 Thomas F. Tropea1,2 Zeeba D. Kabir1,3 Gagandeep Kaur4 Anjali M. Rajadhyaksha1,3 Barry E. Kosofsky1,3*
Thomas F. Tropea1,2 Zeeba D. Kabir1,3 Gagandeep Kaur4 Anjali M. Rajadhyaksha1,3 Barry E. Kosofsky1,3*- 1 Division of Pediatric Neurology, Department of Pediatrics, Weill Cornell Medical College, New York, NY, USA
- 2 College of Osteopathic Medicine, University of New England, Biddeford, ME, USA
- 3 Graduate Program in Neurosciences, Weill Cornell Medical College, New York, NY, USA
- 4 School of Environmental and Biological Sciences, Rutgers, The State University of New Jersey, New Brunswick, NJ, USA
Previous work from our group and others utilizing animal models have demonstrated long-lasting structural and functional alterations in the meso-cortico-striatal dopamine pathway following prenatal cocaine (PCOC) treatment. We have shown that PCOC treatment results in augmented D1-induced cyclic AMP (cAMP) and cocaine-induced immediate-early gene expression in the striatum of adult mice. In this study we further examined basal as well as cocaine or D1-induced activation of a set of molecules known to be mediators of neuronal plasticity following psychostimulant treatment, with emphasis in the dorsal striatum (Str) and nucleus accumbens (NAc) of adult mice exposed to cocaine in utero. Basally, in the Str of PCOC treated mice there were significantly higher levels of (1) CREB and Ser133 P-CREB (2) Thr34 P-DARPP-32 and (3) GluA1 and Ser 845 P-GluA1 when compared to prenatal saline (PSAL) treated mice. In the NAc there were significantly higher basal levels of (1) CREB and Ser133 P-CREB, (2) Thr202/Tyr204 P-ERK2, and (3) Ser845 P-GluA1. Following acute administration of cocaine (15 mg/kg, i.p.) or D1 agonist (SKF 82958; 1 mg/kg, i.p.) there were significantly higher levels of Ser133 P-CREB, Thr34 P-DARPP-32, and Thr202/Tyr204 P-ERK2 in the Str that were evident in all animals tested. However, these cocaine-induced increases in phosphorylation were significantly augmented in PCOC mice compared to PSAL mice. In sharp contrast to the observations in the Str, in the NAc, acute administration of cocaine or D1 agonist significantly increased P-CREB and P-ERK2 in PSAL mice, a response that was not evident in PCOC mice. Examination of Ser 845 P-GluA1 revealed that cocaine or D1 agonist significantly increased levels in PSAL mice, but significantly decreased levels in the PCOC mice in both the Str and NAc. We also examined changes in brain-derived neurotrophic factor (BDNF). Our studies revealed significantly higher levels of the BDNF precursor, pro-BDNF, and one of its receptors, TrkB in the Str of PCOC mice compared to PSAL mice. These results suggest a persistent up-regulation of molecules critical to D1 and BDNF signaling in the Str of adult mice exposed to cocaine in utero. These molecular adaptations may underlie components of the behavioral deficits evident in exposed animals and a subset of exposed humans, and may represent a therapeutic target for ameliorating aspects of the PCOC-induced phenotype.
Introduction
Over the past 25 years since crack cocaine became a drug commonly abused by pregnant women, multiple clinical, and preclinical studies have identified alterations in fetal brain development with lasting consequences on brain structure and function resulting from prenatal cocaine (PCOC) exposure (Kosofsky et al., 1994; reviewed in Trask and Kosofsky, 2000; Kosofsky and Hyman, 2001). Identification of a prenatal drug-induced phenotype uniquely attributable to intrauterine cocaine exposure has been elusive. Specifically, only a subset of exposed infants and children demonstrate persistent deficits, and when they do, may manifest ongoing behavioral abnormalities in subtle neurobehavioral domains including deficits in “Affect, Attention, Arousal, and Action” (the 4A’s: see Lester, 1998; Bada et al., 2007). Specifically, PCOC exposure has been shown to result in subtle reductions in IQ and cognitive development (Alessandri et al., 1998; Lester et al., 1998), delayed language development (Beeghly et al., 2006), and impairments in tasks requiring sustained attention (Accornero et al., 2007). Such studies support the idea that intrauterine exposure to cocaine most profoundly alters attention, arousal, and reactivity, functions that may negatively impact learning and memory in exposed offspring (Mayes et al., 1998). The implications for public policy are far reaching, as when such deficits are evident in PCOC-exposed individuals they may require longer perinatal hospitalizations and associated increments in healthcare costs (Behnke et al., 1997), as well as increased special education needs and associated expenses (Lester et al., 1998; Levine et al., 2008), making prevention of prenatal exposure to cocaine, and early identification and treatment of resulting adverse outcomes a high priority.
As the primary molecular targets of cocaine action are the uptake pumps for the monoamines dopamine, serotonin, and to a lesser extent norepinephrine (Uhl et al., 2002), neurochemical systems which mediate cocaine-induced behaviors, persistent alterations in aminergic function have been suggested as contributing to the PCOC-induced phenotype (Mayes, 2002). Animal models, including work performed in mice (Wilkins et al., 1998), rats (Spear et al., 2002), rabbits (Harvey, 2004), and non-human primates (Lidow and Song, 2001) have been particularly helpful in identifying the independent contribution of cocaine to such neurobehavioral deficits, as well as in understanding the basic mechanisms underlying such changes (Malanga, 1999). In particular, rodent models have demonstrated persistent alterations in dopaminergic (DA) signaling, primarily via the D1 receptor, in adult animals following PCOC treatment (Friedman and Wang, 1998; Unterwald et al., 2003; Stanwood and Levitt, 2007; Malanga et al., 2008; Tropea et al., 2008a).
The cascade of molecular events initiated in the striatum (Str) and nucleus accumbens (NAc) following acute exposure of adult animals to cocaine has been well characterized (reviewed in McGinty et al., 2008). Specifically, a wealth of experimental data identifies a rapid and robust activation of D1-like cell surface receptors activating intracellular signaling pathways to affect specific patterns of gene expression (Self et al., 1996), and alterations thereof in mice genetically engineered to be deficient in D1 mediated signal transduction in the Str (Drago et al., 1996). High throughput array-based methods have identified sets of genes activated in the Str and NAc following acute cocaine exposure that are distinguishable from those following repeated cocaine exposures (Renthal et al., 2009), emphasizing the persistent molecular adaptations, in part via recurrent D1-mediated neuronal stimulation, in contributing to the “addicted state” (Chao and Nestler, 2004).
One phenomenon that has been extensively investigated in animal models has been the process of sensitization, by which prior psychostimulant exposure augments the subsequent response to a challenge dose of drug (reviewed in Kalivas et al., 1998). Work from our lab and others has identified that signaling via second messenger molecules such as (P-)CREB, (P-)DARPP-32, (P-)ERK, and (P-)GluA1 in the Str and NAc are persistently altered following recurrent psychostimulant exposure, and may underlie aspects of the “sensitized state.” These data raise the possibility that following PCOC exposure, such signaling pathways may similarly demonstrate persistent dysregulation, and may render adult animals susceptible to altered behavioral responses to subsequent administration of drugs of abuse (reviewed in Crozatier et al., 2003; Malanga and Kosofsky, 2003).
