 Atsushi Fukushima1*
Atsushi Fukushima1* Michimi Nakamura2
Michimi Nakamura2 Hideyuki Suzuki3
Hideyuki Suzuki3 Mami Yamazaki2
Mami Yamazaki2 Eva Knoch1
Eva Knoch1 Tetsuya Mori1
Tetsuya Mori1 Naoyuki Umemoto1
Naoyuki Umemoto1 Masaki Morita4Go Hirai4,5Mikiko Sodeoka4,5
Masaki Morita4Go Hirai4,5Mikiko Sodeoka4,5 Kazuki Saito1,2*
Kazuki Saito1,2*- 1RIKEN Center for Sustainable Resource Science, Yokohama, Japan
- 2Graduate School of Pharmaceutical Sciences, Chiba University, Chiba, Japan
- 3Department of Biotechnology Research, Kazusa DNA Research Institute, Chiba, Japan
- 4Synthetic Organic Chemistry Laboratory, RIKEN, Saitama, Japan
- 5RIKEN Center for Sustainable Resource Science, Saitama, Japan
The genus Physalis in the Solanaceae family contains several species of benefit to humans. Examples include P. alkekengi (Chinese-lantern plant, hôzuki in Japanese) used for medicinal and for decorative purposes, and P. peruviana, also known as Cape gooseberry, which bears an edible, vitamin-rich fruit. Members of the Physalis genus are a valuable resource for phytochemicals needed for the development of medicines and functional foods. To fully utilize the potential of these phytochemicals we need to understand their biosynthesis, and for this we need genomic data, especially comprehensive transcriptome datasets for gene discovery. We report the de novo assembly of the transcriptome from leaves of P. alkekengi and P. peruviana using Illumina RNA-seq technologies. We identified 75,221 unigenes in P. alkekengi and 54,513 in P. peruviana. All unigenes were annotated with gene ontology (GO), Enzyme Commission (EC) numbers, and pathway information from the Kyoto Encyclopedia of Genes and Genomes (KEGG). We classified unigenes encoding enzyme candidates putatively involved in the secondary metabolism and identified more than one unigenes for each step in terpenoid backbone- and steroid biosynthesis in P. alkekengi and P. peruviana. To measure the variability of the withanolides including physalins and provide insights into their chemical diversity in Physalis, we also analyzed the metabolite content in leaves of P. alkekengi and P. peruviana at five different developmental stages by liquid chromatography-mass spectrometry. We discuss that comprehensive transcriptome approaches within a family can yield a clue for gene discovery in Physalis and provide insights into their complex chemical diversity. The transcriptome information we submit here will serve as an important public resource for further studies of the specialized metabolism of Physalis species.
Introduction
The specialized or secondary metabolism in plants is an important source for fine chemicals including drugs, dyes, vitamins, and other chemical materials. The genus Physalis, the largest genera in the Solanoideae subfamily, contains the most economically important genera, e.g., Solanum tuberosum (potato), S. lycopersicum (tomato), and Capsicum annuum (red pepper; Martinez, 1998). Some plants in approximately 90 species have a long history of cultivation. P. alkekengi var. franchetii (Chinese-lantern, hôzuki in Japanese) has been used as a medicinal plant and for decorative purposes. P. peruviana, also known as Cape gooseberry, has an edible fruit that contains many vitamins and antioxidants (Ramadan and Morsel, 2003).
Members of the genus Physalis produce bioactive metabolites such as steroidal lactones withanolides (Eich, 2008). P. peruviana can produce withanolides and, P. alkekengi physalins, a different subgroup of withanolides. The structures of physalins A and B were first determined in 1969 (Matsuura and Kawai, 1969; Matsuura et al., 1969) and subsequently more than 30 physalins were isolated. Studies using spectroscopic methods isolated 3 new- and 7 known steroids including physalins and demonstrated that physalin B exhibited the most significant cytotoxic activities against HeLa human cervical cells (Kawai et al., 2002; Li et al., 2014). Physalin B or F inhibited NF-kappa B activation (Jacobo-Herrera et al., 2006; Wu et al., 2012) and both right- and left-sided partial structures were proposed to play a significant role in their mode of action (Ozawa et al., 2013). 4β-Hydroxywithanolide E derived from P. peruviana inhibited the growth of a human non-small lung cancer cell line (Yen et al., 2010).
High-throughput DNA sequencing with a next-generation sequencer (NGS) is useful for genome assembly, the detection of single nucleotide polymorphisms (SNPs) and genetic variations, and for transcriptome characterization (Wang et al., 2009; Garber et al., 2011). RNA sequencing (RNA-seq) on NGSs can be used for both model and non-model plants (Gongora-Castillo and Buell, 2013; Fukushima and Kusano, 2014; Weber, 2015). Successful transcriptome studies on non-model plants, e.g., crop plants such as the eggplant (Ramesh et al., 2016), pepper (Gordo et al., 2012) and tobacco (Lei et al., 2014) and medicinal plants, e.g., Withania somnifera (Gupta et al., 2013; Senthil et al., 2015) and P. peruviana (Garzon-Martinez et al., 2012) from the Solanaceae family have been published. Resources for transcriptome data from medicinal plants, e.g., the Medicinal Plant Genomics Resource (MPGR1) and the Medicinal Plants Transcriptomics (Medplants2; Gongora-Castillo et al., 2012a,b; Yeo et al., 2013) are available. To take full advantage of the potential of important phytochemicals we need to elucidate their biosynthesis. For this we need genomic data, especially comprehensive transcriptome datasets for gene discovery.
