
- 1 Department of Psychiatry and Neurochemistry, Addiction Biology Unit, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 2 Department of Pharmacology, Merck Sharp & Dohme, Newhouse, Lanarkshire, UK
- 3 Beroendekliniken, Sahlgrenska University Hospital, Gothenburg, Sweden
Accumbal glycine modulates basal and ethanol-induced dopamine levels in the nucleus accumbens (nAc) as well as voluntary ethanol consumption. Also, systemic administration of the glycine transporter-1 inhibitor Org25935 elevates dopamine levels in nAc, prevents a further ethanol-induced dopamine elevation and robustly and dose-dependently decreases ethanol consumption in rats. Here we investigated whether Org25935 applied locally in nAc modulates dopamine release, and whether accumbal glycine receptors or NMDA receptors are involved in this tentative effect. We also addressed whether Org25935 and ethanol applied locally in nAc interact with dopamine levels, as seen after systemic administration. We used in vivo microdialysis coupled to HPLC-ED in freely moving male Wistar rats to monitor dopamine output in nAc after local perfusion of Org25935 alone, with ethanol, or Org25935-perfusion after pre-treatment with the glycine receptor antagonist strychnine or the NMDA receptor glycine site antagonist L-701.324. Local Org25935 increased extracellular dopamine levels in a subpopulation of rats. Local strychnine, but not systemic L-701.324, antagonized the dopamine-activating effect of Org25935. Ethanol failed to induce a dopamine overflow in the subpopulation responding to Org25935 with a dopamine elevation. The study supports a role for accumbal glycine receptors rather than NMDA receptor signaling in the dopamine-activating effect of Org25935. The results further indicate that the previously reported systemic Org25935–ethanol interaction with regard to accumbal dopamine is localized to the nAc. This adds to the growing evidence for the glycine receptor as an important player in the dopamine reward circuitry and in ethanol’s effects within this system.
Introduction
Alcohol addiction is a major public health problem with enormous socioeconomic and negative health consequences. Alcohol addiction is described as compulsive alcohol use with chronic relapses and with an underlying enduring pathology that may begin with a dysregulation of brain hedonic systems (Koob, 2009). The mesolimbic dopamine pathway is the central substrate for reward, reinforcement, and motivational learning (Koob, 1992; Wise, 1998; Gonzales et al., 2004) and consists of cell bodies in the ventral tegmental area (VTA) that primarily project to the nucleus accumbens (nAc). There is considerable evidence from animal studies and humans that acute alcohol administration activates this system (Di Chiara and Imperato, 1988; Boileau et al., 2003; Volkow et al., 2007). Moreover, chronic exposure to alcohol is known to result in allostatic and functional changes in the VTA–nAc pathway that is linked to development of addiction (Hyman et al., 2006; Koob and Volkow, 2010).
The activity of the mesolimbic dopamine system is controlled by several neurotransmitter systems that involve primary targets of alcohol, e.g., the N-methyl-D-aspartate receptor (NMDAR) and the glycine receptor (GlyR); yet the molecular mechanisms by which ethanol activates the system is not clear. Recently studies have identified the GlyR in nAc as an access point for ethanol to the brain reward system (Molander and Söderpalm, 2005a). It was further shown that glycine perfusion in the nAc increases accumbal dopamine levels and reduces ethanol intake, whereas perfusion with the GlyR antagonist strychnine instead decreases dopamine levels and increases ethanol intake (Molander et al., 2005). It was hypothesized that accumbal GlyRs provide tonic regulation of dopaminergic output via a feed-back circuitry, where GlyRs control GABAergic medium spiny neurons projecting from nAc to the VTA. By activation, these GlyRs hyperpolarize the GABAergic neurons and reduce the inhibitory tone on ventral tegmental dopamine neurons and/or on incoming cholinergic neurons; thereby allowing the dopamine neurons to fire (Söderpalm et al., 2009).
Extracellular glycine levels are regulated by high affinity glycine transporters (GlyTs) and two distinct subtypes referred to as GlyT-1 and GlyT-2 have been characterized. GlyT-2 displays high neuronal expression in caudal CNS regions, whereas GlyT-1 is expressed throughout most regions of the CNS, mainly by glial cells (Zafra et al., 1995; Jursky and Nelson, 1996). GlyT-2 has a specialized role in reuptake of glycine into nerve terminals for vesicular storage, whereas GlyT-1 is the principal regulator of extracellular glycine levels (Gomeza et al., 2006). Within the above described feed-back mechanism, GlyT-1 inhibition may elevate extracellular glycine concentrations and possibly protect the GlyR from ethanol-induced activation. We have examined the selective GlyT-1 blocker cis-N-methyl-N-(6-methoxy-1-phenyl-1,2,3,4- tetrahydronaphtalen-2-ylmethyl)amino-methylcarboxylic acid hydrochloride (Org25935), which easily passes the blood–brain barrier and elevates extracellular glycine levels after systemic administration (Ge et al., 2001; Lidö et al., 2009). In a two- bottle free choice model Org25935 produced a robust and dose-dependent decrease in ethanol consumption in rats (Molander et al., 2007). It was further demonstrated that systemic Org25935 increases dopamine levels in nAc in a similar manner as local glycine and counteracts accumbal dopamine elevations provoked by systemic ethanol administration (Lidö et al., 2009). Yet the actual involvement of accumbal GlyRs in the effects of Org25395 on dopamine levels remains to be determined.
Apart from being a GlyR agonist, glycine serves as an obligatory co-agonist to glutamate at the NMDAR and along with other ligand-gated ion channels, also the NMDAR is a primary substrate for ethanol in the brain (Gass and Olive, 2008). The nAc is richly innervated by glutamatergic inputs and ethanol is reported to interact with NMDAR in this brain region (Nie et al., 1993; Moghaddam and Bolinao, 1994). In the progression from substance use to addiction, a shift from a dopamine-based to a more glutamate-based behavior is described (Ross and Peselow, 2009), and glutamate is implicated in addiction processes such as relapse (Vengeliene et al., 2005). It was therefore judged of interest to look into the mechanisms by which Org25935 modulates accumbal dopamine release. To this end, we investigated if Org25935 applied locally in nAc elevates accumbal dopamine levels, and, if so, whether the effect observed involves accumbal GlyRs or NMDAR signaling. The study also addressed whether Org25935 and ethanol applied locally in the nAc interact with dopamine in a similar manner as previously observed after systemic administration.
Materials and Methods
Animals
A total number of 120 male Wistar rats weighing 240–260 g were purchased from Beekay (Stockholm, Sweden). The rats were housed five per cage at constant room temperature (22°C) and humidity (65%) and kept under regular light–dark conditions (lights on at 7.00 AM and off at 7.00 PM) with free access to standard rat feed (Harlan Teklad Europe, UK) and tap water. The rats were allowed minimum 1 week of adaptation before the experiment was performed. The study was approved by the Ethics Committee for Animal Experiments in Gothenburg in Sweden (diary number 5/04).