Consistent with this thinking, we have focused our attention on the effect of PCOC treatment on persistent dysregulation of a set of target genes known to mediate aspects of synaptic plasticity, including growth factors (e.g., brain-derived neurotrophic factor, BDNF), immediate-early genes (e.g., zif-268), and synaptic scaffolding proteins (e.g., homer 1a). Previous work from our group analyzing the Str and NAc has focused on the role of dopamine D1-mediated cyclic AMP (cAMP) regulation, and demonstrated increased cocaine-mediated induction of both zif-268 and homer 1a mRNA in the Str, but not the NAc of adult PCOC treated vs. prenatal saline (PSAL) treated mice (Tropea et al., 2008a). Here we extend that work to identify that an additional set of signaling molecules activated via D1 stimulation including (P-)CREB, (P-)DARPP-32, (P-)ERK, and (P)GluA1 are differentially activated in the Str and NAc of adult PCOC vs. PSAL mice. We found that following acute administration of cocaine (15 mg/kg, i.p.) or D1 agonist (SKF 82958; 1 mg/kg, i.p.) there were significantly higher levels of Ser133 P-CREB, Thr34 P-DARPP-32, and Thr202/Tyr204 P-ERK2 evident in the Str in both prenatal treatment groups. However, this increase was significantly augmented in PCOC vs. PSAL mice. In sharp contrast, neither acute cocaine nor SKF 82958-induced phosphorylation of CREB or ERK2 in the NAc of PCOC mice, but did in the NAc of PSAL mice. Following acute administration of cocaine or D1 agonist there were significantly increased levels of Ser845 P-GluA1 in both the Str and NAc of PSAL mice, in contrast to significantly decreased levels of Ser845 P-GluA1 in both the Str and NAc of PCOC mice. In parallel we have additionally identified that the growth factor pro-BDNF, and TrkB, a BDNF receptor, are upregulated in the Str but not NAc of adult PCOC mice.
Taken together our data identifies region-specific patterns (i.e., Str vs. NAc) in the constitutive expression of a set of proteins and phospho-proteins, as well as their pattern of expression following acute administration of cocaine or the D1 agonist SKF 82958, which distinguish PCOC from PSAL mice. The differential pattern of constitutive as well as inducible proteins and phospho-proteins that we have identified suggest a persistent molecular memory in PCOC mice evidenced as a cocaine-induced augmentation in CREB and ERK phosphorylation in the Str, blunting of CREB and ERK phosphorylation in the NAc, and de-phosphorylation of GluA1 in both the Str and NAc, all via D1 mechanisms. Such data extends the idea that recurrent drug exposure induces abnormal synaptic learning and memory (Berke and Hyman, 2000; Hyman and Malenka, 2001; Hyman, 2005) in a developmental context such that adaptations in Str and NAc neuronal function established in the womb may “feed forward” to induce alterations in dopaminergic neurotransmission and associated behaviors in adulthood.
Materials and Methods
Prenatal Cocaine Treatment
Prenatal treatments were performed as previously described (Tropea et al., 2008b). Briefly, timed-pregnant Swiss Webster dams (Taconic Labs, New York) were assigned to one of two treatment groups and received twice-daily subcutaneous (SC) injections (at 7:00 AM and 7:00 PM) from embryonic (E) day E8 to E17, inclusive, of cocaine HCl (Sigma-Aldrich, St. Louis, MO, USA; 20 mg/kg/injection, SC, dissolved in saline) totaling 40 mg/kg per day (offspring referred to as PCOC for prenatal cocaine treated) or 0.9% saline (offspring referred to as PSAL for prenatal saline treated). All pups were surrogate fostered to control dams (Black Swiss Webster; Taconic Labs), which had delivered within the previous 48 h. Litters were culled to a maximum of 10 pups per dam. Animals were weaned at 28 days in to same sex cages, at which point female animals were euthanized. Only one male animal per litter was used for any of the studies reported, thereby avoiding the problem of litter effects resulting in “oversampling.” As a result, the individual animal’s data was the unit of statistical measure, and represented the “litter mean” for that data point. All experimental protocols were approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee, and were in accordance with NIH directives for animal studies.
Western Blot Analyses
Western blot analysis was performed as previously described (Tropea et al., 2008b). Briefly, adult (P60) male PSAL and PCOC treated mice were injected with saline, cocaine (15 mg/kg, i.p.), or the D1 agonist SKF 82958 (1 mg/kg, i.p.) followed 15 min later by rapid decapitation, brain dissection and freezing at −40°C in isopentane. All brains were serially cut rostro-caudally in a freezing cryostat to obtain bilateral punches of the dorsal striatum (Str; A/P +1.7 to +1.2; Paxinos and Franklin, 2003), the NAc (A/P stereotactic coordinates +1.7 to +1.2), bilateral 0.5 mm deep tissue punches of somato-sensory cortex (CTX; A/P +1.7 to +1.2 mm), medial prefrontal cortex (mPFC; A/P +1.98 to +1.54 mm), and unilateral ventral tegmental area (VTA; A/P −3.16 to −3.64 mm) punches. All tissue punches were obtained with a 17-gage stainless steel stylet.
For pro- and mature BDNF, TrkB, and p75 Western blot analyses, tissue from the NAc, Str, mPFC, and VTA, of untreated PSAL and PCOC animals was used. Tissue was sonicated in SDS sample buffer (1% SDS in TE pH 7.4) containing protease and phosphatase inhibitors and 25 μg of protein was separated on a 15% gel along with a Kaleidoscope-prestained standard (Bio-Rad, Hercules, CA, USA). For all other protein analyses, protein lysates were isolated on a 12% gel. Blots were incubated in primary antibody [CREB (1:850), Ser133 P-CREB (1:850), DARPP-32 (1:1000), Thr34 P- DARPP-32 (1:500), Thr75 P-DARPP-32 (1:500), Thr202/Tyr204 P-ERK1/2 (1:1000), ERK1/2 (1:1000), Ser 845 P-GluA1 (1:850), GluA1 (1:1000), Cell Signaling, Danvers, MA, USA; BDNF N-20 (1:200), Santa Cruz Biotechnology, Santa Cruz, CA, USA; TrkB (1:500), Upstate Cell Signaling Solutions, Lake Placid, NY, USA; p75 (1:1000), NR2B (1:1000), actin (1:20,000), Chemicon, Temecula, CA, USA] for 12–48 h at 4°C. Secondary antibody incubations were performed at room temperature in blocking buffer for 1 h (horseradish peroxidase-linked IgG conjugated goat anti-rabbit 1:5000 for CREB, P-CREB, DARPP-32, Thr34 P-DARPP-32, BDNF, TrkB, and NR2B and 1:10,000 for p75, or horse anti-mouse 1:30,000 for actin, Vector Laboratories, Burlingame, CA, USA). Membranes were visualized with Western Lightning Chemiluminescence solution (Perkin Elmer Life Science, Boston, MA, USA). Optical density from films was analyzed using NIH Image (NIH, Bethesda, MD, USA). For BDNF, pro-BDNF bands were analyzed at 30 kDa, while mature BDNF bands were analyzed at 14 kDa. To confirm the identity of these bands, striatal cell lysate and recombinant BDNF protein (generously supplied by Dr. Francis Lee, Weill Cornell Medical College, New York, NY, USA) was analyzed as shown in Figure 3A.
Statistical Analyses
Gestational data were analyzed using t-test, while western blot data were analyzed by one-way ANOVA, and when significant at p < 0.05 level, post hoc comparisons (Bonferroni–Dunn) between treatment groups was performed.
Results
Gestational Data
The average percentage weight gain of dams from E8 to E17 and the number of live pups per litter for each prenatal treatment group were recorded. PCOC dams gained less weight during pregnancy (p < 0.0001), and gave birth to less live pups per litter (p < 0.001) as compared to PSAL dams (Table 1).

Table 1. Effect of prenatal cocaine treatment on dam weight gain and offspring number.
Altered Protein Phosphorylation Levels in the Str of PCOC Mice
To examine protein phosphorylation levels, adult PSAL and PCOC mice were administered saline (PSAL sal and PCOC sal), cocaine (PSAL coc and PCOC coc) or the dopamine agonist, SKF 82958 (SKF; PSAL skf and PCOC skf). Fifteen minutes later mice were rapidly decapitated and tissue was isolated in a cryostat for Western blot analysis of the Str (Figure 1) and NAc (Figure 2). To evaluate the effect of PCOC exposure on basal differences in protein levels of CREB/Ser133 P-CREB, DARPP-32/Thr34 and Thr75 P-DARPP-32, ERK2/Thr202/Tyr204 P-ERK, and GluA1/Ser 845 P-GluA1 we compared PCOC sal vs. PSAL sal mice. The effect of PCOC exposure on cocaine and dopamine D1 signaling was evaluated by comparing cocaine- and D1 agonist, SKF 82958-induced changes in phospho-protein levels (P-CREB, Thr34- and Thr75-DARPP-32, P-ERK, and P-GluA1) in PCOC coc vs. PSAL coc, and PCOC skf vs. PSAL skf, respectively.