Our main aim of this study is to describe the whole transcriptome map of P. alkekengi and P. peruviana leaves and to inventory the basic information about a wide range of the metabolism including the withanolide biosynthesis pathway in leaves (Figure 1). Here we present a de novo assembly transcriptome from the leaves of P. alkekengi and P. peruviana. We employed Illumina RNA-seq techniques and demonstrate that these resources can provide the means for a better understanding of chemical diversity in Physalis species. We annotated the assembled unigenes in P. alkekengi and P. peruviana and classified unigenes encoding enzyme candidates putatively involved in secondary metabolism. Our approaches provide a basis for future researches on bio-engineering of Physalis plants and they represent a shortcut to gene discovery in these species.
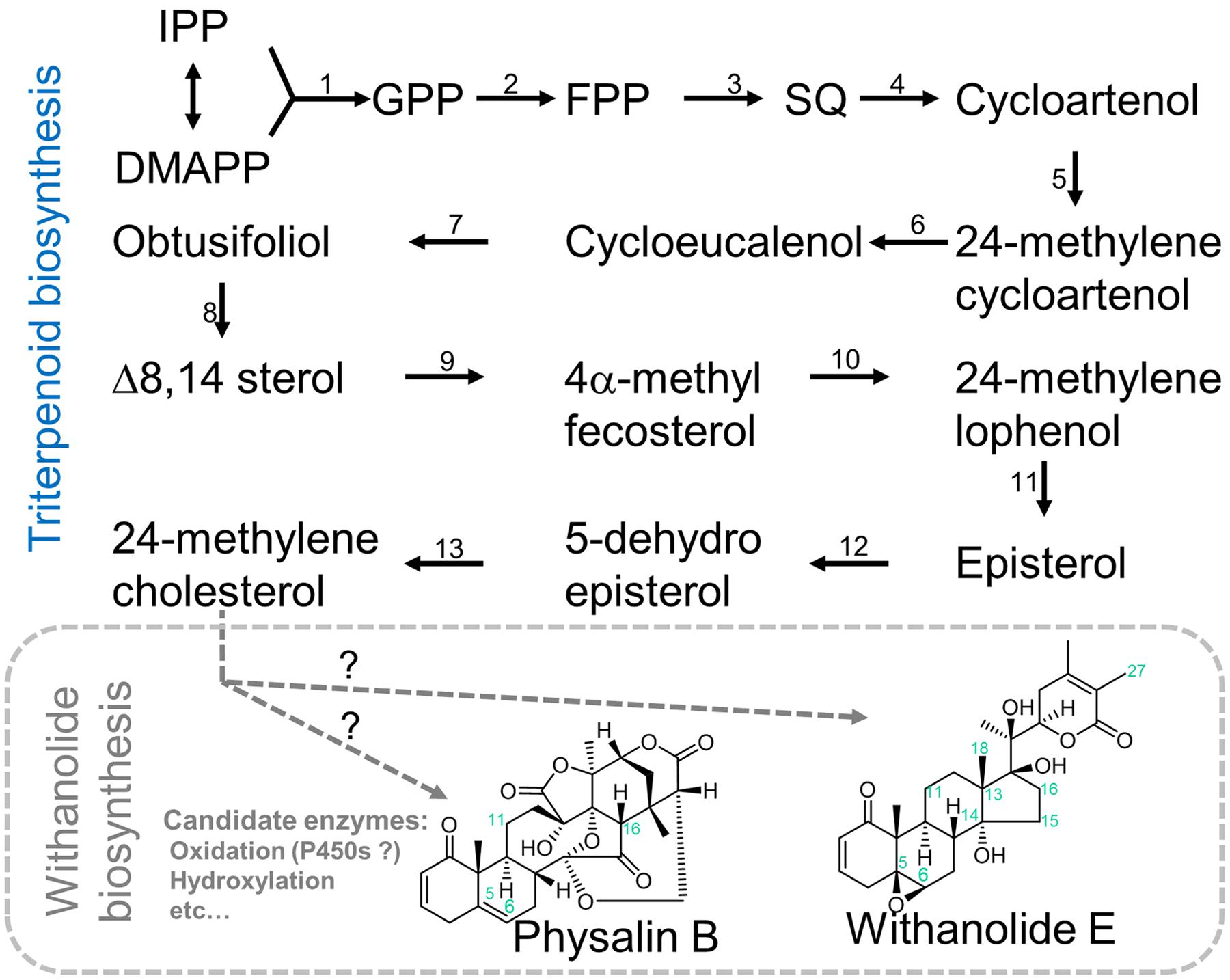
FIGURE 1. Possible pathways of withanolide biosynthesis. Our main aim of this study is to describe the whole transcriptome map of P. alkekengi and P. peruviana leaves and to inventory the basic information about a wide range of the metabolism including the withanolide biosynthesis pathway in leaf tissues. Although the assembled unigenes are hypothetic/proposed candidate genes, comparative transcriptome analysis using other Physalis species is a promising way to study and identify species-specific and evolutionary conserved pathways involved in highly complex biosynthesis of withanolides like physalin. Single black arrows show one step, two or more gray arrows show multiple unknown steps. Abbreviation: IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; GPP, geranyl pyrophosphate; FPP, farnesyl pyrophosphate; and SQ, squalene. Enzyme abbreviation: (1) Geranyl diphosphate Synthase (GPPS, EC 2.5.1.1); (2) Farnesyl diphosphate Synthase (FPPS, EC 2.5.1.10); (3), Squalene Synthase (SQS, EC 2.5.1.21); (4) Cycloartenol Synthase (CAS, EC 5.4.99.8); (5) Cycloartenol C-24 methyltransferase (SMT1, EC 2.1.1.41); (6) Sterol-4a-methyl oxidase (SMO, EC 1 1.14.13.72); (7) Cycloeucalenol cycloisomerase (CECI, EC 5.5.1.9); (8) obtusifoliol 14-demethylase (CYP51G1, EC 1.14.13.70); (9) D14-sterol reductase (FK, EC 1.3.1.70); (10) C-7,8 sterol isomerase (HYDI, EC 5.3.3.5); (11) Sterol-4a-methyl oxidase 2 (SMO2, EC 1.14.13.72); (12) C-5 sterol desaturase (STE1, EC 1.14.21.6); and (13) Sterol Δ7 reductase (DWF5, EC 1.3.1.21).
Materials and Methods
Plant Materials, RNA Isolation, and cDNA Synthesis
Physalis alkekengi var. franchetii and P. peruviana (Ground Cherry or Golden Berry) plants were grown in the experimental gardens of the RIKEN Center for Sustainable Resource Science, Wako, Saitama, Japan. They are not an endangered or protected species. The third fresh leaves were collected from healthy plants (Supplementary Image 1). RNA isolation and cDNA synthesis for sequencing were carried out as previously reported (Fukushima et al., 2015; Han et al., 2015a,b). We used unreplicated data for one sample per species.
Illumina Sequencing
For the generation of cDNA libraries we employed an Illumina HiSeq 2000 sequencer (Illumina Inc., San Diego, CA, USA). We sequenced 100-bp paired-end (PE) reads as described (Fukushima et al., 2015; Han et al., 2015a,b). Our short-read data in FASTQ file format were produced by Casava 1.8 (Illumina, Inc. San Diego, CA, USA). Short reads that did not pass Illumina’s standard quality filter were eliminated. The process yielded clean reads from the mRNA pool isolated from P. alkekengi and P. peruviana, respectively (Table 1).
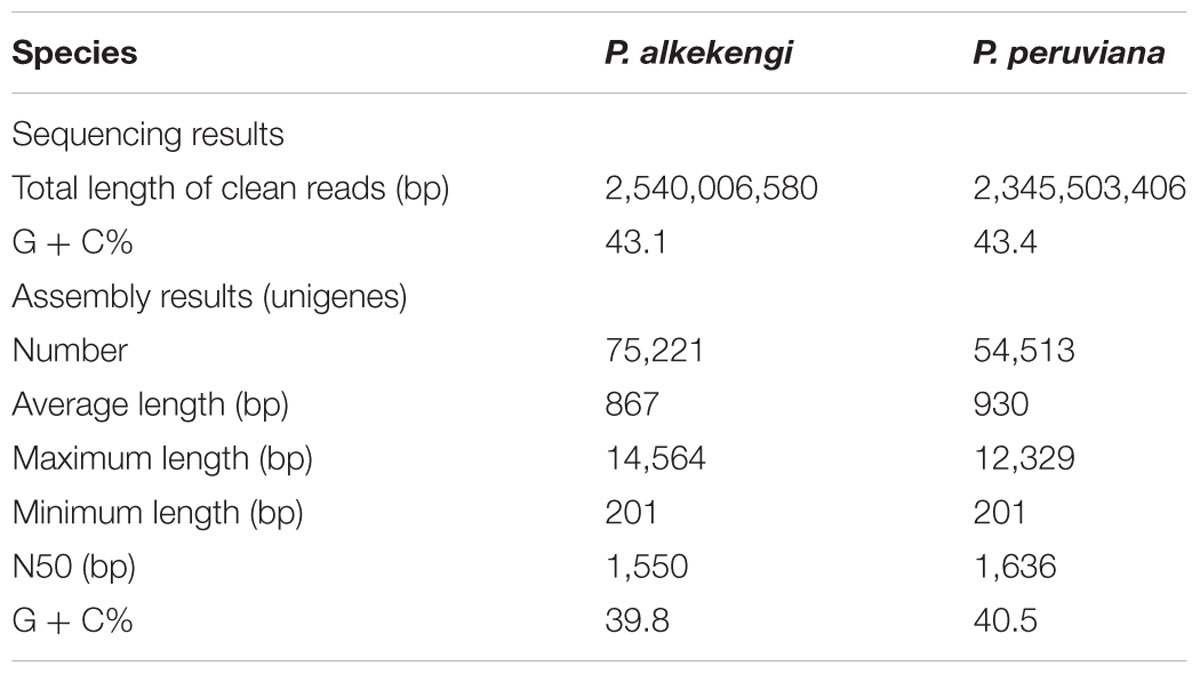
TABLE 1. Summary of sequencing and assembly results after Illumina sequencing of P. alkekengi and P. peruviana.
Data Pre-processing and De novo Transcriptome Assembly
We performed de novo transcriptome assembly using the Trinity program (Grabherr et al., 2011). We then subjected the assembled unigenes to read alignment and transcript abundance estimation with Bowtie (Langmead et al., 2009) and RSEM (Li and Dewey, 2011). To estimate transcript abundance we used the Fragments Per Kilobase of exon per Million mapped fragments (FPKM) method. We identified 75,221 unigenes in P. alkekengi and 54,513 unigenes in P. peruviana. The length and G + C% distribution of all unigenes are shown in Figures 2A,B. The G + C% and basic statistics were calculated with the custom Ruby/Bioruby script (Goto et al., 2010), “Biostrings” (Pages et al., 2009), and the R/Bioconductor package “ShortRead” (Morgan et al., 2009). For quality control we used the FASTX-Toolkit3 and the FastQC package4. Default settings were used in all calculations in this section.
