
- Max Planck Institute of Psychiatry, Munich, Germany
Comprehensive clinical studies show that adverse conditions in early life can severely impact the developing brain and increase vulnerability to mood disorders later in life. During early postnatal life the brain exhibits high plasticity which allows environmental signals to alter the trajectories of rapidly developing circuits. Adversity in early life is able to shape the experience-dependent maturation of stress-regulating pathways underlying emotional functions and endocrine responses to stress, such as the hypothalamo–pituitary–adrenal (HPA) system, leading to long-lasting altered stress responsivity during adulthood. To date, the study of gene–environment interactions in the human population has been dominated by epidemiology. However, recent research in the neuroscience field is now advancing clinical studies by addressing specifically the mechanisms by which gene–environment interactions can predispose individuals toward psychopathology. To this end, appropriate animal models are being developed in which early environmental factors can be manipulated in a controlled manner. Here we will review recent studies performed with the common aim of understanding the effects of the early environment in shaping brain development and discuss the newly developing role of epigenetic mechanisms in translating early life conditions into long-lasting changes in gene expression underpinning brain functions. Particularly, we argue that epigenetic mechanisms can mediate the gene–environment dialog in early life and give rise to persistent epigenetic programming of adult physiology and dysfunction eventually resulting in disease. Understanding how early life experiences can give rise to lasting epigenetic marks conferring increased risk for mental disorders, how they are maintained and how they could be reversed, is increasingly becoming a focus of modern psychiatry and should pave new guidelines for timely therapeutic interventions.
Introduction
The close relationship between the quality of early life and mental health in later life is a longstanding certainty (Gluckman et al., 2008). Many studies in humans, primates, and rodents illustrate that aspects of the early environment can lead to dramatic changes in physical and mental development compromising cardiovascular and metabolic diseases, altered cognition, mood, and behavior. In particular, adverse conditions during early life periods of development can shape individual differences in vulnerability to stress-related disorders throughout life (for review see Heim and Nemeroff, 2002), dependent on the degree of “match” and “mismatch” between early and later life environments.
These findings raise the intriguing question of how these experiences become incorporated at the cellular and molecular level in the brain architecture leading to long-term alterations in various functions ultimately culminating in an increased risk to mental disease. Developmental plasticity defines an organism’s ability to adapt to the environment during early life and to implement long-lasting changes in sets of key biological programs on the assumption that the environmental conditions during this early period will persist throughout later life (for review see Hochberg et al., 2010). Adverse maternal experience during early life might profoundly influence the experience-dependent maturation of structures underlying emotional functions and endocrine responses to stress, such as the hypothalamo– pituitary– adrenal (HPA) system – an integral component of the body’s stress response – leading to increased stress responsivity in adulthood (for review see Seckl and Meaney, 2004). Indeed, depressed patients with a history of childhood abuse or neglect are often characterized by hyperactivity of the HPA axis (for review see Heim and Nemeroff, 2002).
To understand gene–environment interactions in human populations, and to elucidate the pathways through which programming in response to early life experiences is mediated, researchers in the neuroscience field rely on animal models in which the early environment can be manipulated in a controlled fashion. Current work suggests that so-called epigenetic mechanisms of gene regulation, which alter the activity of genes without changing the order of their DNA sequence, could explain how early life experiences can leave indelible chemical marks on the brain and influence both physical and mental health later in life even when the initial trigger is long gone (for review see Dudley et al., 2011).
In this review we highlight recent animal and human studies addressing epigenetic regulation of gene expression in sustaining the effects of early life experiences. Hereby, we focus on clinical and animal studies that have investigated how biological stress systems, particularly the HPA axis, are shaped by adversity and then provide a description of what we know about the function of epigenetic systems and their roles in brain development and disease. The dynamic nature of epigenetic mechanisms may have important implications when considering the possibility of therapeutic interventions, wherefore we conclude on current evidences of this new research field for the treatment of mental diseases.
Childhood Decides
Sigmund Freud postulated that the trauma of birth – disrupting the physical symbiosis between fetus and mother – becomes a central force in our adult life. This fear and experience of abandonment is a deep-rooted subject in psychology. According to Bowlby (1982), attachment processes are central to understanding anxiety, as illustrated in his decades long studies of children and their attachments to their caregivers, where infants demonstrate distress upon impending departure of the mother as soon as they are old enough to sense signs that she is leaving – around 6–9 months (Sartre, 1964).
It has been suggested that a mother’s external regulation of the infant’s developing immature emotional systems during selected critical periods may represent the essential factor influencing the experience-dependent growth of brain areas and structures important in regulating mood and behavior. Attachment behaviors could therefore be considered a biological system, interrelated with the fear and stress system, evolved specifically to increase the likelihood of infant–parent proximity which, in turn ensures increased chances of survival of the infant. Indeed, the attachment system is activated especially in times of stress, and the availability of an attachment figure such as the mother, has a great influence in reducing a child’s fearfulness (for review see Schore, 2000). However, frequent or prolonged bouts of abandonment can lead to the stress becoming a part of the infant’s and, later, the adult’s personality. Unfortunately, such situations are all too common where infants are inhibited from forming attachments by either being raised without the stimulation and attention of a regular caregiver, or suffering abuse or extreme neglect; conservative estimates suggest that each year in the United States, more than 1,000,000 children are exposed to such conditions (Sedlak and Broadhurst, 1996). The possible short-term effects of this are anger, despair, detachment, and temporary delay in intellectual development while long-term effects can include psychosomatic disorders, and increased risk of depression or anxiety.
Studies of institutionally reared children have been instrumental in understanding the long-term consequences of childhood social deprivation and have revealed the presence of cognitive, social, and physical deficits (e.g., Rutter, 1998), with good reason to consider emotional neglect as the major precipitating factor (for review see Tarullo and Gunnar, 2006). These observations are consistent with the view that early social interactions play a significant role in the development of basic affective processes, supporting learning through connections between cues, situations, and emotional experiences. In support, post-institutionalized children demonstrate significant difficulty matching appropriate facial expressions to happy, sad, and fearful scenarios (Fries and Pollak, 2004). Reports on post-institutionalized children from Romania reveal the development of difficulties in forming emotional attachments to adoptive caregivers, providing further validation for the attachment disorder construct; moreover, these children display irregular glucose metabolic rates in brain regions controlling cognitive and emotional functions, and an increased HPA axis activity associated with the length of time spent in this type of postnatal environment (e.g., O’Connor and Rutter, 2000). These findings are supported by longitudinal studies of abuse and neglect that indicate increased risk of cognitive impairment, social and emotional difficulties, and risk of mental and physical disease, with those later manifesting posttraumatic stress disorder showing smaller cerebellar and cerebral volumes correlating with earlier onsets and increased durations of abuse (e.g., De Bellis and Kuchibhatla, 2006). Furthermore, in adulthood, the experience of childhood abuse or neglect is tightly associated with increased HPA axis activity (e.g., Carpenter et al., 2010).
Early Life Adversity Shapes the HPA Axis
On exposure to a stressor a body activates stress systems to prepare for events that may threaten well-being or survival. The autonomic nervous system initiates a rapid and relatively short-lived “ fight-or-flight” response while the HPA axis is slower, instigating a more protracted response. The HPA axis is therefore core to the long-term regulation of systems controlling stress responsivity.
Following exposure to a stressor, the neuropeptides corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) are released from the paraventricular nucleus (PVN) of the hypothalamus into the portal vessel system. These neuropeptides bind to and activate specific receptors (the CRHR1 and V1b receptors for CRH and AVP, respectively) on anterior pituitary corticotroph cells stimulating the release of adrenocorticotrophic hormone (ACTH) which then acts on the adrenal cortex to synthesize and release glucocorticoid hormones (cortisol in primates and corticosterone in rats). Glucocorticoids mobilize glucose from energy stores and increase cardiovascular tone, among further widespread effects. Feedback loops, primarily mediated by the PVN and pituitary corticotroph cells, through glucocorticoid receptors (GR) restrain responsiveness of the HPA axis to reset the system to baseline activity (Figure 1; for review see De Kloet et al., 1998). In short, the forward loop prepares the organism to anticipate and respond optimally to a threat, while the feedback loop ensures returning efficiently to a homeostatic balance when it is no longer challenged.
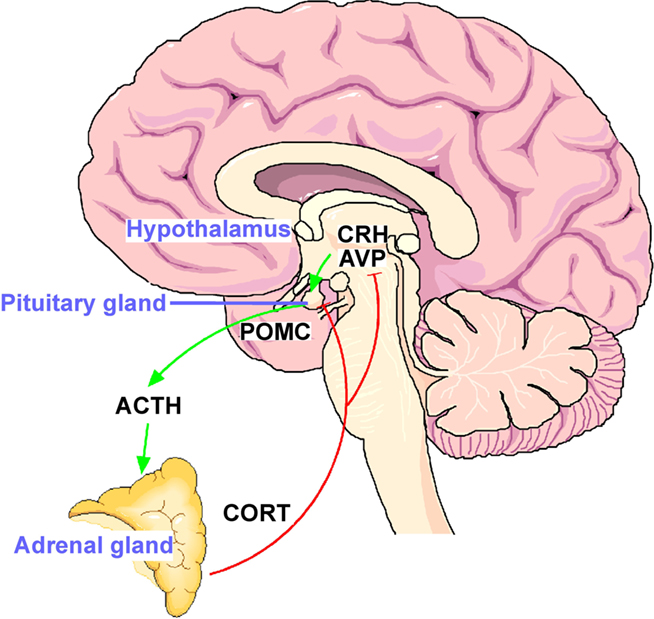
Figure 1. The hypothalamo–pituitary–adrenal (HPA) stress axis. The neuropeptides corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) are expressed in the parvocelluar neurons of the hypothalamic nucleus paraventricularis. The joint release of CRH and AVP into the portal blood vessels leads to potent stimulation of anterior pituitary ACTH secretion and in turn of corticosterone from the adrenal glands. The activational effects of the HPA axis are counteracted by the inhibitory effects of glucocorticoid receptors expressed in the hippocampus, hypothalamus, and anterior pituitary.
Following periods of sustained stress, negative feedback control of the HPA axis can become dysregulated and this may underlie the development of disorders such as major depression (for review see Holsboer, 2000). Indeed, altered activity of this circuit is one of the most commonly observed neuroendocrine symptoms in patients suffering from major depressive disorder (MDD) and dysregulation of cortisol secretion can be found in as many as 80% of depressed patients if subjects are clustered into different age ranges (Heuser et al., 1994).