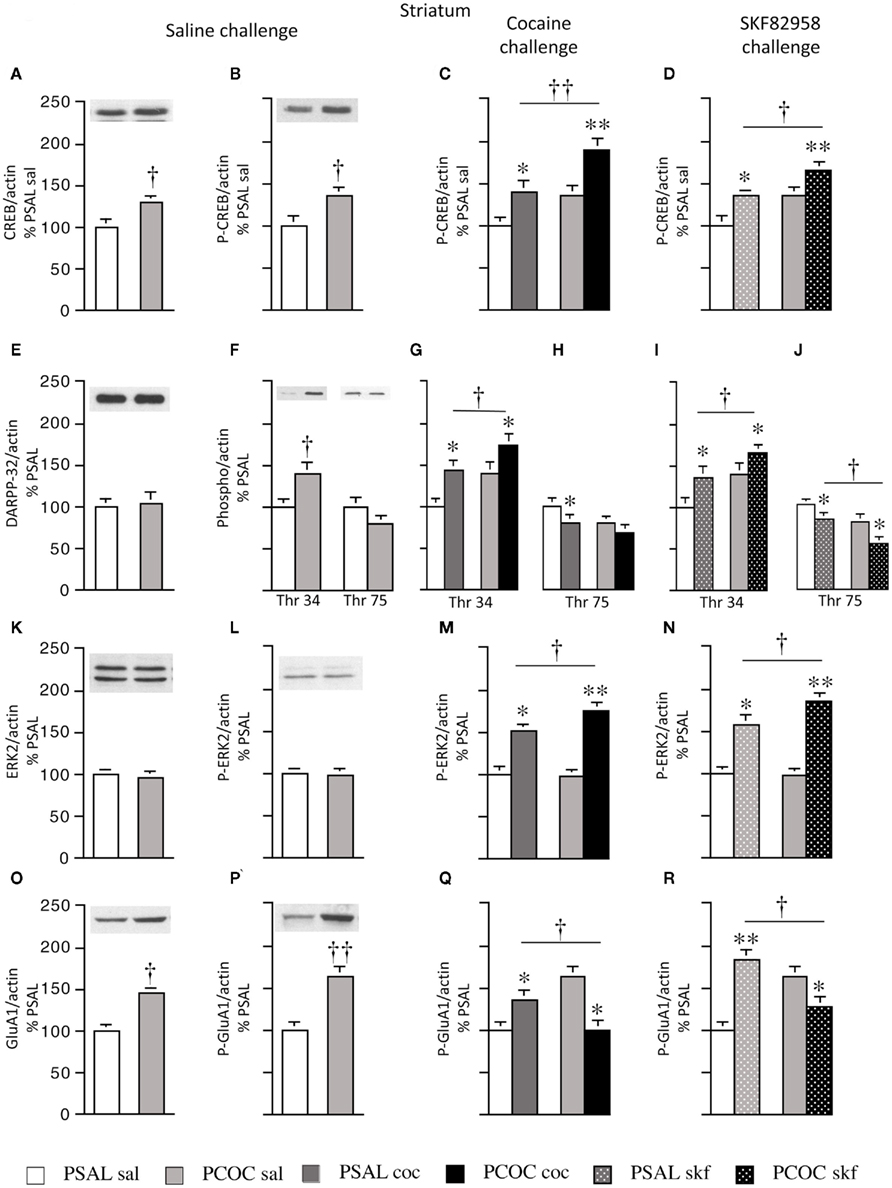
Figure 1. Effect of prenatal cocaine treatment on basal, cocaine- and SKF 82958-induced protein phosphorylation in the striatum of adult mice. Representative immunoblots and quantitative analysis of protein [(A) CREB; (E) DARPP-32; (K) ERK2; (O) GluA1] and phospho-protein [(B–D) P-CREB; (F–J) Thr 34 and Thr 75 P-DARPP-32; (L–N) P-ERK2; (P–R) P-GluA1] levels normalized to actin (mean optical density ± SEM). Protein levels were measured in prenatal saline treated (PSAL) and prenatal cocaine (PCOC) treated mice administered normal saline (sal), cocaine (15 mg/kg; coc) or D1 agonist, SKF 82958 (1 mg/kg; skf) as adults. Data are represented as percentage of PSAL mice treated with saline (PSAL sal). †p < 0.05, ††p < 0.01 PCOC pretreatment groups vs. PSAL pretreatment groups. *p < 0.05, **p < 0.01, coc or skf treated mice vs. sal treated mice within the same prenatal treatment. Error bars represent ± SEM. N = 6–8 mice/group.
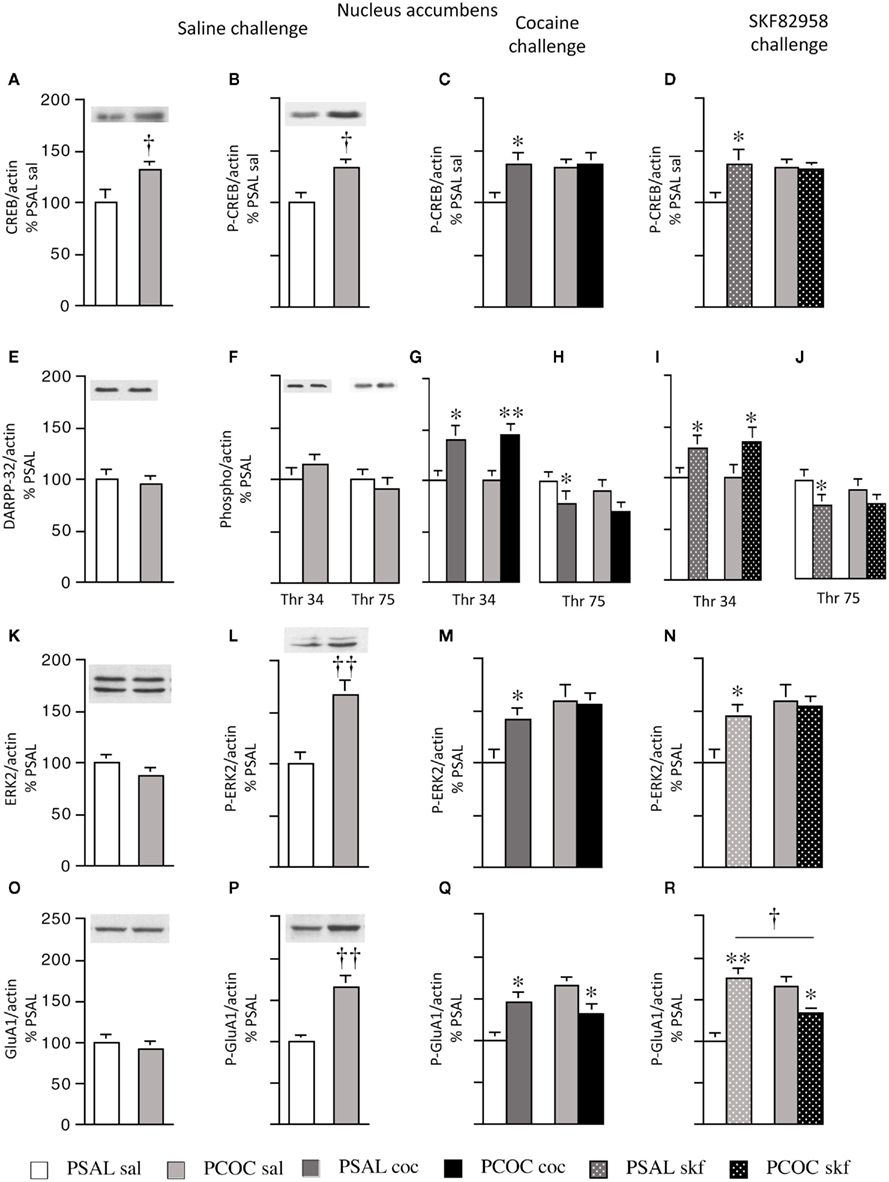
Figure 2. Effect of prenatal cocaine treatment on basal, cocaine- and SKF 82958-induced protein phosphorylation in the nucleus accumbens of adult mice. Representative immunoblots and quantitative analysis of protein [(A) CREB; (E) DARPP-32; (K) ERK2; (O) GluA1] and phospho-protein [(B–D) P-CREB; (F–J) Thr 34 and Thr 75 P-DARPP-32; (L–N) P-ERK2; (P–R) P-GluA1] levels normalized to actin (mean optical density ± SEM). Protein levels were measured in prenatal saline (PSAL) treated and prenatal cocaine (PCOC) treated mice administered normal saline (sal), cocaine (15 mg/kg; coc) or D1 agonist, SKF 82958 (1 mg/kg; skf) as adults. Data are represented as percentage of PSAL mice treated with saline (PSAL sal). †p < 0.05, ††p < 0.01 PCOC pretreatment groups vs. PSAL pretreatment groups. *p < 0.05, **p < 0.01, coc or skf treated mice vs. sal treated mice within the same prenatal treatment. Error bars represent ± SEM. N = 6–8 mice/group.