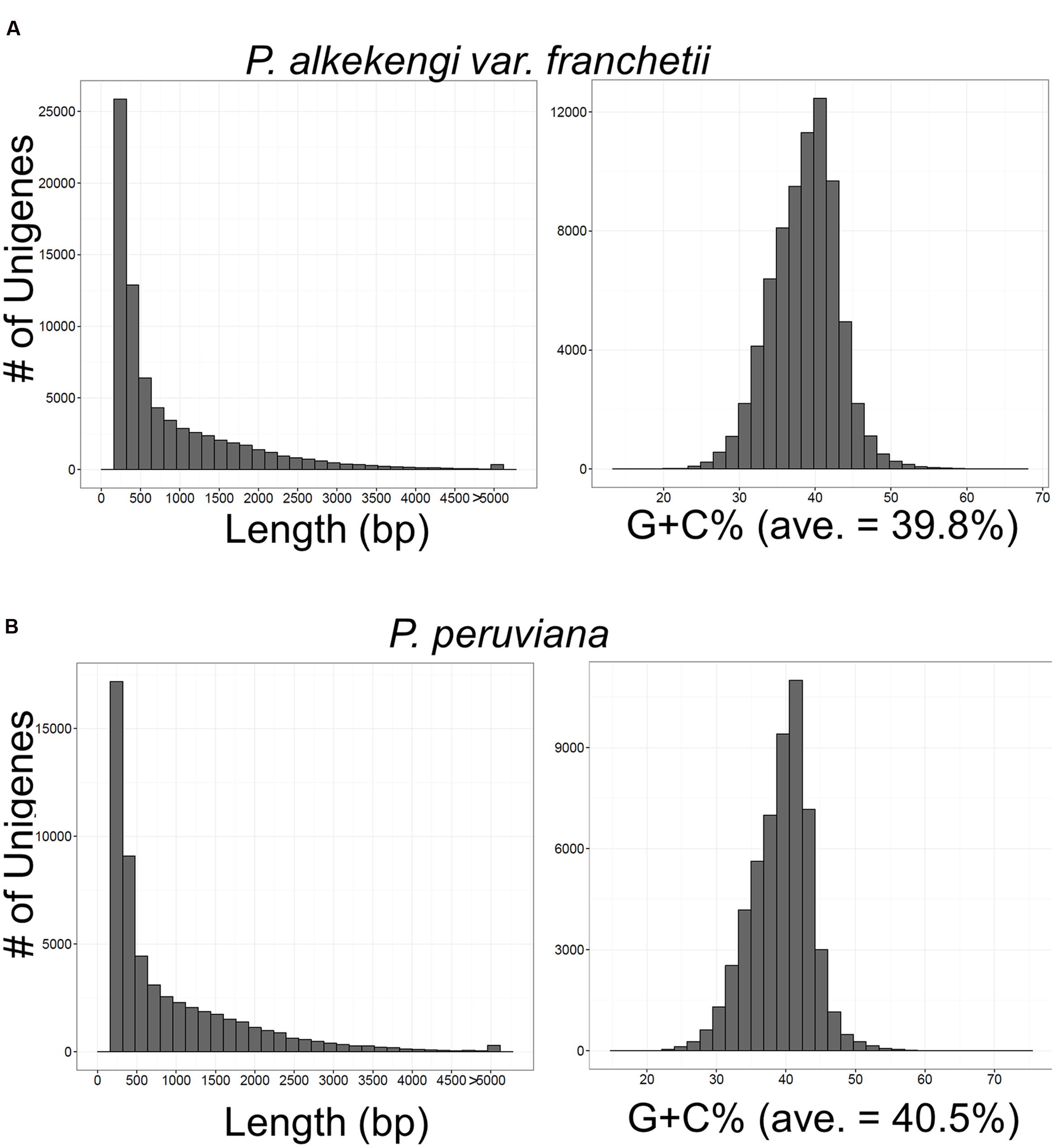
FIGURE 2. Overview of the de novo transcriptome assembly in P. alkekengi (A) and P. peruviana (B). Length and G + C% distribution of unigenes assembled from high-quality clean reads by the Trinity program (Grabherr et al., 2011).
Functional Annotation and Classification of Unigenes
We performed functional annotation of all unigenes with a BLASTx search (Altschul et al., 1990) against the NCBI NR database5 (formatted on April 7, 2014); our cutoff E-value was <1E-5. We applied the Blast2GO program v 2.7.1 (Conesa et al., 2005) to assign GO categories, an EC number, and KEGG pathways (Kanehisa et al., 2014) based on the BLAST results with default settings. Visualization of the GO functional category of all unigenes and of the distribution of gene functions in the different species was with the BGI WEGO program (Ye et al., 2006). We identified microsatellites in the unigenes using the microsatellite identification tool (MISA)6 (Thiel et al., 2003). The default parameters (unit size-minimum repeats: 1-10, 2-6, 3-5, 4-5, 5-5, and 6-5) were used.
Authentic Samples and Extraction of Withanolides and LC-QTOF-MS Analysis
The authentic samples of withanolides were isolated from plants as described in our previous paper (Ozawa et al., 2013). The leaves of P. alkekengi and P. peruviana were collected at five different developmental stages of the growing season before bearing fruit, because our pilot study showed to detect withanolides of P. alkekengi and P. peruviana at these stages (data not shown). Fresh samples of leaves at five different developmental stages were extracted with 5 μl of 80% MeOH containing 2.5 μM lidocaine (internal standard) per mg fresh weight using a mixer mill with zirconia beads (7 min at 18 Hz and 4°C). After 10-min centrifugation, the supernatant was filtered using an HLB μElution plate (Waters). The extracts (1 μl) were analyzed with LC-QTOF-MS (LC, Waters Acquity UPLC system; MS, Waters Xevo G2 Q-Tof). The analytical conditions for metabolite profiling were as described in elsewhere (Tamura et al., 2014). The polarity of electrospray ionization was applied in positive ionization mode.
Results and Discussion
Sample Preparation, Illumina Sequencing, and De novo Transcriptome Assembly
For the transcriptome analysis of P. alkekengi and P. peruviana, total RNA samples were isolated from leaves (Supplementary Image 1). We performed DNase treatment and confirmed RNA integrity using a bioanalyzer (see Materials and Methods). Total RNA was used in mRNA preparation, fragmentation, and cDNA synthesis. Illumina sequencing produced clean reads (in total 2,540,006,580 bp and 2,345,503,406 bp) from the mRNA pool isolated from P. alkekengi and P. peruviana, respectively (Table 1). The short reads exhibited mean quality scores of 35.0 in P. alkekengi and 34.9 in P. peruviana, confirming that our sequencing was sufficient for de novo assembly. After the removal of adaptor-, ambiguous- and low-quality reads, we assembled all clean reads into unigenes using the Trinity program (Grabherr et al., 2011). There were 75,221 and 54,513 total transcripts in leaves of P. alkekengi and P. peruviana, respectively. The unigenes in P. alkekengi had an average length of 867 basepairs (bp) and an N50 of 1,550 bp; in P. peruviana the average length was 930 and 1,636 bp (N50). The length and the G + C% distribution for all unigenes in P. alkekengi and P. peruviana are shown in Figure 2. In total, 39,709 unigenes were less than 500 bp in length. In P. alkekengi 2,732 unigenes were longer than 3,000 bp. In P. peruviana 26,991 unigenes were less than 500 bp and 2,283 were longer than 3,000 bp (Supplementary Data Sheet 1). The average G + C content of unigenes in P. alkekengi and P. peruviana was 39.8 and 40.5%, respectively. The G + C content in P. peruviana is consistent with what has been reported previously (Garzon-Martinez et al., 2012).
Functional Annotation of Physalis Unigenes
To assess and annotate the assembled unigenes, we performed a sequence homology search against the GenBank non-redundant (NR) database using BLASTx (E-value < 1E-5; Altschul et al., 1990). We found that 40,090 and 33,183 unigene sequences in P. alkekengi and P. peruviana, respectively, had BLAST hits to annotated sequences in the NR database (Figure 3). Further analysis of the similarity distributions showed that 72.5 and 74.7% of matched sequences in P. alkekengi and P. peruviana, respectively, manifested alignment identities greater than 80%. For P. alkekengi a large number of the best/top-hits matched the sequences of S. tuberosum (61.5%) and S. lycopersicum (26.3%); other hits were detected within the reference protein databases of Vitis vinifera (1.5%) and S. demissum (1.2%; Figure 3A). The P. peruviana transcriptome showed that the hits matched the sequences of S. tuberosum (61.8%) and S. lycopersicum (27.4%); other hits were detected within the reference protein databases of V. vinifera (1.2%) and Nicotiana tabacum (1.0%; Figure 3B). These observations indicate that the distribution of the top BLAST hits for the obtained unigenes from two different Physalis species was similar. Unigenes with no BLAST hits may imply the presence of additional genes that do not exist in the annotated sequence databases or of sequences too short for BLAST hits.
