 Xiu Huang1
Xiu Huang1 Hai-Dong Yan1
Hai-Dong Yan1 Xin-Quan Zhang1Jian Zhang2*Taylor P. Frazier3De-Jun Huang2Lu Lu4
Xin-Quan Zhang1Jian Zhang2*Taylor P. Frazier3De-Jun Huang2Lu Lu4 Lin-Kai Huang1*Wei Liu1Yan Peng1
Lin-Kai Huang1*Wei Liu1Yan Peng1 Xiao Ma1Yan-Hong Yan1
Xiao Ma1Yan-Hong Yan1- 1Department of Grassland Science, Animal Science and Technology College, Sichuan Agricultural University, Chengdu, China
- 2Herbivorous Livestock Research Institute, Chongqing Academy of Animal Sciences, Chongqing, China
- 3Department of Horticulture, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA
- 4Department of Biochemistry and Molecular Biology, Biosciences Faculty, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, Spain
Hemarthria R. Br. is an important genus of perennial forage grasses that is widely used in subtropical and tropical regions. Hemarthria grasses have made remarkable contributions to the development of animal husbandry and agro-ecosystem maintenance; however, there is currently a lack of comprehensive genomic data available for these species. In this study, we used Illumina high-throughput deep sequencing to characterize of two agriculturally important Hemarthria materials, H. compressa “Yaan” and H. altissima “1110.” Sequencing runs that used each of four normalized RNA samples from the leaves or roots of the two materials yielded more than 24 million high-quality reads. After de novo assembly, 137,142 and 77,150 unigenes were obtained for “Yaan” and “1110,” respectively. In addition, a total of 86,731 “Yaan” and 48,645 “1110” unigenes were successfully annotated. After consolidating the unigenes for both materials, 42,646 high-quality SNPs were identified in 10,880 unigenes and 10,888 SSRs were identified in 8330 unigenes. To validate the identified markers, high quality PCR primers were designed for both SNPs and SSRs. We randomly tested 16 of the SNP primers and 54 of the SSR primers and found that the majority of these primers successfully amplified the desired PCR product. In addition, high cross-species transferability (61.11–87.04%) of SSR markers was achieved for four other Poaceae species. The amount of RNA sequencing data that was generated for these two Hemarthria species greatly increases the amount of genomic information available for Hemarthria and the SSR and SNP markers identified in this study will facilitate further advancements in genetic and molecular studies of the Hemarthria genus.
Introduction
Hemarthria R. Br. is one of the most important genera of the Poaceae family. This genus consists of about 20 species that are distributed across wide geographic regions. Among these species, the two most important and widely studied are Hemarthria compressa and Hemarthria altissima. H. compressa, also known as whipgrass, is mainly distributed in China and is one of the most significant and widely utilized forage grasses in southern China. Three commercial hexaploid (2n = 6x = 54) H. compressa cultivars (“Yaan,” “Guangyi” and “Chonggao”) have been released by the Sichuan Agricultural University (Yang et al., 2003; Guo et al., 2014). H. altissima, commonly known as limpograss, is native to Africa; however, it was later introduced into the southeastern part of the United States where its germplasms has since been evaluated. Four commercial H. altissima cultivars, including the diploids (2n = 2x = 18) “Greenalta” and “Redalta” and the tetraploids (2n = 4x = 36) “Bigalta” and “Floralta,” have been released (Huang et al., 2014d). These two species of Hemarthria have been extensively used and commercially grown in subtropical and tropical areas due to their fast growth, high forage yield, good quality, and tolerance to poorly drained soils. In addition, these two Hemarthria species are known to play an essential role in local animal husbandry and agro-ecosystem maintenance (Rumball and Lambert, 1981; Yang et al., 2003; Huang et al., 2014a,d).
Previous molecular studies on Hemarthria concentrated on estimating genetic diversity within naturally occurring germplasm, constructing DNA fingerprints, and associating molecular markers with agronomically important traits (Chen et al., 2011a; Guo et al., 2014; Huang et al., 2014a). A variety of molecular markers have been developed for Hemarthria, particularly for H. compressa and H. altissima, based on anonymous DNA sequences (Chen et al., 2005, 2011b; Liu et al., 2006; Huang et al., 2008, 2014b,c). In addition, only 19 PopSet (sequence sets from phylogenetic and population studies) and 37 nucleotide sequences of Hemarthria have been deposited in GenBank. Hemarthria species are some of the most economically and ecologically important forage crops and have been used for grazing, modulating hay, making silage, soil and water conservation, and greening the environment. Despite its singnificance, there is a serious lack of available genomic information available for Hemarthria spieces, which has greatly limited progress with breeding studies in this genus. Therefore, additional high quality genomic resources of Hemarthria are urgently needed in order to understand the molecular mechanisms underlying desirable agronomic traits in this important forage species.
High-throughput next-generation sequencing (NGS) technologies are rapidly transforming the fields of ecology, evolution, and genetics by increasing the amount of large-scale genomic and transcriptomic data that is available for non-model plant species (Rokas and Abbot, 2009; Renaut et al., 2010). NGS technologies can efficiently produce enormous amounts of sequence data at a significantly reduced cost. There are three main commercial NGS platforms including the Applied Biosystems SOliD System, Roche/454 GS FLX Instrument, and the Illumina/Solexa Genome Analyzer that can be widely applied to generate massively parallel DNA sequencing reads. Among these technologies, the Illumina sequencing platform is a short-read based technology that utilizes reversible terminator chemistry (Trick et al., 2009). Though the Illumina platform produces shorter reads than the other platforms, it offers several advantages over the others including a high level of accuracy, throughput, and cost effectiveness. In addition, a plethora of powerful bioinformatics tools have been developed for the analysis of Illumina sequencing data, which have addressed the short-comings of the sequencing technology. Currently, this sequencing-by-synthesis platform has been successfully used in whole genome sequencing (Li et al., 2010, 2013), digital gene expression analysis (Nagalakshmi et al., 2008), DNA-protein interaction profiling (Johnson et al., 2007), small RNA identification (Vidal et al., 2013), and the sequencing of transcriptomes (Sablok et al., 2014; Yates et al., 2014; Xie et al., 2015).
With the development of NGS technologies, RNA sequencing (RNA-Seq) has emerged as a powerful tool for transcriptome analysis. RNA sequencing can largely and precisely quantify gene expression levels with a high degree of sensitivity. Additionally, RNA-Seq has a high level of reproducibility for both technical and biological replicates. With minimal RNA input, RNA-Seq can also accelerate the assembly of transcriptomes and the identification of expressed genes including gene isoforms and alternatively spliced gene products (Cloonan et al., 2008; Nagalakshmi et al., 2008; Wang et al., 2009, 2010; Zhang et al., 2010; Vijay et al., 2013). RNA-Seq technology can extensively and accurately sequence expressed genes. It can also detect and characterize nucleotide variations such as SNPs and SSRs (Kaur et al., 2012). Finally, RNA-Seq does not rely on existing genomic sequences, which makes RNA-Seq especially useful for the analysis of non-model species that possess large nuclear genomes such as polyploids (Wang et al., 2009). To date, numerous RNA-Seq analyses have been reported for non-model plant species (Toledo-Silva et al., 2013; Wang et al., 2013; Yates et al., 2014); however, no RNA-Seq experiments have been performed for any Hemarthria species.
In this study, we used the Illumina HiSeq™2500 platform to perform a large-scale transcriptome analysis of two different Hemarthria species, H. compressa “Yaan” and H. altissima “1110.” H. compressa “Yaan” is one of three registered cultivars of H. compressa and is tall, light green in color, and has a high yield. H. altissima “1110,” a wild material of H. altissima that is derived from KwaZulu-Nata, South Africa, is also tall and high-producing. The main objectives of this study were to enrich the genomic resources available for Hemarthria species and to develop, characterize, and validate SNP and SSR molecular markers for the two materials. The results of this study will provide valuable genomic resources that contribute to (1) understanding the relationships among germplasm within the Hemarthria genus, (2) identifing Hemarthria varieties, (3) associating molecular markers with agronomically important traits of H. compressa and H. altissima, and (4) facilitating further advancements in marker-assisted selection (MAS), comparative transcriptomic studies, and candidate genes research in Hemarthria species.
Materials and Methods
Ethics Statement
This study was approved by the Department of Grassland Science, Animal Science and Technology College, Sichuan Agricultural University. H. compressa “Yaan” and H. altissima “1110” are not endangered or protected materials.