Drugs
Strychnine and L-701.324 were purchased from Sigma Aldrich (Stockholm, Sweden) and ethanol was purchased from Kamet AB (Haninge, Sweden). The selective GlyT-1 inhibitor Org25935 was kindly provided by Organon Labs. Ltd. (now Merck Sharp & Dohme, Newhouse, Lanarkshire, UK). Strychnine, Org25935, and ethanol were dissolved in Ringer solution (140 mM NaCl, 1.2 mM CaCl2, 3.0 mM KCl, and 1.0 mM MgCl2) for perfusion into nAc by reverse microdialysis. Due to insolubility in Ringer, L-701.324 was dissolved in Polysorbate 80, diluted in 0.9% NaCl to 10%, and administered i.p. in a volume of 2 ml/kg. With reverse microdialysis it is difficult to determine the exact concentration of the perfused drug in the extracellular space outside the probe. However in our laboratory the recovery of dopamine in vitro is ∼5–10% of the actual concentration outside the probe. In vivo, we estimate the recovery/excovery to be ∼1–5% depending on the molecular size and charge and the area of the active space of the probe. We therefore estimate that perfusion of 100 μM Org25935 equals a concentration of 1–5 μM in the extracellular space. The concentration of Org25935 used was selected based on data obtained from Organon Labs. Ltd. The concentrations of the GlyR antagonist strychnine (10 and 20 μM) were selected based on results obtained in previous studies (Molander and Söderpalm, 2005b; Ericson et al., 2006; Chau et al., 2010b). The dose of L-701.324, 5 mg/kg i.p., was chosen based on studies showing antagonistic effects on relapse-like drinking in rats (Vengeliene et al., 2005). Finally, to be able to compare the present results to those obtained after systemic administration of both Org25935 and ethanol we used an ethanol concentration (300 mM) producing a dopamine elevation (∼30%) comparable to that observed after 2.5 g/kg i.p. (Blomqvist et al., 1993; Ericson et al., 2003; Lof et al., 2007).
Microdialysis Technique
The microdialysis experiments were performed in awake and freely moving rats. Rats were anesthetized by isoflurane (Baxter, Sweden), mounted into a stereotaxic instrument (David Kopf Instruments, AgnTho’s AB, Lidingö, Sweden) and put on a heating pad to prevent hypothermia during the surgery. Holes were drilled for placement of two anchoring screws and a dialysis probe custom made in the laboratory. The probe with 2 mm active space was lowered into the nAc monolaterally; A/P: +1.85, M/L: −1.4, relative to bregma, and D/V: −7.8 mm relative to dura (Paxinos and Watson, 2007), coordinates corresponding to a core–shell borderline region. A 2.5% ketoprofen gel was applied on the injured tissue to relieve inflammation and pain. The dialysis probe and screws were fixed to the scull with Harward cement (DAB Dental AB; Gothenburg, Sweden). Animals were given 2 ml saline i.p. post surgery to prevent dehydration and allowed to recover for 48 h prior to the experiment. On the experimental day, the sealed inlet and outlet of the probe were cut open and connected to a microperfusion pump (U-864 Syringe Pump, AgnTho’s, Sweden) via a swivel allowing the animal to move freely. The probe was perfused with Ringer solution at a rate of 2 μl/min and dialysate (40 μl) was collected every 20 min. Before sampling begun, rats were perfused with Ringer solution for 1 h to obtain a balanced fluid exchange. Animals were killed immediately after the experiment and brains were removed. The positions of the probes were verified by visual examination of brain slices, cut by a vibroslicer (Campden Instruments Ltd., Leicester, UK) and visualized in Figure 1. A number of 20 rats were excluded from the study after probe verification, due to hemorrhage or incorrect probe placement.
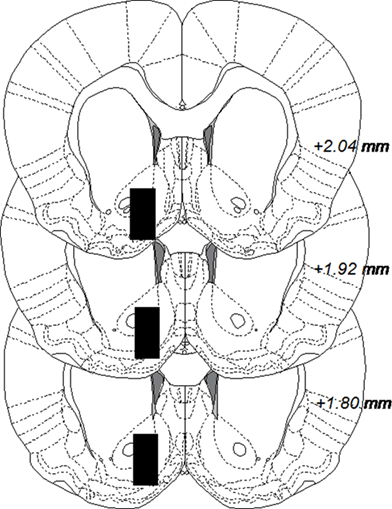
Figure 1. Coronal rat brain sections displaying black boxes which represent accepted areas for probe placements in nucleus accumbens. The number in each section indicates millimeters anterior from bregma. The figure is adapted from Paxinos and Watson (2007).
Experimental Procedure and Neurochemical Assay
A high-pressure liquid chromatography system with electrochemical detection was used for the separation and detection of dopamine content of the dialysate samples, as previously described in Lidö et al. (2009). An external standard containing 3.25 fmol/μl of dopamine was used to identify the dopamine peak. For all experiments, microdialysis was performed during continuous monitoring of accumbal dopamine efflux. When three stable dopamine values were obtained (±10%), the mean of these was set to 100% for each animal, the time point was set to 0 min, and all percentages throughout the experiments were related to this. In Experiment 1, Org25935 (100 μM) was perfused into nAc from time point 40 min while Ringer perfusion was maintained throughout the experiment in the control group. Experiment 2 studied the influence of strychnine (10 and 20 μM) and L-701.324 (5 mg/kg i.p.) pre-treatment on the effect of Org25935. Therefore, perfusion of strychnine was initiated at time point 0 min as was injection of L-701.324, while perfusion of Org25935 started 40 min later, at time point 40 min. In Experiment 3 we examined the influence of Org25935 on ethanol-induced dopamine increase, and Org25935 (100 μM) was perfused from time point 0 min while ethanol (300 mM) was perfused alone or co-perfused with Org25935 from time point 100 min and throughout the experiment.
Statistics
All values are expressed as means ± SEM and a probability value (p) less than 0.05 was considered statistically significant. Due to the interaction design where we studied effects of drugs/drug combinations over different time intervals, ANOVA with repeated measures was performed over relevant time periods for respective drug perfusions, and not over the time period as a whole. As the study was clearly hypothesis-driven and since Org25935- and ethanol-evoked dopamine responses could be anticipated due to previous studies, the more liberal Fisher’s protected least significant difference (PLSD) post hoc test was used in order to reduce risk of obtaining type 2 errors. Due to the biological variability in the ability of Org25935 to elicit an accumbal dopamine response, separate time courses for the responding, and non-responding subgroups were analyzed in Experiment 1 and Experiment 3 in addition to the group as a whole. The criteria for being a responder was set to >10% increase in dopamine output, while remaining animals were classified as non-responders.