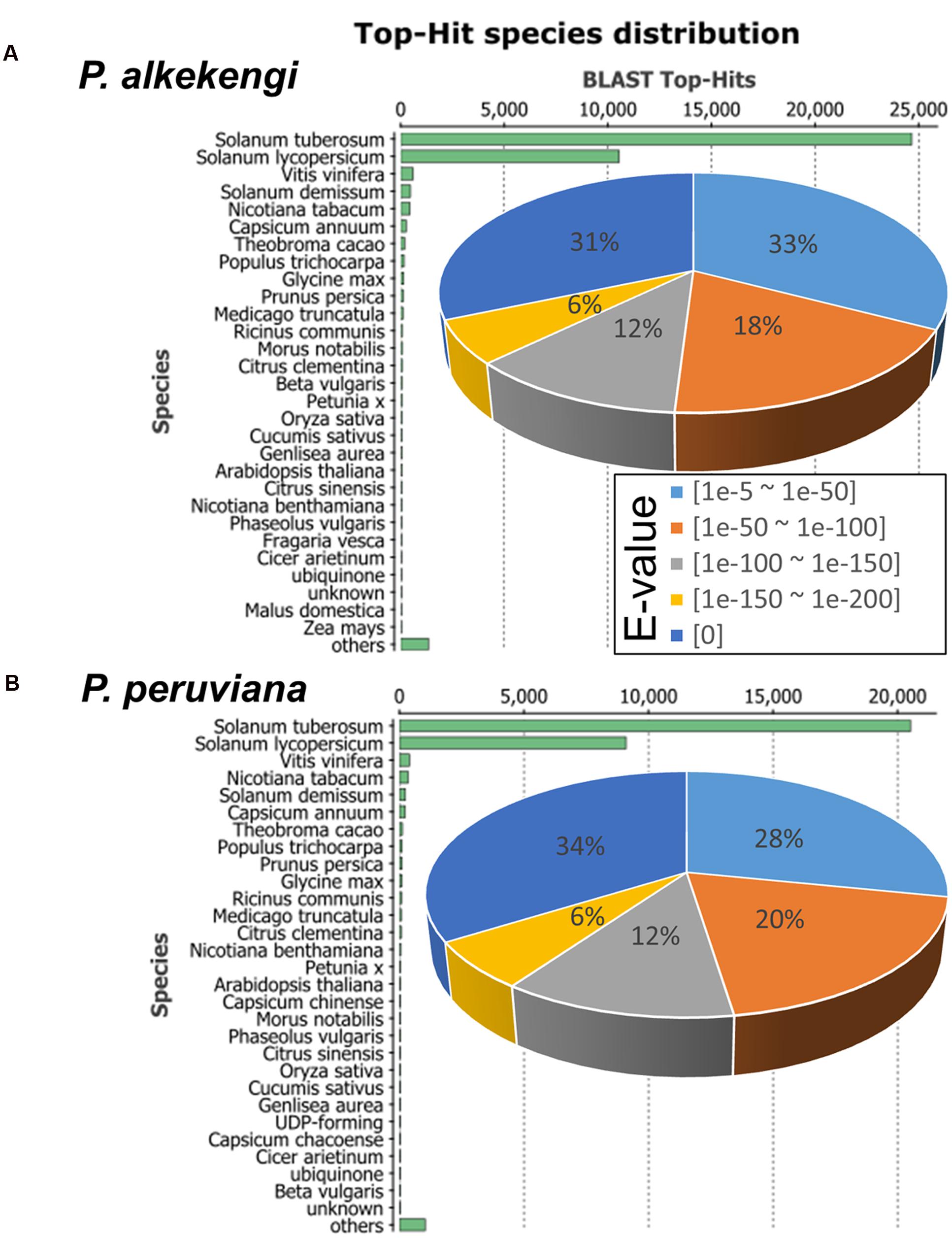
FIGURE 3. Characterization of the assembled unigenes based on a non-redundant (NR) protein database search in Physalis. (A) Top-hit species- and E-value-distributions of P. alkekengi. The E-value distribution of BLAST hits for the assembled unigenes with a cutoff of E-value < 1E-5. (B) Top-hit species- and E-value-distributions of P. peruviana. Top-hit distribution was calculated based on only the best/top sequence alignment with the lowest E-value for our BLAST result.
The gene ontology (GO), classification of standardized gene functions, is useful for annotating gene functions and gene products in any organism. GO contains three main independent categories: cellular component, molecular function, and biological process (The Gene Ontology Consortium, 2014). We used Blast2GO software (Conesa et al., 2005) to analyze the GO functional categories of the assembled unigenes and then applied WEGO program (Ye et al., 2006) to visualize the results of GO functional classifications. WEGO maps all annotated unigenes to GO categories and detect the number of unigenes associated with each GO category. Based on NR annotation, 30,689 and 25,751 unigenes in P. alkekengi and P. peruviana, respectively, were assigned to three main categories (Figure 4). Further analysis of the GO categories showed that the dominant categories were “cell,” “cell part,” “binding,” “catalytic,” “metabolic process,” and “cellular process” (black arrows in Figure 4). We observed that within the biological process group, most unigenes were putatively involved in “cellular process” and “metabolic process.” Most unigenes were assigned to the GO categories “binding” and “catalytic activity” in the molecular function group, and to “cells” and “cell parts” in the cellular component. Please note that these observation depends on the layer of the GO categories.
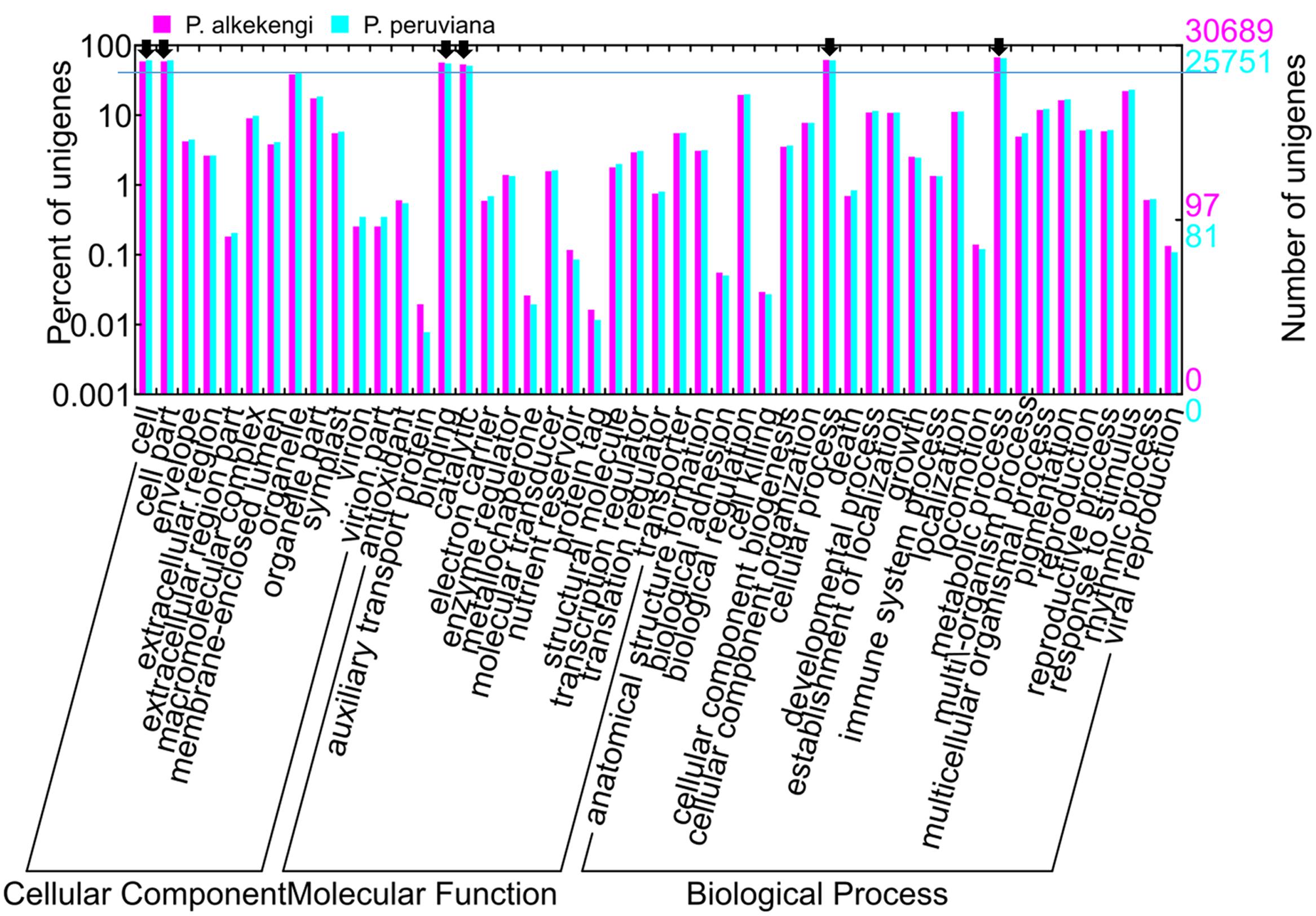
FIGURE 4. GO assignments for all assembled unigenes in P. alkekengi and P. peruviana. The results are summarized in sets of three functional categories: cellular component, molecular function, and biological process. 30,689 and 25,751 unigenes from P. alkekengi (magenta) and P. peruviana (cyan), respectively, were categorized by GO categories. The GO categories were displayed using WEGO (http://wego.genomics.org.cn; Ye et al., 2006).
KEGG (Kanehisa et al., 2014) is a database resource for various levels of molecules in biological systems. It provides useful information for exploring the functional characteristics of genes. To further elucidate the function of the Physalis transcriptomes, the unigenes were annotated by the KEGG pathway. A total of 3,819 and 3,669 unigenes in P. alkekengi and P. peruviana was assigned into 134 and 139 KEGG pathways, respectively (Figure 5 and Supplementary Data Sheet 2). The top-five ranking pathways in P. alkekengi were purine- (683 unigenes), starch and sucrose- (344 unigenes), thiamine- (258 unigenes), pyrimidine- (244 unigenes), and glycerolipid metabolism (190 unigenes). The top-five ranking pathways in P. peruviana were similar to P. alkekengi except for glycolysis/gluconeogenesis (178 unigenes in P. peruviana; Supplementary Data Sheet 2). These results agree with a previous study that performed the P. peruviana leaf transcriptome (Garzon-Martinez et al., 2012). In addition, we observed a higher number of the classified unigenes putatively involved in starch and sucrose- and glycolysis metabolism in P. peruviana than that of P. alkekengi. Such gene diversity derived from genomic architecture may reflect different source-to-sink balance between the both species, causing different photosynthetic rate and the carbon partitioning and allocation [for example, see (Osorio et al., 2014)]. This analysis implies that the pathway-based approach (see reviews by Ramanan et al., 2012; Fukushima et al., 2014) is useful for a better understanding of biological functions, gene interactions, and specific processes in Physalis species.