Striatum
Examination of basal levels of total and phospho-proteins revealed significantly higher levels of CREB and P-CREB in PCOC vs. PSAL mice (Figures 1A,B, respectively). Examination of DARPP-32 revealed no effect of PCOC on basal DARPP-32 levels (Figure 1E). However, there were significantly higher basal levels of Thr34 P-DARPP-32 in PCOC mice compared to PSAL mice (Figure 1F) with a trend toward lower levels of Thr75 P-DARPP-32 (Figure 1G). Examination of ERK2 revealed no change in the basal levels of ERK2 or P-ERK2 in PCOC mice (Figures 1K,L, respectively). Examination of GluA1 revealed significantly higher levels of basal GluA1 and P-GluA1 in PCOC mice compared to PSAL mice (Figures 1O,P, respectively).
Examination of cocaine or SKF 82958-induced changes in phospho-protein levels revealed that cocaine or SKF 82958 significantly increased P-CREB in PSAL and PCOC mice compared to saline treated mice (Figure 1C, PSAL coc vs. PSAL sal and PCOC coc vs. PCOC sal and Figure 1D, PSAL skf vs. PSAL sal and PCOC skf vs. PCOC sal, respectively). Furthermore, the increase in P-CREB observed in PCOC mice was significantly augmented compared to PSAL mice (Figure 1C, PCOC coc vs. PSAL coc; Figure 1D, PCOC skf vs. PSAL skf). Similarly cocaine or SKF 82958 treatment significantly increased Thr34 P-DARPP-32 levels in PSAL and PCOC mice (Figures 1G,I, respectively) with significantly augmented levels evident in PCOC mice compared to that observed in PSAL mice (Figure 1G, PCOC coc vs. PSAL coc; Figure 1I, PCOC skf vs. PSAL skf). Cocaine or SKF 82958 treatment significantly decreased Thr75 P-DARPP-32 levels in PSAL mice (Figures 1H,J, respectively). In PCOC mice, cocaine treatment had no effect on Thr75 P-DARPP-32 levels (Figure 1H) whereas SKF 82958 significantly decreased Thr75 P-DARPP-32 levels (Figure 1J) to levels that were significantly lower than that seen in PSAL mice (Figure 1J, PCOC skf vs. PSAL skf). Examination of P-ERK2 levels revealed that cocaine or SKF 82958 treatment significantly increased P-ERK2 levels in both PSAL and PCOC mice (Figures 1M,N, respectively) with significantly augmented levels evident in PCOC mice compared to PSAL mice (Figure 1M, PCOC coc vs. PSAL coc; Figure 1N, PCOC skf vs. PSAL skf). Examination of P-GluA1 levels revealed that cocaine or SKF 82958 treatment significantly increased P-GluA1 levels in PSAL mice (Figures 1Q,R, respectively). However, interestingly in PCOC mice, cocaine or SKF 82958 treatment significantly decreased P-GluA1 levels (Figures 1Q,R, respectively), and these levels were significantly lower than that observed in PSAL mice (Figure 1Q, PCOC coc vs. PSAL coc; Figure 1R, PCOC skf vs. PSAL skf).
Nucleus accumbens
Examination of basal levels of total and phospho-proteins revealed significantly higher levels of CREB and P-CREB in the NAc of PCOC mice compared to PSAL mice (Figures 2A,B, respectively). Examination of basal DARPP-32 levels revealed no difference in DARPP-32, Thr34 P-DARPP-32 or Thr75 P-DARPP-32 between PSAL and PCOC mice (Figures 2E,F, respectively). Examination of ERK2 revealed no change in basal ERK2 in PCOC mice compared to PSAL mice (Figure 2K), but significantly higher P-ERK2 levels in PCOC mice compared to PSAL mice (Figure 2L). Similarly, examination of GluA1 levels revealed no difference in basal GluA1 levels (Figure 2O) between prenatal treatment groups. However, Ser 845 P-GluA1 levels were significantly higher in the NAc of PCOC mice compared to PSAL mice (Figure 2P).
Examination of cocaine or SKF 82958-induced changes in phospho-protein levels revealed that cocaine or SKF 82958 treatment significantly increased P-CREB levels in PSAL mice compared to saline treated mice (Figure 2C, PSAL coc vs. PSAL sal and Figure 2D, PSAL SKF vs. PSAL sal), a response that was not evident in PCOC mice (Figures 2C,D, respectively). Cocaine or SKF 82958 treatment significantly increased Thr34 P-DARPP-32 levels in PSAL and PCOC mice with no difference in levels between the two prenatal treatment groups (Figures 2G,I, respectively). Cocaine or SKF 82958 administration significantly decreased Thr75 P-DARPP-32 levels in PSAL mice, with a trend toward lower levels in PCOC mice evident (Figures 2H,J, respectively). Examination of P-ERK2 levels revealed that cocaine or SKF 82958 treatment significantly increased P-ERK2 levels in PSAL mice compared to saline treated mice (Figures 2M, PSAL coc vs. PSAL sal and Figures 2N, PSAL SKF vs. PSAL sal), a response that was not evident in PCOC mice (Figures 2M,N, respectively. Examination of P-GluA1 revealed that cocaine or SKF 82958 administration increased P-GluA1 in PSAL mice (Figures 2Q,R, respectively) while either cocaine or SKF 82958 treatment significantly decreased P-GluA1 in PCOC mice (Figures 2Q,R, respectively). SKF 82958-induced P-GluA1 levels were significantly lower in PCOC mice compared to PSAL mice (Figure 2R, PCOC skf vs. PSAL skf).
Altered BDNF in the Str of PCOC Mice
We next examined levels of pro- and mature BDNF in the Str and NAc of PCOC vs. PSAL mice, along with levels in the mPFC and VTA, anatomical regions where BDNF is synthesized and transported to those targets (Conner et al., 1997; Altar and DiStefano, 1998). To identify the precise protein bands that correspond to pro- vs. mature BDNF we first compared BDNF Western blots containing striatal protein lysates with recombinant BDNF protein lysates (Figure 3A). A pro-BDNF protein band at 30 kDa and mature BDNF protein band at 14 kDa was used to compare levels of the two proteins in protein lysates obtained from mPFC, Str, NAc, and VTA tissue of PCOC vs. PSAL mice. Western blots revealed significantly higher levels of pro-BDNF in the Str of PCOC mice compared to PSAL mice (Figure 3B). No significant differences in pro-BDNF levels were observed for any other regions sampled. Examination of mature BDNF levels revealed no significant differences between PCOC and PSAL mice in any of the regions sampled (Figure 3C).