Plant Material and RNA Isolation
Hemarthria species are infertile grasses that propagate through rhizomes. In this study, the rhizomes of H. compressa “Yaan” and H. altissima “1110” were used for further research. Rhizomes of each material were collected from individual clones and were cultivated in Erlenmeyer flasks (200 mL volume) containing Hoagland's nutrient solution (Hoagland and Arnon, 1950). The Erlenmeyer flasks containing the rhizomes were placed in a temperature-controlled growth chamber programmed at 28°C/19°C (average day/night temperature) under a light/dark photoperiod of 16 h/8 h. The nutrient solution was changed every 3 days. After 2 months of growth, the leaves and roots of two individuals per material were collected separately and immediately frozen in liquid nitrogen. The tissue was stored at −80°C until RNA isolation.
To isolate RNA, equal amounts of leaf or root tissue from individual clones of the same material were pooled together to create one sample. The samples were labeled as follows: T01 = the mixed leaves of “Yaan,” T02 = the mixed roots of “Yaan,” T03 = the mixed leaves of “1110,” and T04 = the mixed roots of “1110.” Total RNA was extracted using an RNAprep pure Plant Kit (Tiangen Biotech, China) in accordance with the manufacturer's protocol. The purity and concentration of total RNA was detected using a Nanodrop 2000 UV-Vis spectrophotometer (Thermo Fisher Scientific Inc., USA) and a Qubit® 2.0 Fluorometer (Life Technologies, USA). The RNA Integrity Number (RIN) for the four RNA samples was examined with an Agilent 2100 Bioanalyzer (Agilent Technologies, USA).
cDNA Library Construction and Illumina Sequencing
The four total RNA samples from the two Hemarthria species were sent to Biomarker Technologies Co., Ltd. (Beijing, China) for the construction of cDNA libraries and for Illumina sequencing reactions. After the total RNA was extracted, the mRNA in each of the samples was purified and enriched using magnetic oligo(dT)-rich beads. Next, a specialized buffer and high temperatures were used to chemically break the mRNA into fragments. Using reverse transcriptase and random hexamer-primers, the cleaved mRNA fragments were then used as templates to synthesize first-strand cDNA. Next, second-strand cDNA was synthesized in a buffer containing dNTPs, DNA polymerase I, and RNaseH. After purification of the double-stranded cDNA with Agencourt AMPure XP beads, the purified cDNA ends were repaired using T4 DNA polymerase and Klenow DNA polymerase, which adds a single A base to the end of the sequence. The repaired cDNA fragments were then ligated to sequencing adapters. Agencourt AMPure XP beads were used to select suitable length fragments that functioned as sequencing templates for downstream analyses. Next, PCR amplification was then performed using the Phusion High-Fidelity DNA polymerase in order to enrich the purified cDNA template. To ensure that the quality of the library was sufficient for sequencing, the concentration and insert size of the library was detected using a Qubit® 2.0 Fluorometer and Agilent 2100 Bioanalyzer, respectively. Additionally, the effective concentration of the library was accurately quantified using quantitative PCR (Q-PCR). Finally, the qualified cDNA library was sequenced on the Illumina HiSeq™ 2500 sequencing platform using paired-end technology in a single run.
De novo Transcriptome Assembly and Annotation
The abundant raw reads that were obtained from the Illumina sequencing platform (NCBI SRA: SRP058845) were initially processed in order to ensure the accuracy of de novo assembly and subsequent analyses. The pre-processing guidelines included trimming reads with adaptors and eliminating low quality reads (reads with ambiguous “N” bases >5% and more than 10% Q < 20 bases). The clean reads from each library were evaluated for GC-content, N-content, Q20, CycleQ20, and Q30. Next, Trinity (http://trinityrnaseq.sourceforge.net/) software, which is specific for high-throughput transcript assembly of RNA-Seq data without a reference genome (Grabherr et al., 2011), was used to separately assemble the clean reads from each library into unigene sequences with the parameters set at K-mer length of 25, a similarity of 80%, and the other parameters set to their default values. The Trinity software used three independent modules: Inchworm, Chrysalis and Butterfly, which first combined all the clean reads with a certain length of overlap to form contigs (longer contiguous fragments without N). Then, the software let the clean reads map back to the contigs and used paired end reads to calculate the distance and relation among these contigs. Next, Trinity connected these contigs to obtain consensus sequences that could not be extended on either end were called transcripts. Finally, the transcripts were then further clustered into unigenes.
The program BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to assign putative functions to the assembled unigenes (Altschul et al., 1997). All of the unigene sequences were aligned using BLASTx (e < 1e-5) to the following publicly available protein databases: National Center for Biotechnology Information (NCBI) non-redundant protein (Nr), Swiss-Prot protein, Protein family (Pfam), Eukaryotic Orthologous Groups of proteins (KOG), Gene Ontology (GO), and the Kyoto Encyclopedia of Genes and Genomes (KEGG). In addition, a BLASTn search was performed against the NCBI non-redundant nucleotide sequence (Nt) database with a cut-off E-value of 10−5. Next, functions were assigned to the unigenes with priority given to the seven databases in the following order: Nr, GO, KEGG, Swiss-Prot, Pfam, KOG, and Nt (Moriya et al., 2007; Finn et al., 2008) The best BLAST alignments from this study were selected in order to determine the sequence direction of the unigenes, as well as to predict the coding regions (CDSs). Next, the CDS regions of the unigenes were translated into their protein sequences according to standard genetic codes. For the unigenes that did not align to any of the above databases, the Getorf program (http://emboss.sourceforge.net/apps/cvs/emboss/apps/getorf.html) was used to predict their open reading frames (ORFs). The longest open-ended ORF was extracted as the most probable translated region for each unigene.
Identification and Validation of SNPs
To identify SNPs in H. compressa and H. altissima that could be developed as molecular markers, the sequencing data from “Yaan” and “1110” were compared to the unigene database using SOAPsnp software (http://soap.genomics.org.cn/soapsnp; Li et al., 2009). The thresholds for SNP identification were as follows: the prior probability values for novel heterozygous SNPs and the prior probability values for novel homozygous SNPs were set at 0.0001 and 0.0005, respectively; the sequencing depth was between 10X and 100X; and the consensus base quality score had to be ≥30 (quality score 30 represents 99.9% accuracy of a base call).
Twenty Hemarthria materials were used to validate a subset of the SNP markers identified in this study (Table S2). The same amount of leaf tissue was harvested for each accession and DNA was extracted using a genomic DNA extraction kit (Tiangen Biotech, China) following the manufacturer's protocol. SNP primers were randomly selected from primer sets that were generated in our study (Table S3). PCR amplification reactions were performed in 30 μL volumes and contained 2 μL (20 ng/μL) of DNA template, 15 μL Premix Taq (TakaRa Taq Version 2.0 plus dye; TakaRa Bio Inc., China), 2 μL (10 pmol/μL) each of the forward and reverse primers, and 9 μL ddH2O. The PCR cycling conditions for SNP detection were as follows: 5 min pre-denaturation at 94°C, followed by 35 cycles of 1 min denaturation at 94°C, 1 min annealing at 53°C, 1 min extension at 72°C, and a final 10 min extension at 72°C. PCR products were separated on 1.5% agarose gels at 130 V for 30 min and then visualized using UV light. The PCR products that were the correct size were then purified by gel extraction and sent to the Beijing Genomics Institute for gene sequencing.
Identification, Validation, and Cross-Species Transferability of SSRs
The assembled sequences of all four data sets that were longer than 1 kb were merged for microsatellite mining using MISA software (http://pgrc.ipk-gatersleben.de/misa/). The sequences were searched for perfect mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs with a minimum of 10, six, five, five, five, and five repeats, respectively. We also searched for compound SSRs with a minimum distance of less than 100 nt between two single SSRs. Based on the results from the MISA software, Primer 3 v 2.23 (http://primer3.sourceforge.net) was used to design PCR primers in the flanking regions of the SSRs. To acquire a high probability of amplification, the primer design parameters were set as follows: the primer length range was 18–23 nt, with the optimal length at 21 nt; the PCR products were between 100 and 300 bp long; the annealing temperature range was between 52 and 58°C, with 55°C as the optimum; and the GC percentage ranged from 40 to 60%, with an optimal GC content of 50%.