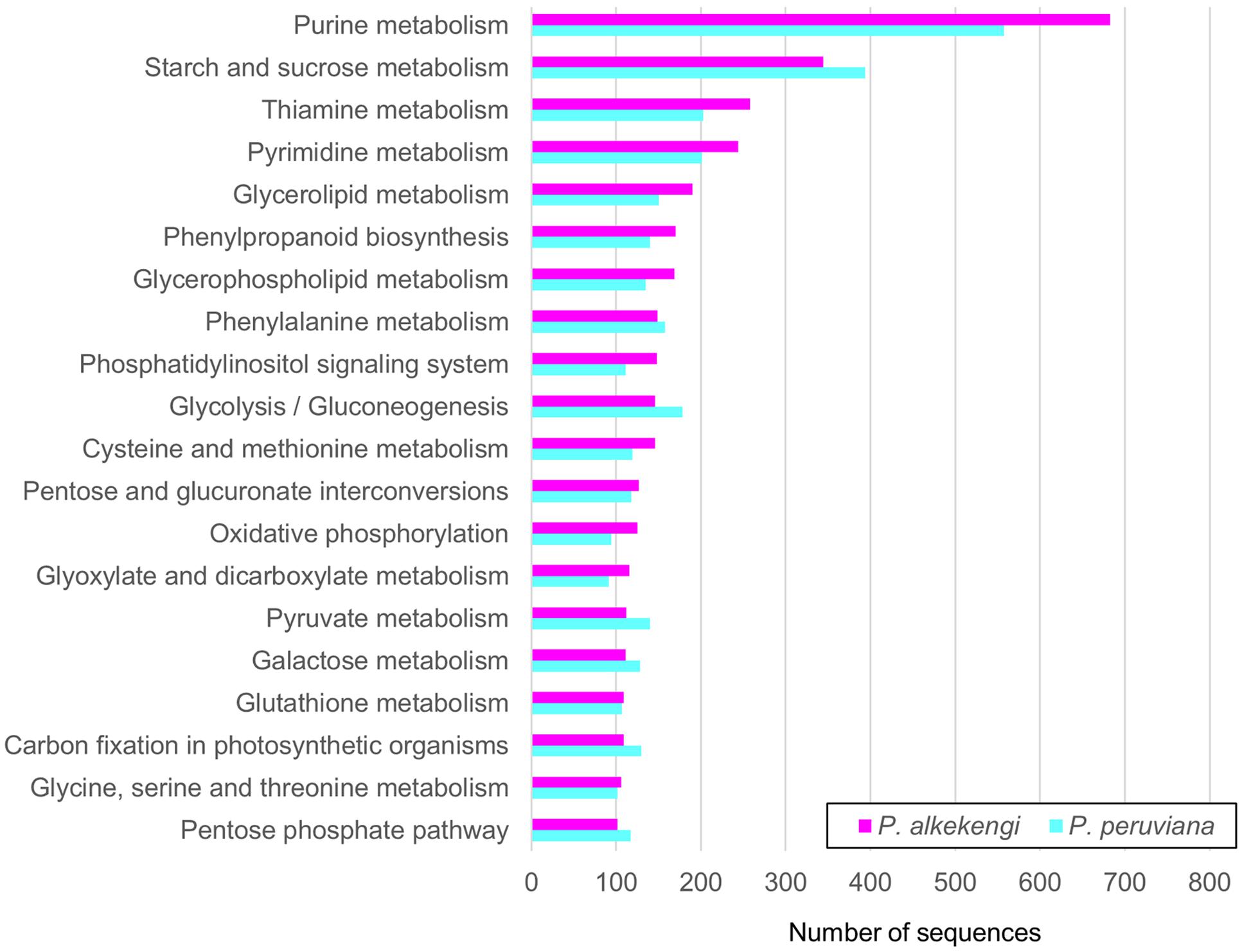
FIGURE 5. Pathway enrichment analysis of assembled unigenes in Physalis. Annotated unigenes were classified into 134 and 139 KEGG pathways in P. alkekengi and P. peruviana, respectively. The top 20 pathways including unigenes in P. alkekengi and the corresponding pathways in P. peruviana are displayed.
Candidate Gene Families Associated with Withanolide Biosynthesis
To visualize diversity in secondary metabolism, we classified unigenes into gene families (Supplementary Data Sheet 2C). The resultant table shows unigenes encoding enzyme candidates putatively involved in the biosynthesis of secondary metabolites such as alkaloids, terpenoids, steroids, and flavonoids. For terpenoid backbone- and steroid biosynthesis we identified more than one unigene for each step in P. alkekengi and P. peruviana. For example, unigenes encoding enzyme candidates putatively involved in the biosynthesis of 24-methylene cholesterol from isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) [the synthesis requires 13 reaction steps (Gupta et al., 2013, 2015)] contained Δ7 desaturase (EC:1.14.21.6), 24-C-methyltrasferase (EC:2.1.1.41), squalene epoxidase (EC: 1.14.13.132→1.14.14.17), squalene synthase (EC: 2.5.1.21), cholestenol delta-isomerase (EC: 5.3.3.5), and sterol Δ7-reductase (EC:1.3.1.21). Sterol Δ7-reductase catalyzes the biosynthesis of 24-methylene cholesterol, a central precursor in withanolide biosynthesis (Lockley et al., 1976). Gupta et al. demonstrated that silencing of sterol Δ7-reductase in W. somnifera elicited a reduction in the level of the major withanolide withaferin A in leaves (Gupta et al., 2015).
The specific withanolide contents in leaves of P. alkekengi and P. peruviana at different stages of the leaf development remain largely unknown. To monitor the variability of the withanolides including physalins and provide insights into the chemical diversity of Physalis, we analyzed the metabolite content in leaves of P. alkekengi and P. peruviana at five different developmental stages by liquid chromatography-quadrupole time-of-flight-mass spectrometry (LC-QTOF-MS), focusing on six withanolide metabolites, i.e., physalin B, D, F, withanolide E and F, and perulactone B (Supplementary Image 2). We found that P. alkekengi produced high levels of physalin B, D, and F. In P. peruviana we only detected withanolide E and F, and perulactone B (Figure 6). Low levels of withanolide E, F, and perulactone B in P. peruviana were observed at the early developmental stages, while the highest levels of withanolide E and F were detected in the young leaves at day 65 (t-test, p < 0.01). A possible explanation for the difference in metabolite levels could be that there are sequence diversity in candidate genes putatively involved in various secondary transformations, including hydroxylation, glycosylation, methylation, and oxidation/reduction, to produce species-specific and development-specific withanolides from 24-methylene cholesterol as a substrate.