In clinical studies early life stress has been shown to be a strong predictor of impaired inhibitory feedback regulation of the HPA axis (Heim et al., 2008) as deduced by dexamethasone/CRH challenge tests. Clinical findings give strong evidence to assume that depression is characterized by a hypothalamic overdrive of CRH and/or AVP systems. Postmortem studies of brain tissue reveal elevated CRH (Raadsheer et al., 1994) and AVP (e.g., Meynen et al., 2006) in the hypothalamus of depressed individuals. In cerebrospinal fluid increased concentrations of CRH have been reported to associate with depression, PTSD and obsessive–compulsive disorder (e.g., Bremner et al., 1997;) while higher plasma levels of AVP have been noted in depression(e.g., Goekoop et al., 2006). Receptors for the hormones also appear to be altered in depression with reduced CRH binding sites being detected in the frontal cortex (Nemeroff et al., 1988) and reduced ACTH responses to CRH (Rupprecht et al., 1989). A recent study further suggested that variations in the CRH receptor1 gene moderated the effect of childhood maltreatment on cortisol responses to a dexamethasone/CRH test (Tyrka et al., 2009) and mutation screens have linked polymorphisms in the V1b and AVP genes with childhood-onset depression (e.g., Dempster et al., 2009), supporting previous preclinical work in which selective inbreeding of rodents for high levels of anxiety-like behavior inadvertently enriches for alterations in the AVP gene (Murgatroyd et al., 2004; Kessler et al., 2007; Bunck et al., 2009).
Studies in rodent models demonstrate that exposure to chronic stress can dramatically alter the balance between AVP and CRH control of the HPA axis. For instance, repeated exposure to a stressor leads to enhanced expression of AVP in the PVN, an increase in the number of CRH-containing neurons that co-express AVP and elevation of V1b receptor mRNA in the pituitary – data consistent with the hypothesis that under circumstances of sustained stress AVP becomes the driving force in regulation of the HPA axis (Aguilera and Rabadan-Diehl, 2000). In further support, in vitro studies have shown that stimulation of ACTH release by AVP is less sensitive to negative feedback control by glucocorticoids in comparison with ACTH responses to CRH (e.g., Bilezikjian et al., 1987). Conclusively, these data suggest a reduction in the sensitivity of GR-mediated negative feedback regulation of the HPA axis in depression, which would appear to result from a shift toward increased AVP secretion.
Modeling Early Adverse Experiences
Given that early life stress may lead to enduring dysregulation of the HPA axis and the close link between HPA dysfunction and major depression it has been suggested that early life stress may predispose individuals to psychiatric diseases in later life. Evidently there are ethical limitations to conducting prospective early life stress experiments in human participants. Therefore, animal models are invaluable in gaining insight into the behavioral and physiological mechanisms underlying the long-term effects of early experiences on emotional reactivity and the stress response.
Rodents are particularly well suited to studies of the effects of early life experience as it is possible to rear them in large numbers and under controlled environmental conditions. Even quite subtle alterations in the experience of rats during the early postnatal or prenatal periods can provoke long enduring consequences in behavioral and endocrine phenotypes (for review see Holmes et al., 2005). Acknowledging attachment theories, many of these models investigate adverse early life factors by focusing on the preeminent mother–infant relationship.
Early Life Stress
One of the most widely studied models for early life stress is maternal separation (MS) in which rodents are separated for around 3 h per day for the first 2 weeks of life. Whether the exact psychological cause of the effects of postnatal MS stress results as a direct effect of the pup itself, or indirectly though the manipulation of the mother is still not completely understood: either simulating maternal stroking and feeding during the separation period or providing the dams with a foster litter during the period of separation from their own pups can both dampen the effects of MS on later stress hyper-reactivity (e.g., van Oers et al., 1999; Huot et al., 2004). This procedure can result in persistent increases in anxiety-related behaviors and life-long hyperactivity of the HPA axis in response to stressors (for review see Holmes et al., 2005). Early life stress can also induce long-term effects on hippocampal associated cognitive function and memory (e.g., Eiland and McEwen, 2010) with growing evidence suggesting that the paradigm can disrupt development of neural systems mediating reward-related behaviors, as evident from increases in voluntary ethanol consumption and exaggerated responses to psychostimulants (e.g., Lopez et al., 2010), supporting the concept that maternal and infant attachment is a reward-mediated behavior (e.g., Moles et al., 2004).
Elucidation of neuroendocrine changes underlying the persistent effects of early life stress in rodents has become a major research area. A growing number of reports has documented permanent increases in neurotransmission and hypothalamic expression of CRH (e.g., Vazquez et al., 2006) and AVP (e.g., Veenema et al., 2006) following early life stress. In addition, the ability of hippocampal GRs to attenuate HPA axis may be persistently disrupted; rats subjected to postnatal MS exhibit significantly reduced expression of forebrain GRs and an impairment of the synthetic glucocorticoid dexamethasone to suppress HPA axis activity in adults (e.g., Ladd et al., 2004).
Early Life Care
Shorter periods of separation in rodents (e.g., 15 min) or “handling,” appear to evoke qualitative changes in maternal care and have consequently been found to elicit effects on behavioral and stress-related responses that are opposite to MS, i.e., reduced anxiety and attenuated HPA axis responses to stress when tested as adults (for review see Fernández-Teruel et al., 2002). This result would support the so called “maternal mediation” hypothesis (Smotherman et al., 1974) whereby the “emotional state” of the mother imprints the one of the offspring.
Indeed, there appears to be a direct relationship between variations in the levels of maternal care and the development of individual differences in the behavioral and neuroendocrine responses to stress of the offspring. In particular, high levels of maternal care appear to be directly associated with reduced behavioral and neuroendocrine responses to novelty in the offspring (Liu et al., 1997). Data from rodent models indicate that the long-term effects of handling appear to depend upon changes in the differentiation of those neurons known to curtail the stress response (Meaney et al., 1996). These alterations include increases in GR expression in the hippocampus, a region strongly implicated in glucocorticoid feedback regulation and reduced levels of hypothalamic CRH (Plotsky and Meaney, 1993).
Higher Primate Models
Primates are particularly interesting for studying the role of environmental influences during early life; most psychopathology revolves around social functions and, compared to rodents, non-human primates display complex social behaviors and structures resembling humans. The mother–infant bond in primates is the most fundamental early relationship and, as such, primate infants demonstrate remarkable similarities to humans upon separation from their mother, with chronic and sustained separation in infanthood leading to anxiety-like behaviors, cognitive impairments and long-term alterations in the HPA axis (e.g., Sanchez et al., 2010). It is also possible to deploy more subtle manipulations in early life experience in primates. One study, increased stress experiences of the mother through unpredictable disruption of food availability during foraging. Such hardship led to infants developing increased concentrations of cerebrospinal fluid CRH, reduced cortisol levels and fearful behaviors when compared to control infants. Furthermore, when followed into young adulthood, concentrations of CRH remained elevated, indicating that even relatively brief disturbances of the maternal–infant relationship can establish long-lasting changes in stress response systems in monkeys (Coplan et al., 2001).
New Answers to Old Questions
The aforementioned studies demonstrate that various aspects of the early environment can lead to dramatic changes in neurodevelopmental trajectories and lead to differential risk of physical and psychiatric disorders. However, the question remains as to how early life exposure to stress is able to evoke such persistent alterations in neuronal substrates, hormonal regulation, and behavioral responses in the adult?
Cells of a multicellular organism are genetically identical but structurally and functionally distinct owing to the differential expression of genes. Many of these differences in gene expression arise during development and are subsequently retained through mitosis. The term epigenetics is now commonly used to describe the study of stable alterations in gene expression potential that arise during development, differentiation and under the influence of the environment (for review see Jaenisch and Bird, 2003). In contrast to DNA sequence that is identical in all tissues, the patterns of epigenetic marks are tissue-specific. Hence, a genome can be considered to contain two layers of information: the DNA sequence inherited from our parents which are conserved throughout life and mostly identical in all cells and tissues of our body, and epigenetic marks (i.e., chromatin and DNA methylation patterns) which are cell- and tissue-specific.
Epigenetic regulation of gene expression therefore allows the integration of intrinsic and environmental signals in the genome, thus facilitating the adaptation of an organism to changing environment through alterations in gene activity. In this way, epigenetics could be thought of as conferring additional plasticity to the hard-coded genome. In the context of the early life environment, epigenetic changes offer a plausible mechanism by which early experiences could be integrated into the genome to program adult hormonal and behavioral responses.
The epigenome refers to the ensemble of coordinated epigenetic marks that govern accessibility of the DNA to the machinery driving gene expression; inaccessible genes become silenced whereas accessible genes are actively transcribed. Although the understanding of the interplay between epigenetic modifications is still evolving, the modification of core histones that package the DNA into chromatin and methylation of DNA at the cytosine side-chain in cytosine– guanine (CpG) dinucleotides represent the best-understood epigenetic marks (Figure 2).
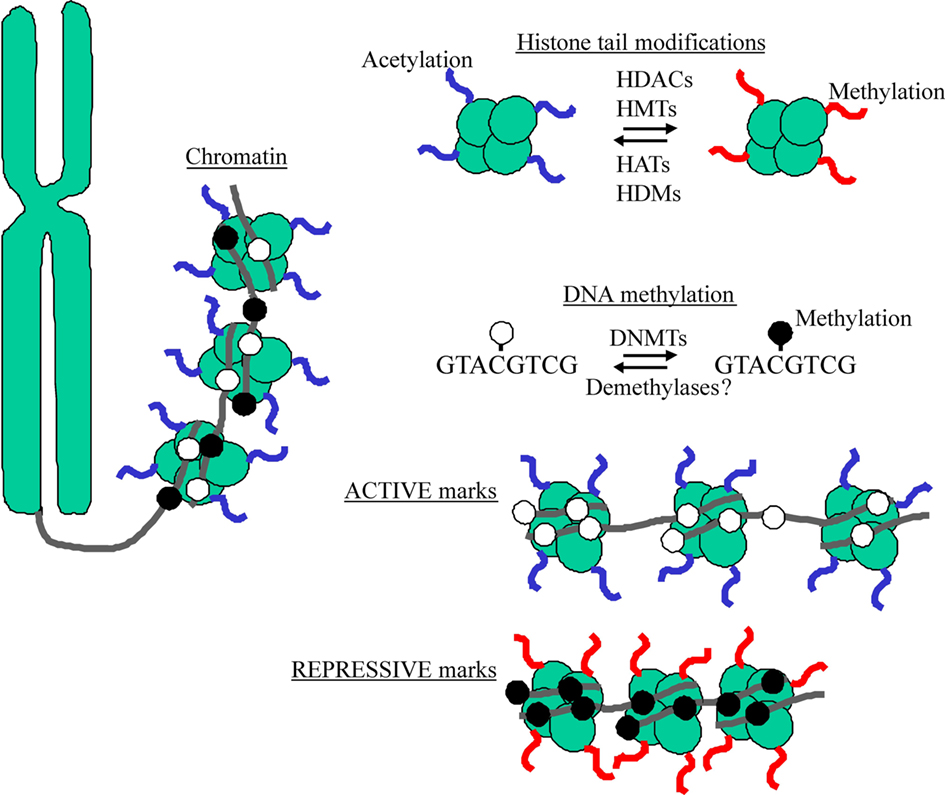
Figure 2. DNA wraps around histones to form a complex referred to as chromatin. The N-terminal tails of histones serve as sites for epigenetic marking by acetylation and methylation via chromatin remodeling enzymes (e.g., HDACs, HATs, HMTs, and HDMs). DNA methylation refers to the chemical transfer of methyl groups to CpG acceptor sites through a class of enzymes known DNA methyltransferases (DNMTs). Active marks (e.g., histone acetylation and DNA hypomethylation) characterize “open” chromatin, while repressive marks (e.g., histone methylation and DNA hypermethylation) occur at “closed” chromatin.