Forty-four Hemarthria materials were used to validate the SSR markers identified in this study (Table S2). In addition, we explored the transferability of the identified SSR makers to other four Poaceae genera (Lolium, Pennisetum, Miscanthus, and Dactylis) and analyzed their presence/absence in seven Lolium multiflorum cultivars, five Pennisetum varieties, four wild Miscanthus sinensis accessions, and six Dactylis glomerata cultivars (Table S5). The genomic DNA of these materials was extracted from young leaves using a genomic DNA extraction kit (Tiangen Biotech, China) in accordance with the manufacturer's protocol. To evaluate SSR polymorphic differences and transferability within the Poaceae family, we randomly chose 54 SSR primers from our list of self-designed SSR primers (the final selected SSR primer sequences for validation is shown in Table S4). For PCR amplification of the SSR primers, reactions were performed in 15 μL total volume and included 1.5 μL (20 ng/μL) of genomic DNA, 7.5 μL of 2X Reaction Mix (Tiangen, Biotech, China), 0.6 μL (10 pmol/μL) of each forward and reverse primer, 0.3 μL of Golden DNA Polymerase (Tiangen Biotech, China), and 4.5 μL of distilled water. The PCR amplification program included a pre-denaturation at 94°C for 5 min, followed by 35 cycles of 94°C for 30 s, 52–56°C for 45 s, and 72°C for 1 min with a final extension step at 72°C for 10 min. The obtained PCR products were examined using the method described by Huang et al. (2014d).
Results and Discussion
Sequence Analysis and Assembly
Illumina RNA-Seq technology has the ability to generate billions of reads and has been successfully used for transcriptome sequencing and de novo transcriptome assembly. As such, RNA-Seq has emerged as an important tool for gene discovery and molecular marker development. In this study, we used the Illumina HiSeq™ 2500 platform to generated more than 24,528,820 raw reads (~4.95 Gb) for each of the four Hemarthria RNA samples. More than 24,240,920 high-quality reads (~4.89 Gb clean data) were obtained for each sample after quality control trimming and filtering. The GC content, reads with “N” proportion, and cycleQ20 proportion of the clean data was over 54.61, 0.04, and 100.00%, respectively, for each of the four samples. More than 91.90% of the cycles had an average Phred score greater than 20, and the average base quality value was 30 or greater (≥30) for more than 85.46% of the cycles (Table S1). These results reflect the quality of the clean data reads and demonstrate that the clean reads were sufficient for subsequent analysis.
Using the Trinity de novo assembly software, 9,660,044 and 7,707,533 contigs were obtained for “Yaan” and “1110,” respectively. Using clustering and local assembly analyses, the contigs from “Yaan” were assembled into 269,972 transcripts with a mean length of 892.91 nt, and the contigs from “1110” were assembled into 170,550 transcripts with an average size of 1059.80 nt. The other characteristics of the contigs are given in Table 1. For the cultivar “Yaan,” these 269,972 transcripts were then further clustered into 137,142 unigenes with a mean length of 566.31 nt and an N50 length of 826 bp. For H. altissima “1110,” the 170,550 transcripts were clustered into 77,150 unigenes with a mean length of 679.26 nt and an N50 length of 1189 bp. After de novo assembly, the total transcriptome size was estimated to be 74.07 Mb and 49.98 Mb for “Yaan” and “1110,” respectively. These results indicate that the two Hemarthria materials used have complex genomes. The larger transcriptome size of “Yaan” could be attributed to the larger genome size of H. compressa compared to H. altissima, which is consistent with previous reports (Yang et al., 2003; Guo et al., 2014). In “Yaan,” 62,639 or 45.67% of the unigenes were 200–300 nt in size followed by 23,145 unigenes (16.88%) that were 300–400 nt in size. Similarly, in the “1110” material 30,958 unigenes or 40.13% were 200–300 nt size, followed by 11,936 unigenes (15.47%) that were 300–400 nt long (Table 2). Interestingly, a high proportion of the unigenes (40–45%) assembled in this study were shorter than 300 nt, indicating that these transcripts were fragmented. This could be explained by the fact that the clean sequencing data of young leaves and roots of the same material were used in the assembly process. Additionally, the short unigenes could be attributed to insufficient sequencing depth (~4.95 Gb). Despite the high percentage, the proportion of short unigenes generated for the two Hemarthria materials was similar to those observed in other non-model organisms, such as Camellia sinensis (44.36% < 300 bp) and Oenanthe javanica (40.34% < 300 bp; Jiang et al., 2014; Wu et al., 2014).
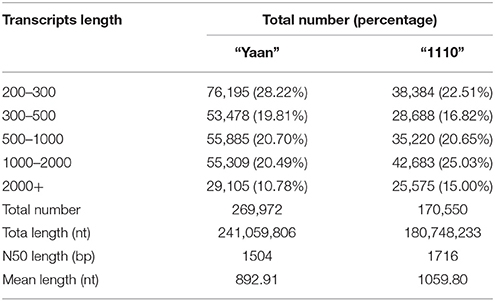
Table 1. Summary of the transcripts for the two Hemarthria materials: H. compressa “Yaan” and H. altissima “1110”.
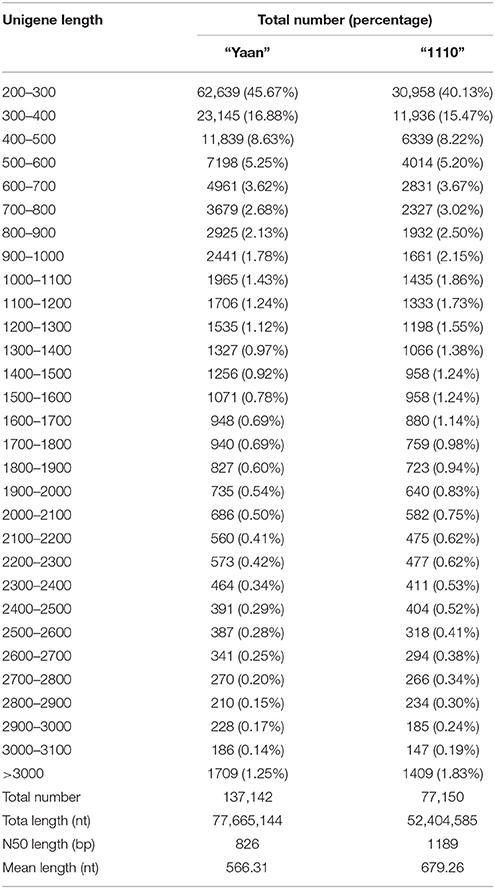
Table 2. Summary of unigenes for the two Hemarthria materials: H. compressa “Yaan” and H. altissima “1110”.
Although Hemarthria species are economically and ecologically important forage crops, there is a serious lack of available genomic information available for them. No EST database currently exists for Hemarthria species. In this study, we have obtained a large number of representative transcript sequences of Hemarthria genes using Illumina RNA-Seq technology. The sequencing reads provide valuable genomic information for Hemarthria spieces, which can be utilized not merely for gene discovery but also for molecular marker (SSRs and SNPs) identification.
Sequence Annotation
Functional annotation and classification of transcriptomes can shed light on intracellular metabolic pathways and biological behaviors of genes (Tang et al., 2014). To predict the potential functions of the assembled unigenes, the obtained unigene sequences for the two Hemarthria materials were aligned using BLAST (e < 1e-5) to various databases, including Nt, Nr, Swiss-Prot, Pfam, GO, KOG, and KEGG. Amongst the 137,142 unigenes obtained from “Yaan,” 58,661 (42.77%) had significant hits in the Nt database, 75,478 (55.04%) in Nr, 46,906 (34.20%) in Swiss-Prot, 44,285 (32.29%) in Pfam, 50,638 (36.92%) in GO, 42,047 (30.66%) in KOG, and 18,148 (13.23%) in KEGG. For the 77,150 unigenes assembled for “1110,” 40,408 (52.38%) were a significant match to Nt, 41,262 (53.48%) to Nr, 28,513 (36.96%) to Swiss-Prot, 25,190 (32.65%) to Pfam, 30,538 (39.58%) to GO, 21,867 (28.34%) to KOG, and 8434 (10.93%) to KEGG. Thus, the number of “1110” unigenes that were matched to each database was only about one-half of the number of unigenes that were matched to the databases for “Yaan.” In total, 86,731 unigenes (63.24%) and 48,645 unigenes (63.05%) were successfully annotated for “Yaan” and “1110,” respectively (Table 3). The ratios of annotated unigenes of the two Hemarthria materials analyzed in our study were comparable to the range of previously reported annotated unigenes in other non-model organisms (Li et al., 2012; Lu et al., 2012). The remaining unigenes (36.76% in “Yaan” and 36.95% in “1110”) did not align with any known genes. This could be attributed to either a large number (40–45%) of unigenes that had lengths shorter than 300 nt, or a lack of relevant genetic data. For both the unigenes identified in the “Yaan” and “1110,” the most frequent and significant annotation hits in the databases were to four well-annotated monocot plant species: Sorghum bicolor, Zea mays, Setaria italic, and Oryza sativa Japonica Group. These matched species all belong to the Poaceae family, supporting that the sequences obtained in our study were annotated properly.