Results
Experiment 1: Org25935 in nAc Increases Accumbal Dopamine Levels in a Subpopulation of Rats
Figure 2A displays accumbal dopamine output after perfusion of Org25935 (100 μM) and Ringer solution in the nAc. A repeated measures ANOVA (time period 40–100 min) revealed a significant group effect [F(1,16) = 4.682, p = 0.046], but no time effect [F(3,48) = 1.116, p = 0.333] or group by time interaction [F(3,48) = 0.790, p = 0.506]. The peak level of dopamine was 26%, observed after perfusion for 40 min. Figure 2B displays accumbal dopamine output after Org25935 perfusion in the responding (n = 7) and non-responding subgroups (n = 6). A repeated measures ANOVA (time period 40–100 min) revealed a significant group effect [F(1,8) = 6.861, p = 0.030], but no effect of time [F(3,24) = 1.056, p = 0.386] or group by time interaction [F(3,24) = 0.479, p = 0.700]. Responders had a maximum of 36% increase in dopamine levels, observed after 40 min.
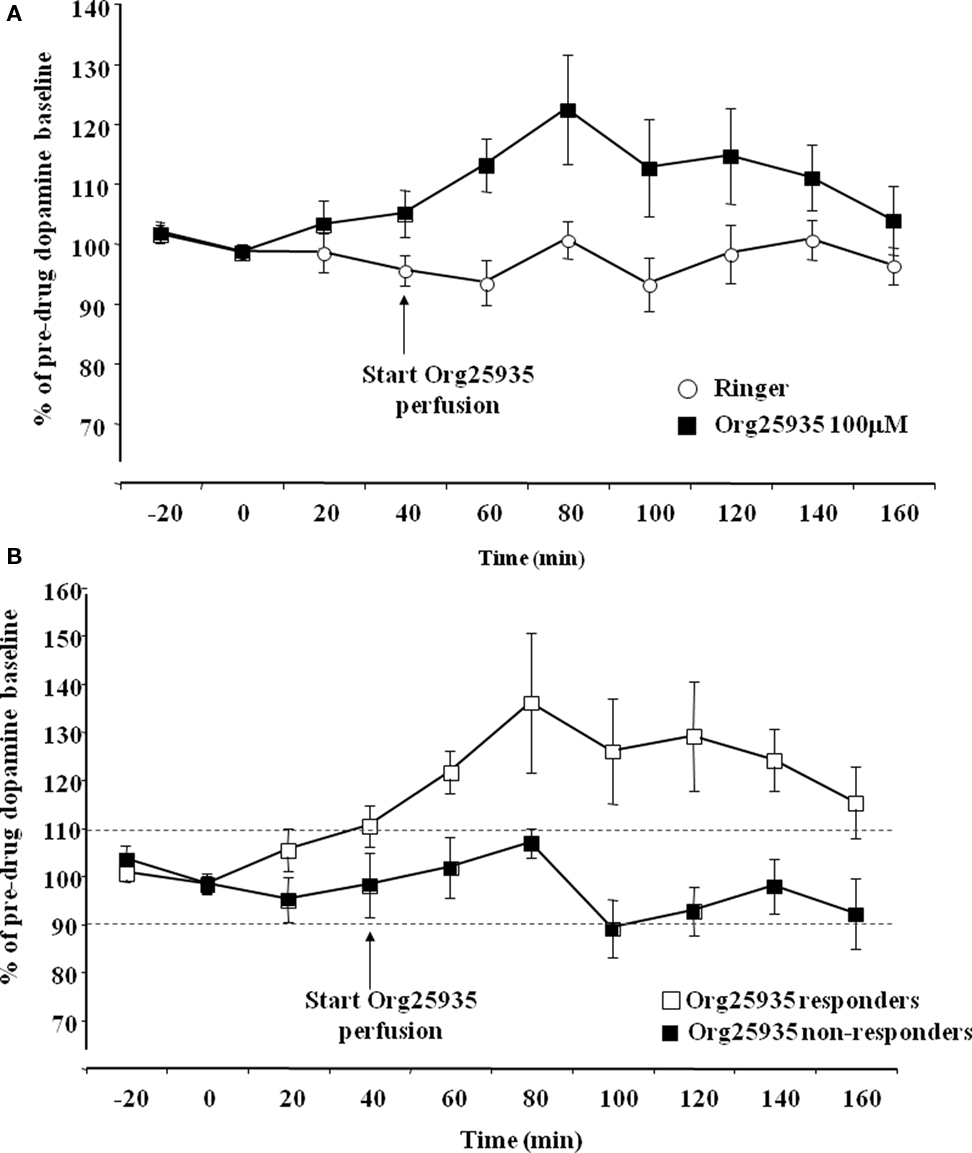
Figure 2. (A) Accumbal dopamine levels as measured by in vivo microdialysis after perfusion of 100 μM Org25935 (n = 13) and Ringer (n = 8) into nAc, shown as means ± SEM. Drug perfusion started at time point 40 min as indicated by the arrow. Basal dopamine concentrations (mean ± SEM) for the Org25935 and vehicle group were 2.66 ± 0.66 and 2.02 ± 0.55 nM, respectively. For statistics: See Section “Results, Experiment 1.” (B) Accumbal dopamine levels as measured by in vivo microdialysis after perfusion of 100 μM Org25935 into nAc in the responding (n = 7) and non-responding (n = 6) subgroup, shown as means ± SEM. Drug perfusion started at time point 40 min as indicated by arrow. Basal dopamine concentrations (mean ± SEM) for responders and non-responders were 3.02 ± 1.24 and 2.37 ± 0.55 nM, respectively. For statistics: See Section “Results, Experiment 1.”
Experiment 2: Strychnine but Not L-701.324 Inhibits the Org25935-Induced Elevation of Accumbal Dopamine Levels
Figure 3A displays accumbal dopamine levels after (1) 100 μM Org25935, (2) 10 μM strychnine, and after (3) pre- and co- perfusion of strychnine with Org25935 in nAc. A repeated measures ANOVA (time period 60–140 min) demonstrated a group effect [F(2,22) = 4.687, p = 0.020], but no time effect [F(4,88) = 0.717, p = 0.583] or group by time interaction [F(8,88) = 1.407, p = 0.205]. A Fisher’s PLSD test further demonstrated a significant difference between the Org25935 group and the strychnine–Org25935 group (p = 0.006) but not between the strychnine–Org25935 and the strychnine group (p = 0.187). Strychnine (10 μM) per se, prior to Org25935 challenge, did no significantly affect dopamine output (repeated measures ANOVA over time period 0–40 min, group effect [F(1,13) = 0.009, p = 0.926], time effect [F(2,26) = 0.169, p = 0.845], and interaction effect [F(2,26) = 0.140, p = 0.870]).