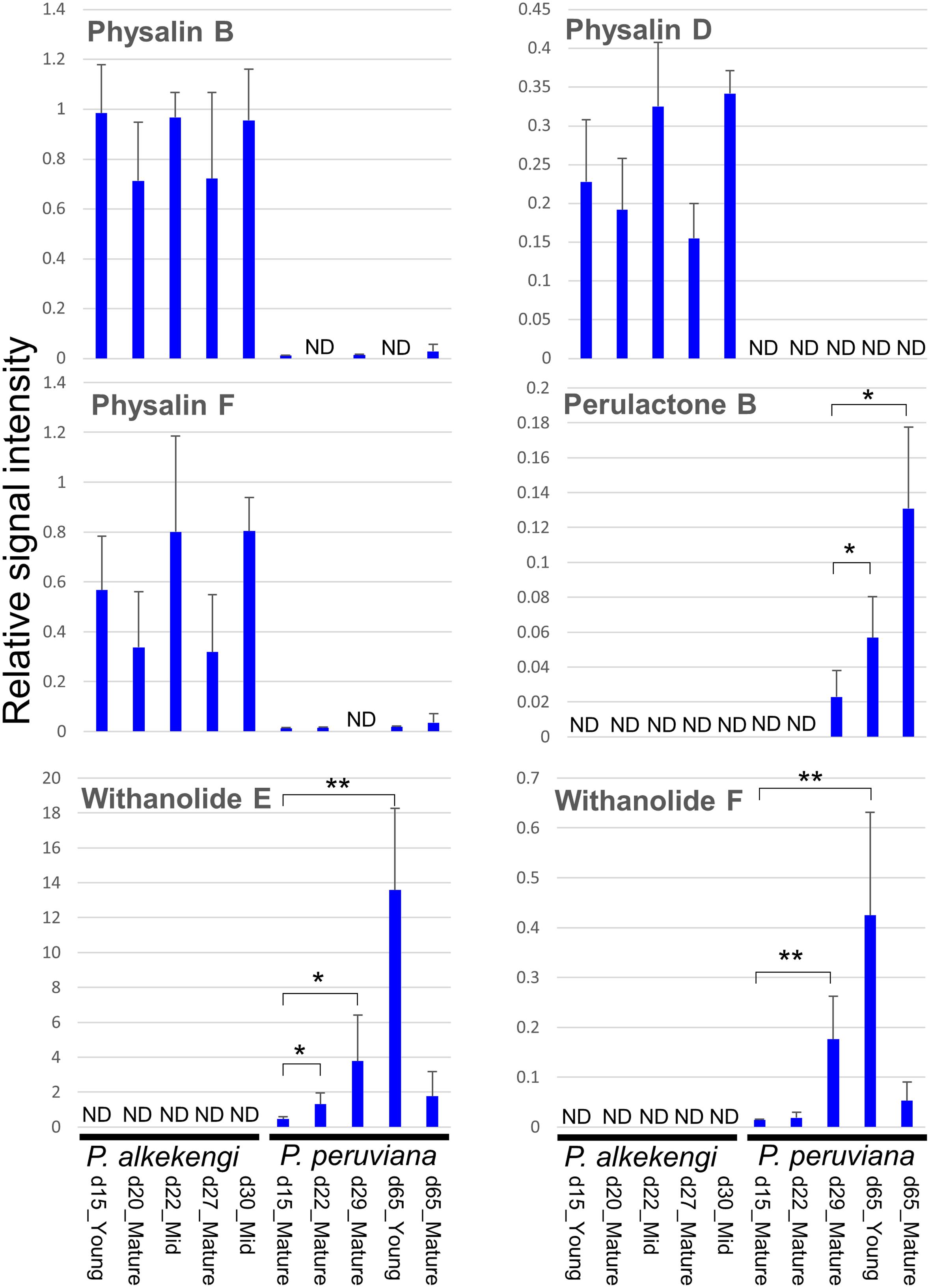
FIGURE 6. Measurement of six withanolides of the leaves from P. alkekengi and P. peruviana. Different developmental stages and leaf ages were used for comparison between P. alkekengi and P. peruviana. The analysis was performed with 3–6 biological replicates for each tissue/leaf age. An error bar indicates standard deviation. The authentic compounds used in this analysis were chemically synthesized as described in our previous paper (Ozawa et al., 2013). The limit of detection of each sample was 0.011923 (P. alkekengi, d15_Young), 0.010910 (P. alkekengi, d20_Mature), 0.013556 (P. alkekengi, d22_Mid), 0.010820 (P. alkekengi, d27_Mature), 0.010884 (P. alkekengi, d30_Mid), 0.012211 (P. peruviana, d15_Mature), 0.012456 (P. peruviana, d22_Mature), 0.012366 (P. peruviana, d29_Mature), 0.014794 (P. peruviana, d65_Young), and 0.013141 (P. peruviana, d65_Mature). Asterisks represent statistically significant differences from the sample of the youngest stage (i.e., d29_Mature for perulactone B and d15_Mature for withanolide E and F; t-test, ∗ p < 0.05 and ∗∗ p < 0.01). Abbreviation: d, day; Mid, middle; and ND, not detected.
To further narrow down the candidate genes responsible for the biosynthesis of withanolides, we sought to identify unigenes that are uniquely expressed in either species. Such unigenes may play important roles in the difference of metabolic phenotypes. To this end we performed reciprocal best-hit BLAST search to identify homologous unigenes in P. alkekengi and P. peruviana with BLASTn (E-value < 1E-14) resulting in 25,670 homologous unigenes. Of these unigenes, we identified unigenes expressed uniquely in either of these species (Supplementary Data Sheet 3). We uncovered 234 unigenes that are exclusive to P. alkekengi (FPKM > 1). Of these, a cytochrome p450 chloroplastic-like protein (unigene-ID: c13295_g2_i2) and oxidoreductase-like protein (c16207_g5_i1), are possible candidate genes for the oxidations at the C-15 and C-18 positions of the steroid backbone required in the synthesis of physalis (Figure 1). In contrast, 355 unigenes found in P. peruviana were identified through this approach. There were unigenes encoding sterol reductase (c27112_g1_i1), methyltransferase family proteins, and transcription factors like MYB as candidates of specialized metabolites that are likely produced in P. peruviana. We had expected some obvious, differentially expressed candidates from the comparative transcriptomics, as was reported by (Yamazaki et al., 2008). However, the list of genes expressed exclusively in either P. alkekengi or P. peruviana provided only few candidates. The transcriptomes of both P. alkekengi and P. peruviana contain many cytochrome p450 monooxygenases and dioxygenases that could be modify both withanolides and physalins. However, none of these stand out as predominant in either Physalis species. We expect the biosynthesis of physalins to include cleavage of the C13-C14 bond as well as crosslinking between C14 and C27. Currently we have no good candidate enzymes for these reactions. Cleavage of the C13-C14 bond may occur through Grob fragmentation by an oxidosqualene cyclase type enzyme as described by Xiong et al. (2006).
Although there was no convincing evidence for significant changes in gene expressions encoding any of the key enzymes, we were able to provide a set of the hypothetical candidate genes involved in the pathways. We also recognize the limitation of our metabolite analysis in all its quantitative aspects, and that further assessment of the integration of our transcriptome data with wider metabolite profile is needed, but the findings presented here can provide a relevant resource for future study in Physalis. Our transcriptome dataset can be used for the identification of genes that contribute to the diversification in Physalis species-specific secondary metabolism, such as glycosyltransferases, methyltransferases, and cytochrome P450s.
Identification and Comparison of Single Sequence Repeat (SSR) Markers
Single sequence repeats SSRs are microsatellites, repetitive DNA sequences in eukaryotes. They are useful markers in population genetics research, genetic map construction, and genetic diversity assessment (for example, see reviews Hancock, 1996; Varshney et al., 2005). To detect and compare SSRs in the two different Physalis species we performed in silico SSR marker identification with MISA (Thiel et al., 2003). We identified 11,415 SSRs in 75,221 transcripts of P. alkekengi and 12,180 SSRs in P. peruviana (Table 2 and Supplementary Data Sheet 4). All SSRs can be classified by their repeat-unit sizes. Mono-nucleotide SSRs represented the largest portion (60.9%) of identified SSRs, followed by tri-nucleotide- (19.8%) and di-nucleotide (18.2%) SSRs. Although only a small portion of tetra- (n = 86), penta- (n = 14), and hexa-nucleotide SSRs (n = 19) were detected in P. alkekengi transcripts, it was significant. P. peruviana sequences showed nearly the same tendency and had more SSRs than P. alkekengi, especially mono- and tri-nucleotide SSRs. There were small numbers of tetra-, penta- and hexa-nucleotide SSRs in our transcriptome sequences from the two species, implying that their base patterns were highly complex. The SSRs of both P. alkekengi and P. peruviana may yield potential genetic markers for a wide range of investigations including population genetics-, comparative genomic-, and gene-based association studies aimed at elucidating the genetic control of important traits.
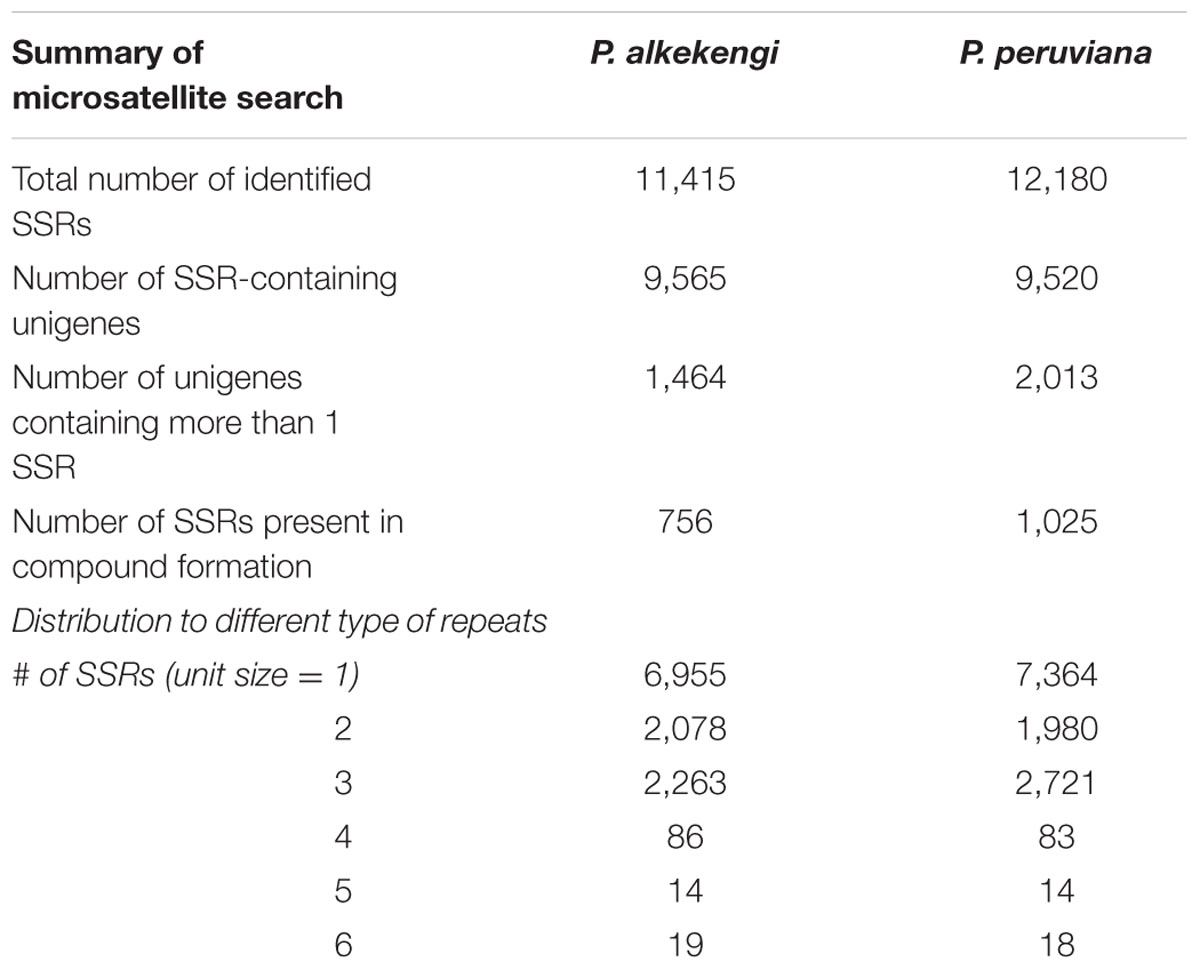
TABLE 2. Statistics of SSRs detected in P. alkekengi and P. peruviana.