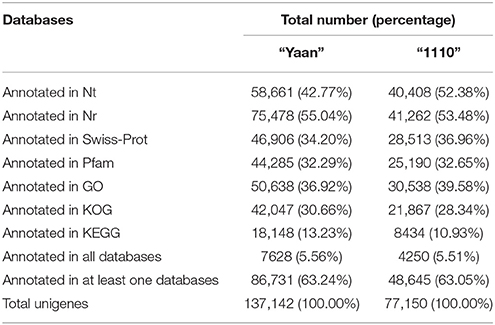
Table 3. Functional annotation of the Hemarthria transcriptome for H. compressa “Yaan” and H. altissima “1110”.
In order to predict CDSs, the unigenes for both “Yaan” and “1110” were consolidated into a single set of sequences. We predicted a total of 180,932 CDSs in our study, of which 99,037 CDSs aligned to the eight previously discussed databases. The largest number of CDSs (23,182, 23.41%) was 100–200 nt long (Figure 1). An additional 81,895 CDSs without BLAST hits were predicted by the Getorf software. Interestingly, 100–200 nt long CDSs were also the highest in number in this group (40,973 CDSs, 50.03%; Figure 2).
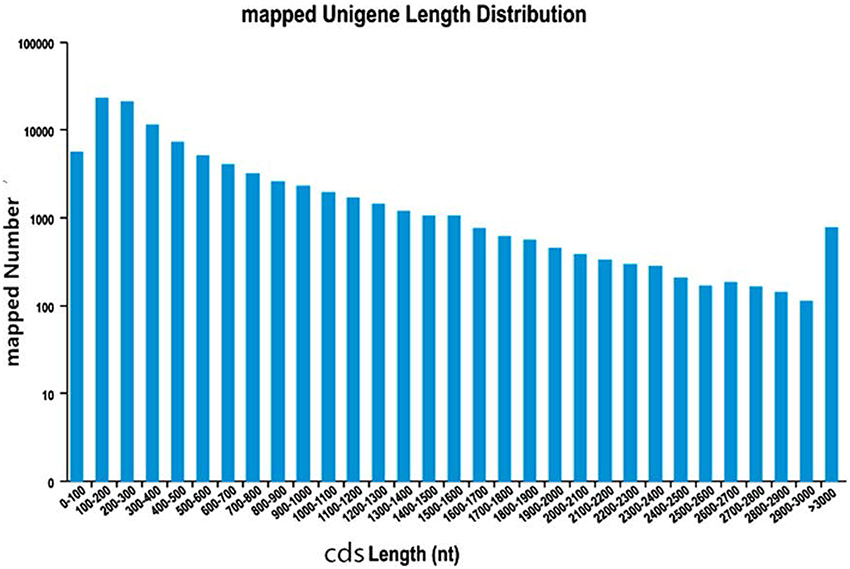
Figure 1. The length distribution of CDSs mapped to know genes.
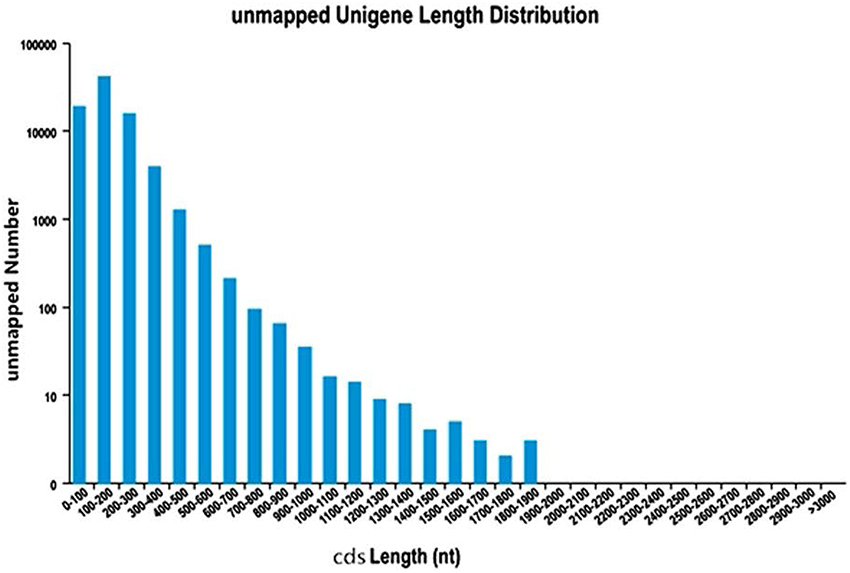
Figure 2. The length distribution of CDSs unmapped to know genes.
The Pfam database is a comprehensive collection of protein domains and families, and its primary use in genome annotation is to quickly identify all genes that have the same protein domains (Bateman et al., 2004). A total of 44,285 and 25,190 unigenes of “Yaan” and “1110” were annotated to 4127 and 3584 Pfam protein families with an average of 10.73 and 7.03 unigenes per protein family, respectively. Of these identified protein families, the highest percentages of unigenes were predicted to contain a protein kinase domain (4.06% in “Yaan” and 4.89% in “1110”), a protein tyrosine kinase domain (3.71% in “Yaan” and 4.77% in “1110”), and protein domains with unknown functions (2.78% in “Yaan” and 3.58% in “1110”). Figure 3 shows the 17 most abundant Pfam protein families that were obtained from the two Hemarthria cultivars.
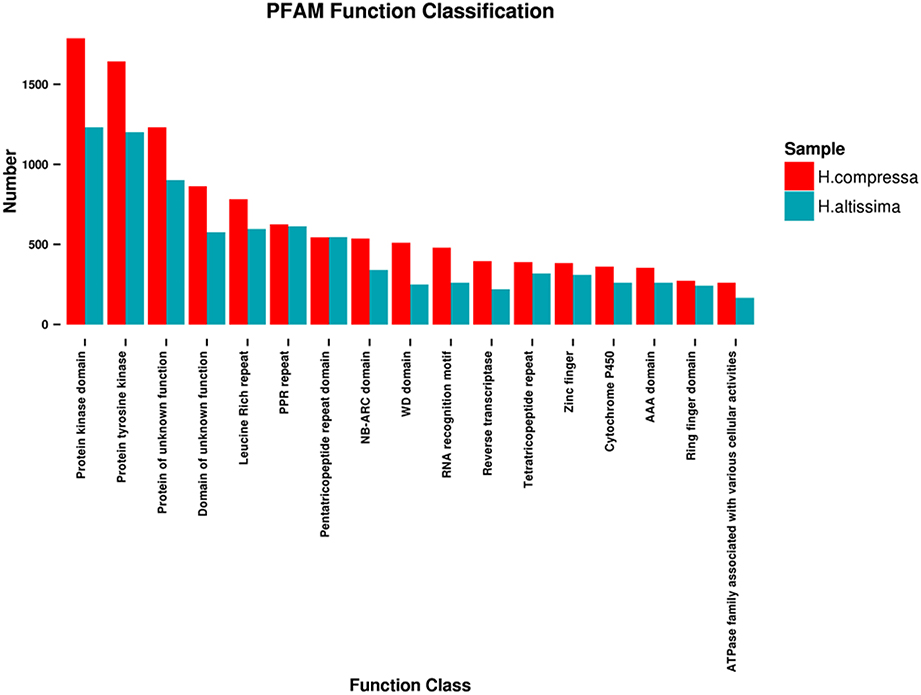
Figure 3. Distribution of the 17 most abundant Pfam function classifications for H. compressa (“Yaan”) and H. altissima (“1110”).
GO annotation is an international gene functional classification system that provides a controlled vocabulary that is able to comprehensively describe properties of the uncharacterized sequences in any organism (Gene Ontology Consortium, 2004; Tang et al., 2014). Based on sequence homology, 50,638 “Yaan” unigenes (36.92% of all the assembled “Yaan” unigenes) and 30,538 “1110” unigenes (39.58% of all the assembled “1110” unigenes) were assigned to at least one GO term and were classified into three groups: biological processes, molecular function, and cellular component. The distribution of GO groups for the two Hemarthria materials was extremely similar. As described in Figure 4, most of the unigenes were classified into the cellular components of cell part, cell, organelle, and membrane. In the molecular function group, binding, catalytic activity, structural molecules and transporter activity were found to be the four most frequent classes. Among 25 different biological process categories, the metabolic processes category was the most represented followed by cellular processes, response to stimuli, and biological regulation. With the help of GO functional classification, a large number of the unigenes were assigned to a wide range of biological processes, molecular functions, and cellular components. This information can provide a valuable resource for gene expression profile analysis, gene location, and gene isolation in Hemarthria species. In addition, the main GO classifications identified in this study pertained to fundamental biological processes. These results are similar to previously reported studies of de novo transcriptome analyses in the taproots of radish and the leaves of young tea plant (Wang et al., 2013; Wu et al., 2014).