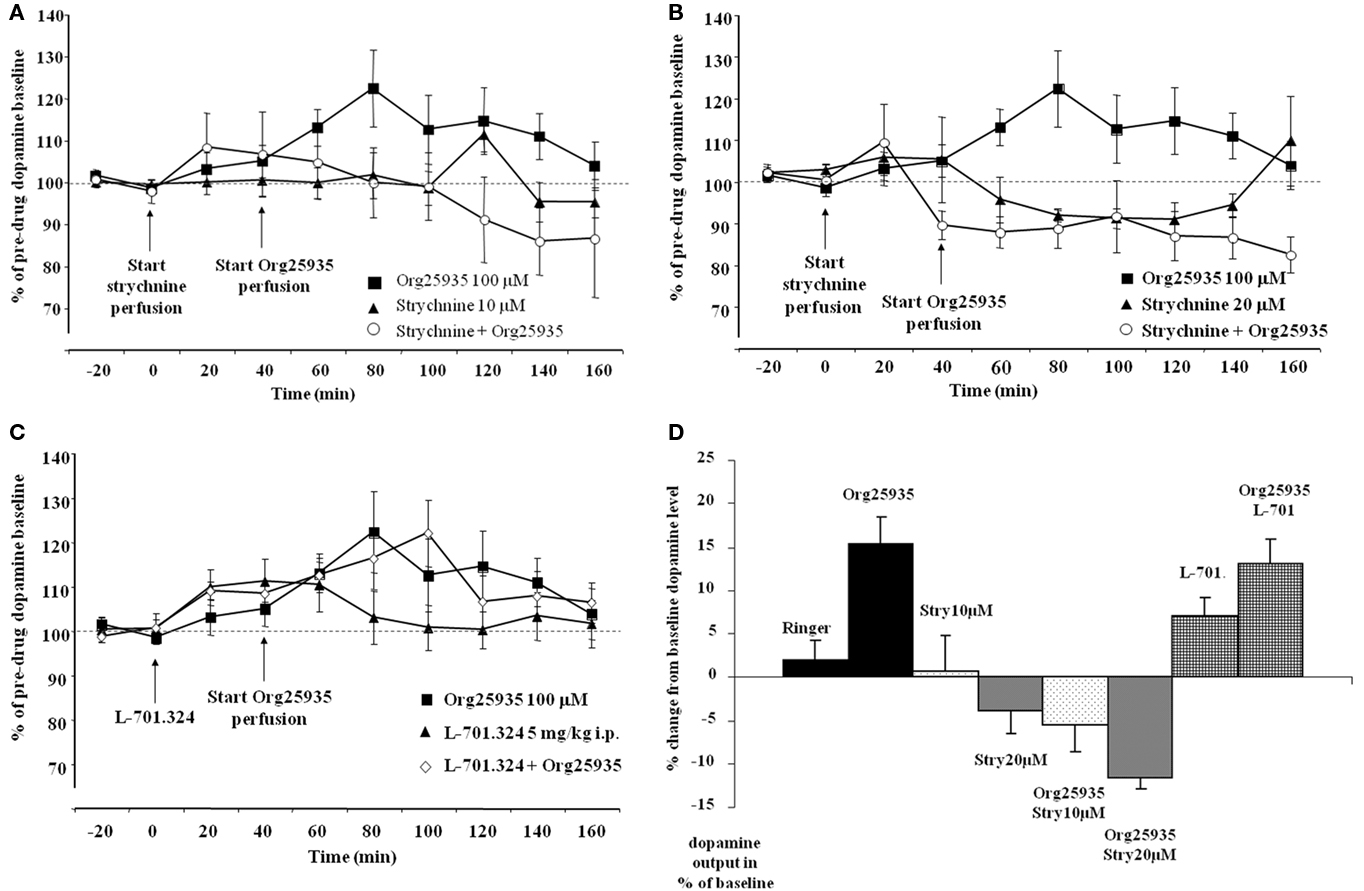
Figure 3. (A) Accumbal dopamine levels as measured by in vivo microdialysis after perfusion of 10 μM strychnine (n = 8), 100 μM Org25935 (n = 13) and after strychnine perfusion followed by Org25935 co-perfusion (n = 11), shown as means ± SEM. Strychnine perfusion started at time point 0 min and Org25935 perfusion started at time point 40 min, as indicated by arrows. Basal dopamine concentrations (mean ± SEM) for Org25935, strychnine, and strychnine–Org25935 were 2.66 ± 0.66, 1.59 ± 0.23, and 1.69 ± 0.21 nM, respectively. For statistics: See Section “Results, Experiment 2.” (B) Accumbal dopamine levels as measured by in vivo microdialysis after perfusion of 20 μM strychnine (n = 8), 100 μM Org25935 (n = 13), and after strychnine perfusion followed by Org25935 co-perfusion (n = 11), shown as means ± SEM. Strychnine perfusion started at time point 0 min and Org25935 perfusion started at time point 40 min, as indicated by arrows. Basal dopamine concentrations (mean ± SEM) for Org25935, strychnine, and strychnine–Org25935 were 2.66 ± 0.66, 1.34 ± 0.57, and 2.26 ± 0.44 nM, respectively. For statistics: See Section “Results, Experiment 2.” (C) Accumbal dopamine levels as measured by in vivo microdialysis after systemic 5 mg/kg i.p. L-701.324 (n = 7), 100 μM Org25935 (n = 13), and after systemic L-701.324 followed by Org25935 perfusion (n = 10), shown as means ± SEM. L-701.324 was administered at time point 0 min and Org25935 perfusion started at time point 40 min, as indicated by arrows. Basal dopamine concentrations (mean ± SEM) for Org25935, L-701.324, and L-701.324–Org25935 were 2.66 ± 0.66, 3.70 ± 0.56, and 2.92 ± 0.70 nM, respectively. For statistics: See Section “Results, Experiment 2.” (D) A summary of the % change in accumbal dopamine levels provoked by the drugs/drug combinations previously displayed in Figures 2A, 3A–C. Each bar represents the mean ± SEM dopamine output from the two or three collected samples during the respective drug perfusion. Further data analyses on these pooled values were not performed.
Figure 3B displays accumbal dopamine levels after (1) 100 μM Org25935, (2) 20 μM strychnine, and after (3) pre- and co- perfusion of strychnine with Org25935 in nAc. A repeated measures ANOVA (time period 60–140 min) demonstrated a group effect [F(2,25) = 8.477, p = 0.002], but no time effect [F(4,100) = 0.702, p = 0.593] or group by time interaction [F(8,100) = 0.710, p = 0.683]. A Fisher’s PLSD test further demonstrated a significant difference between the Org25935 and strychnine–Org25935 (p < 0.001) and between Org25935 and strychnine (p = 0.012) but not between strychnine–Org25935 and strychnine (p = 0.562). Strychnine (20 μM) per se prior to Org25935 challenge did not significantly affect dopamine output (repeated measures ANOVA over time period 0–40 min, group effect [F(2,26) = 0.163, p = 0.851], time effect [F(2,52) = 2.689, p = 0.077], and interaction effect [F(4,52) = 2.125, p = 0.091]).
Figure 3C displays accumbal dopamine levels after (1) perfusion of Org25935, (2) L-701.324 (5 mg/kg, i.p.), and (3) L-701.324 (5 mg/kg, i.p.) prior to perfusion of Org25935. A repeated measures ANOVA (time period 60–140 min) revealed no group effect [F(2,26) = 0.156, p = 0.856], no time effect [F(4,104) = 0.975, p = 0.424], or group by time interaction [F(8,104) = 0.219, p = 0.300]. Repeated measures ANOVA over time period 0–40 min (effect of L-701.324 prior to Org25935 challenge) revealed no group effect [F(2,26) = 0.1343, p = 0.279], a significant effect of time [F(2,52) = 5.485, p = 0.007] but no interaction effect [F(4,52) = 0.752, p = 0.561].