Histone Signatures
Chromatin refers to a complex of DNA wrapped around histone proteins to form nucleosomes. These histone proteins are extensively modified at their N-terminal tails by methylation, phosphorylation, acetylation, and ubiquitination as part of a histone signature serving to define accessibility to the DNA; densely packaged “closed” chromatin is termed heterochromatin while accessible “open” chromatin is termed euchromatin (Jenuwein and Allis, 2001). Histone acetylation is known to be a predominant signal for active chromatin configurations while some specific histone methylation reactions are associated with either gene silencing or activation. Key to this chromatin remodeling are histone-modifying enzymes such as histone acetyltransferases (HAT) that acetylate histone tails, histone deacetylases (HDAC) which deacetylate and histone demethylases that remove methylation. These histone-modifying enzymes are generally recruited through interactions with specific transcription factors that recognize and bind to certain cis-acting sequences in genes. In this way, signaling pathways registering environmental conditions and governing transcription factor activities, could serve as conduits for linking experiences to epigenetic modifications of genes.
DNA Methylation – The Sound of Silence
DNA methylation refers to the chemical transfer of a methyl group to the 5 position of cytosine rings, usually in the context of CpG dinucleotide sequences, and generally results in gene silencing. Since DNA methylation represents a covalent bond, it is thought to be more stable than other epigenetic marks. Such CpG sequences are conspicuously under-represented in mammalian genomes with over 85% of these locating to repetitive sequences scattered throughout the genome and being heavily hypermethylated, possibly playing a crucial role in silencing these elements and thereby maintaining integrity of the genome. The remaining ∼15%, of CpG dinucleotides typically cluster within GC-rich regions known as “CpG islands” that locate within or around the promoters of approximately 40% of genes in the genome (for review see Illingworth and Bird, 2009). Accounting for ∼1% of the genome, these CpG islands are largely unmethylated in the normal somatic cells while regions showing lower CpG density are more frequently methylated (Weber et al., 2007). In general, patterns of methylation tend to correlate with chromatin structure, i.e., active regions of the chromatin are associated with hypomethylated DNA whereas hypermethylated DNA is packaged in inactive chromatin (Razin and Cedar, 1977).
In mitotically active cells DNA methylation is not copied by the DNA replication machinery, but maintained by DNA methyltransferase enzymes (DNMTs) that use S-adenosylmethionine as a methyl donor. The DNA methylation machinery first has to establish new cell-type-specific DNA methylation patterns during development and possibly during adulthood in response to new signals. These patterns have to be renewed once cells undergo division, i.e., duplicate their genome. Of the three DNA methyltransferases identified in mammals, DNMT1 shows preference for hemimethylated DNA in vitro, which is consistent with its role as maintenance DNMT, while DNMT3a and DNMT3b can methylate unmethylated DNA supportive of roles as de novo methylases. The activity of these enzymes is crucial to the vast array of developmental programs in mammals and is, therefore, tightly regulated. During early embryogenesis, epigenetic silencing of genes from either maternal or paternal origins occurs predominantly through the DNA methylation activity of the “maintenance” methyltransferase DNMT1 while further tissue-specific gene expression and DNA methylation during later postnatal development requires also the activity of the “de novo” methyltransferases DNMT3a and DNMT3b (for review see Turek-Plewa and Jagodziński, 2005).
In general, the endpoint of both DNA methylation processes is either long-term silencing or fine-tuning of gene expression potential. Though methylated CpG dinucleotides can directly interfere with transcription factor binding, most effects of repression seem to occur indirectly, via recruitment of methyl-CpG binding domain (MBD) proteins and their associated repressor complexes (for review see Jørgensen and Bird, 2002). The MBD family of proteins, comprising MBD1, MBD2, MBD3, MBD4, and Kaiso in addition to the founding member MeCP2 assemble at methylation marks to provide a platform for the recruitment of corepressor complexes including HDACs that can rewrite histone marks evoking subsequent chromatin compaction and transcriptional suppression. These complexes can additionally recruit DNMTs to promote, maintain and enforce gene repression, compatible with a reciprocal cross talk between DNA methylation and chromatin marks in the regulation of gene transcription.
Although promoter and enhancer regions appear to attract most attention in regard to DNA methylation and gene transcription, it is becoming increasingly clear that methylation at other gene elements play important regulatory roles. For example, increased DNA methylation within the body of genes is typically associated with active transcription (Ball et al., 2009), while DNA methylation at 3′ and intragenic regions may play distinct roles on regulating gene expression (for review see Suzuki and Bird, 2008), possibly controlling activity of intragenic non-coding RNAs. Non-coding RNA are functional RNA molecules that are not translated into a protein and can be involved in a variety of RNA-mediated silencing pathways with increasing evidence indicating that the regulation of these factors could play key roles in neural development and in neuropsychiatric disorders (for review see Qureshi and Mehler, 2010).
As a final point, a new epigenetic mark, 5-hydroxymethylcytosine, has been recently discovered and hailed as the “6th base.” This modification of CpG dinucleotides appears to be particularly abundant in human and mouse brains and to increase during aging in mouse hippocampus and cerebellum (e.g., Song et al., 2011). Considering that it can be generated by enzymatic oxidation of methylated cytosine residues, it appears possible that such modifications might regulate gene expression, though any possible role in brain development or function has yet to be explored in detail.
The Emerging Role of Epigenetics in Mental Health
The billions of neurons in a single brain have the same DNA sequence yet are differentiated for their diverse functions through epigenetic programming during pre- and postnatal development and possibly throughout life. Epigenetic mechanisms are therefore gatekeepers to brain development, differentiation and maturation, and ultimately, associated disease processes.
Genetic Defects in the Epigenetic Machinery
Deregulation of epigenetic pathways can lead to either silencing or inappropriate expression of specific sets of genes manifesting with diseases. A growing understanding in this field is leading to the identification of an expanding number of epigenetic diseases comprising various cancers, neurodegenerative disease, syndromes characterized by chromosomal instabilities, mental retardation, and imprinting disorders (for review see Halusková, 2010).
In the nervous system epigenetic marks govern basic cellular processes such as synaptic plasticity and complex behaviors like memory and learning. Genetic defects in the epigenetic machinery, triggering faulty marking, can lead to severe defects in brain development, and manifest as devastating diseases such as the Rett syndrome, Rubinstein–Taybi syndrome, Fragile X syndrome, Alzheimer’s disease, Huntington’s disease, and psychiatric disorders such as autism, schizophrenia, addiction, and depression (for review see Grafodatskaya et al., 2010).
There are a large number of genes encoding epigenetic regulators that, when mutated, can give rise to mental retardation. Though one might assume that different epigenetic factors would orchestrate the expression of a large number of potentially unrelated genes, disruptions in distinct epigenetic regulators seemingly lead to symptomatically similar mental retardation syndromes (Kramer and van Bokhoven, 2009). Ergo, it is conceivable that mental retardation does not take root in changes of specific target gene(s) but by the inability of concerned neurons to respond adequately to environmental signals under conditions of greatly distorted transcriptional homeostasis (Ramocki and Zoghbi, 2008).
One of the most common causes of mental retardation in females is Rett syndrome, a progressive neurodevelopmental disorder resulting from mutations in the methyl-CpG binding protein MeCP2 located on the X chromosome. Interestingly, less disruptive gene mutations or alterations in expression of this gene also appear to underlie some cases of autism (for review see Gonzales and LaSalle, 2010), along with further members of the MBD family in a small number of cases (Cukier et al., 2010). Another severe X-linked form of mental retardation (ATRX; alpha thalassemia/mental retardation syndrome X-linked) results from mutations of a gene coding for a member of the SNF2 subgroup of a superfamily of proteins involved in chromatin remodeling (Picketts et al., 1996). Further defects affecting proteins involved in histone modification leading to mental retardation are mutations of CBP, a protein with HAT function, underlying Rubinstein–Taybi syndrome (Petrij et al., 1995) and alterations in RSK2, involved in histone phosphorylation and interaction with CBP, leading to Coffin–Lowry syndrome, an X-linked mental retardation disorder characterized by psychomotor and growth retardation (Hanauer and Young, 2002). Defects in proteins important for histone methylation are the H3K4-specific histone demethylase JARIDIC resulting in X-linked mental retardation (Jensen et al., 2005), the nuclear receptor set domain containing protein 1 (NSD1) gene causing Sotos syndrome and Weaver syndrome (Tatton-Brown and Rahman, 2007), the histone lysine methyltransferase GLP/EHMT1 gene characterized by severe mental retardation (Kleefstra et al., 2006) and finally, MLLl, a H3K4-specific methyltransferase involved in hippocampal synaptic plasticity, that might underlie in the cortical dysfunction of some cases of schizophrenia (for review see Akbarian and Huang, 2009). The development of, often severe, mental retardation in these syndromes supports the importance of functional epigenetic machinery in the regulation of early brain development. Following on, it could be expected that perturbations in the regulation and expression of various epigenetic components might then lead to further mental pathologies.
Alterations in Epigenetic Regulation
There are a growing number of studies indicating aberrant epigenetic marks in the development of mental pathologies later in life, though whether changes originate during early development or in later life as a response to an environmental exposure remain important questions. For example, DNA hypomethylation at the promoter of the gene for catechol-O-methyltransferase (COMT), an enzyme regulating the level of dopamine, has been found to associate with schizophrenia and bipolar disorder (Abdolmaleky et al., 2006). At the promoter region of the RELN gene, encoding a protein implicated in long-term memory, a number of studies have evidenced hypermethylation, correlating with reduced expression, in schizophrenia (e.g., Grayson et al., 2005). Within the frontopolar cortex, a further study discovered DNA hypermethylation at the gamma-aminobutyric acid receptor alpha1 subunit (GABA-A) promoter region, in suicide and MDD brains that correlated with altered DNMT mRNA expression (Poulter et al., 2008), while another investigation reported alterations in DNA methylation and mRNA expression of the peptidylprolyl isomerase E-like (PPIEL) gene in monozygotic twins discordant for bipolar disorder (Kuratomi et al., 2008).
Monozygotic twin pairs share a virtually identical genome though may differ to various extents in their pre- and postnatal environments, making them highly informative in understanding how epigenetic variation could affect complex traits (for review see Bell and Spector, 2011). Indeed, twins frequently differ in their prevalence of mental disorders with a number of studies reporting differences in DNA methylation between monozygotic twins discordant for schizophrenia (e.g., Petronis et al., 2003) and a more recent report detecting lower levels of DNA methylation in the temporal cortex of Alzheimer-affected twins (Mastroeni et al., 2009). A further genome-wide screen detected substantial variability in DNA methylation between twins (Kaminsky et al., 2009), which may relate to the earlier findings, in which DNA methylation profiles of young monozygotic twin pairs were shown to be more epigenetically similar than older monozygotic twins (Fraga et al., 2005). This would therefore suggest that epigenetic changes increase with age, following the view that epigenetic mechanisms such as DNA methylation deteriorate with age and may even accelerate the aging process (Murgatroyd et al., 2010b; Rodríguez-Rodero et al., 2010). However, the influence of genetic factors should also be considered as reports have found higher epigenetic variation in dizygotic than in monozygotic twins (Kaminsky et al., 2009) and intra-individual changes in DNA methylation showing degrees of familial clustering (Bjornsson et al., 2008). Finally, the possible influence of random stochastic factors may further complicate such studies with reports that even genetically identical laboratory animals living under the same controlled environmental conditions, nevertheless develop phenotypic and epigenetic differences (e.g., Vogt et al., 2008).