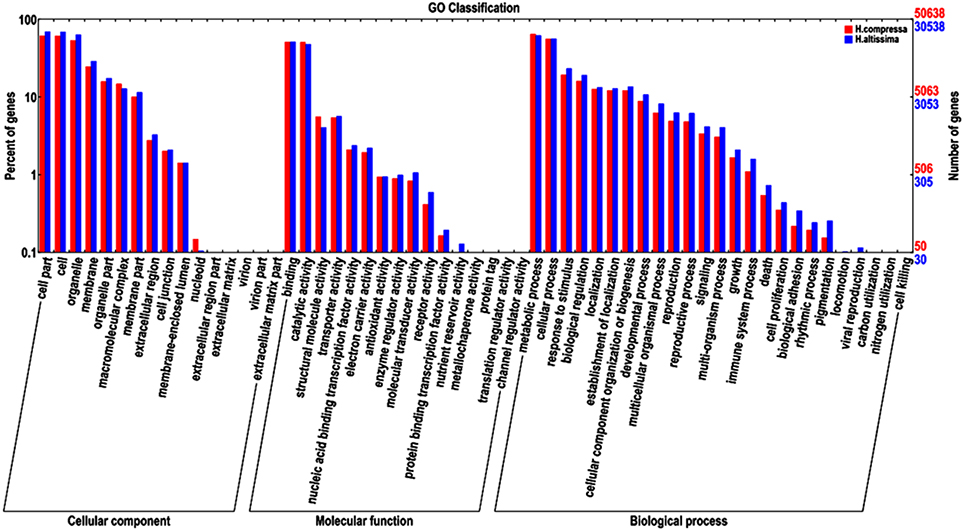
Figure 4. Comparative distribution of GO categories for H. compressa (“Yaan”) and H. altissima (“1110”).
For KOG analyses, 42,047 unigenes from “Yaan” were classified into 25 functional categories under four larger groups (metabolism, cellular processes and signaling, information storage and processing, and poorly characterized). Among these functional categories, the top three largest KOG categories were: 1) the prediction of general function only (6604 unigenes, 15.71%), 2) translation, ribosomal structure, and biogenesis (4730 unigenes, 11.25%), and 3) post-translational modification, protein turnover, and chaperones (4113 unigenes, 9.78%). Interestingly, only a few unigenes were assigned to extracellular structures (232 unigenes, 0.55%), nuclear structure (55 unigenes, 0.13%), and cell motility (20 unigenes, 0.05%). We identified several differences between the results from the KOG analyses for the “1110” unigenes and the “Yaan” unigenes. A total of 21,867 of the “1110” unigenes were distributed into 25 functional categories with the top three categories being: (1) general function prediction only (18.42%), (2) signal transduction mechanisms (8.99%), and (3) post-translational modification, protein turnover, and chaperones (8.88%). In both the “1110” and “Yaan” data, extracellular structures, nuclear structure, and cell motility categories had the smallest number of unigenes (Figure 5).
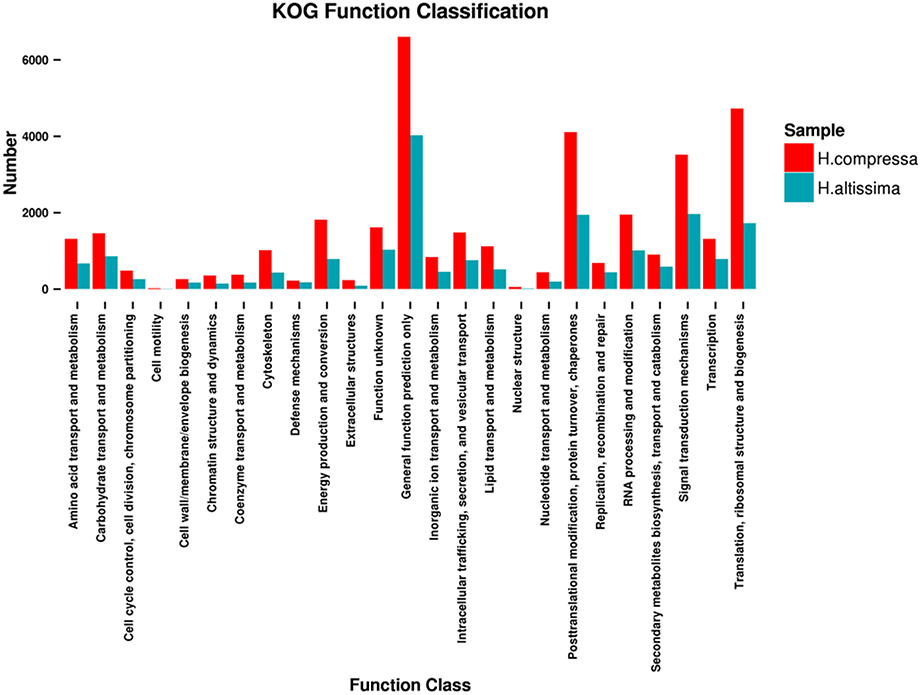
Figure 5. Comparative distributions of KOG categories for H. compressa (“Yaan”) and H. altissima (“1110”).
To systematically understand the biological pathways activated in Hemarthria roots and leaves, a KEGG analysis of the assembled unigenes of “Yaan” and “1110” was performed. A total of 18,148 and 8434 unigenes for “Yaan” and “1110,” respectively, were assigned to 121 biological pathways including transcription and translation, protein processing and modification, biosynthesis of secondary metabolites, signal transduction, replication and repair, transport and catabolism, immune system function, amino acid metabolism, and several others. The six major pathway groups for “Yaan” and “1110” were ribosomal [2832 (15.60%); 994 (11.79%)], oxidative phosphorylation [789 (4.35%); 324 (3.84%)], protein processing in the endoplasmic reticulum [798 (4.40%); 246 (2.92%)], RNA transport [739 (4.07%); 293 (3.47%)], spliceosomal [617 (3.40%); 247 (2.93%)], and glycolysis/gluconeogenesis [469 (2.58%); 234 (2.77%)] (Figure 6). These findings revealed a high level of protein synthesis occurring during the growth and development of Hemarthria species, and reflect the reliability of the KEGG orthology-based annotation. Furthermore, these data may provide reference information for future experiments such as gene expression analyses and gene cloning.
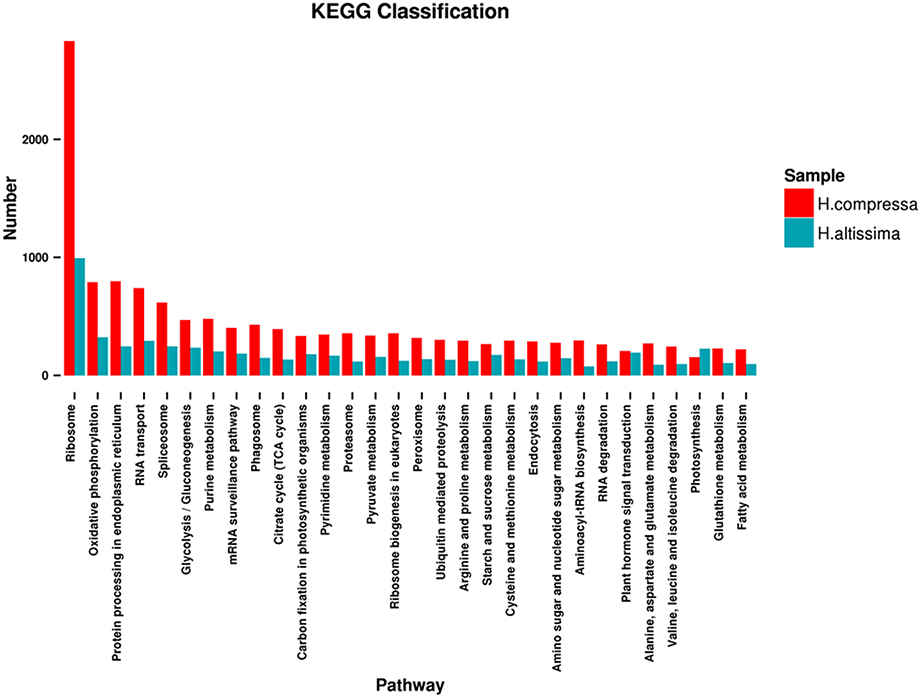
Figure 6. KEGG classification of H. compressa (“Yaan”) unigenes and H. altissima (“1110”) unigenes.