Figure3D illustrates a summary of the % change in accumbal dopamine levels provoked by the drug/drug combinations during the microdialysis experiments previously displayed in Figures 2A and 3A–C. Each bar represents the mean ± SEM of the % change in dopamine output during 40 min following the respective drug perfusions.
Experiment 3: Ethanol-Induced Elevation of Accumbal Dopamine Levels is Prevented by Org25935
Figure 4A displays accumbal dopamine output after (1) 100 μM Org25935 from time point 0 min and 300 mM ethanol perfusion from time point 100 min, (2) ethanol perfusion alone from time point 100 min, and (3) perfusion of Ringer solution. As in Experiment 1, Org25935 produced a dopamine elevation, revealed by a repeated measures ANOVA over the time period for Org25935 perfusion prior to ethanol perfusion (0–100 min, a significant group effect [F(2,25) = 4.291, p = 0.025], no time effect [F(4,100) = 1.345, p = 0.259] but a significant group by time interaction [F(8,100) = 2.408, p = 0.0202]). Fisher’s PLSD test revealed a significant difference between the Org25935–ethanol and the two Ringer-perfused groups during this time interval (p = 0.022 and 0.031, respectively) but not between the two Ringer-perfused groups (p = 0.980). Org25935 maximally increased dopamine levels by 20–22%, detected over the time period 60–100 min. Over the time period for ethanol perfusion (100–180 min), a repeated measures ANOVA revealed no group effect [F(2,24) = 1.620, p = 0.219], no time effect [F(4,96) = 1.730, p = 0.150] but a group by time interaction [F(8,96) = 3.45, p = 0.001]. However ethanol increased dopamine output by maximally 23% compared to the time point before the ethanol challenge (paired t-test 100–120 min, p = 0.038) whereas ethanol did not increase dopamine output in the Org25935 pre-treated group (paired t-test 100–120 min, p = 0.792). Conversely, dopamine output in this group decreased 40 min after start of ethanol perfusion (paired t-test 100–140 min, p = 0.037).
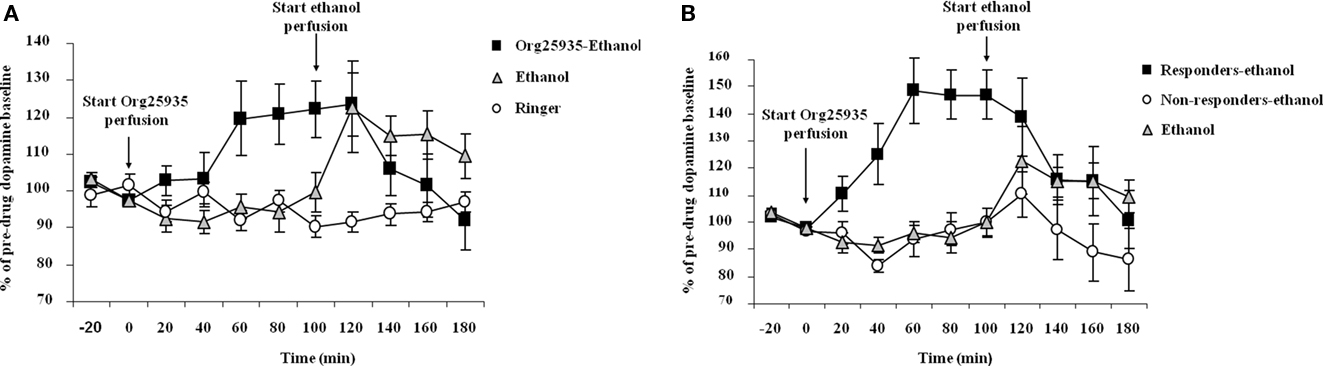
Figure 4. (A) Accumbal dopamine levels as measured by in vivo microdialysis after perfusion of 300 mM ethanol (n = 7), Ringer (n = 8), and after Org25935 perfusion followed by ethanol co-perfusion (n = 17), shown as means ± SEM. Org25935 perfusion started at time point 0 min and ethanol perfusion started at time point 100 min, as indicated by arrows. Basal dopamine concentrations (mean ± SEM) for ethanol, ethanol–Org25935, and Org25935 were 2.91 ± 0.64, 2.14 ± 0.24, and 2.66 ± 0.66 nM, respectively. For statistics: See Section “Results, Experiment 3.” (B) Accumbal dopamine levels as measured by in vivo microdialysis after perfusion of 300 mM ethanol (n = 7) and after Org25935 perfusion followed by ethanol co-perfusion in the responding (n = 8) and non-responding (n = 9) subgroup, shown as means ± SEM. Org25935 perfusion started at time point 0 min and ethanol perfusion started at time point 100 min, as indicated by arrows. Basal dopamine concentrations (mean ± SEM) for ethanol, drug responders and non-responders were 2.91 ± 0.64, 2.13 ± 1.4, and 2.14 ± 0.22 nM, respectively. For statistics: See Section “Results, Experiment 3”.
Figure 4B displays the dopamine output after Org25935 and/or ethanol perfusion, as described above, in the Org25935 responding (n = 8) and non-responding subgroups (n = 9). Prior to ethanol perfusion, a repeated measures ANOVA (time period 0–100 min) revealed a significant group effect [F(2,21) = 19.624, p = 0 < 0.001], time effect [F(4,84) = 5.144, p = 0.001], and group by time interaction [F(8,84) = 5.393, p = 0 < 0.001]. The Fisher’s PLSD test revealed a significant difference between responders and non-responders (p < 0.001) and responders and Ringer-perfused animals (p < 0.001) but not between non-responders and Ringer-perfused animals (p = 0.879). As previously shown in Figure 4A, ethanol increased dopamine output by maximally 23% compared to the time point before the ethanol challenge (paired t-test 100–120 min, p = 0.038). There was a trend for an ethanol-induced dopamine elevation in the Org25935 non-responding group (paired t-test 100–120 min, p = 0.057) whereas ethanol did not elevate dopamine levels in responders (paired t-test 100–120 min, p = 0.374). Instead, dopamine output in responders significantly decreased 40 min after ethanol perfusion (paired t-test 100–140 min, p = 0.024).