Epigenetics and Aging
The relationship between DNA methylation and aging was originally proposed by Berdyshev, who discovered that genomic global DNA methylation decreases with age in spawning humpbacked salmon (Berdyshev et al., 1967). This was later backed-up by further evidence for the presence of gradual global losses of methylation in various mouse, rat, and human tissues (e.g., Fuke et al., 2004). Much of this global hypomethylation may result from the fact that different types of interspersed repetitive sequences, that make up the major bulk of genomes, appear to be targeted at single time-points to varying degrees by age-associated hypomethylation (Jintaridth and Mutirangura, 2010). In general terms, age-associated hypermethylation is thought to preferentially affect loci at CpG islands, while loci devoid of CpG islands lose methylation with age. Although an increase in promoter methylation with aging is generally accepted, such as from studies in human prostate and colon tissues (e.g., Kwabi-Addo et al., 2007), recent evidence from Bjornsson et al. suggest a more complex picture with both increases and decreases in intra-individual global methylation levels over time (Bjornsson et al., 2008). Certainly, epigenetic dysregulation with age appears to be a highly tissue-dependent phenomenon. Furthermore, there appear to be subsets of genes that are tissue-specifically affected by age-associated DNA methylation changes, such as genes involved in metabolism and metabolic regulation in liver and visceral adipose tissues (Thompson et al., 2010) or CpG sites physically close to genes involved in DNA binding and regulation of transcription in various brain regions (Hernandez et al., 2011). This later study provides a particularly strong argument that specific age-related DNA methylation changes may have quite broad impacts on gene expression in the human brain. We may, however, have to readdress some of our preconceptions regarding age and aging in relation to epigenetic changes concerning a new study of various mouse tissues in which a majority of DNA methylation changes found in adulthood were preceded by similar DNA methylation changes in juveniles, and that the expression levels of genes near progressively methylated and demethylated CpG sites were already significantly up- or down-regulated, respectively, in young mice (Takasugi, 2011). This would imply that the epigenome maturates continuously throughout lifespan and that affects of aging emerge gradually over time.
Compatible with this view, a growing number of reports in the literature challenge the conventional view of a locked state for DNA methylation, demonstrating that DNA methylation remains an active process in post-mitotic cells (i.e., neurons). For example, DNMTs are expressed and regulated in the adult brain (e.g., Brown et al., 2008) and evidence continues to mount that epigenetic mechanisms are able to dynamically control changes in gene activity throughout the lifespan as a result of exposure to a variety of environmental factors during aging, as previously described.
Overall, these findings suggest the presence of several mechanisms regulating methylation that are possibly adjusted at different steps during development, maturation and aging by processes that are responsive to numerous environmental and genetic factors.
The Epigenome and Early Life Adversity
Aside from controlling constitutive gene expression, epigenetic mechanisms can also serve to fine-tune gene expression potential in response to environmental cues. DNA methylation has been the most studied in regard to understanding early life experiences and the epigenetic programming of their neurobiological sequelae. Indeed, the stable nature of DNA methylation renders it an ideal template for underpinning sustained gene effects controlling brain function and behavior from early development to old age. Thus we and others have proposed that conditions of early life environment can evoke changes in DNA methylation facilitating epigenetic programming of critical genes involved in regulating stress responsivity that may in turn manifest with neuroendocrine and behavioral symptoms in adulthood (e.g., McGowan et al., 2009; Murgatroyd et al., 2010a; Figure 3).
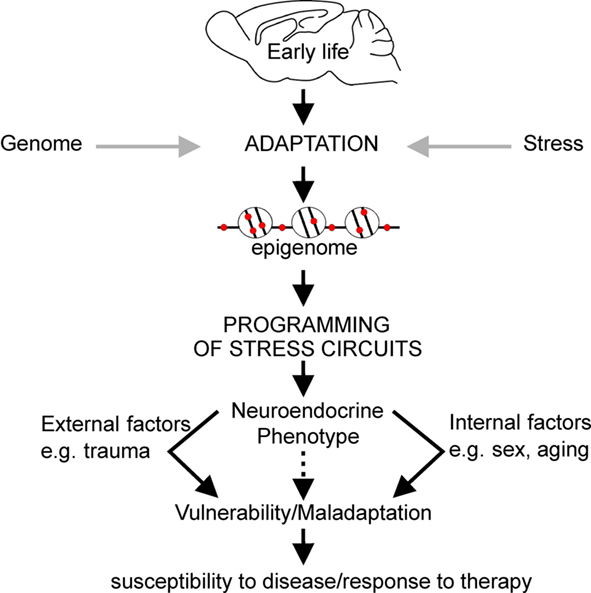
Figure 3. Early life experience can persistently alter expression levels of key genes through epigenetic marking which can underpin changes in behavior, neuroendocrine, and stress responsivity throughout later life. Collectively, this process is referred to as epigenetic programming. The nature of the environment throughout later life, in addition to the impact of biological processes associated with aging and genetic sex, may exacerbate the effects of programming established during early life resulting in increased vulnerability to mood disorders.
Maternal Care and Epigenetic Programming of GR
One well-characterized model established to study the effects of early life environment on stress programming examines variations in the quality of early postnatal maternal care, as measured by levels of licking and grooming. Studies revealed that rat pups receiving high levels of maternal care during early life developed sustained elevations in GR expression within the hippocampus and decreased hippocampal sensitivity to glucocorticoid hormones (Liu et al., 1997). In addition, these rats showed decreased hypothalamic CRH levels and reduced HPA axis responses to stress (Francis et al., 1999) when compared to animals reared by mothers showing low levels of maternal care. Analysis of the molecular mechanisms underlying the long-lasting programming of the GR revealed an important role for epigenetic regulation. The investigators evidenced that the enhanced hippocampal GR expression in animals receiving high levels of maternal care associated with a persistent DNA hypomethylation at specific CpG dinucleotides within the hippocampal GR exon 17 promoter and increased histone acetylation. These epigenetic modifications facilitated binding of the transcriptional activator nerve growth factor-inducible protein A (NGF1a) to this region, providing a plausible mechanism for the epigenetic programming of gene function by maternal care during early life (Weaver et al., 2004). Interestingly, a further study using a different paradigm of MS in another species of rat found that corticosterone release and increased NGF1a expression failed to induce changes in DNA methylation levels at the same hippocampal GR promoter (Daniels et al., 2009), supporting again the idea that variations in the genetic make up, and environmental disparities, are critical determinants in organization of the epigenome. In support of this idea, subsequent studies detected altered GR promoter methylation in human postmortum hippocampal tissue of depressed suicide patients who suffered from a history of early life abuse and neglect (McGowan et al., 2009). In contrast, suicide patients who were not exposed to early life adversity, or patients suffering from major depression only, revealed no epigenetic marking of hippocampal GR (Alt et al., 2010).
Maternal Separation Stress and Epigenetic Programming of AVP
Work in our lab evidenced that MS stress in mice, involving separating pups from their mothers for 3 h each day during the first 10 days of life, induced life-long sustained expression of hypothalamic AVP underlying elevated corticosterone secretion, heightened endocrine responsiveness to subsequent stressors, and altered feedback inhibition of the HPA axis. Starting at 10 days of life, directly following the period of MS, and lasting for at least 1 year, elevated AVP expression was specific to the parvocellular subpopulation of PVN neurons while hypothalamic CRH and hippocampal GR expression remained unaltered. Importantly, this altered expression associated with reduced levels of DNA methylation in the PVN at particular CpG dinucleotides within an enhancer region important for AVP gene activity (Murgatroyd et al., 2009). We further showed that hypomethylation at this region reduced the ability of MeCP2 to bind and recruit repressive histone complexes such as HDACs and DNMTs (Murgatroyd, unpublished), supporting previous evidences for a role of MeCP2 as an epigenetic platform upon which histone deacetylation, H3K9 methylation, and DNA methylation are carried out to confer transcriptional repression and gene silencing (e.g., Murgatroyd et al., 2010a).
We then examined the signals controlling MeCP2 occupancy at this early step. Experience-driven synaptic activity causes membrane depolarization and calcium influx into select neurons, which in turn induce a wide variety of cellular responses, connecting neuronal activity to transcriptional regulation (Greer and Greenberg, 2008). Furthermore, neuronal depolarization has previously been shown to trigger Ca2+-dependent phosphorylation of MeCP2, causing dissociation of MeCP2 from the BDNF promoter and consequently gene derepression (e.g., Martinowich et al., 2003). Subsequent studies revealed that CaMKII (Ca2+/calmodulin-dependent protein kinase II) was able to mediate this phosphorylation of rat MeCP2 at serine 421 (Zhou et al., 2006), and the homologous serine residue 438 in the mouse (Murgatroyd et al., 2009). Analysis of 10-day-old early life stressed mice revealed increased CaMKII and phospho-MeCP2 immunoreactivity that promoted MeCP2 dissociation at the AVP enhancer and depression. However, though early life stressed mice tested in adulthood (6 week) still showed reduced MeCP2 binding at the AVP enhancer, the levels of MeCP2 phosphorylation and CaMKII activation in the PVN no longer differed to those of control animals. Instead, evolving differences in DNA methylation at this region underlie reduced MeCP2 occupancy in early life stressed mice. Taken together, the initial stimulus of early life stress initiates a loss of MeCP2 occupancy that subsequently leads to the hard-coding of the early life experience at the level of DNA methylation (Figure 4). Interestingly, in the control mice an age-associated demethylation also occurred though, critically, the region of the enhancer marked by early life stress was spared, supporting, the importance of this region in AVP regulation (Murgatroyd et al., 2010b).