Identification and Validation of SNPs
Due to its high-throughput, accuracy, and reproducibility, RNA-Seq has become the preferred method of researchers to map and quantify transcriptomes (Vijay et al., 2013). RNA-Seq can be used to identify large numbers of genetic (heterozygous and homozygous) variants such as SNPs and SSRs, which can be further developed into molecular markers (Kaur et al., 2012). This is particularly useful in non-model species where a lack of genomic information hinders molecular marker identification and development. SNPs are the richest type of genetic variation distributed in eukaryotic genomes. They are widely considered the molecular marker of choice, especially in ecological and evolutionary studies, due to their potential for genome-wide coverage, high genotyping efficiency, data quality, automation, and analytical simplicity (Morin et al., 2004; Liu et al., 2011; Dou et al., 2012).
In this study, a total of 42,646 high-quality SNPs were detected in the 10,880 unigenes of the two Hemarthria genotypes, the majority of which (99.40%) were annotated. As summarized in Table 4, out of the 42,646 putative SNPs, the proportions of transversions were: 8.76% A/C; 6.45% A/T; 12.15% C/G, and 8.53% G/T. Additionally, the proportions of transitions were: 31.96% A/G and 32.15% C/T. The percentage of transition variations (64.11%) were approximately 1.79 times more frequent than transversions (35.89%) in these two Hemarthria genotypes. The prevalence of transitions found in these two cultivars was similar to those observed in other plants (Ebersberger et al., 2002; Renaut et al., 2010; Jhanwar et al., 2012).

Table 4. Summary of putative SNPs identified from the two Hemarthria materials: H. compressa “Yaan” and H. altissima “1110”.
Further analysis of the SNPs revealed that 32,166 (75.43%) of the putative SNPs were distributed in sequences with CDSs and the remainder were in non-CDS sequences. The large number of SNPs located in sequences containing CDSs is possibly beneficial for future association with important economic and agronomic traits. These SNPs could be useful for molecular breeding programs and marker-assisted selection practices, as well as for understanding the phenotype differences between Hemarthria species (Huang et al., 2015). Additionally, 39.50% of the SNPs in the CDS regions resulted in non-synonymous substitutions, which corresponded to a change in the amino acid sequence of the translated peptide. These SNPs would be promising candidates for the study of mutations that may alter protein structure and function, and for an association analysis of Hemarthria germplasms.
Fifteen primer pairs were designed to validate putative SNPs that were identified in this study. We tested our primers on a diverse collection of 20 different Hemarthria clonal materials to determine if these SNPs could be used as molecular markers for other Hermarthria species (Table S2). After electrophoresis detection, we selected 10 out of 16 primer pairs that provided a clear, single band of the expected fragment size for sequencing. These 10 primer pairs were used to detect 37 SNPs; however, three of the primer pairs failed during sequencing. Twenty-nine (78.38%) of the 37 putative SNP loci were successfully validated by the remaining seven primer pairs. These SNPs were determined to be polymorphic with a range from two to eight loci per primer pair and an average of 4.1 loci (Table S3). These results suggest that the majority of the putative SNPs identified in this study are expected to be valid. Since the PCR was unable to detect some SNPs, some false SNPs may have been identified; however, we believe the false discovery rate is relatively low. The abundance of SNPs identified in this study will enhance the amount of useful and informative functional genetic marker resources that are available for Hemarthria, and these SNPs can readily be utilized for various maker-based applications in Hemarthria genetics, genomics, and breeding.
Identification, Validation, and Cross-Species Transferability of SSRs
In plant genetics, SSRs are also widely considered to be molecular markers of choice due to their broad range of applications in processes such as MAS, genotype identification, genetic mapping, and the molecular tagging of genes. However, prior to this study, the paucity of available and robust SSRs had restricted the genetic analysis of this important genus. Therefore, there is an urgent need to develop a large set of SSRs for use in Hermarthria genetic studies and breeding programs.
Using a MISA perl script, all 182,842 unigenes from the two Hemarthria genotypes were searched for the presence of microsatellites. We identified a total of 8330 (4.56%) unigenes, 7,701 (92.45%) of which were annotated, that contained 10,888 SSRs with motif lengths ranging from one to six bp. Five hundred and seventy-five SSRs were present in compound types, meaning that they contained stretches of two or more different repeats. Data analysis of the SSR motifs revealed that the mono-nucleotide and tri-nucleotide repeats were the most abundant SSRs detected and accounted for 43.87 and 41.45%, respectively, of the total amount of SSRs. These were followed by the di-, tetra-, penta-, and hexa-nucleotide repeats, which accounted for 12.74, 1.43, 0.26, and 0.25% of the total number of SSRs, respectively (Table 5).

Table 5. Summary of simple sequence repeats (SSRs) identified from the combined Hemarthria materials.
Due to a high probability of homopolymers existing as a quality problem associated with sequencing and genotyping errors, mono-nucleotides were excluded in the subsequent analysis (Gilles et al., 2011). In our study, tri-nucleotide repeat units were predominant, which is consistent with the results of similar studies in other plant genera (Jhanwar et al., 2012; Kaur et al., 2012; Yates et al., 2014). The prevalence of tri-nucleotide expansions within translated regions, in comparison to other nucleotide repeats, could be explained by slippage of the DNA replication machinery and the overall need to keep the integrity of the reading-frame.
The number of replications in a given repeat unit of SSRs ranged from five to >10, of which five reiterations were the most abundant. As the number of nucleotides in the microsatellite increased, the frequency of the given SSR structure (di-, tri-, tetra-, penta-, and hexa-nucleotide repeats) in our dataset progressively decreased (Table 6). This could be attributed to the relative probability of replication slippage events (Kaur et al., 2012). We also examined the repeat motif types of the SSR di- and tri-nucleotides. The results showed that the AG/TC motif had the largest frequency of up to 49.68% in the four types of di-nucleotide repeat motifs, and that the CCG/GGC motif was the most abundant in the tri-nucleotide SSRs, accounting for 48.19% (Table 7). Interestingly, the AG/TC of di-nucleotide SSRs was also found to be the most abundant motif in cereal crops (Temnykh et al., 2000; Kantety et al., 2002). A possible explanation for this is that the inverse GA/CT motif can represent multiple codons (such as GAG, AGA, UCU, and CUC) when transcribed into mRNA, and can translate into different amino acids (Arg, Glu, Ala, and Leu), among which Ala and Leu separately occupy a high proportion (8 and 10%) in proteins (Kantety et al., 2002). The most abundant tri-nucleotide motif in the genomes of other plants was also the inverse GGC/CCG motif (Nicot et al., 2004).
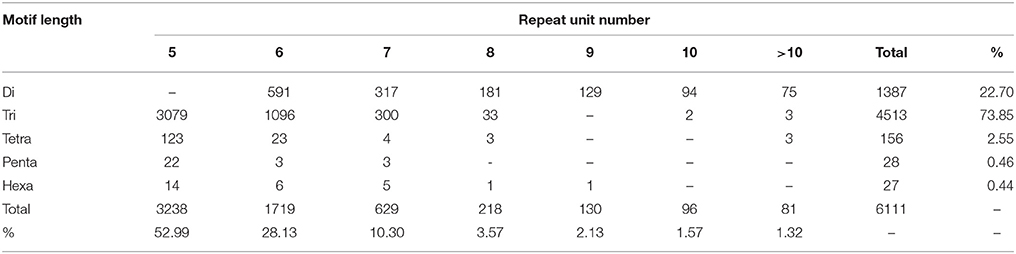
Table 6. Summary information on frequencies of different SSR repeat motif types related to variation of repeat unit numbers in Hemarthria SSRs.
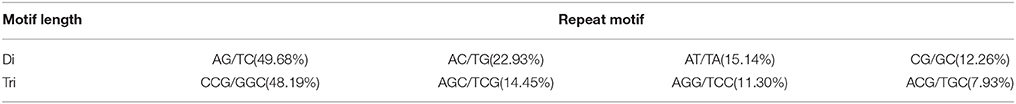
Table 7. Statistics of repeat motifs.
For forage breeding, SSRs located in the CDS regions of the genome may be of more interest than SSRs that reside in non-CDS sequences because of the association of with important economic and agronomic traits. Among the 6111 SSRs (not including mono-nucleotide repeat SSRs) identified in this study, there were 1303 (21.32%) and 4808 (78.68%) SSRs identified in CDSs and non-CDSs sequences, respectively.