Discussion
This study shows that local administration of the GlyT-1 blocker Org25935 in nAc increases accumbal dopamine output in the rat. The effect was reversed by the GlyR antagonist strychnine but not by antagonism of the glycine site on the NMDAR, suggesting that the GlyT-1 blocker modulates the accumbal dopamine tone via accumbal GlyRs rather than via NMDAR signaling. This is in line with previous findings, where accumbal glycine perfusion elevated dopamine levels by ∼50% (Molander and Söderpalm, 2005b), and where taurine and β-alanine, also endogenous GlyR agonists, increased dopamine levels in the same manner (Ericson et al., 2006, 2009). The dopamine responses provoked by glycine, taurine, and β-alanine were reversed by strychnine, supporting the GlyR involvement also in the dopamine response to Org25935. Next, the study reports that Org25935 prevents ethanol from increasing accumbal dopamine levels, thus underlining the interaction with ethanol seen after systemic administration (Lidö et al., 2009). That the drugs were perfused locally in nAc and yet produced results almost identical to those observed after systemic administration, suggests that the Org25935–ethanol interaction takes place in the nAc. This corroborates previous results pointing to the GlyR in nAc as not only a modulator of basal dopamine release in nAc, but as an access point for ethanol to the mesolimbic dopamine system (Molander and Söderpalm, 2005a; Molander et al., 2005). In addition to a robust prevention of ethanol-induced dopamine output in Org25935-responding animals, Org25935 mitigated ethanol’s effect among the non-responding animals, as also seen after systemic administration. Thus the interaction with ethanol may take place regardless of the variability in the strength of the Org25935-induced dopamine response as such. Since ethanol allosterically modulates the GlyR (Mihic et al., 1997; Perkins et al., 2008), excessive glycine availability provoking a conformational change in the GlyR may in turn obstruct the binding of ethanol and the following rise in dopamine levels.
Due to an inconsistent dopamine response provoked by Org25935, rats were divided into responders and non-responders. Org25935 increased accumbal dopamine output by 26% in all rats, consisting of seven responders, displaying a 36% increase, and six non-responders. This is comparable to systemic Org25935, which increased dopamine levels by 13% in all rats, consisting of 11 responders, displaying a 29% increase, and 10 non-responders (Lidö et al., 2009). A similar biological variability in dopamine response was observed after accumbal glycine perfusion (Molander and Söderpalm, 2005a,b). As Org25935 produced a consistent increase in glycine levels, equal among dopamine responders, and non-responders (Lidö et al., 2009), we proposed that the variability lies in the response to glycine as such, and not in glycine removal capacity. This may be linked to a recent study reporting no association of alcohol dependence with GlyT-1gene polymorphism (Koller et al., 2009). Instead, differences in, e.g., GlyR desensitization or internalization phenomena could account for the variance in dopamine response. However a GlyR expression study using genetically high versus low alcohol-preferring rats reported very few differences in mRNA expression despite of the distinct drinking behavior, and few differences after 1 month of ethanol exposure (Jonsson et al., 2009). Also, daily Org25935 injections for 19 days produced no changes in GlyR subunits mRNA nor in GlyT-1 mRNA in several brain regions including the nAc (unpublished results). Thus the GlyR does not seem to be prone to rapid compensatory alterations.
Contrary to the inconsistent dopamine response, Org25935 produced a consistent anti-alcohol intake effect in ethanol high- and medium-preferring rats (Molander et al., 2007). The consistent decrease in voluntary ethanol consumption in all rats contrasts to the effect of glycine perfusion, which decreased voluntary ethanol consumption in animals responding to glycine with a dopamine elevation, but not in the non-responders (Molander et al., 2005). Moreover, an overrepresentation of glycine–dopamine responders in ethanol high-preferring rats compared to naïve rats was reported, suggesting that a powerful glycine–dopamine interaction may be linked to high ethanol preference. Similarly, the selected rats with a high ethanol liking used in the alcohol consumption study with Org25935 may constitute a group of mostly dopamine responsive subjects. Alternatively, as sarcosine-based GlyT-1 inhibitors like Org25935 exhibit a non-competitive mode of action, i.e., independent of glycine concentration (Mezler et al., 2008), this independency of variations in endogenous glycine levels may underlie the consistent anti-alcohol intake effect of Org25935. Nevertheless, both the behavioral and neurochemical results suggest that Org25935 employs a combined substitution and antagonism mechanism, where GlyT-1 inhibition elevates and/or stabilizes extracellular glycine levels, which in turn elevate dopamine in Org25935 responders or preserve/stabilize dopamine levels in non-responding animals. This results in a blocking/dampening mechanism preventing additional ethanol-induced GlyR activation and dopamine elevation. These events are possibly mediated via the proposed GABAergic feed-back circuitry (reviewed in detail in Söderpalm et al., 2009). Given that extracellular glycine modulates ethanol consumption and that Org25935 displays excellent anti-alcohol drinking effects in experimental animals (Molander et al., 2007; Vengeliene et al., 2010), the present results further supports GlyT-1 inhibition as a new concept for pharmacotherapy of alcohol addiction.
Besides serving as a GlyR agonist glycine is also an essential co-agonist to glutamate at the NMDAR, and glycine uptake inhibitors display NMDAR potentiating effects (Shim et al., 2008). Glutamate signaling is implicated in alcohol addiction and withdrawal from chronic alcohol is characterized by elevated glutamate levels in nAc (Dahchour and De Witte, 1999). A negative NMDAR modulation is proposed to be useful for alcohol addiction pharmacotherapy (Nagy, 2004) and various NMDAR antagonists decreases ethanol intake (Vengeliene et al., 2005; Gass and Olive, 2008). This view is not in concordance with a tentative NMDAR mechanism by Org25935, which potentiates instead of antagonizes the NMDAR. The present result with L-701.324 suggests that NMDAR signaling is not involved in the dopamine response to Org25935. Unfortunately, due to solubility problems L-701.324 could not be perfused via the dialysis probe, which weakens the comparison with strychnine in this respect. L-701.324 in the present dose prevents relapse-like drinking behavior (Vengeliene et al., 2005) and ethanol-induced conditioned place preference (Biala and Kotlinska, 1999). However, NMDAR effects on mesolimbic dopamine are not clear-cut and the precise NMDAR contribution to dopamine release in nAc is not established (Spanagel, 2009). Moreover, low doses of glycine (5–20 μM) potentiates NMDAR currents, whereas glycine in the 100 μM range or higher have been found to prime NMDARs for internalization (Nong et al., 2003). In contrast, different GlyR agonists consistently elevate dopamine whereas the GlyR antagonist strychnine concentration-dependently reduces accumbal dopamine levels (Ericson et al., 2006, 2009). As a parallel also acamprosate, a drug structurally related to taurine and in clinical use for treating alcohol dependence, also promotes dopamine release in nAc via the GlyR rather than the NMDAR (Chau et al., 2010a,b). In summary, most evidence support a GlyR mechanism rather than an NMDAR mechanism being involved in alcohol’s interference with accumbal dopamine levels. We therefore suggest that the Org25935-induced dopamine response here observed primarily involves accumbal GlyR. As also other neurotransmitters and receptors are implied in alcohol intake behavior, activation of both NMDARs and GlyRs may be important for the alcohol intake-reducing effect of the GlyT-1 blocker. This view is reinforced by recent findings that Org25935 treatment persistently reduces relapse-like drinking behavior, and that this durable effect may result from restoration of both glycinergic and glutamatergic signaling (Vengeliene et al., 2010).