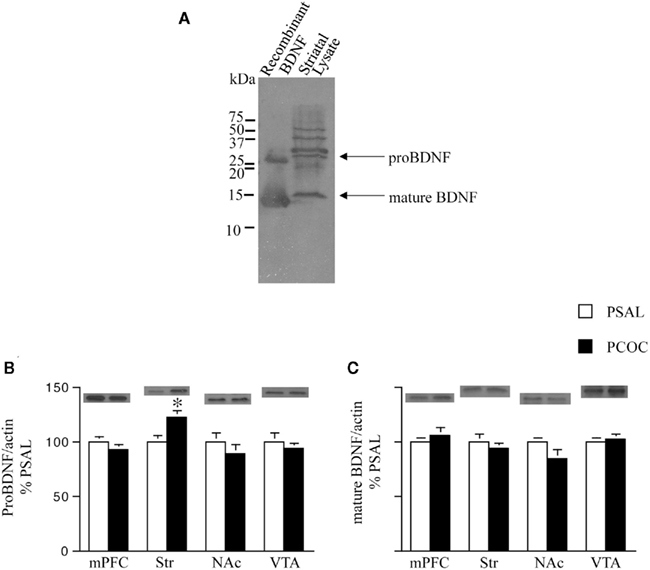
Figure 3. Pro-BDNF protein levels are higher in the striatum of prenatal cocaine treated mice. (A) Immunoblot showing pro- and mature brain-derived neurotrophic factor (BDNF) from recombinant protein and striatal protein lysates. Pro-BDNF was detected at 30 kDa and mature BDNF at 14 kDa; (B,C) Pro- and mature BDNF levels were measured in the medial prefrontal cortex (mPFC), dorsal striatum (Str), nucleus accumbens (NAc), and ventral tegmental area (VTA) of adult mice prenatally treated with saline (PSAL) or cocaine (PCOC). Protein levels were normalized to actin. Striatum of PCOC mice contained significantly higher amounts of pro-BDNF (B) compared to PSAL mice (*p < 0.05) with no difference in levels of mature BDNF (C). All other regions showed no difference in the levels of pro- or mature BDNF. Error bars represent the mean ± SEM. N = 4–6 mice/group.
We next examined the effect of prenatal treatment on the levels of the BDNF receptors TrkB, p75, and NR2B in the Str and NAc of PCOC vs. PSAL mice (Figure 4). TrkB levels were significantly higher in the striatum of PCOC compared to PSAL mice (Figure 4A), while in the NAc there was no significant difference in TrkB levels between the two prenatal treatment groups. No significant differences were observed in p75 protein levels in PCOC vs. PSAL mice in the Str or NAc (Figure 4B). Similarly, examination of NR2B, a gene regulated by the pro-BDNF pathway (Woo et al., 2005) revealed no significant differences between PCOC and PSAL mice in either brain region (Figure 4C).
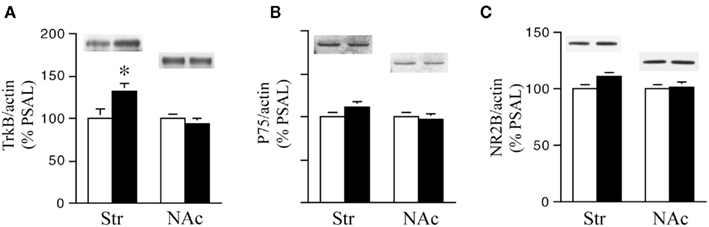
Figure 4. TrkB receptor protein levels are higher in the striatum of prenatal cocaine (PCOC) treated mice. Total protein levels of (A) tyrosine kinase B (TrkB), (B) p75 receptors and (C) NR2B receptor subunits were analyzed in the dorsal striatum (Str) and nucleus accumbens (NAc) of prenatal saline (PSAL) treated vs. PCOC treated adult mice. (A) TrkB receptor levels were significantly higher in the striatum of PCOC mice compared to PSAL mice (*p < 0.05), while no difference was observed in the NAc. No differences in the levels of p75 (B) or NR2B (C) were observed in the Str or NAc of PCOC mice compared to PSAL mice. Error bars represent ± SEM. N = 5–6 mice/group.
Discussion
DA Signaling in the Str and NAc of PCOC Mice
We, like others, find that acute cocaine administration increases protein phosphorylation of CREB, DARPP-32, ERK2, and GluA1 in the Str and NAc of adult mice via a D1 mechanism (Fienberg et al., 1998; Zhang et al., 2002a,b; Gerfen et al., 2008; Guan et al., 2009). Specifically, following acute administration of cocaine (15 mg/kg, i.p.) or D1 agonist (SKF 82958; 1 mg/kg, i.p.) there were significantly higher levels of Ser133 P-CREB, Thr34 P-DARPP-32, Thr202/Tyr 204 P-ERK2, and Ser845 P-GluA1, as well as lower levels of Thr75 P-DARPP-32 evident in the Str and NAc of PSAL mice. Interestingly, in the Str of PCOC mice, administration of cocaine or D1 agonist further augmented the phosphorylation of CREB, DARPP-32 at Thr34, and ERK, but led to a de-phosphorylation of DARPP-32 at Thr75 and of GluA1. The augmented activation of this signaling cascade is a likely mechanism for the increased expression of both zif-268 and homer 1a mRNA observed in the striatum of PCOC mice following acute cocaine administration (see Figure 5), which may also be attributable to a persistent enhancement in the coupling of D1 with cAMP (Tropea et al., 2008a). These data are discrepant with those reported in a rabbit model of PCOC exposure, in which there is demonstration of attenuated D1 activation via uncoupling of GalphaS subunits from D1 receptors, resulting in enhanced internalization of D1 subunits (Wang et al., 1995; Jones et al., 2000; Stanwood and Levitt, 2007). While such data from rabbits suggests attenuated dopaminergic activation following PCOC exposure, this same rabbit model has additionally provided evidence of enhanced DARPP-32 phosphorylation at Thr34 (Zhen et al., 2001), data concordant with our current findings in mice. Results from different models of PCOC exposure may differ as a result of species (e.g., mice vs. rabbits), route (SC vs. IV), dose and gestational timing of cocaine exposure, or brain regions studied (e.g., Str/NAc vs. Cingulate Cortex). Further studies should be directed at elucidating the cause of such differences, and the extent to which they adequately model aspects of the clinical problem.
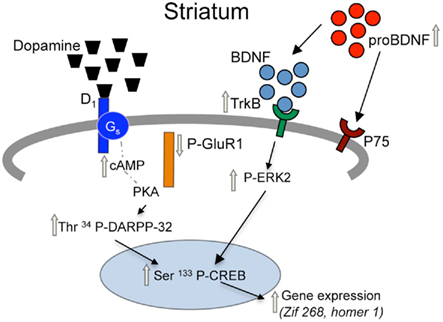
Figure 5. Proposed model for adaptations in D1 receptor and BDNF signaling pathways in the striatum of prenatal cocaine treated mice. Prenatal cocaine treatment results in enhanced signaling via dopamine D1 and TrkB receptors in the striatum of adult mice via persistent adaptations in a coordinately regulated set of pre-synaptic, synaptic, and post-synaptic molecules. Gray arrows represent long-term adaptations seen in adult mice following prenatal cocaine treatment.
We also found significant differences in PCOC mice when contrasting the phosphorylation of both CREB and ERK in the Str vs. NAc following administration of cocaine or D1 agonist; there was enhanced phosphorylation of CREB and ERK evident in the Str of PCOC mice, in contrast to blunted phosphorylation of CREB and ERK in the NAc of PCOC mice. It is possible that the constitutive increase in P-ERK identified in the NAc of PCOC mice, which was not evident in the Str, prevented the subsequent phosphorylation of ERK (and perhaps CREB) in the NAc. The blunted phosphorylation of at least one of these proteins may be related to our previous observation that acute cocaine administration did not increase either zif-268 or homer 1a mRNA expression in the NAc of PCOC mice (Tropea et al., 2008a).