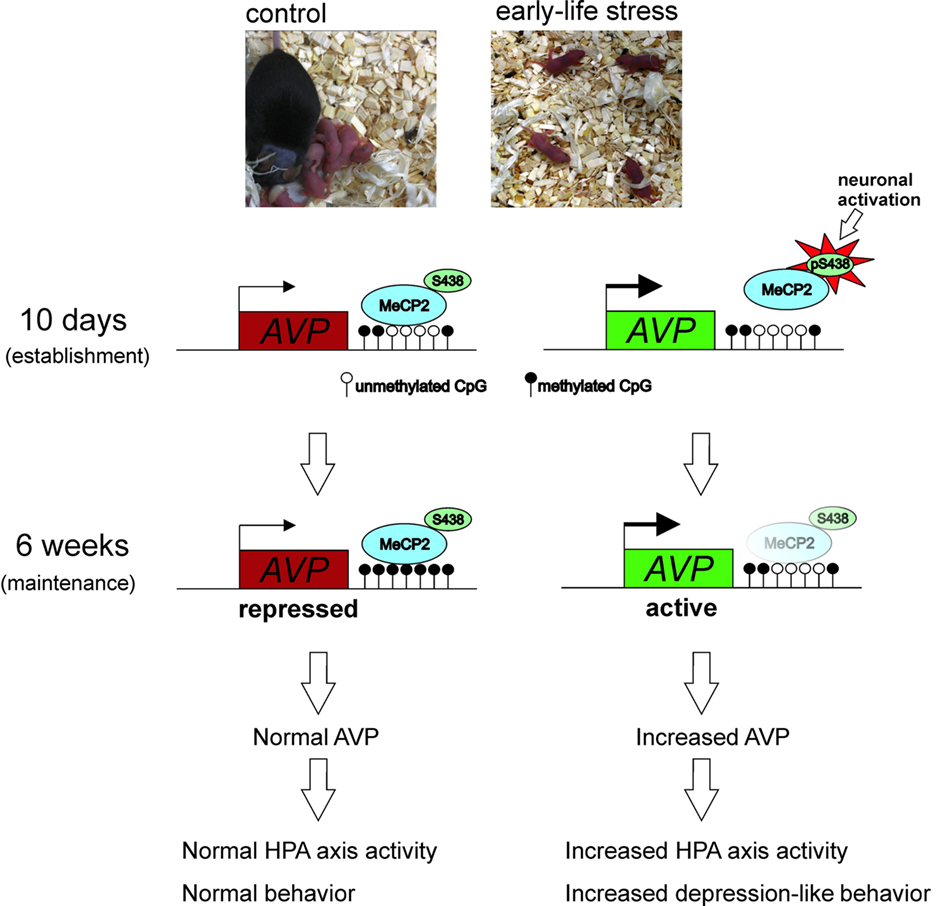
Figure 4. Experience-dependent activation of neurons in the PVN during early life inhibits MeCP2 binding to and repressing the AVP gene in stressed mice. This further favors DNA hypomethylation underpinning reduced MeCP2 occupancy at the AVP enhancer and maintaining epigenetic control of AVP expression into later life.
Additional Targets for Epigenetic Programming of the HPA Axis
A growing number of studies support the role of epigenetic modifications in the control of genes regulating the HPA axis. Two recent ones demonstrate epigenetic programming of hypothalamic CRH in response to stress in either the prenatal period or during adulthood. Pregnant mice subjected to chronic variable stressors during early gestation revealed a hypomethylation at the CRH promoter in the hypothalamus of the offspring in adulthood (Mueller and Bale, 2008). Moreover, chronic social stress in adult mice caused a hypomethylation of the CRH promoter in the PVN of a subset of animals displaying subsequent social avoidance (Elliott et al., 2010).
A number of studies describe epigenetic regulation of the POMC gene (pro-opiomelanocortin, coding for ACTH), which is a downstream target of AVP and CRH signaling. Hypermethylation of the promoter region leads to silencing of expression that appears to be regulated by cortisol (e.g., Mizoguchi et al., 2007). Recent studies suggest epigenetic programming of POMC by nutritional cues such as overfeeding or anorexia (e.g., Ehrlich et al., 2010), while other research suggested an association with alcohol craving (Muschler et al., 2010). These preliminary findings warrant further studies to address the question of whether epigenetic programming of the HPA axis by early life experiences extends to the pituitary gland (POMC) in addition to hippocampal (GR) and hypothalamic (AVP and CRH) tissues.
Epigenetic Programming Beyond the HPA Axis
Aside from the HPA axis, additional pathways and epigenetic targets of early life stress seem to exist. For example, rats reared in a hostile postnatal environment exhibited promoter hypermethylation associating with reduced expression of brain-derived neutrotrophic factor (BDNF) in the prefrontal cortex in adulthood (Roth et al., 2009). BDNF is a neurotrophin that regulates neurodevelopment, neuroplasticity, and neuronal functions. It is further involved in the pathogenesis of psychiatric and neurodegenerative disorders and there is a growing body of literature linking epigenetic gene regulation of the BDNF gene with brain plasticity and cognitive function (e.g., Lubin et al., 2008).
Neuroendocrine systems associated with female sexual and maternal behavior and the development of the GABA system also appear to be influenced by alterations in the early life environment. Female rat offspring of high-maternal care mothers developed a hypomethylation of the estrogen receptor-alpha (ERα) promoter in the medial preoptic area correlating with increased binding of the transcription factor STAT5 and elevated ERα expression (Champagne et al., 2006). This study indicates that epigenetic programming can take place in a sex-specific manner which could shed new light on the long standing question of a sex bias in numerous diseases (for review see Menger et al., 2010) Additionally, rat pups receiving high-maternal care showed enhanced hippocampal glutamic acid decarboxylase 1 (GAD1) mRNA expression, decreased cytosine methylation, and increased histone 3-lysine 9 acetylation (H3K9ac) of the GAD1 promoter (Zhang et al., 2010). Furthermore, DNMT1 expression was also significantly lower in the offspring of those rats that received high-maternal care (Zhang et al., 2010). This is interesting, as a number of studies suggest the regulation of components of the epigenetic machinery in response to stressors. For example, recent work has shown that chromatin remodeling in various brain regions, including the hippocampus, is associated with the effects of stress in a variety of models (e.g., Hunter et al., 2009).
Digressing from the literature of early life induced epigenetic changes; one recent study found that traumatic stress during adulthood can also drive epigenetic changes in the brain. Adult rats exposed to severe chronic psychosocial stress showed increased BDNF promoter methylation in the hippocampal regions associating with reduced expression of this gene (Roth et al., 2011). Another neurotrophin known to be important for the regulation of brain plasticity and implicated in depression, is glial cell-derived neurotrophic factor (GDNF). Examining two genetically distinct mouse strains exhibiting different behavioral responses to chronic stress, researches recently discovered GDNF in the nucleus accumbens to be epigenetically silenced following adulthood stress only in the stress-vulnerable mice (Uchida et al., 2011), again supporting the importance of dynamic epigenetic changes in response to environmental cues lasting into adulthood.
Clinical Studies of Epigenetic Programming by Early Life Adversity
Evidence is beginning to emerge that the findings of epigenetic programming in animal models may extrapolate to human studies and psychopathologies associated with severe abuse or neglect during childhood. McGowan et al. (2008) found that suicide subjects who had a history of early childhood neglect or abuse exhibited with hypermethylation at the ribosomal RNA (rRNA) promoter. rRNA genes encode ribosomal RNA, important for protein synthesis, leading to the consideration that such reductions in rRNA levels might reflect a reduced capacity for protein synthesis in the hippocampus of suicide victims. Though how this may contribute to the pathology is yet to be determined. Subsequent studies of postmortum tissue by the same researchers further evidenced a hypermethylation of the GR gene promoter among suicide victims with a history of abuse in childhood, but not among controls or suicide victims who did not suffer such early life stress (McGowan et al., 2009). This data appear consistent with previous studies demonstrating that the epigenetic status of the homologous GR gene promoter is regulated by parental care during early postnatal development in rats (Weaver et al., 2004). Although indirect, this correlation in the human study suggests that epigenetic mechanisms, thought to play a role in vulnerability to stress-related conditions in rodents, might extrapolate to humans. However, it should be noted that further work is necessary to establish a causal link for this epigenetic modification to general depressive behavior in humans. Furthermore, an independent follow-up study failed to identify similar DNA methylation at the GR promoter or alterations in major depression (Alt et al., 2010), again pointing to the importance of inter-individual genetic and environmental variations.
Overall, these studies suggest that epigenetic processes could mediate the effects of the early environment on gene expression and that stable epigenetic marks such as DNA methylation might then persist into adulthood and influence vulnerability for psychopathology through effects on intermediate levels of function such as activity of the HPA axis.
Future Research
With the advent of new high-throughput sequencing technologies research is now beginning to move on from the single-gene scale to epigenome-wide analyses of all epigenetic marks throughout a genome in a specific tissue. Given that epigenetic modifications are sensitive to changes in the environment, it might be anticipated that these efforts will identify epigenomic signatures for mental disorders and molecular dysregulations resulting from early life stress (Albert, 2010). While such a strategy appears highly attractive in the field of cancer research addressing clonally expanded cell populations, rather than heterogenous tissues, the field of epigenetic association studies in mental illnesses might be more challenging. Epigenetic modifications reported to date in animal models and postmortem brain tissues are seldom “all or none” but gradual, and seem to occur in a highly cell-type-specific manner. In this view, the extraordinary complexity and heterogeneity of neural tissues poses a major hurdle to derive epigenetic biomarkers in psychiatric disease.
Epigenetic Biomarkers
Ultimately, the identification of “epigenetic biomarkers” in distinct genomic regions may provide important information for the understanding of biological processes underlying mental diseases and thus allow for the development and design of new therapeutics. Though expression and DNA methylation changes in the brain are more obviously relevant to changes in behavior, comparable changes in blood might provide a clinically valuable surrogate, given the easy access to this tissue in patients. Some early studies have shown partial correlations in gene expression between various brain regions and blood (e.g., Brown et al., 2001), which have been supported by following data demonstrating epigenetic differences in lymphocytes associating with the brain in Rett syndrome and Alzheimer’s disease (e.g., Wang et al., 2008). Recently it was described that chronic corticosterone exposure in mice stimulated parallel increases in FKBP5 expression between brain tissues and blood together with some, generally subtle, alterations in DNA methylation at the promoter of this gene (Lee et al., 2010). However, the other candidate HPA axis genes tested in this study – namely, NR3C1, HSP90, CRH, and CRHR1 – failed to show such effects while further studies in this area of research also describe little correlation (e.g., Yuferov et al., 2011). Consequently, the prospect of diagnostic epigenetic testing for mental diseases using markers in the blood appears so far unresolved, and certainly further research is required.
A Matter of Timing
Many of the studies surveyed in this review highlight the importance of the temporal nature of brain developmental processes in relation to the effects that environmental stressors can have on epigenetic programming of long-term changes in neurodevelopment and behavior. Developing brain regions typically pass through critical “windows” of sensitivity that stretch over different perinatal periods. Therefore, it stands to reason that the impact of stressors at different time-points will confer more pronounced and long-lasting effects within brain regions that are actively developing at that particular time. For example, late prenatal and early postnatal life is the critical period for hippocampal development, possibly explaining why environmental exposures during this time strongly associate with cellular, morphological and epigenetic changes within these structures (for review see McCrory et al., 2010). In contrast, environmental exposures during later life tend to confer their phenotypic effects by altering other areas of the brain. For example, repeated restraint stress in adult rats fails to cause long-term hippocampal-related effects such as those observed following similar stress exposure during prenatal and early postnatal life (Conrad et al., 1999). On the other hand, substantial cortical development and differentiation is known to continue well into adolescence (e.g., Wang and Gao, 2009), possibly explaining why these regions are more susceptible to epigenetic changes in response to later life environmental factors.