Conclusion
We present the comprehensive transcriptome of the leaves of P. alkekengi and P. peruviana and provide a large-scale resource for assembled and functionally annotated gene candidates. Deep transcriptome analysis provided 75,221 unigenes in P. alkekengi and 54,513 in P. peruviana, respectively. A BLAST search of these sequences identified 40,090 and 33,183 unigenes in P. alkekengi and P. peruviana as annotated proteins, respectively. We discussed our findings on the gene candidates putatively involved in withanolide biosynthesis and their transcript- and metabolite profiles. Our detection of 11,415 SSRs in P. alkekengi and 12,180 SSRs in P. peruviana provides a useful resource for population genetics studies, genetic map construction, and genetic diversity assessment in Physalis. Because the Physalis genus is valuable for producing medicines and functional foods, more genomic data on other members are needed. Our study suggests that comprehensive approaches applied within the family can provide a clue to gene discovery in Physalis and yield insights into their complex diversity. Given the insufficient knowledge on the molecular mechanisms controlling the biosynthetic pathways involved in various bioactive metabolites like withanolides, the transcriptome information reported here represents an important public resource for further study on the specialized metabolism of Physalis species.
Data Accessibility
All raw read sequences in FASTQ format can be downloaded from the DDBJ Sequence Read Archive (Kosuge et al., 2014) under accession number DRA004085. Our assembled transcripts were deposited as a Transcriptome Shotgun Assembly (TSA) in GenBank/EMBL/DDBJ under the accession IABG01000001-IABG01075221 (75221 entries for P. alkekengi) and IABH01000001-IABH01054513 (54513 entries for P. peruviana).
Author Contributions
Conceived and designed the experiments: AF, HS, MY, MS, and KS. Performed the experiments: MN, HS, MY, TM, MM, GH, and MS. Analyzed the data: AF, EK, NU, TM, and HS. Contributed reagents/materials/analysis tools: MN, HS, MY, TM, MM, GH, and MS. Wrote the paper: AF, EK, and KS. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported, in part, by Studies on enhancement of ’Comprehensive Medicinal Plant Database’ aiming for cultivation of medicinal plants and industrial development, a Health and Labour Sciences Research Grant, a Japan Agency for Medical Research and Development Sciences Research Grant, the Japan Advanced Plant Science Research Network, and Strategic Priority Research Promotion Program of Chiba University. Research by EK was financially supported by the Carlsberg Foundation. We thank Dr. Masaaki Ozawa (RIKEN Center for Sustainable Resource Science) for experimental assistance, Ms. Ursula Petralia and Prof. Miyako Kusano (University of Tsukuba) for editorial assistance, and Dr. Tetsuya Sakurai and Mr. Yutaka Yamada (RIKEN Center for Sustainable Resource Science) for computational assistance. We also thank Ms. Sayaka Shinpo (Kazusa DNA Research Institute) for technical support in Illumina sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01883/full#supplementary-material
IMAGE 1 | Schematic workflow of this study. We identified 75,221 and 54,513 transcripts in leaves of P. alkekengi and P. peruviana with the Trinity program (Grabherr et al., 2011). Assembled unigenes were annotated by Blast2GO program (Conesa et al., 2005).
IMAGE 2 | Structures of complex and oxidized steroidal constituents isolated from Physalis plants. We analyzed the metabolite content in leaf tissues of P. alkekengi and P. peruviana at five different developmental stages by liquid chromatography-quadrupole time-of-flight-mass spectrometry (LC-QTOF-MS), focusing on these six withanolide metabolites.
Footnotes
- ^ http://medicinalplantgenomics.msu.edu/
- ^ http://medplants.ncgr.org/
- ^ http://hannonlab.cshl.edu/fastx_toolkit/
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://www.ncbi.nlm.nih.gov
- ^ http://pgrc.ipk-gatersleben.de/misa/
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Fukushima, A., Kanaya, S., and Nishida, K. (2014). Integrated network analysis and effective tools in plant systems biology. Front. Plant Sci. 5:598. doi: 10.3389/fpls.2014.00598
Fukushima, A., and Kusano, M. (2014). A network perspective on nitrogen metabolism from model to crop plants using integrated ’omics’ approaches. J. Exp. Bot. 65, 5619–5630. doi: 10.1093/jxb/eru322
Fukushima, A., Nakamura, M., Suzuki, H., Saito, K., and Yamazaki, M. (2015). High-throughput sequencing and de novo assembly of red and green forms of the perilla frutescens var. crispa transcriptome. PLoS ONE 10:e0129154. doi: 10.1371/journal.pone.0129154
Garber, M., Grabherr, M. G., Guttman, M., and Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 8, 469–477. doi: 10.1038/nmeth.1613
Garzon-Martinez, G. A., Zhu, Z. I., Landsman, D., Barrero, L. S., and Marino-Ramirez, L. (2012). The Physalis peruviana leaf transcriptome: assembly, annotation and gene model prediction. BMC Genomics 13:151. doi: 10.1186/1471-2164-13-151
Gongora-Castillo, E., and Buell, C. R. (2013). Bioinformatics challenges in de novo transcriptome assembly using short read sequences in the absence of a reference genome sequence. Nat. Prod. Rep. 30, 490–500. doi: 10.1039/c3np20099j
Gongora-Castillo, E., Childs, K. L., Fedewa, G., Hamilton, J. P., Liscombe, D. K., Magallanes-Lundback, M., et al. (2012a). Development of transcriptomic resources for interrogating the biosynthesis of monoterpene indole alkaloids in medicinal plant species. PLoS ONE 7:e52506. doi: 10.1371/journal.pone.0052506
Gongora-Castillo, E., Fedewa, G., Yeo, Y., Chappell, J., Dellapenna, D., and Buell, C. R. (2012b). Genomic approaches for interrogating the biochemistry of medicinal plant species. Methods Enzymol. 517, 139–159. doi: 10.1016/B978-0-12-404634-4.00007-3
Gordo, S. M., Pinheiro, D. G., Moreira, E. C., Rodrigues, S. M., Poltronieri, M. C., De Lemos, O. F., et al. (2012). High-throughput sequencing of black pepper root transcriptome. BMC Plant Biol. 12:168. doi: 10.1186/1471-2229-12-168
Goto, N., Prins, P., Nakao, M., Bonnal, R., Aerts, J., and Katayama, T. (2010). BioRuby: bioinformatics software for the Ruby programming language. Bioinformatics 26, 2617–2619. doi: 10.1093/bioinformatics/btq475
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gupta, P., Goel, R., Agarwal, A. V., Asif, M. H., Sangwan, N. S., Sangwan, R. S., et al. (2015). Comparative transcriptome analysis of different chemotypes elucidates withanolide biosynthesis pathway from medicinal plant Withania somnifera. Sci. Rep. 5:18611. doi: 10.1038/srep18611
Gupta, P., Goel, R., Pathak, S., Srivastava, A., Singh, S. P., Sangwan, R. S., et al. (2013). De novo assembly, functional annotation and comparative analysis of Withania somnifera leaf and root transcriptomes to identify putative genes involved in the withanolides biosynthesis. PLoS ONE 8:e62714. doi: 10.1371/journal.pone.0062714
Han, R., Takahashi, H., Nakamura, M., Bunsupa, S., Yoshimoto, N., Yamamoto, H., et al. (2015a). Transcriptome analysis of nine tissues to discover genes involved in the biosynthesis of active ingredients in sophora flavescens. Biol. Pharm. Bull. 38, 876–883. doi: 10.1248/bpb.b14-00834
Han, R., Takahashi, H., Nakamura, M., Yoshimoto, N., Suzuki, H., Shibata, D., et al. (2015b). Transcriptomic landscape of Pueraria lobata demonstrates potential for phytochemical study. Front. Plant Sci. 6:426. doi: 10.3389/fpls.2015.00426
Hancock, J. M. (1996). Simple sequences in a ”minimal’ genome. Nat. Genet. 14, 14–15. doi: 10.1038/ng0996-14
Jacobo-Herrera, N. J., Bremner, P., Marquez, N., Gupta, M. P., Gibbons, S., Munoz, E., et al. (2006). Physalins from Witheringia solanacea as modulators of the NF-kappaB cascade. J. Nat. Prod. 69, 328–331. doi: 10.1021/np050225t
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kawai, M., Makino, B., Yamamura, H., Araki, S., Butsugan, Y., and Ohya, J. (2002). Cytotoxic activity of physalins and related compounds against HeLa cells. Pharmazie 57, 348–350.