To validate the SSRs identified in this study, we successfully designed at least one primer pair for 4846 (79.30%) of the 6111 SSRs. A subset of 54 SSR primer pairs was selected for validation. Approximately 44 different Hemarthria clonal materials from different genetic backgrounds were utilized to test if these primers could be applied to other Hemarthria species (Table S2). The SSR primers were evaluated for their potential to amplify the target sequence and detect polymorphisms. In 34 (62.96%) cases, PCR products containing rich polymorphisms could be amplified from genomic DNA. The remaining 20 SSR primer pairs resulted in either weak or no amplification and were eliminated from further analysis. As seen in Table S4, these 34 SSR primer pairs covered di-, tri-, and tetra-nucleotide motifs and detected a total of 441 alleles. The number of PCR products for these primer pairs ranged from seven to 19 with an average of 12.97. Of the total number of alleles detected, 420 (95.24%) were determined to be polymorphic. The primer pairs for these alleles amplified six to 19 PCR products, with an average number of 12.35 PCR products per primer pair. The polymorphism information content (PIC) of the SSRs ranged from 0.5227 to 0.9496 with an equilibration of 0.7136, and the average Nei's gene diversity (H) and Shannon's information index of diversity (I) was calculated to be 0.2553 and 0.3991, respectively. These results revealed that the Hemarthria SSR markers developed in the current study have a high level of polymorphism, and that they may prove to be a valuable tool for population genetic studies, variety identification, association analysis, and MAS of Hemarthria species.
To further explore the transferability of SSR makers from Hemarthria to other Poaceae species, the 54 SSR primers used in the preceding part of the study were also employed to amplify SSRs from M. sinensis (awn), D. glomerata (orchardgrass), Pennisetum, and L. multiflorum (Italian rygrass). High levels of cross-species transferability were observed in all four species. The transferability from Hemarthria to L. multiflorum was the highest, at 87.04% (47/54), and to Pennisetum, M. sinensis, and D. glomerata at 61.11% (33/54), 83.33% (45/54), and 77.78% (42/54), respectively. In general, the ratio of transferability of the SSR markers roughly reflects the genetic relationships among the different species; a higher transferability indicates a closer genetic relationship (Zhang et al., 2014). In our study, the highest transferability was from Hemarthria to L. multiflorum, indicating that Hemarthria has a close genetic relationship with L. multiflorum, followed by M. sinensis, D. glomerata, and Pennisetum. These results are inconsistent with the taxonomic relationships of Hemarthria species, M. sinensis, and Pennisetum which belong to the same subfamily, while L. multiflorum and D. glomerata do not. It is possible that these relationships were seen because of the different amounts of materials used, which resulted in detecting varying degrees of nucleotide variation. In other words, the more materials from a species, the more nucleotide variation could be found. Additionally, the samples of each of the other Poaceae species may not accurately represent the corresponding species. Even so, transfer of the SSR markers could be utilized in other Poaceae species that also lack genomic resources.
Conclusions
In this study, we performed Illumina RNA sequencing, as well as de novo transcriptome assembly and annotation, of two Hemarthria cultivars, H. compressa “Yaan” and H. altissima “1110.” Based on the newly assembled unigenes, we successfully identified, designed primers, and validated a large number of potential genetic SNP and SSR markers. A selected subset of these SNP and SSR primers were able to be applied to a number of genetically diverse H. compressa and H. altissima materials. Additionally, we were able to utilize the SSR primers designed for Hemarthria to detect SSRs in other Poaceae species, supporting the quality of the polymorphisms detected in this study and their transferability to other closely related species. The results of this study help us to gain a better understanding of Hermarthria genetics, and they can be directly used in future genomic studies aimed at improving this important forage crop.
Data Accessibility
Raw Illumina reads: NCBI SRA: SRP058845.
Author Contributions
XH performed experiments, analyzed data, and wrote the manuscript. DHY guided and edited the writing. KLH conceived the experiments, conducted the bioinformatics analysis, and guided the writing. KLH, JZ, and JDH organized the funding and collected the materials. DHY, JZ, LL, QXZ, WL, YP, XM, and HYY participated in one or more processes of materials cultivation, samples collection, data analysis and manuscript editing. All authors approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Basic Research Program of China (973 Program) (2014CB138705), Strategic Planning and Management Innovation of Science and Technology Support Demonstration Project of Chongqing Modern Cattle and Sheep Industry (cstc2014zktjccxbx0021), Modern Agro-industry Technology Research System (CARS-35-05), and the Sichuan Agricultural University Excellent Master's Degree Papers Breeding (YS2014003).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00496
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., et al. (2004). The Pfam protein families database. Nucl Acids Res 32, D138–D141. doi: 10.1093/nar/gkh121
Chen, Y. X., Zhang, X. Q., Ma, X., and Xie, W. G. (2011a). Association analysis between SSR molecular markers and agronomic characters of Hemarthria compressa. Hubei Agr. Sci. 50, 1494–1498. doi: 10.3969/j.issn.0439-8114.2011.07.057
Chen, Y. X., Zhang, X. Q., Xie, W. G., Ma, X., and Liu, Y. (2011b). Genetic diversity of Hemarthria compressa germplasms from southwestern China based on EST-SSR markers. Acta Pratacul. Sin. 20, 245–253.
Chen, Y. X., Zhang, X. Q., Yang, C. H., and Liu, J. P. (2005). Analysis of peroxidase isozyme of wild Hemarthria compressa. in Sichuan province. J. Sichuan Grassland 15–16. doi: 10.3969/j.issn.1673-8403.2005.04.005
Cloonan, N., Forrest, A. R., Kolle, G., Gardiner, B. B., Faulkner, G. J., Brown, M. K., et al. (2008). Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5, 613–619. doi: 10.1038/nmeth.1223
Dou, J., Zhao, X., Fu, X., Jiao, W., Wang, N., Zhang, L., et al. (2012). Reference-free SNP calling: improved accuracy by preventing incorrect calls from repetitive genomic regions. Biol. Direct 7:17. doi: 10.1186/1745-6150-7-17
Ebersberger, I., Metzler, D., Schwarz, C., and Pääbo, S. (2002). Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70, 1490–1497. doi: 10.1086/340787
Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, S. J., Hotz, H. R., et al. (2008). The Pfam protein families database. Nucleic Acids Res. 36, D281–D288. doi: 10.1093/nar/gkm960
Gene Ontology Consortium (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. doi: 10.1093/nar/gkh036
Gilles, A., Meglécz, E., Pech, N., Ferreira, S., Malausa, T., and Martin, J. F. (2011). Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics 12:245. doi: 10.1186/1471-2164-12-245
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guo, Z. H., Fu, K. X., Zhang, X. Q., Bai, S. Q., Fan, Y., Peng, Y., et al. (2014). Molecular insights into the genetic diversity of Hemarthria compressa germplasm collections native to southwest China. Molecules 19, 21541–21559. doi: 10.3390/molecules191221541
Hoagland, D. R., and Arnon, D. I. (1950). The water-culture method for growing plants without soil. Circ. California Agr. Exp. Station 347, 32. Record Number: 19500302257.