In addition to the temporal nature of brain development, we should also consider the temporal and gene-specific manner in which epigenetic marks are established. This will be of crucial importance in understanding how experience-dependent epigenetic marks may undergo the transition from a preliminary labile state to a hard-coded stable print. Indeed, in our studies epigenetic marking of the methylation landmarks in the AVP enhancer persisted under vasopressin receptor blockade in adult (3 month) mice consistent with the concept that the early life stress had already engraved a lasting cellular memory (Murgatroyd et al., 2010a). The question however remains of whether critical “windows” for timely psychotherapeutic and pharmacological interventions following exposure to severe trauma might exist at periods prior to the establishment of stable epigenetic marks. In this view, therapeutic interventions might then re-stimulate or inhibit those same neurons and genes epigenetically programmed earlier in life and reverse these changes. Apparently, timing, quality and duration of such environmental stimulations might be highly specific and context-dependent in regard to the exact nature of the early life stressor.
Nevertheless, studies in rats have demonstrated that environmental enrichment during puberty can alleviate some of the anxiety-related effects of ELS (e.g., Imanaka et al., 2008). This result appears consistent with observations from the clinic. For example, infants who have experienced previous maltreatment, if placed in environments in which positive parenting strategies are being used, show significant improvements in behavior and cortisol regulation (e.g., Fisher et al., 2007). A further study in foster children has revealed that a relational-based intervention focused more on social interactions with caregivers proved to be far more efficient in reducing cortisol levels when compared to an intervention focused more on enhancing cognitive skills (Dozier et al., 2008), bringing us back to the importance of the early life attachment system. Considering these observations, further preclinical studies addressing the effects of such environmental enrichment following early life stress could focus on whether critical time-windows exist for heightened sensitivity to stress, bearing in mind the temporal aspects of brain development and epigenetic mechanisms, and adaptability to ameliorative effects of an enriched environment.
A further option to modify the effects of early life stress could be to target DNMTs and chromatin remodeling enzymes. Though such histone-modifying and DNA methylation-targeting drugs are proving highly attractive to cancer therapeutics, it remains to be seen whether they have any real potential in the realm of psychiatric diseases. Nonetheless, it is notable that the frequently used mood stabilizer valproate (VPA) has been shown to modulate the epigenome by inhibiting HDACs and can additionally promote demethylation in brain cells (e.g., Perisic et al., 2010). However, much caution centers on the fact that most currently available “epigenetic drugs” suffer from a lack of specificity at both the levels of tissue and the genome. This field may yet advance with research targeting specific histone-modifying enzymes, such as G9a, that may be selective enough to ameliorate addiction or mental illness (Maze et al., 2010). Another interesting approach to regulate methylation relies on treatment with the methyl donor l-methionine, or folic acid supplementation during gestation. Intriguingly, there are a number of recent reports linking low folate levels during pregnancy with behavioral symptoms in children (e.g., Schlotz et al., 2010). Although effects in gene expression and behavior have been reported (e.g., Weaver et al., 2005), the mechanisms of how such treatment could be able to target and induce such effects in specific regions of the brain is still yet not properly understood.
In sum, understanding how early life experiences can give rise to lasting epigenetic memories conferring increased risk for mental disorders is emerging at the epicenter of modern psychiatry. Whether suitable social or pharmacological interventions could reverse deleterious epigenetic programming triggered by adverse conditions during early life, should receive highest priority on future research agendas. Progress in this field will further garner public interest, a general understanding and appreciation of the consequences of childhood abuse and neglect for victims in later life.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the European Union (CRESCENDO – European Union Contract number LSHM-CT-2005-018652).
References
Abdolmaleky, H. M., Cheng, K. H., Faraone, S. V., Wilcox, M., Glatt, S. J., Gao, F., Smith, C. L., Shafa, R., Aeali, B., Carnevale, J., Pan, H., Papageorgis, P., Ponte, J. F., Sivaraman, V., Tsuang, M. T., and Thiagalingam, S. (2006). Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum. Mol. Genet. 15, 3132–3145.
Aguilera, G., and Rabadan-Diehl, C. (2000). Vasopressinergic regulation of the hypothalamic-pituitary-adrenal axis: implications for stress adaptation. Regul. Pept. 96, 23–29.
Akbarian, S., and Huang, H. S. (2009). Epigenetic regulation in human brain-focus on histone lysine methylation. Biol. Psychiatry 65, 198–203.
Albert, P. R. (2010). Epigenetics in mental illness: hope or hype? J. Psychiatry Neurosci. 35, 366–368.
Alt, S. R., Turner, J. D., Klok, M. D., Meijer, O. C., Lakke, E. A., Derijk, R. H., and Muller, C. P. (2010). Differential expression of glucocorticoid receptor transcripts in major depressive disorder is not epigenetically programmed. Psychoneuroendocrinology 35, 544–556.
Ball, M. P., Li, J. B., Gao, Y., Lee, J. H., LeProust, E. M., Park, I. H., Xie, B., Daley, G. Q., and Church, G. M. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–368.
Bell, J. T., and Spector, T. D. (2011). A twin approach to unraveling epigenetics. Trends Genet. 27, 116–125.
Berdyshev, G. D., Korotaev, G. K., Boiarskikh, G. V., and Vaniushin, B. F. (1967). Nucleotide composition of DNA and RNA from somatic tissues of humpback and its changes during spawning. Biokhimiia 32, 988–993.
Bilezikjian, L. M., Blount, A. L., and Vale, W. W. (1987). The cellular actions of vasopressin on corticotrophs of the anterior pituitary: resistance to glucocorticoid action. Mol. Endocrinol. 1, 451–458.
Bjornsson, H. T., Sigurdsson, M. I., Fallin, M. D., Irizarry, R. A., Aspelund, T., Cui, H., Yu, W., Rongione, M. A., Ekstrom, T. J., Harris, T. B., Launer, L. J., Eiriksdottir, G., Leppert, M. F., Sapienza, C., Gudnason, V., and Feinberg, A. P. (2008). Intra-individual change over time in DNA methylation with familial clustering. JAMA 299, 2877–2883.
Bowlby, J. (1982). Attachment and loss: retrospect and prospect. Am. J. Orthopsychiatry 52, 664–678.
Bremner, J. D., Licinio, J., Darnell, A., Krystal, J. H., Owens, M. J., Southwick, S. M., Nemeroff, C. B., and Charney, D. S. (1997). Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am. J. Psychiatry 154, 624–629.
Brown, S. E., Weaver, I. C. G., Meaney, M. J., and Szyf, M. (2008). Regional-specific global cytosine methylation and DNA methyltransferase expression in the adult rat hippocampus. Neurosci. Lett. 440, 49–53.
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., Jin, X., Feng, Y., Wilkinson, K. D., Keene, J. D., Darnell, R. B., and Warren, S. T. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487.
Bunck, M., Czibere, L., Horvath, C., Graf, C., Frank, E., Kessler, M. S., Murgatroyd, C., Müller-Myhsok, B., Gonik, M., Weber, P., Pütz, B., Muigg, P., Panhuysen, M., Singewald, N., Bettecken, T., Deussing, J. M., Holsboer, F., Spengler, D., and Landgraf, R. (2009). A hypomorphic vasopressin allele prevents anxiety-related behavior. PLoS ONE 4, e5129. doi: 10.1371/journal.pone.0005129
Carpenter, L. L., Shattuck, T. T., Tyrka, A. R., Geracioti, T. D., and Price, L. H. (2010). Effect of childhood physical abuse on cortisol stress response. Psychopharmacology (Berl.) 214, 367–375.
Champagne, F., Weaver, I., Diorio, J., Dymov, S., Szyf, M., and Meaney, M. J. (2006). Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology 147, 2909–2915.
Conrad, C. D., LeDoux, J. E., Magariños, A. M., and McEwen, B. S. (1999). Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav. Neurosci. 113, 902–913.
Coplan, J. D., Smith, E. L., Altemus, M., Scharf, B. A., Owens, M. J., Nemeroff, C. B., Gorman, J. M., and Rosenblum, L. A. (2001). Variable foraging demand rearing: sustained elevations in cisternal cerebrospinal fluid corticotropin-releasing factor concentrations in adult primates. Biol. Psychiatry 50, 200–204.
Cukier, H. N., Rabionet, R., Konidari, I., Rayner-Evans, M. Y., Baltos, M. L., Wright, H. H., Abramson, R. K., Martin, E. R., Cuccaro, M. L., Pericak-Vance, M. A., and Gilbert, J. R. (2010). Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics 11, 291–303.
Daniels, W. M., Fairbairn, L. R., van Tilburg, G., McEvoy, C. R., Zigmond, M. J., Russell, V. A., and Stein, D. J. (2009). Maternal separation alters nerve growth factor and corticosterone levels but not the DNA methylation status of the exon 1(7) glucocorticoid receptor promoter region. Metab. Brain Dis. 24, 615–627.
De Bellis, M. D., and Kuchibhatla, M. (2006). Cerebellar volumes in pediatric maltreatment-related posttraumatic stress disorder. Biol. Psychiatry 60, 697–703.
De Kloet, E. R., Vreugdenhil, E., Oitzl, M. S., and Joëls, M. (1998). Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 19, 269–301.
Dempster, E. L., Burcescu, I., Wigg, K., Kiss, E., Baji, I., Gadoros, J., Tamás, Z., Kapornai, K., Daróczy, G., Kennedy, J. L., Vetró, A., Kovacs, M., Barr, C. L., and International Consortium for Childhood-Onset Mood Disorders. (2009). Further genetic evidence implicates the vasopressin system in childhood-onset mood disorders. Eur. J. Neurosci. 30, 1615–1619.
Dozier, M., Peloso, E., Lewis, E., Laurenceau, J. P., and Levine, S. (2008). Effects of an attachment-based intervention on the cortisol production of infants and toddlers in foster care. Dev. Psychopathol. 20, 845–859.
Dudley, K. J., Li, X., Kobor, M. S., Kippin, T. E., and Bredy, T. W. (2011). Epigenetic mechanisms mediating vulnerability and resilience to psychiatric disorders. Neurosci. Biobehav. doi: 10.1016/j.neubiorev.2010.12.016. [Epub ahead of print].
Ehrlich, S., Weiss, D., Burghardt, R., Infante-Duarte, C., Brockhaus, S., Muschler, M. A., Bleich, S., Lehmkuhl, U., and Frieling, H. (2010). Promoter specific DNA methylation and gene expression of POMC in acutely underweight and recovered patients with anorexia nervosa. J. Psychiatr. Res. 44, 827–833.
Eiland, L., and McEwen, B. S. (2010). Early life stress followed by subsequent adult chronic stress potentiates anxiety and blunts hippocampal structural remodeling. Hippocampus. doi: 10.1002/hipo.20862. [Epub ahead of print].
Elliott, E., Ezra-Nevo, G., Regev, L., Neufeld-Cohen, A., and Chen, A. (2010). Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 13, 1351–1353.