Kosuge, T., Mashima, J., Kodama, Y., Fujisawa, T., Kaminuma, E., Ogasawara, O., et al. (2014). DDBJ progress report: a new submission system for leading to a correct annotation. Nucleic Acids Res. 42, D44–D49. doi: 10.1093/nar/gkt1066
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lei, B., Lu, K., Ding, F., Zhang, K., Chen, Y., Zhao, H., et al. (2014). RNA sequencing analysis reveals transcriptomic variations in tobacco (Nicotiana tabacum) leaves affected by climate, soil, and tillage factors. Int. J. Mol. Sci. 15, 6137–6160. doi: 10.3390/ijms15046137
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, X., Zhao, J., Yang, M., Liu, Y., Li, Z., Li, R., et al. (2014). Physalins and withanolides from the fruits of Physalis alkekengi L. var. franchetii (Mast.) Makino and the inhibitory activities against human tumor cells. Phytochem. Lett. 10, 95–100. doi: 10.1016/j.phytol.2014.08.004
Lockley, W. J. S., Rees, H. H., and Goodwin, T. W. (1976). Biosynthesis of steroidal withanolides in Withania somnifera. Phytochemistry 15, 937–939. doi: 10.1016/S0031-9422(00)84374-5
Martinez, M. (1998). Revision of Physalis section epiteiorhiza (Solanaceae). Anal. Inst. Biol. Serie Bot. 69, 71–117.
Matsuura, T., and Kawai, M. (1969). Bitter principles of physalis alkekengi var francheti: structure of physalin B. Tetrahedron Lett. 10, 1765–1766. doi: 10.1016/S0040-4039(01)88006-0
Matsuura, T., Kawai, M., Nakashima, R., and Butsugan, Y. (1969). Bitter principles of Physalis alkekengi var francheti: structure of physalin a. Tetrahedron Lett. 10, 1083–1086. doi: 10.1016/S0040-4039(01)88006-0
Morgan, M., Anders, S., Lawrence, M., Aboyoun, P., Pages, H., and Gentleman, R. (2009). ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608. doi: 10.1093/bioinformatics/btp450
Osorio, S., Ruan, Y. L., and Fernie, A. R. (2014). An update on source-to-sink carbon partitioning in tomato. Front. Plant Sci. 5:516. doi: 10.3389/fpls.2014.00516
Ozawa, M., Morita, M., Hirai, G., Tamura, S., Kawai, M., Tsuchiya, A., et al. (2013). Contribution of cage-shaped structure of physalins to their mode of action in inhibition of NF-kappaB activation. ACS Med. Chem. Lett. 4, 730–735. doi: 10.1021/ml400144e
Pages, H., Aboyoun, P., Gentleman, R., and Debroy, S. (2009). Biostrings: String Objects Representing Biological Sequences, and Matching Algorithms. R Package Version 2.32.1. Available at: http://bioconductor.org/packages/Biostrings/
Ramadan, M. F., and Morsel, J. T. (2003). Oil goldenberry (Physalis peruviana L.). J. Agric. Food Chem. 51, 969–974. doi: 10.1021/jf020778z
Ramanan, V. K., Shen, L., Moore, J. H., and Saykin, A. J. (2012). Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet. 28, 323–332. doi: 10.1016/j.tig.2012.03.004
Ramesh, K. R., Hemalatha, R., Vijayendra, C. A., Arshi, U. Z., Dushyant, S. B., and Dinesh, K. B. (2016). Transcriptome analysis of Solanum melongena L. (eggplant) fruit to identify putative allergens and their epitopes. Gene 576, 64–71. doi: 10.1016/j.gene.2015.09.064
Senthil, K., Jayakodi, M., Thirugnanasambantham, P., Lee, S. C., Duraisamy, P., Purushotham, P. M., et al. (2015). Transcriptome analysis reveals in vitro cultured Withania somnifera leaf and root tissues as a promising source for targeted withanolide biosynthesis. BMC Genomics 16:14. doi: 10.1186/s12864-015-1214-0
Tamura, M., Tsuji, Y., Kusunose, T., Okazawa, A., Kamimura, N., Mori, T., et al. (2014). Successful expression of a novel bacterial gene for pinoresinol reductase and its effect on lignan biosynthesis in transgenic Arabidopsis thaliana. Appl. Microbiol. Biotechnol. 98, 8165–8177. doi: 10.1007/s00253-014-5934-x
The Gene Ontology Consortium (2014). Gene ontology consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. doi: 10.1093/nar/gku1179
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Varshney, R. K., Graner, A., and Sorrells, M. E. (2005). Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 23, 48–55. doi: 10.1016/j.tibtech.2004.11.005
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Weber, A. P. (2015). Discovering new biology through sequencing of RNA. Plant Physiol. 169, 1524–1531. doi: 10.1104/pp.15.01081
Wu, S. Y., Leu, Y. L., Chang, Y. L., Wu, T. S., Kuo, P. C., Liao, Y. R., et al. (2012). Physalin F induces cell apoptosis in human renal carcinoma cells by targeting NF-kappaB and generating reactive oxygen species. PLoS ONE 7:e40727. doi: 10.1371/journal.pone.0040727
Xiong, Q., Wilson, W. K., and Matsuda, S. P. (2006). An Arabidopsis oxidosqualene cyclase catalyzes iridal skeleton formation by grob fragmentation. Angew. Chem. Int. Ed. Engl 45, 1285–1288. doi: 10.1002/anie.200503420
Yamazaki, M., Shibata, M., Nishiyama, Y., Springob, K., Kitayama, M., Shimada, N., et al. (2008). Differential gene expression profiles of red and green forms of Perilla frutescens leading to comprehensive identification of anthocyanin biosynthetic genes. FEBS J. 275, 3494–3502. doi: 10.1111/j.1742-4658.2008.06496.x
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Yen, C. Y., Chiu, C. C., Chang, F. R., Chen, J. Y., Hwang, C. C., Hseu, Y. C., et al. (2010). 4beta-Hydroxywithanolide E from Physalis peruviana (golden berry) inhibits growth of human lung cancer cells through DNA damage, apoptosis and G2/M arrest. BMC Cancer 10:46. doi: 10.1186/1471-2407-10-46
Yeo, Y. S., Nybo, S. E., Chittiboyina, A. G., Weerasooriya, A. D., Wang, Y. H., Gongora-Castillo, E., et al. (2013). Functional identification of valerena-1,10-diene synthase, a terpene synthase catalyzing a unique chemical cascade in the biosynthesis of biologically active sesquiterpenes in Valeriana officinalis. J. Biol. Chem. 288, 3163–3173. doi: 10.1074/jbc.M112.415836
Keywords: Physalis, de novo transcriptome assembly, deep sequencing, marker development, secondary metabolism, physalin, withanolide
Citation: Fukushima A, Nakamura M, Suzuki H, Yamazaki M, Knoch E, Mori T, Umemoto N, Morita M, Hirai G, Sodeoka M and Saito K (2016) Comparative Characterization of the Leaf Tissue of Physalis alkekengi and Physalis peruviana Using RNA-seq and Metabolite Profiling. Front. Plant Sci. 7:1883. doi: 10.3389/fpls.2016.01883
Received: 13 July 2016; Accepted: 29 November 2016;
Published: 20 December 2016.
Edited by:
Xiaowu Wang, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Erli Pang, Beijing Normal University, ChinaZhonghua Zhang, Chinese Academy of Agricultural Sciences, China
Copyright © 2016 Fukushima, Nakamura, Suzuki, Yamazaki, Knoch, Mori, Umemoto, Morita, Hirai, Sodeoka and Saito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Fukushima, atsushi.fukushima@riken.jp Kazuki Saito, kazuki.saito@riken.jp