Substances that stimulate dopamine release in nAc may be associated with risk of abuse. Org25935 elevates dopamine levels in nAc after systemic administration but several points argue against an addictive potential. Org25935 has a mild and indirect mode of increasing dopamine levels that probably rather affects tonic dopamine release. This contrasts to the rapid increase in dopamine levels that is associated with a reported “high” in humans, and which in turn is considered an important feature of a potentially addictive drug working via the dopamine system (Volkow et al., 1999). Org25936 may have ethanol-like effects on dopamine, but the brain perceives ethanol through several known primary targets and not only through the GlyR, and the interoceptive cues of Org25935 and ethanol in the brain are most likely not the same. Preclinical information obtained from Organon Labs. Ltd. (now MSD) reported no hedonic profile of Org25935 on intra-cranial self administration and no effect on locomotion. Org25935 has previously been tested in clinical trials with no reportage of a positive hedonic profile in man. In reports studying the antipsychotic effects of glycine, glycine agonists, and GlyT-1 inhibitors in man, the question of tentative addictive properties of GlyT-1 blockers was never raised (Shim et al., 2008). Chronic elevation of extracellular glycine in humans appears safe in the clinic (Patel et al., 1990; Shoham et al., 2001; Yang and Svensson, 2008) and overall, the abuse liability risk associated with Org25935 is low.
This is the first study to demonstrate a role for accumbal GlyRs in the dopamine-modulating effects of a GlyT-1 blocker, and it appears that the GlyR rather than NMDAR signaling is involved in the Org25935-evoked dopamine elevation. The study suggests that the Org25935–ethanol interaction takes place in nAc, since Org25935 locally in nAc increases basal dopamine levels and counteracts ethanol-induced activation of dopamine. We propose a partial substitution and antagonism mechanism of Org25935. The GlyT-1 blocker elevates and stabilizes extracellular glycine levels, which via the GlyR elevate dopamine levels (in responders) or preserve stable dopamine levels (in non-responders), which in turn prevents additional ethanol-mediated GlyR activation and dopamine elevation. This partial agonistic mechanism may offer great potential for GlyT-1 inhibition as a new concept for decreasing alcohol intake, and GlyT-1 inhibition as a tentative “anti-alcohol drinking therapy” is emphasized by Org25935’s excellent ability to decrease ethanol consumption in experimental animals. The study adds to the growing evidence for the GlyR as an important player in the dopamine reward circuitry and in ethanol’s effects within this system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Merck Sharp & Dohme, Newhouse, Lanarkshire, UK, for providing the Org25935 compound. The authors are grateful to Anne Fagerberg and PeiPei Chau for technical assistance. The work is financial supported by the Organon Research Foundation (now Merck Sharp & Dohme), the Swedish Medical Research Council (no. 4477 and no. 6385), government support under the LUA/ALF agreement, Bror Gadelius Foundation, Sigurd and Elsa Goljes Foundation, Wilhelm and Martina Lundgrens Scientific Foundation, Lars Hiertha foundation, and Gunnar and Märta Bergendahl Foundation. Helga Höifödt Lidö performed the studies, prepared the manuscript and the study design, Mia Ericson and Bo Söderpalm were responsible for the study concept and design Hugh Marston, Mia Ericson, and Bo Söderpalm, provided critical revision of the manuscript.
References
Biala, G., and Kotlinska, J. (1999). Blockade of the acquisition of ethanol-induced conditioned place preference by N-methyl-D-aspartate receptor antagonists. Alcohol. Alcohol. 34, 175–182.
Blomqvist, O., Engel, J. A., Nissbrandt, H., and Soderpalm, B. (1993). The mesolimbic dopamine-activating properties of ethanol are antagonized by mecamylamine. Eur. J. Pharmacol. 249, 207–213.
Boileau, I., Assaad, J. M., Pihl, R. O., Benkelfat, C., Leyton, M., Diksic, M., Tremblay, R. E., and Dagher, A. (2003). Alcohol promotes dopamine release in the human nucleus accumbens. Synapse 49, 226–231.
Chau, P., Hoifodt-Lido, H., Lof, E., Soderpalm, B., and Ericson, M. (2010a). Glycine receptors in the nucleus accumbens involved in the ethanol intake-reducing effect of Acamprosate. Alcohol. Clin. Exp. Res. 34, 39–45.
Chau, P., Stomberg, R., Fagerberg, A., Soderpalm, B., and Ericson, M. (2010b). Glycine receptors involved in acamprosate’s modulation of accumbal dopamine levels: an in vivo microdialysis study. Alcohol. Clin. Exp. Res. 34, 32–38.
Dahchour, A., and De Witte, P. (1999). Effect of repeated ethanol withdrawal on glutamate microdialysate in the hippocampus. Alcohol. Clin. Exp. Res. 23, 1698–1703.
Di Chiara, G., and Imperato, A. (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. U.S.A. 85, 5274–5278.
Ericson, M., Clarke, R. B., Chau, P., Adermark, L., and Soderpalm, B. (2009). β-Alanine elevates dopamine levels in the rat nucleus accumbens: antagonism by strychnine. Amino Acids 38, 1051–1055.
Ericson, M., Molander, A., Lof, E., Engel, J. A., and Soderpalm, B. (2003). Ethanol elevates accumbal dopamine levels via indirect activation of ventral tegmental nicotinic acetylcholine receptors. Eur. J. Pharmacol. 467, 85–93.
Ericson, M., Molander, A., Stomberg, R., and Soderpalm, B. (2006). Taurine elevates dopamine levels in the rat nucleus accumbens; antagonism by strychnine. Eur. J. Neurosci. 23, 3225–3229.
Gass, J. T., and Olive, M. F. (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochem. Pharmacol. 75, 218–265.
Ge, J., Hamilton, M., Shahid, D., Hill, R., and Walker, G. (2001). The effects of Org25935 on the extracellular levels of glycine in brain regions of freely moving rats using microdialysis. Br. J. Pharmacol. 133, 135.
Gomeza, J., Armsen, W., Betz, H., and Eulenburg, V. (2006). Lessons from the knocked-out glycine transporters. Handb. Exp. Pharmacol. 175, 457–483.
Gonzales, R. A., Job, M. O., and Doyon, W. M. (2004). The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol. Ther. 103, 121–146.
Hyman, S. E., Malenka, R. C., and Nestler, E. J. (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 29, 565–598.
Jonsson, S., Kerekes, N., Hyytia, P., Ericson, M., and Soderpalm, B. (2009). Glycine receptor expression in the forebrain of male AA/ANA rats. Brain Res. 1305(Suppl.), S27–S36.
Jursky, F., and Nelson, N. (1996). Developmental expression of the glycine transporters GLYT1 and GLYT2 in mouse brain. J. Neurochem. 67, 336–344.