P-GluA1 Signaling in the Str and NAc
In both the Str and NAc of PCOC mice, where increased constitutive expression of P-GluA1 was evident, administration of cocaine or SKF 82958 resulted in decreased GluA1 phosphorylation. This is in sharp contrast to PSAL mice, in which administration of cocaine or SKF 82958 resulted in increased expression of P-GluA1. Again, it is possible that the constitutive increase in P-GluA1 identified in both the Str and NAc of PCOC mice, prevented subsequent phosphorylation of GluA1 in both regions. Recent work has suggested that increased P-GluA1 sequesters this receptor in the cytoplasm, thereby preventing insertion of a functional receptor into the membrane, a phenomena that has been correlated with the sensitized state (for review see Mazzucchelli et al., 2002; Wolf and Ferrario, 2010). The mechanism that contributes to the constitutive increase in P-ERK evident in the Str of PCOC mice is presumably different than the mechanism that contributes to the constitutive increase in P-GluA1 evident in both the Str and NAc of PCOC mice, but both may be mediated by epigenetic mechanisms.
BDNF and TrkB Signaling in the Str vs. NAc of PCOC Mice
We see increases in the constitutive expression of pro-BDNF and TrkB in the Str, but not in the NAc of PCOC mice. However, we do not see changes in the expression of mature BDNF, p75, or NR2B receptor subunits, identifying a regional as well as molecular specificity in the BDNF signaling pathway that is persistently altered in PCOC mice. Work from others (Yang et al., 2009) suggests that pro-BDNF preferentially binds the p75 receptor, whereas mature BDNF preferentially binds the TrkB receptor. We are therefore pursuing additional experiments to identify the functional relevance of the increased constitutive expression of pro-BDNF and TrkB in the adult Str, which may be a result of enhanced cortico-striatal projections, which are the predominant source of striatal BDNF (Conner et al., 1997; Altar and DiStefano, 1998). Interestingly, recent data obtained from ex vivo cultures of embryonic mouse brains suggests that the tangential migration of GABAergic neurons from their site of origin in the ganglionic eminence to their cortical destination is delayed in the forebrain of mice prenatally exposed to cocaine, and that supplementation of those cultures with exogenous BDNF normalized this migration (McCarthy et al., 2011). Furthermore, cocaine has distinct acute and long-term effects on BDNF transcription and expression in striatum and frontal cortex (Liu et al., 2006), which is further complicated by post-transcriptional alterations in the isoforms of BDNF expressed (Jiang et al., 2009). Taken together the data suggests that perturbations in the level of BDNF at specific developmental periods can have immediate as well as long-lasting implications for neuronal migration and maturation, with impact on brain function that can persist into adulthood.
Implications of Our Molecular Findings on Brain Function
What is unknown is whether the differential adaptations in dopaminergic signaling that persist in the Str and NAc of PCOC mice evident following acute administration of cocaine we have reported will enhance their liability for addiction following recurrent cocaine exposure as adults. Previous experiments from our group contrasting PCOC and PSAL mice have identified alterations in cocaine-induced brain stimulation reward (Malanga et al., 2008), self-administration (Rocha et al., 2002), conditioned place preference (Malanga et al., 2007), and locomotor sensitization (Crozatier et al., 2003), as well as dopamine release in the Str and NAc during that same locomotor sensitization regimen (Malanga et al., 2009). However, in each study while the PCOC mice could be distinguished from the PSAL mice, the phenotype did not dramatically demonstrate an enhanced liability toward addiction. Such complexity could be attributable to the differential adaptations in PCOC vs. PSAL mice that we report here in the Str vs. NAc. This may preclude the progression of habit learning associated with recurrent drug exposure which is thought to require the expanded recruitment of successively more dorsal striatal circuits following the initial activation of the NAc (Everitt and Robbins, 2005; Belin and Everitt, 2008; Haber, 2008). In addition, the liability for addiction in humans is critically dependent on genetic as well as environmental factors, which may be significantly enhanced in offspring prenatally exposed to cocaine, and may be powerfully interactive with adaptations in Str and NAc neuronal function as we have described in our mouse model. As the generation of young adults prenatally exposed to cocaine initiate their own experiences with drug experimentation, they may be at greater risk for the fifth “A” – addiction.
Human imaging studies can help to identify the structural and functional correlates of the behavioral and molecular aberrations seen in animal models of PCOC exposure (reviewed in Roussotte et al., 2010). Whole brain MRI has provided evidence for reductions in parietal and occipital cortical gray matter volumes and a cocaine dose-dependant reduction in white matter of the corpus callosum in humans exposed to cocaine in utero (Dow-Edwards et al., 2006; Rivkin et al., 2008). Callosal volume loss was corroborated in a rodent model as well (Ma et al., 2009). Attenuated white matter integrity on DTI imaging of the left frontal callosal and right frontal projection fibers suggests suboptimal white matter development in those areas (Warner et al., 2006). Similarly, studies in opiate-exposed offspring show that white matter integrity seems to be most susceptible to damage in areas undergoing earlier CNS development (Walhovd et al., 2010). Analyses of subcortical structures have revealed a persistent decrease in caudate volume following prenatal cocaine exposure (Avants et al., 2007). Functional studies using fMRI provide evidence of a 10% reduction in cerebral blood flow most prominent in posterior and inferior brain regions of adolescents (Rao et al., 2007). Sheinkopf et al. (2009) have shown that performance in a go-no go task adolescents who were previously exposed to cocaine in utero showed a greater activation of right inferior frontal and striatal regions compared to controls who activated fusiform gyrus and occipital cortex more prominently, suggesting differences in cognition and attention in the PCOC-exposed group. Correlations between reduced frontal white matter and visuo-spatial and executive functioning tests (Warner et al., 2006), right parietal volume loss with visual attention, sensori-motor tasks, and syntax construction, and left occipital volume loss with poor performance in visual attention, recognition, and visuomotor tasks (Dow-Edwards et al., 2006) suggest PCOC affects visual, sensori-motor, and executive functions.
A deeper appreciation of the relevance of the persistent molecular adaptations evident in animal models, including that which we report here, to the results obtained in structural and functional imaging studies performed in humans, will require a better understanding of the mechanisms by which such molecular changes are interactive with genetic factors including common polymorphisms for genes such as BDNF, which independent of PCOC exposure may confer enhanced vulnerability vs. resilience to addiction. Such gene X (fetal) environment interactions may contribute to aspects of the PCOC phenotype demonstrated in humans by others, including some of those reported in this monograph. Conceptualized this way, intrauterine cocaine exposure can be thought of as a pharmacologic means of inducing a state of “fetal reprogramming” (Barker, 1995) by which molecular pathways underlying ongoing brain development are permanently altered, thereby enhancing an individual’s vulnerability to subsequent disease, in this case addiction. Like with other diseases, early detection of such enhanced vulnerabilities will provide a rational starting point for behavioral and perhaps pharmacologic interventions to prevent expression of disease, which in the case of prenatal drug exposure may help prevent the problem from begetting itself.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Francis Lee for his generous donation of recombinant BDNF protein.
References
Accornero, V. H., Amado, A. J., Morrow, C. E., Xue, L., Anthony, J. C., and Bandstra, E. S. (2007). Impact of prenatal cocaine exposure on attention and response inhibition as assessed by continuous performance tests. J. Dev. Behav. Pediatr. 28, 195–205.
Alessandri, S. M., Bendersky, M., and Lewis, M. (1998). Cognitive functioning in 8- to 18-month-old drug-exposed infants. Dev. Psychol. 34, 565–573.
Altar, C. A., and DiStefano, P. S. (1998). Neurotrophin trafficking by anterograde transport. Trends Neurosci. 21, 433–437.
Avants, B. B., Hurt, H., Giannetta, J. M., Epstein, C. L., Shera, D. M., Rao, H., Wang, J., and Gee, J. C. (2007). Effects of heavy in utero cocaine exposure on adolescent caudate morphology. Pediatr. Neurol. 37, 275–279.