Fernández-Teruel, A., Giménez-Llort, L., Escorihuela, R. M., Gil, L., Aguilar, R., Steimer, T., and Tobeña, A. (2002). Early-life handling stimulation and environmental enrichment: are some of their effects mediated by similar neural mechanisms? Pharmacol. Biochem. Behav. 73, 233–245.
Fisher, P. A., Stoolmiller, M., Gunnar, M. R., and Burraston, B. O. (2007). Effects of a therapeutic intervention for foster preschoolers on diurnal cortisol activity. Psychoneuroendocrinology 32, 892–905.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., Heine-Suñer, D., Cigudosa, J. C., Urioste, M., Benitez, J., Boix-Chornet, M., Sanchez-Aguilera, A., Ling, C., Carlsson, E., Poulsen, P., Vaag, A., Stephan, Z., Spector, T. D., Wu, Y. Z., Plass, C., and Esteller, M. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609.
Francis, D., Diorio, J., Liu, D., and Meaney, M. J. (1999). Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science 286, 1155–1158.
Fries, A. B., and Pollak, S. D. (2004). Emotion understanding in postinstitutionalized Eastern European children. Dev. Psychopathol. 16, 355–369.
Fuke, C., Shimabukuro, M., Petronis, A., Sugimoto, J., Oda, T., Miura, K., Miyazaki, T., Ogura, C., Okazaki, Y., and Jinno, Y. (2004). Age related changes in 5-methylcytosine content in human peripheraleukocytes and placentas: an HPLC-based study. Ann. Hum. Genet. 68, 196–204.
Gluckman, P. D., Hanson, M. A., Cooper, C., and Thornburg, K. L. (2008). Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 359, 61–73.
Goekoop, J. G., de Winter, R. P., de Rijk, R., Zwinderman, K. H., Frankhuijzen-Sierevogel, A., and Wiegant, V. M. (2006). Depression with above-normal plasma vasopressin: validation by relations with family history of depression and mixed anxiety and retardation. Psychiatry Res. 141, 201–211.
Gonzales, M. L., and LaSalle, J. M. (2010). The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 12, 127–134.
Grafodatskaya, D., Chung, B., Szatmari, P., and Weksberg, R. (2010). Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry 49, 794–809.
Grayson, D. R., Jia, X., Chen, Y., Sharma, R. P., Mitchell, C. P., Guidotti, A., and Costa, E. (2005). Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 102, 9341–9346.
Greer, P. L., and Greenberg, M. E. (2008). From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860.
Hanauer, A., and Young, I. D. (2002). Coffin-Lowry syndrome: clinical and molecular features. J. Med. Genet. 39, 705–713.
Heim, C., Mletzko, T., Purselle, D., Musselman, D. L., and Nemeroff, C. B. (2008). The dexamethasone/corticotropin-releasing factor test in men with major depression: role of childhood trauma. Biol. Psychiatry 63, 398–405.
Heim, C., and Nemeroff, C. B. (2002). Neurobiology of early life stress: clinical studies. Semin. Clin. Neuropsychiatry 7, 147–159.
Hernandez, D. G., Nalls, M. A., Gibbs, J. R., Arepalli, S., van der Brug, M., Chong, S., Moore, M., Longo, D. L., Cookson, M. R., Traynor, B. J., and Singleton, A. B. (2011). Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 20, 1164–1172.
Heuser, I., Yassouridis, A., and Holsboer, F. (1994). The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J. Psychiatr. Res. 28, 341–356.
Hochberg, Z., Feil, R., Constancia, M., Fraga, M., Junien, C., Carel, J. C., Boileau, P., Le Bouc, Y., Deal, C. L., Lillycrop, K., Scharfmann, R., Sheppard, A., Skinner, M., Szyf, M., Waterland, R. A., Waxman, D. J., Whitelaw, E., Ong, K., and Albertsson-Wikland, K. (2010). Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 32, 159–224.
Holmes, A., le Guisquet, A. M., Vogel, E., Millstein, R. A., Leman, S., and Belzung, C. (2005). Early life genetic, epigenetic and environmental factors shaping emotionality in rodents. Neurosci. Biobehav. Rev. 29, 1335–1346.
Holsboer, F. (2000). The corticosteroid receptor hypothesis of depression. Neuropsychopharma 23, 477–501.
Hunter, R. G., McCarthy, K. J., Milne, T. A., Pfaff, D. W., and McEwen, B. S. (2009). Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc. Natl. Acad. Sci. U.S.A. 106, 20912–20917.
Huot, R. L., Gonzalez, M. E., Ladd, C. O., Thrivikraman, K. V., and Plotsky, P. M. (2004). Foster litters prevent hypothalamic-pituitary-adrenal axis sensitization mediated by neonatal maternal separation. Psychoneuroendocrinology 29, 279–289.
Imanaka, A., Morinobu, S., Toki, S., Yamamoto, S., Matsuki, A., Kozuru, T., and Yamawaki, S. (2008). Neonatal tactile stimulation reverses the effect of neonatal isolation on open-field and anxiety-like behavior, and pain sensitivity in male and female adult Sprague-Dawley rats. Behav. Brain Res. 186, 91–97.
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals? Nat. Genet. 33, 245–254.
Jensen, L. R., Amende, M., Gurok, U., Moser, B., Gimmel, V., Tzschach, A., Janecke, A. R., Tariverdian, G., Chelly, J., Fryns, J. P., Van Esch, H., Kleefstra, T., Hamel, B., Moraine, C., Gecz, J., Turner, G., Reinhardt, R., Kalscheuer, V. M., Ropers, H. H., and Lenzner, S. (2005). Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 76, 227–236.
Jintaridth, P., and Mutirangura, A. (2010). Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol. Genomics PMID: 20145203. 41, 194–200.
Jørgensen, H. F., and Bird, A. (2002). MeCP2 and other methyl-CpG binding proteins. Ment. Retard. Dev. Disabil. Res. Rev. 8, 87–93.
Kaminsky, Z. A., Tang, T., Wang, S. C., Ptak, C., Oh, G. H., Wong, A. H., Feldcamp, L. A., Virtanen, C., Halfvarson, J., Tysk, C., McRae, A. F., Visscher, P. M., Montgomery, G. W., Gottesman, I. I., Martin, N. G., and Petronis, A. (2009). DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 41, 240–245.
Kessler, M. S., Murgatroyd, C., Bunck, M., Czibere, L., Frank, E., Jacob, W., Horvath, C., Muigg, P., Holsboer, F., Singewald, N., Spengler, D., and Landgraf, R. (2007). Diabetes insipidus and, partially, low anxiety-related behaviour are linked to a SNP-associated vasopressin deficit in LAB mice. Eur. J. Neurosci. 26, 2857–2864.
Kleefstra, T., Brunner, H. G., Amiel, J., Oudakker, A. R., Nillesen, W. M., Magee, A., Geneviève, D., Cormier-Daire, V., van Esch, H., Fryns, J. P., Hamel, B. C., Sistermans, E. A., de Vries, B. B., and van Bokhoven, H. (2006). Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2, 370–377.
Kramer, J. M., and van Bokhoven, H. (2009). Genetic and epigenetic defects in mental retardation. Int. J. Biochem. Cell Biol. 41, 96–107.
Kuratomi, G., Iwamoto, K., Bundo, M., Kusumi, I., Kato, N., Iwata, N., Ozaki, N., and Kato, T. (2008). Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol. Psychiatry 13, 429–441.
Kwabi-Addo, B., Chung, W., Shen, L., Ittmann, M., Wheeler, T., Jelinek, J., and Issa, J. P. (2007). Age-related DNA methylation changes in normal human prostate tissues. Clin. Cancer Res. 13, 3796–3802.
Ladd, C. O., Huot, R. L., Thrivikraman, K. V., Nemeroff, C. B., and Plotsky, P. M. (2004). Long-term adaptations in glucocorticoid receptor and mineralocorticoid receptor mRNA and negative feedback on the hypothalamo-pituitary-adrenal axis following neonatal maternal separation. Biol. Psychiatry 55, 367–375.
Lee, R. S., Tamashiro, K. L., Yang, X., Purcell, R. H., Harvey, A., Willour, V. L., Huo, Y., Rongione, M., Wand, G. S., and Potash, J. B. (2010). Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 151, 4332–4343.
Liu, D., Diorio, J., Tannenbaum, B., Caldji, C., Francis, D., Freedman, A., Sharma, S., Pearson, D., Plotsky, P. M., and Meaney, M. J. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 277, 1659–1662.
Lopez, M. F., Doremus-Fitzwater, T. L., and Becker, H. C. (2010). Chronic social isolation and chronic variable stress during early development induce later elevated ethanol intake in adult C57BL/6J mice. Alcohol. doi: 10.1016/j.alcohol.2010.08.017. [Epub ahead of print].
Lubin, F. D., Roth, T. L., and Sweatt, J. D. (2008). Epigenetic regulation of bdnf gene transcription in the consolidation of fear memory. J. Neurosci. 28, 10576–10586.
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., Fan, G., and Sun, Y. E. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893.
Mastroeni, D., McKee, A., Grover, A., Rogers, J., and Coleman, P. D. (2009). Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 4, e6617. doi: 10.1371/journal.pone.0006617
Maze, I., Covington, H. E., III Dietz, D. M., LaPlant, Q., Renthal, W., Russo, S. J., Mechanic, M., Mouzon, E., Neve, R. L., Haggarty, S. J., Ren, Y., Sampath, S. C., Hurd, Y. L., Greengard, P., Tarakhovsky, A., Schaefer, A., and Nestler, E. J. (2010). Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216.
McCrory, E., De Brito, S. A., and Viding, E. (2010). Research review: the neurobiology and genetics of maltreatment and adversity. J. Child Psychol. Psychiatry 51, 1079–1095.
McGowan, P. O., Sasaki, A., D’Alessio, A. C., Dymov, S., Labonte, B., Szyf, M., Turecki, G., and Meaney, M. J. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348.
McGowan, P. O., Sasaki, A., Huang, T. C. T., Unterberger, A., Suderman, M., Ernst, C., Meaney, M. J., Turecki, G., and Szyf, M. (2008). Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS ONE 3, e2085. doi: 10.1371/journal.pone.0002085
Meaney, M. J., Diorio, J., Francis, D., Widdowson, J., LaPlante, P., Caldji, C., Sharma, S., Seckl, J. R., and Plotsky, P. M. (1996). Early environmental regulation of forebrain glucocorticoid receptor gene expression: implications for adrenocortical responses to stress. Dev. Neurosci. 18, 49–72.
Menger, Y., Bettscheider, M., Murgatroyd, C., and Spengler, D. (2010). Sex differences in brain epigenetics. Epigenomics 2, 807–821.
Meynen, G., Unmehopa, U. A., van Heerikhuize, J. J., Hofman, M. A., Swaab, D. F., and Hoogendijk, W. J. (2006). Increased arginine vasopressin mRNA expression in the human hypothalamus in depression: a preliminary report. Biol. Psychiatry 60, 892–895.
Mizoguchi, Y., Kajiume, T., Miyagawa, S., Okada, S., Nishi, Y., and Kobayashi, M. (2007). Steroid-dependent ACTH-produced thymic carcinoid: regulation of POMC gene expression by cortisol via methylation of its promoter region. Horm. Res. 67, 257–262.
