156. doi: 10.1038/361153a0
Han, L., Li, G.-J., Yang, K.-Y., Mao, G., Wang, R., Liu, Y., et al. (2010). Mitogen-activated protein kinase 3 and 6 regulate Botrytis cinerea-induced ethylene production in Arabidopsis. Plant J. 64, 114–127. doi: 10.1111/j.1365-313x.2010.04318.x
Harpaz-Saad, S., Yoon, G. M., Mattoo, A. K., and Kieber, J. J. (2012). The Formation of ACC and competition between Polyamines and Ethylene for SAM. Annu.Plant Rev. 44, 53–81. doi: 10.1002/9781118223086.ch3
Howe, G. A. (2001). Cyclopentenone signals for plant defense: Remodeling the jasmonic acid response. Proc. Natl. Acad. Sci. U.S.A. 98, 12317–12319. doi: 10.1073/pnas.231480898
Howlett, B. J. (2006). Secondary metabolite toxins and nutrition of plant pathogenic fungi. Curr. Opin. Plant Biol. 9, 371–375. doi: 10.1016/j.pbi.2006.05.004
Huang, X., and Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Jeandet, P., Bessis, R., and Gautheron, B. (1991). The production of Resveratrol (3,5,4′-Trihydroxystilbene) by Grape Berries in different developmental stages. Am. J. Enol. Vitic. 42, 41–46.
Katsir, L., Chung, H. S., Koo, A. J. K., and Howe, G. A. (2008a). Jasmonate signaling: a conserved mechanism of hormone sensing. Curr. Opin. Plant Biol. 11, 428–435. doi: 10.1016/j.pbi.2008.05.004
Katsir, L., Schilmiller, A. L., Staswick, P. E., He, S. Y., and Howe, G. A. (2008b). COI1 is a critical component of a receptor for jasmonate and the bacterial virulence factor coronatine. Proc. Natl. Acad. Sci. U.S.A. 105, 7100–7105. doi: 10.1073/pnas.0802332105
Kazan, K., and Manners, J. M. (2013). MYC2: the master in action. Mol. Plant 6, 686–703. doi: 10.1093/mp/sss128
Kende, H. (1993). Ethylene biosynthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 44, 283–307. doi: 10.1146/annurev.pp.44.060193.001435
Kim, J.-E., Oh, S.-K., Lee, J.-H., Lee, B.-M., and Jo, S.-H. (2014). Genome-Wide SNP calling using next generation sequencing data in tomato. Mol. Cells 37, 36–42. doi: 10.14348/molcells.2014.2241
Koning-Boucoiran, C. F. S., Esselink, G. D., Vukosavljev, M., van 't Westende, W. P. C., Gitonga, V. W., Krens, F. A., et al. (2015). Using RNA-Seq to assemble a rose transcriptome with more than 13,000 full-length expressed genes and to develop the WagRhSNP 68k Axiom SNP array for rose (Rosa L.). Front. Plant Sci. 6:249. doi: 10.3389/fpls.2015.00249
Koskela, S., Söderholm, P. P., Ainasoja, M., Wennberg, T., Klika, K. D., Ovcharenko, V. V., et al. (2011). Polyketide derivatives active against Botrytis cinerea in Gerbera hybrida. Planta 233, 37–48. doi: 10.1007/s00425-010-1277-8
Kuang, Q., Li, L., Peng, J., Sun, S., and Wang, X. (2013). Transcriptome Analysis of Gerbera hybrida Ray Florets: putative genes associated with Gibberellin metabolism and signal transduction. PLoS ONE 8:e57715. doi: 10.1371/journal.pone.0057715
Laitinen, R. A. E., Immanen, J., Auvinen, P., Rudd, S., Alatalo, E., Paulin, L., et al. (2005). Analysis of the floral transcriptome uncovers new regulators of organ determination and gene families related to flower organ differentiation in Gerbera hybrida (Asteraceae). Genome Res. 15, 475–486. doi: 10.1101/gr.3043705
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maule, A. J., and Ride, J. P. (1976). Ammonia-lyase and O-methyl transferase activities related to lignification in wheat leaves infected with Botrytis. Phytochemistry 15, 1661–1664. doi: 10.1016/S0031-9422(00)97448-X
Maule, A. J., and Ride, J. P. (1983). Cinnamate 4-hydroxylase and hydroxycinnamate: CoA ligase in wheat leaves infected with Botrytis cinerea. Phytochemistry 22, 1113–1116. doi: 10.1016/0031-9422(83)80202-7
Moyer, C., and Peres, N. A. (2008). “Evaluation of biofungicides for control of powdery mildew of gerbera daisy,” in Paper Presented at the Proceedings of the Florida State Horticultural Society (FSHS) (Fort Lauderdale, FL), 389–394.