Bada, H. S., Das, A., Bauer, C. R., Shankaran, S., Lester, B., LaGasse, L., Hammond, J., Wright, L. L., and Higgins, R. (2007). Impact of prenatal cocaine exposure on child behavior problems through school age. Pediatrics 119, e348–e359.
Beeghly, M., Martin, B., Rose-Jacobs, R., Cabral, H., Heeren, T., Augustyn, M., Bellinger, D., and Frank, D. A. (2006). Prenatal cocaine exposure and children’s language functioning at 6 and 9.5 years: moderating effects of child age, birthweight, and gender. J. Pediatr. Psychol. 31, 98–115.
Behnke, M., Eyler, F. D., Conlon, M., Casanova, O. Q., and Woods, N. S. (1997). How fetal cocaine exposure increases neonatal hospital costs. Pediatrics 99, 204–208.
Belin, D., and Everitt, B. J. (2008). Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron 57, 432–441.
Berke, J. D., and Hyman, S. E. (2000). Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25, 515–532.
Chao, J., and Nestler, E. J. (2004). Molecular neurobiology of drug addiction. Annu. Rev. Med. 55, 113–132.
Conner, J. M., Lauterborn, J. C., Yan, Q., Gall, C. M., and Varon, S. (1997). Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J. Neurosci. 17, 2295–2313.
Crozatier, C., Guerriero, R. M., Mathieu, F., Giros, B., Nosten-Bertrand, M., and Kosofsky, B. E. (2003). Altered cocaine-induced behavioral sensitization in adult mice exposed to cocaine in utero. Brain Res. Dev. Brain Res. 147, 97–105.
Dow-Edwards, D. L., Benveniste, H., Behnke, M., Bandstra, E. S., Singer, L. T., Hurd, Y. L., and Stanford, L. R. (2006). Neuroimaging of prenatal drug exposure. Neurotoxicol. Teratol. 28, 386–402.
Drago, J., Gerfen, C. R., Westphal, H., and Steiner, H. (1996). D1 dopamine receptor-deficient mouse: cocaine-induced regulation of immediate-early gene and substance P expression in the striatum. Neuroscience 74, 813–823.
Everitt, B. J., and Robbins, T. W. (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 8, 1481–149.
Fienberg, A. A., Hiroi, N., Mermelstein, P. G., Song, W., Snyder, G. L., Nishi, A., Cheramy, A., O’Callaghan, J. P., Miller, D. B., Cole, D. G., Corbett, R., Haile, C. N., Cooper, D. C., Onn, S. P., Grace, A. A., Ouimet, C. C., White, F. J., Hyman, S. E., Surmeier, D. J., Girault, J., Nestler, E. J., and Greengard, P. (1998). DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science 281, 838–842.
Friedman, E., and Wang, H. Y. (1998). Prenatal cocaine exposure alters signal transduction in the brain D1 dopamine receptor system. Ann. N. Y. Acad. Sci. 846, 238–247.
Gerfen, C. R., Paletzki, R., and Worley, P. (2008). Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J. Neurosci. 28, 7113–7120.
Guan, X., Tao, J., and Li, S. (2009). Dopamine D1 receptor, but not dopamine D2 receptor, is a critical regulator for acute cocaine-enhanced gene expression. Neurol. Res. 31, 17–22.
Haber, S. (2008). Parallel and integrative processing through the Basal Ganglia reward circuit: lessons from addiction. Biol. Psychiatry 64, 173–174.
Harvey, J. A. (2004). Cocaine effects on the developing brain: current status. Neurosci. Biobehav. Rev. 27, 751–764.
Hyman, S. E., and Malenka, R. C. (2001). Addiction and the brain: the neurobiology of compulsion and its persistence. Nat. Rev. Neurosci. 2, 695–703.
Jiang, X., Zhou, J., Mash, D. C., Marini, A. M., and Lipsky, R. H. (2009). Human BDNF isoforms are differentially expressed in cocaine addicts and are sorted to the regulated secretory pathway independent of the Met66 substitution. Neuromolecular Med. 11, 1–12.
Jones, L. B., Stanwood, G. D., Reinoso, B. S., Washington, R. A., Wang, H. Y., Friedman, E., and Levitt, P. (2000). In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J. Neurosci. 20, 4606–4614.
Kalivas, P. W., Pierce, R. C., Cornish, J., and Sorg, B. A. (1998). A role for sensitization in craving and relapse in cocaine addiction. J. Psychopharmacol. (Oxford) 12, 49–53.
Kosofsky, B. E., and Hyman, S. E. (2001). No time for complacency: the fetal brain on drugs. J. Comp. Neurol. 435, 259–262.
Kosofsky, B. E., Wilkins, A. S., Gressens, P., and Evrard, P. (1994). Transplacental cocaine exposure: a mouse model demonstrating neuroanatomic and behavioral abnormalities. J. Child Neurol. 9, 234–241.
Lester, B. M., LaGasse, L. L., and Seifer, R. (1998). Cocaine exposure and children: the meaning of subtle effects. Science 282, 633–634.
Levine, T. P., Liu, J., Das, A., Lester, B., Lagasse, L., Shankaran, S., Bada, H. S., Bauer, C. R., and Higgins, R. (2008). Effects of prenatal cocaine exposure on special education in school-aged children. Pediatrics 122, e83–e91.
Lidow, M. S., and Song, Z. M. (2001). Primates exposed to cocaine in utero display reduced density and number of cerebral cortical neurons. J. Comp. Neurol. 435, 263–275.
Liu, Q. R., Lu, L., Zhu, X. G., Gong, J. P., Shaham, Y., and Uhl, G. R. (2006). Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 1067, 1–12.
Ma, L., Hasan, K. M., Steinberg, J. L., Narayana, P. A., Lane, S. D., Zuniga, E. A., Kramer, L. A., and Moeller, F. G. (2009). Diffusion tensor imaging in cocaine dependence: regional effects of cocaine on corpus callosum and effect of cocaine administration route. Drug Alcohol Depend. 104, 262–267.
Malanga, C. J. III, and Kosofsky, B. E. (1999). Mechanisms of action of drugs of abuse on the developing fetal brain. Clin. Perinatol. 26, 17–37, v–vi.
Malanga, C. J., and Kosofsky, B. E. (2003). Does drug abuse beget drug abuse? Behavioral analysis of addiction liability in animal models of prenatal drug exposure. Brain Res. Dev. Brain Res. 147, 47–57.
Malanga, C. J., Pejchal, M., and Kosofsky, B. E. (2007). Prenatal exposure to cocaine alters the development of conditioned place-preference to cocaine in adult mice. Pharmacol. Biochem. Behav. 87, 462–471.
Malanga, C. J., Ren, J. Q., Guerriero, R. M., and Kosofsky, B. E. (2009). Augmentation of cocaine-sensitized dopamine release in the nucleus accumbens of adult mice following prenatal cocaine exposure. Dev. Neurosci. 31, 76–89.
Malanga, C. J., Riday, T. T., Carlezon, W. A. Jr., and Kosofsky, B. E. (2008). Prenatal exposure to cocaine increases the rewarding potency of cocaine and selective dopaminergic agonists in adult mice. Biol. Psychiatry 63, 214–221.
Mayes, L. C. (2002). A behavioral teratogenic model of the impact of prenatal cocaine exposure on arousal regulatory systems. Neurotoxicol. Teratol. 24, 385–395.
Mayes, L. C., Grillon, C., Granger, R., and Schottenfeld, R. (1998). Regulation of arousal and attention in preschool children exposed to cocaine prenatally. Ann. N. Y. Acad. Sci. 846, 126–143.
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., Welzl, H., Wolfer, D. P., Pages, G., Valverde, O., Marowsky, A., Porrazzo, A., Orban, P. C., Maldonado, R., Ehrengruber, M. U., Cestari, V., Lipp, H. P., Chapman, P. F., Pouyssegur, J., and Brambilla, R. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34, 807–820.