Koller, G., Zill, P., Fehr, C., Pogarell, O., Bondy, B., Soyka, M., and Preuss, U. W. (2009). No association of alcohol dependence with SLC6A5 and SLC6A9 glycine transporter polymorphisms. Addict. Biol. 14, 506–508.
Koob, G. F. (2009). Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsychiatry 42(Suppl. 1), S32–S41.
Koob, G. F., and Volkow, N. D. (2010). Neurocircuitry of addiction. Neuropsychopharmacology 35, 217–238.
Lidö, H. H., Stomberg, R., Fagerberg, A., Ericson, M., and Soderpalm, B. (2009). The glycine reuptake inhibitor org 25935 interacts with basal and ethanol-induced dopamine release in rat nucleus accumbens. Alcohol. Clin. Exp. Res. 33, 1151–1157.
Lof, E., Ericson, M., Stomberg, R., and Soderpalm, B. (2007). Characterization of ethanol-induced dopamine elevation in the rat nucleus accumbens. Eur. J. Pharmacol. 555, 148–155.
Mezler, M., Hornberger, W., Mueller, R., Schmidt, M., Amberg, W., Braje, W., Ochse, M., Schoemaker, H., and Behl, B. (2008). Inhibitors of GlyT1 affect glycine transport via discrete binding sites. Mol. Pharmacol. 74, 1705–1715.
Mihic, S. J., Ye, Q., Wick, M. J., Koltchine, V. V., Krasowski, M. D., Finn, S. E., Mascia, M. P., Valenzuela, C. F., Hanson, K. K., Greenblatt, E. P., Harris, R. A., and Harrison, N. L. (1997). Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 389, 385–389.
Moghaddam, B., and Bolinao, M. L. (1994). Biphasic effect of ethanol on extracellular accumulation of glutamate in the hippocampus and the nucleus accumbens. Neurosci. Lett. 178, 99–102.
Molander, A., Lidö, H. H., Löf, E., Ericson, M., and Söderpalm, B. (2007). The glycine reuptake inhibitor org 25935 decreases ethanol intake and preference in male wistar rats. Alcohol. Alcohol. 42, 11–18.
Molander, A., Löf, E., Stomberg, R., Ericson, M., and Söderpalm, B. (2005). Involvement of accumbal glycine receptors in the regulation of voluntary ethanol intake in the rat. Alcohol. Clin. Exp. Res. 29, 38–45.
Molander, A., and Söderpalm, B. (2005a). Accumbal strychnine-sensitive glycine receptors: an access point for ethanol to the brain reward system. Alcohol. Clin. Exp. Res. 29, 27–37.
Molander, A., and Söderpalm, B. (2005b). Glycine receptors regulate dopamine release in the rat nucleus accumbens. Alcohol. Clin. Exp. Res. 29, 17–26.
Nagy, J. (2004). Renaissance of NMDA receptor antagonists: do they have a role in the pharmacotherapy for alcoholism? IDrugs 7, 339–350.
Nie, Z., Yuan, X., Madamba, S. G., and Siggins, G. R. (1993). Ethanol decreases glutamatergic synaptic transmission in rat nucleus accumbens in vitro: naloxone reversal. J. Pharmacol. Exp. Ther. 266, 1705–1712.
Nong, Y., Huang, Y. Q., Ju, W., Kalia, L. V., Ahmadian, G., Wang, Y. T., and Salter, M. W. (2003). Glycine binding primes NMDA receptor internalization. Nature 422, 302–307.
Patel, J., Zinkand, W. C., Thompson, C., Keith, R., and Salama, A. (1990). Role of glycine in the N-methyl-D-aspartate-mediated neuronal cytotoxicity. J. Neurochem. 54, 849–854.
Paxinos, G., and Watson, C. (2007). The Rat Brain in Stereotaxic Coordinates. London: Academic Press Inc.
Perkins, D. I., Trudell, J. R., Crawford, D. K., Alkana, R. L., and Davies, D. L. (2008). Targets for ethanol action and antagonism in loop 2 of the extracellular domain of glycine receptors. J. Neurochem. 106, 1337–1349.
Ross, S., and Peselow, E. (2009). The neurobiology of addictive disorders. Clin. Neuropharmacol. 32, 269–276.
Shim, S. S., Hammonds, M. D., and Kee, B. S. (2008). Potentiation of the NMDA receptor in the treatment of schizophrenia: focused on the glycine site. Eur. Arch. Psychiatry Clin. Neurosci. 258, 16–27.
Shoham, S., Javitt, D. C., and Heresco-Levy, U. (2001). Chronic high-dose glycine nutrition: effects on rat brain cell morphology. Biol. Psychiatry 49, 876–885.
Söderpalm, B., Lof, E., and Ericson, M. (2009). Mechanistic studies of ethanol’s interaction with the mesolimbic dopamine reward system. Pharmacopsychiatry 42(Suppl. 1), S87–S94.
Spanagel, R. (2009). Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol. Rev. 89, 649–705.
Vengeliene, V., Bachteler, D., Danysz, W., and Spanagel, R. (2005). The role of the NMDA receptor in alcohol relapse: a pharmacological mapping study using the alcohol deprivation effect. Neuropharmacology 48, 822–829.
Vengeliene, V., Leonardi-Essmann, F., Sommer, W. H., Martson, H. M., and Spanagel, R. (2010). Glycine transporter-1 blockade leads to persistently reduced relapse-like alcohol drinking in rats. Biol. Psychiatry 68, 704–711.
Volkow, N. D., Fowler, J. S., Wang, G. J., Swanson, J. M., and Telang, F. (2007). Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch. Neurol. 64, 1575–1579.
Volkow, N. D., Wang, G. J., Fowler, J. S., Logan, J., Gatley, S. J., Wong, C., Hitzemann, R., and Pappas, N. R. (1999). Reinforcing effects of psychostimulants in humans are associated with increases in brain dopamine and occupancy of D(2) receptors. J. Pharmacol. Exp. Ther. 291, 409–415.
Yang, C. R., and Svensson, K. A. (2008). Allosteric modulation of NMDA receptor via elevation of brain glycine and D-serine: the therapeutic potentials for schizophrenia. Pharmacol. Ther. 120, 317–332.
Keywords: dopamine, ethanol, glycine receptors, GlyT-1 inhibition, in vivo microdialysis, nucleus accumbens
Citation: Lidö HH, Ericson M, Marston H and Söderpalm B (2011) A role for accumbal glycine receptors in modulation of dopamine release by the glycine transporter-1 inhibitor Org25935. Front. Psychiatry 2:8. doi: 10.3389/fpsyt.2011.00008
Received: 01 December 2010;
Paper pending published: 16 December 2010;
Accepted: 21 February 2011;
Published online: 07 March 2011.
Edited by:
Eero Vasar, University of Tartu, EstoniaCopyright: © 2011 Lidö, Ericson, Marston and Söderpalm. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Helga Höifödt Lidö, Department of Psychiatry and Neurochemistry, Addiction Biology Unit, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, P.O. Box 410, 405 30 Gothenburg, Sweden. e-mail: helga.lido@neuro.gu.se