Nijveen, H., van Kaauwen, M., Esselink, D. G., Hoegen, B., and Vosman, B. (2013). QualitySNPng: a user-friendly SNP detection and visualization tool. Nucleic Acids Res. 41, W587–W590. doi: 10.1093/nar/gkt333
Oliver, R. P., and Ipcho, S. V. S. (2004). Arabidopsis pathology breathes new life into the necrotrophs-vs.-biotrophs classification of fungal pathogens. Mol. Plant Pathol. 5, 347–352. doi: 10.1111/j.1364-3703.2004.00228.x
Poinssot, B., Vandelle, E., Bentéjac, M., Adrian, M., Levis, C., Brygoo, Y., et al. (2003). The Endopolygalacturonase 1 from Botrytis cinerea activates grapevine defense reactions unrelated to its enzymatic activity. Mol. Plant Microbe Inter. 16, 553–564. doi: 10.1094/MPMI.2003.16.6.553
Rowe, H. C., Walley, J. W., Corwin, J., Chan, E. K. F., Dehesh, K., and Kliebenstein, D. J. (2010). Deficiencies in jasmonate-mediated plant defense reveal quantitative variation in botrytis cinerea pathogenesis. PLoS Pathog. 6:e1000861. doi: 10.1371/journal.ppat.1000861
Seo, K.-I., Lee, G.-A., Park, S.-K., Yoon, M.-S., Ma, K.-H., Lee, J.-R., et al. (2012). Genome shotgun sequencing and development of microsatellite markers for gerbera (Gerbera hybrida H.) by 454 GS-FLX. Afr. J. Biotechnol. 11, 7388–7396. doi: 10.5897/AJB11.4064
Shahin, A., van Gurp, T., Peters, S. A., Visser, R. G., van Tuyl, J. M., and Arens, P. (2012). SNP markers retrieval for a non-model species: a practical approach. BMC Res. Notes 5:79. doi: 10.1186/1756-0500-5-79
Simpson, B. B. (2009). “Economic Importance of Compositae,” in Systematics, Evolution, and Biogeography of Compositae, eds V. A. Funk, A. Susanna, T. F. Stuessy, and R. J. Bayer (Vienna: International Association for Plant Taxonomy), 45–58.
Smeds, L., and Künstner, A. (2011). ConDeTri - a content dependent read trimmer for illumina data. PLoS ONE 6:e26314. doi: 10.1371/journal.pone.0026314
Smulders, M. J. M., Vukosavljev, M., Shahin, A., van de Weg, W. E., and Arens, P. (2012). “High throughput marker development and application in horticultural crops,” in 7th International Symposium on In Vitro Culture and Horticultural Breeding, Vol. 961, ed D. Geelen (Ghent: Acta Horticulturae), 547–551.
Tamura, K.-I., Yonemaru, J.-I., and Yamada, T. (2013). “Insertion-deletion marker targeting for intron polymorphisms,” in Diagnostics in Plant Breeding, eds T. Lübberstedt and R. K. Varshney (Dordrecht: Springer), 211–228.
Teeri, T. H., Elomaa, P., Kotilainen, M., and Albert, V. A. (2006). Mining plant diversity: Gerbera as a model system for plant developmental and biosynthetic research. Bioessays 28, 756–767. doi: 10.1002/bies.20439
Thomma, B. P. H. J., Eggermont, K., Tierens, K. F. M. J., and Broekaert, W. F. (1999). Requirement of functional ethylene-insensitive 2 gene for efficient resistance of Arabidopsis to infection by Botrytis cinerea. Plant Physiol. 121, 1093–1101. doi: 10.1104/pp.121.4.1093
Thomma, B. P. H. J., Penninckx, I. A. M. A., Cammue, B. P. A., and Broekaert, W. F. (2001). The complexity of disease signaling in Arabidopsis. Curr. Opin. Immunol. 13, 63–68. doi: 10.1016/S0952-7915(00)00183-7
Timperio, A. M., D'Alessandro, A., Fagioni, M., Magro, P., and Zolla, L. (2012). Production of the phytoalexins trans-resveratrol and delta-viniferin in two economy-relevant grape cultivars upon infection with Botrytis cinerea in field conditions. Plant Physiol. Biochem. 50, 65–71. doi: 10.1016/j.plaphy.2011.07.008
Treutter, D. (2005). Significance of flavonoids in plant resistance and enhancement of their biosynthesis. Plant Biol. 7, 581–591. doi: 10.1055/s-2005-873009
Trotel-Aziz, P., Couderchet, M., Vernet, G., and Aziz, A. (2006). Chitosan Stimulates defense reactions in grapevine leaves and inhibits development of botrytis cinerea. Eur. J. Plant Pathol. 114, 405–413. doi: 10.1007/s10658-006-0005-5
van Baarlen, P., Legendre, L., and van Kan, J. A. L. (2007). “Plant defence compounds against botrytis infection,” in Biology, Pathology and Control, eds Y. Elad, B. Williamson, P. Tudzynski, and N. Delen (Botrytis: Springer), 143–161.
Wang, K. L. C., Li, H., and Ecker, J. R. (2002). Ethylene biosynthesis and signaling networks. Plant Cell 14, S131–S151. doi: 10.1105/tpc.001768
Wasternack, C. (2007). Jasmonates: an update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 100, 681–697. doi: 10.1093/aob/mcm079
Keywords: EST, candidate gene, annotation, disease pathway, gerbera gray mold
Citation: Fu Y, Esselink GD, Visser RGF, van Tuyl JM and Arens P (2016) Transcriptome Analysis of Gerbera hybrida Including in silico Confirmation of Defense Genes Found. Front. Plant Sci. 7:247. doi: 10.3389/fpls.2016.00247
Received: 04 September 2015; Accepted: 14 February 2016;
Published: 01 March 2016.
Edited by:
Thomas Debener, Leibniz University Hannover, GermanyReviewed by:
Kathryn Kamo, United States Department of Agriculture, USAMichael H. Walter, Leibniz Institute of Plant Biochemistry, Germany
Copyright © 2016 Fu, Esselink, Visser, van Tuyl and Arens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Arens, paul.arens@wur.nl