Moles, A., Kieffer, B. L., and D’Amato, F. R. (2004). Deficit in attachment behavior in mice lacking the mu-opioid receptor gene. Science 304, 1983–1986.
Mueller, B. R., and Bale, T. L. (2008). Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28, 9055–9065.
Murgatroyd, C., Patchev, A. V., Wu, Y., Micale, V., Bockmühl, Y., Fischer, D., Holsboer, F., Wotjak, C. T., Almeida, O. F., and Spengler, D. (2009). Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 12, 1559–1566.
Murgatroyd, C., Wigger, A., Frank, E., Singewald, N., Bunck, M., Holsboer, F., Landgraf, R., and Spengler, D. (2004). Impaired repression at a vasopressin promoter polymorphism underlies overexpression of vasopressin in a rat model of trait anxiety. J. Neurosci. 24, 7762–7770.
Murgatroyd, C., Wu, Y., Bockmühl, Y., and Spengler, D. (2010a). Genes learn from stress: How infantile trauma programs us for depression? Epigenetics 5, 194–199.
Murgatroyd, C., Wu, Y., Bockmühl, Y., and Spengler, D. (2010b). The Janus face of DNA methylation in aging. Aging (Albany, NY) 2, 107–110.
Muschler, M. A., Hillemacher, T., Kraus, C., Kornhuber, J., Bleich, S., and Frieling, H. (2010). DNA methylation of the POMC gene promoter is associated with craving in alcohol dependence. J. Neural Transm. 117, 513–519.
Nemeroff, C. B., Owens, M. J., Bissette, G., Andorn, A. C., and Stanley, M. (1988). Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch. Gen. Psychiatry 45, 577–579.
O’Connor, T. G., and Rutter, M. (2000). Attachment disorder behavior following early severe deprivation: extension and longitudinal follow-up. English and Romanian adoptees study team. J. Am. Acad. Child Adolesc. Psychiatry 39, 703–712.
Perisic, T., Zimmermann, N., Kirmeier, T., Asmus, M., Tuorto, F., Uhr, M., Holsboer, F., Rein, T., and Zschocke, J. (2010). Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology 35, 792–805.
Petrij, F., Giles, R. H., Dauwerse, H. G., Saris, J. J., Hennekam, R. C., Masuno, M., Tommerup, N., van Ommen, G. J., Goodman, R. H., and Peters, D. J. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348–351.
Petronis, A., Gottesman, I. I., Kan, P., Kennedy, J. L., Basile, V. S., Paterson, A. D., and Popendikyte, V. (2003). Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophr. Bull. 29, 169–178.
Picketts, D. J., Higgs, D. R., Bachoo, S., Blake, D. J., Quarrell, O. W., and Gibbons, R. J. (1996). ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum. Mol. Genet. 5, 1899–1907.
Plotsky, P. M., and Meaney, M. J. (1993). Early, postnatal experience alters hypothalamic corticotropin-releasing factor (CRF) mRNA, median eminence CRF content and stress-induced release in adult rats. Brain Res. Mol. Brain Res. 18, 195–200.
Poulter, M. O., Du, L., Weaver, I. C., Palkovits, M., Faludi, G., Merali, Z., Szyf, M., and Anisman, H. (2008). GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol. Psychiatry 64, 645–652.
Qureshi, I. A., and Mehler, M. F. (2010). Genetic and epigenetic underpinnings of sex differences in the brain and in neurological and psychiatric disease susceptibility. Prog. Brain Res. 186, 77–95.
Raadsheer, F. C., Hoogendijk, W. J., Stam, F. C., Tilders, F. J., and Swaab, D. F. (1994). Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 60, 436–444.
Ramocki, M. B., and Zoghbi, H. Y. (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455, 912–918.
Razin, A., and Cedar, H. (1977). Distriubtion of 5-methylcytosine in chromatin. Proc. Natl. Acad. Sci. U.S.A. 74, 2725–2728.
Rodríguez-Rodero, S., Fernández-Morera, J. L., Fernandez, A. F., Menéndez-Torre, E., and Fraga, M. F. (2010). Epigenetic regulation of aging. Discov. Med. 10, 225–233.
Roth, T. L., Lubin, F. D., Funk, A. J., and Sweatt, J. D. (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 65, 760–769.
Roth, T. L., Zoladz, P. R., Sweatt, J. D., and Diamond, D. M. (2011). Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J. Psychiatr. Res. doi: 10.1016/j.psychires.2011.01.013. [Epub ahead of print].
Rupprecht, R., Lesch, K. P., Muller, U., Beck, G., Beckmann, H., and Schulte, H. M. (1989). Blunted adrenocorticotropin but normal beta-endorphin release after human corticotropin-releasing hormone administration in depression. J. Clin. Endocrinol. Metab. 69, 600–603.
Rutter, M. (1998). Developmental catch-up, and deficit, following adoption after severe global early privation English and Romanian adoptees (ERA) study team. J. Child Psychol. Psychiatry 39, 465–476.
Sanchez, M. M., McCormack, K., Grand, A. P., Fulks, R., Graff, A., and Maestripieri, D. (2010). Effects of sex and early maternal abuse on adrenocorticotropin hormone and cortisol responses to the corticotropin-releasing hormone challenge during the first 3 years of life in group-living rhesus monkeys. Dev. Psychopathol. 22, 45–53.
Schlotz, W., Jones, A., Phillips, D. I., Gale, C. R., Robinson, S. M., and Godfrey, K. M. (2010). Lower maternal folate status in early pregnancy is associated with childhood hyperactivity and peer problems in offspring. J. Child Psychol. Psychiatry 51, 594–602.
Seckl, J. R., and Meaney, M. J. (2004). Glucocorticoid programming. Ann. N.Y. Acad. Sci. 1032, 63–84.
Sedlak, A. J., and Broadhurst, D. D. (1996). Third National Incidence Study of Child Abuse and Neglect. Washington, DC: U.S. Department of Health and Human Services, National Center on Child Abuse and Neglect.
Smotherman, W. P., Bell, R. W., Starzec, J., Elias, J., and Zachman, T. A. (1974). Maternal responses to infant volalizations and olfactory cues in rats and mice. Behav. Biol. 15, 55–66.
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., Li, Y., Chen, C. H., Zhang, W., Jian, X., Wang, J., Zhang, L., Looney, T. J., Zhang, B., Godley, L. A., Hicks, L. M., Lahn, B. T., Jin, P., and He, C. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72.
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476.
Takasugi, M. (2011). Progressive age-dependent DNA methylation changes start before adulthood in mouse tissues. Mech. Ageing Dev. 132, 65–71.
Tarullo, A. R., and Gunnar, M. R. (2006). Child maltreatment and the developing HPA axis. Horm. Behav. 50, 632–639.
Thompson, R. F., Atzmon, G., Gheorghe, C., Liang, H. Q., Lowes, C., Greally, J. M., and Barzilai, N. (2010). Tissue-specific dysregulation of DNA methylation in aging. Aging Cell 9, 506–518.
Turek-Plewa, J., and Jagodziński, P. P. (2005). The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol. Biol. Lett. 10, 631–647.
Tyrka, A. R., Price, L. H., Gelernter, J., Schepker, C., Anderson, G. M., and Carpenter, L. L. (2009). Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity. Biol. Psychiatry 66, 681–685.
Uchida, S., Hara, K., Kobayashi, A., Otsuki, K., Yamagata, H., Hobara, T., Suzuki, T., Miyata, N., and Watanabe, Y. (2011). Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 69, 359–372.
van Oers, H. J., de Kloet, E. R., and Levine, S. (1999). Persistent effects of maternal deprivation on HPA regulation can be reversed by feeding and stroking, but not by dexamethasone. J. Neuroendocrinol. 11, 581–588.
Vazquez, D. M., Bailey, C., Dent, G. W., Okimoto, D. K., Steffek, A., López, J. F., and Levine, S. (2006). Brain corticotropin-releasing hormone (CRH) circuits in the developing rat: effect of maternal deprivation. Brain Res. 1121, 83–94.
Veenema, A. H., Blume, A., Niederle, D., Buwalda, B., and Neumann, I. D. (2006). Effects of early life stress on adult male aggression and hypothalamic vasopressin and serotonin. Eur. J. Neurosci. 24, 1711–1720.
Vogt, G., Huber, M., Thiemann, M., van den Boogaart, G., Schmitz, O. J., and Schubart, C. D. (2008). Production of different phenotypes from the same genotype in the same environment by developmental variation. J. Exp. Biol. 211, 510–523.
Wang, H.-X., and Gao, W.-J. (2009). Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology 34, 2028–2040.
Wang, S. C., Oelze, B., and Schumacher, A. (2008). Age-specific epigenetic drift in late-onset alzheimer’s disease. PLoS ONE 3, e2698. doi: 10.1371/journal.pone.0002698
Weaver, I. C., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., Dymov, S., Szyf, M., and Meaney, M. J. (2004). Epigenetic programming by maternal behaviour. Nat. Neurosci. 7, 847–854.
Weaver, I. C., Champagne, F. A., Brown, S. E., Dymov, S., Sharma, S., Meaney, M. J., and Szyf, M. (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J. Neurosci. 25, 11045–11054.
Weber, M., Hellmann, I., Stadler, M. B., Ramos, L., Pääbo, S., Rebhan, M., and Schübeler, D. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466.
Yuferov, V., Nielsen, D. A., Levran, O., Randesi, M., Hamon, S., Ho, A., Morgello, S., and Kreek, M. J. (2011). Tissue-specific DNA methylation of the human prodynorphin gene in post-mortem brain tissues and PBMCs. Pharmacogenet. Genomics. 21, 185–196.
Zhang, T. Y., Hellstrom, I. C., Bagot, R. C., Wen, X., Diorio, J., and Meaney, M. J. (2010). Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J. Neurosci. 30, 13130–13137.
Zhou, Z., Hong, E. J., Cohen, S., Zhao, W. N., Ho, H. Y., Schmidt, L., Chen, W. G., Lin, Y., Savner, E., Griffith, E. C., Hu, L., Steen, J. A., Weitz, C. J., and Greenberg, M. E. (2006). Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269.
Keywords: epigenetics, DNA methylation, chromatin, hypothalamo–pituitary–adrenal axis, brain, early life
Citation: Murgatroyd C and Spengler D (2011) Epigenetics of early child development. Front. Psychiatry 2:16. doi: 10.3389/fpsyt.2011.00016
Received: 22 February 2011;
Paper pending published: 12 March 2011;
Accepted: 04 April 2011;
Published online: 18 April 2011.
Edited by:
Josephine Johns, University of North Carolina at Chapel Hill, USAReviewed by:
Ronald De Kloet, Leiden University, NetherlandsBen Nephew, Tufts University Cummings School of Veterinary Medicine, USA
Copyright: © 2011 Murgatroyd and Spengler. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Chris Murgatroyd, Max Planck Institute of Psychiatry, Kraepelinstrasse 2-10, 80804 Munich, Germany. e-mail: murgatroyd@mpipsykl.mpg.de