McCarthy, D. M., Zhang, X., Darnell, S. B., Sangrey, G. R., Yanagawa, Y., Sadri-Vakili, G., and Bhide, P. G. (2011). Cocaine alters BDNF expression and neuronal migration in the embryonic mouse forebrain. J. Neurosci. 31(38), 13400–13411.
McGinty, J. F., Shi, X. D., Schwendt, M., Saylor, A., and Toda, S. (2008). Regulation of psychostimulant-induced signaling and gene expression in the striatum. J. Neurochem. 104, 1440–1449.
Paxinos, G., and Franklin, K. (2003). The Mouse Brain in Stereotaxc Coordinates, 2nd Edn. New York: Academic Press.
Rao, H., Wang, J., Giannetta, J., Korczykowski, M., Shera, D., Avants, B. B., Gee, J., Detre, J. A., and Hurt, H. (2007). Altered resting cerebral blood flow in adolescents with in utero cocaine exposure revealed by perfusion functional MRI. Pediatrics 120, e1245–e1254.
Renthal, W., Kumar, A., Xiao, G., Wilkinson, M., Covington, H. E. III, Maze, I., Sikder, D., Robison, A. J., LaPlant, Q., Dietz, D. M., Russo, S. J., Vialou, V., Chakravarty, S., Kodadek, T. J., Stack, A., Kabbaj, M., and Nestler, E. J. (2009). Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62, 335–348.
Rivkin, M. J., Davis, P. E., Lemaster, J. L., Cabral, H. J., Warfield, S. K., Mulkern, R. V., Robson, C. D., Rose-Jacobs, R., and Frank, D. A. (2008). Volumetric MRI study of brain in children with intrauterine exposure to cocaine, alcohol, tobacco, and marijuana. Pediatrics 121, 741–750.
Rocha, B. A., Mead, A. N., and Kosofsky, B. E. (2002). Increased vulnerability to self-administer cocaine in mice prenatally exposed to cocaine. Psychopharmacology (Berl.) 163, 221–229.
Roussotte, F., Soderberg, L., and Sowell, E. (2010). Structural, metabolic, and functional brain abnormalities as a result of prenatal exposure to drugs of abuse: evidence from neuroimaging. Neuropsychol. Rev. 20, 376–397.
Self, D. W., Barnhart, W. J., Lehman, D. A., and Nestler, E. J. (1996). Opposite modulation of cocaine-seeking behavior by D1- and D2-like dopamine receptor agonists. Science 271, 1586–1589.
Sheinkopf, S. J., Lester, B. M., Sanes, J. N., Eliassen, J. C., Hutchison, E. R., Seifer, R., Lagasse, L. L., Durston, S., and Casey, B. J. (2009). Functional MRI and response inhibition in children exposed to cocaine in utero. Preliminary findings. Dev. Neurosci. 31, 159–166.
Spear, L. P., Silveri, M. M., Casale, M., Katovic, N. M., Campbell, J. O., and Douglas, L. A. (2002). Cocaine and development: a retrospective perspective. Neurotoxicol. Teratol. 24, 321–327.
Stanwood, G. D., and Levitt, P. (2007). Prenatal exposure to cocaine produces unique developmental and long-term adaptive changes in dopamine D1 receptor activity and subcellular distribution. J. Neurosci. 27, 152–157.
Trask, C. L., and Kosofsky, B. E. (2000). Developmental considerations of neurotoxic exposures. Neurol. Clin. 18, 541–562.
Tropea, T. F., Guerriero, R. M., Willuhn, I., Unterwald, E. M., Ehrlich, M. E., Steiner, H., and Kosofsky, B. E. (2008a). Augmented D1 dopamine receptor signaling and immediate-early gene induction in adult striatum after prenatal cocaine. Biol. Psychiatry 63, 1066–1074.
Tropea, T. F., Kosofsky, B. E., and Rajadhyaksha, A. M. (2008b). Enhanced CREB and DARPP-32 phosphorylation in the nucleus accumbens and CREB, ERK, and GluR1 phosphorylation in the dorsal hippocampus is associated with cocaine-conditioned place preference behavior. J. Neurochem. 106, 1780–1790.
Uhl, G. R., Hall, F. S., and Sora, I. (2002). Cocaine, reward, movement and monoamine transporters. Mol. Psychiatry 7, 21–26.
Unterwald, E. M., Ivkovic, S., Cuntapay, M., Stroppolo, A., Guinea, B., and Ehrlich, M. E. (2003). Prenatal exposure to cocaine decreases adenylyl cyclase activity in embryonic mouse striatum. Brain Res. Dev. Brain Res. 147, 67–75.
Walhovd, K. B., Westlye, L. T., Moe, V., Slinning, K., Due-Tonnessen, P., Bjornerud, A., van der Kouwe, A., Dale, A. M., and Fjell, A. M. (2010). White matter characteristics and cognition in prenatally opiate- and polysubstance-exposed children: a diffusion tensor imaging study. AJNR Am. J. Neuroradiol. 31, 894–900.
Wang, H. Y., Runyan, S., Yadin, E., and Friedman, E. (1995). Prenatal exposure to cocaine selectively reduces D1 dopamine receptor-mediated activation of striatal Gs proteins. J. Pharmacol. Exp. Ther. 273, 492–498.
Warner, T. D., Behnke, M., Eyler, F. D., Padgett, K., Leonard, C., Hou, W., Garvan, C. W., Schmalfuss, I. M., and Blackband, S. J. (2006). Diffusion tensor imaging of frontal white matter and executive functioning in cocaine-exposed children. Pediatrics 118, 2014–2024.
Wilkins, A. S., Genova, L. M., Posten, W., and Kosofsky, B. E. (1998). Transplacental cocaine exposure. 1: a rodent model. Neurotoxicol. Teratol. 20, 215–226.
Wolf, M. E., and Ferrario, C. R. (2010). AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci. Biobehav. Rev. 35, 185–211.
Woo, N. H., Teng, H. K., Siao, C. J., Chiaruttini, C., Pang, P. T., Milner, T. A., Hempstead, B. L., and Lu, B. (2005). Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat. Neurosci. 8, 1069–1077.
Yang, J., Siao, C. J., Nagappan, G., Marinic, T., Jing, D., McGrath, K., Chen, Z. Y., Mark, W., Tessarollo, L., Lee, F. S., Lu, B., and Hempstead, B. L. (2009). Neuronal release of proBDNF. Nat. Neurosci. 12, 113–115.
Zhang, D., Zhang, L., Lou, D. W., Nakabeppu, Y., Zhang, J., and Xu, M. (2002a). The dopamine D1 receptor is a critical mediator for cocaine-induced gene expression. J. Neurochem. 82, 1453–1464.
Zhang, J., Zhang, D., and Xu, M. (2002b). Identification of chronic cocaine-induced gene expression through dopamine d1 receptors by using cDNA microarrays. Ann. N. Y. Acad. Sci. 965, 1–9.
Keywords: prenatal cocaine, striatum, nucleus accumbens, D1, TrkB, BDNF, CREB, GluA1
Citation: Tropea TF, Kabir ZD, Kaur G, Rajadhyaksha AM and Kosofsky BE (2011) Enhanced dopamine D1 and BDNF signaling in the adult dorsal striatum but not nucleus accumbens of prenatal cocaine treated mice. Front. Psychiatry 2:67. doi: 10.3389/fpsyt.2011.00067
Received: 30 June 2011;
Paper pending published: 19 July 2011;
Accepted: 12 November 2011;
Published online: 07 December 2011.
Edited by:
Linda Mayes, Yale University, USAReviewed by:
Katerina Maniadaki, Technological Educational Institute of Athens, GreeceDiana Dow-Edwards, State University of New York, USA
Copyright: © 2011 Tropea, Kabir, Kaur, Rajadhyaksha and Kosofsky. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Barry E. Kosofsky, Division of Child Neurology, Weill Cornell Medical College/New York Presbyterian Hospital, 525 East 68th Street, Box 91, New York, NY 10021, USA. e-mail: bar2009@med.cornell.edu