Huang, L. K., Huang, X., Yan, H. D., Yin, G. H., Zhang, X. Q., Tian, Y., et al. (2014a). Constructing DNA fingerprinting of Hemarthria cultivars using EST-SSR and SCoT markers. Genet. Resour. Crop. Ev. 61, 1047–1055. doi: 10.1007/s10722-014-0107-4
Huang, L. K., Yan, H. D., Zhao, X. X., Zhang, X. Q., Wang, J., Frazier, T., et al. (2015). Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Mol. Ecol. Resour. 15, 1497–1509. doi: 10.1111/1755-0998.12418
Huang, L. K., Zhang, X. Q., Ma, X., Liu, W., Li, F., and Zeng, B. (2008). Genetic differentiation among Hemarthria compressa populations in south China and its genetic relationship with H. japonica. Hereditas 145, 84–91. doi: 10.1111/j.2008.0018-0661.02031.x
Huang, L. K., Zhang, Y., Zhang, J., Zhang, X. Q., Xie, W. G., Jiang, X. M., et al. (2014b). Genetic stability and DNA fingerprinting of the Hemarthria compressa cultivar “Guangyi”. Biochem. Syst. Ecol. 55, 310–316. doi: 10.1016/j.bse.2014.03.027
Huang, X., Zeng, J., Nie, G., Liu, H., Huang, L. K., Zhang, X. Q., et al. (2014c). Construction of fingerprinting and genetic diversity analysis of Hemarthia Cultivars by EST-SSR. J. Trop. Subtrop. Bot. 22, 165–171. doi: 10.3969/j.issn.1005–3395.2014.02.009
Huang, X., Zhang, X. Q., Huang, L. K., Ma, Y. m., Yin, G. H., Lee, S., et al. (2014d). Genetic diversity of Hemarthria altissima and its related species by EST-SSR and SCoT markers. Biochem. Sys. Ecol. 57, 338–344. doi: 10.1016/j.bse.2014.09.016
Jhanwar, S., Priya, P., Garg, R., Parida, S. K., Tyagi, A. K., and Jain, M. (2012). Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol. J. 10, 690–702. doi: 10.1111/j.1467-7652.2012.00712.x
Jiang, Q., Wang, F., Tan, H. W., Li, M. Y., Xu, Z. S., Tan, G. F., et al. (2014). De novo transcriptome assembly, gene annotation, marker development, and miRNA potential target genes validation under abiotic stresses in Oenanthe javanica. Mol. Genet. Genomics 290, 671–683. doi: 10.1007/s00438-014-0953-y
Johnson, D. S., Mortazavi, A., Myers, R. M., and Wold, B. (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502. doi: 10.1126/science.1141319
Kantety, R. V., La Rota, M., Matthews, D. E., and Sorrells, M. E. (2002). Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 48, 501–510. doi: 10.1023/A:1014875206165
Kaur, S., Pembleton, L. W., Cogan, N. O., Savin, K. W., Leonforte, T., Paull, J., et al. (2012). Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13:104. doi: 10.1186/1471-2164-13-104
Li, H., Dong, Y., Yang, J., Liu, X., Wang, Y., Yao, N., et al. (2012). De novo transcriptome of safflower and the identification of putative genes for oleosin and the biosynthesis of flavonoids. PLoS ONE 7:e30987. doi: 10.1371/journal.pone.0030987
Li, M., Tian, S., Jin, L., Zhou, G., Li, Y., Zhang, Y., et al. (2013). Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438. doi: 10.1038/ng.2811
Li, R., Fan, W., Tian, G., Zhu, H., He, L., Cai, J., et al. (2010). The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317. doi: 10.1038/nature08696
Li, R., Li, Y., Fang, X., Yang, H., Wang, J., Kristiansen, K., et al. (2009). SNP detection for massively parallel whole-genome resequencing. Genome Res. 19, 1124–1132. doi: 10.1101/gr.088013.108
Liu, J. P., Zhang, X. Q., Peng, Y., and Yang, C. H. (2006). AFLP analysis about genetic diversity of Hemarthria compressa in southwest region of China. J. Anhui. Agric. Sci. 12, 46–47. doi: 10.3969/j.issn.1007-7731.2006.13.020
Liu, S., Zhou, Z., Lu, J., Sun, F., Wang, S., Liu, H., et al. (2011). Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array. BMC Genomics 12:53. doi: 10.1186/1471-2164-12-53
Lu, L. H., Xiao, Y., Pei, S., Wen, T., and and, H. S. Q. (2012). The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLoS ONE 7:e38653. doi: 10.1371/journal.pone.0038653
Morin, P. A., Luikart, G., and Wayne, R. K. (2004). SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 19, 208–216. doi: 10.1016/j.tree.2004.01.009
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Nagalakshmi, U., Wang, Z., Waern, K., Shou, C., Raha, D., Gerstein, M., et al. (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. doi: 10.1126/science.1158441
Nicot, N., Chiquet, V., Gandon, B., Amilhat, L., Legeai, F., Leroy, P., et al. (2004). Study of simple sequence repeat (SSR) markers from wheat expressed sequence tags (ESTs). Theor. Appl. Genet. 109, 800–805. doi: 10.1007/s00122-004-1685-x
Renaut, S., Nolte, A. W., and Bernatchez, L. (2010). Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs (Coregonus spp. Salmonidae). Mol. Ecol. 19 (suppl. 1), 115–131. doi: 10.1111/j.1365-294X.2009.04477.x
Rokas, A., and Abbot, P. (2009). Harnessing genomics for evolutionary insights. Trends Ecol. Evol. 24, 192–200. doi: 10.1016/j.tree.2008.11.004
Rumball, P. J., and Lambert, J. P. (1981). Plant introduction trials: performance of some subtropical grasses in Northland. New Zealand J. Exp. Agr. 9, 79–83. doi: 10.1080/03015521.1981.10427807
Sablok, G., Fu, Y., Bobbio, V., Laura, M., Rotino, G. L., Bagnaresi, P., et al. (2014). Fuelling genetic and metabolic exploration of C3 bioenergy crops through the first reference transcriptome of Arundo donax L. Plant Biotechnol. J. 12, 554–567. doi: 10.1111/pbi.12159
Tang, X. Q., Xiao, Y. H., Lv, T. T., Wang, F. Q., Zhu, Q. H., Zheng, T. Q., et al. (2014). High-throughput sequencing and de novo assembly of the Isatis indigotica transcriptome. PLoS ONE 9:e102963. doi: 10.1371/journal.pone.0102963
Temnykh, S., Park, W. D., Ayres, N., Cartinhour, S., Hauck, N., Lipovich, L., et al. (2000). Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 100, 697–712. doi: 10.1007/s001220051342
Toledo-Silva, G., Cardoso-Silva, C. B., Jank, L., and Souza, A. P. (2013). De novo transcriptome assembly for the tropical grass Panicum maximum Jacq. PLoS ONE 8:e70781. doi: 10.1371/journal.pone.0070781
Trick, M., Long, Y., Meng, J., and Bancroft, I. (2009). Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 7, 334–346. doi: 10.1111/j.1467-7652.2008.00396.x
Vidal, E. A., Moyano, T. C., Krouk, G., Katari, M. S., Tanurdzic, M., McCombie, W. R., et al. (2013). Integrated RNA-Seq and sRNA-Seq analysis identifies novel nitrate-responsive genes in Arabidopsis thaliana roots. BMC Genomics 14:701. doi: 10.1186/1471-2164-14-701
Vijay, N., Poelstra, J. W., Kunstner, A., and Wolf, J. B. (2013). Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-Seq experiments. Mol. Ecol. 22, 620–634. doi: 10.1111/mec.12014
Wang, B., Guo, G., Wang, C., Lin, Y., Wang, X., Zhao, M., et al. (2010). Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 38, 5075–5087. doi: 10.1093/nar/gkq256
Wang, Y., Pan, Y., Liu, Z., Zhu, X., Zhai, L., Xu, L., et al. (2013). De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genomics 14:836. doi: 10.1186/1471-2164-14-836
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wu, Z. J., Li, X. H., Liu, Z. W., Xu, Z. S., and Zhang, J. (2014). De novo assembly and transcriptome characterization: novel insights into catechins biosynthesis in Camellia sinensis. BMC Plant Biol. 2014:277. doi: 10.1186/s12870-014-0277-4
Xie, Q., Niu, J., Xu, X. L., Xu, L. X., Zhang, Y. B., Fan, B., et al. (2015). De novo assembly of the Japanese lawngrass (Zoysia japonica Steud.) root transcriptome and identification of candidate unigenes related to early responses under salt stress. Front. Plant Sci. 6:610. doi: 10.3389/fpls.2015.00610
Yang, C. H., Zhang, X. Q., Li, X. L., and Wu, Y. Q. (2003). Hemarthria germplasm resources and breeding. Acta Pratacul. Sin. 13, 7–12. doi: 10.3321/j.issn:1004-5759.2004.02.002
Yates, S. A., Swain, M. T., Hegarty, M. J., Chernukin, I., Lowe, M., Allison, G. G., et al. (2014). De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics 15:453. doi: 10.1186/1471-2164-15-453
Zhang, G., Guo, G., Hu, X., Zhang, Y., Li, Q., Li, R., et al. (2010). Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 20, 646–654. doi: 10.1101/gr.100677.109
Keywords: de novo assembly, marker development, Hemarthria R. Br., RNA-Seq, transcriptome
Citation: Huang X, Yan H-D, Zhang X-Q, Zhang J, Frazier TP, Huang D-J, Lu L, Huang L-K, Liu W, Peng Y, Ma X and Yan Y-H (2016) De novo Transcriptome Analysis and Molecular Marker Development of Two Hemarthria Species. Front. Plant Sci. 7:496. doi: 10.3389/fpls.2016.00496
Received: 04 October 2015; Accepted: 29 March 2016;
Published: 18 April 2016.
Edited by:
Ramin Yadegari, The University of Arizona, USAReviewed by:
Ana Elena Dorantes-Acosta, Universidad Veracruzana, MexicoChuang Ma, Northwest Agricultural and Forestry University, China
Copyright © 2016 Huang, Yan, Zhang, Zhang, Frazier, Huang, Lu, Huang, Liu, Peng, Ma and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin-Kai Huang, huanglinkai@sicau.edu.cn;
Jian Zhang, zjturf@163.com