
- Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, Charlottesville, VA, USA
Paucity of sensitive biomarkers to quantify transient changes in alcohol consumption level remains a critical barrier for the development of efficacious therapeutic agents to treat alcoholism. Recently, in an 11-week, randomized, placebo-controlled, double-blind trial of 283 alcohol-dependent individuals, we demonstrated that ondansetron was efficacious at reducing the severity of drinking (measured as drinks per drinking day; DDD) in alcoholics carrying the LL compared with the LS/SS genotype of the serotonin transporter gene, 5′-HTTLPR. Using peripheral blood samples from a cohort of 41 of these subjects, we determined whether there was a relationship between mRNA expression level of the 5′-HTTLPR genotypes (measured at weeks 0, 4, and 11) and self-reported alcohol consumption following treatment with either ondansetron (4 μg/kg twice daily; N = 19) or placebo (N = 22). Using a mixed-effects linear regression model, we analyzed the effects of DDD and 5′-HTTLPR genotypes on mRNA expression levels within and between the ondansetron and placebo groups. We found a significant three-way interaction effect of DDD, 5′-HTTLPR genotypes, and treatment on mRNA expression levels (p = 0.0396). Among ondansetron but not placebo recipients, there was a significant interaction between DDD and 5′-HTTLPR genotype (p = 0.0385 and p = 0.7938, respectively). In the ondansetron group, DDD was associated positively with mRNA levels at a greater rate of expression alteration per standard drink in those with the LL genotype (slope = +1.1698 in ln scale). We suggest that the combination of the LL genotype and 5′-HTTLPR mRNA expression levels might be a promising and novel biomarker to quantify drinking severity in alcoholics treated with ondansetron.
Introduction
Severe drinking is an important risk factor for the development of alcohol dependence, an important cause of morbidity and mortality in the US and worldwide (Heinz et al., 2003; Bierut et al., 2010; Paula et al., 2010). Hence, it is important to find not only efficacious medicines to treat alcohol dependence but also sensitive methods for quantifying objectively transient changes in drinking.
Several converging lines of scientific evidence have proposed that variations in the expression of the serotonin transporter gene in the human brain might be associated with differences in the severity of alcohol consumption (Heinz et al., 1998; Gorwood et al., 2000; Matsushita et al., 2001; Barr et al., 2004; Barr and Goldman, 2006).
Indeed, dysregulation of the bio-molecular control of alcohol consumption can predispose an individual to alcohol dependence (Heinz et al., 2003). For instance, carriers of the homozygous high-activity long allele (LL) of the serotonin transporter-linked polymorphic region (5′-HTTLPR) have been associated with reduced intoxication and a higher risk of developing alcohol dependence (Schuckit and Smith, 1996; Barr et al., 2003; Hinckers et al., 2006; Buchmann et al., 2009; Laucht et al., 2009). Furthermore, the severity of current drinking has been associated with greater uptake of serotonin into human platelets in alcohol-dependent individuals carrying the L allele but not in SS homozygotes (Heinz et al., 2000; Johnson et al., 2008a). Also, it has been shown that the expression of the LL homozygous form of the 5′-HTTLPR genotype is influenced differentially by severe drinking (Heinz et al., 2000; Philibert et al., 2008; Ho et al., 2011). Based on this knowledge, we have hypothesized that the interaction between LL genotype and alcohol consumption would be associated with upregulation of post-synaptic receptors, and that the blockade of upregulated 5-HT3 receptors by ondansetron would be associated with a significant decrease in severe drinking (Johnson, 2000).
Consistent with our hypothesis, we found, in a recent proof-of-concept pharmacogenetic study, that ondansetron, a 5-HT3 antagonist, was significantly more efficacious in reducing drinking severity in alcohol-dependent individuals carrying the LL genotype of the 5′-HTTLPR compared with the LS or SS genotype (Johnson et al., 2011). Based on these previous findings, we hypothesized further that the statistical interaction detected between LL genotype and ondansetron treatment response might have been mediated through the specific regulatory effects of varying alcohol amounts on transcription rates of serotonin transporter mRNA expression. As an exploratory extension of this hypothesis, in this proof-of-concept pilot study, we examined whether alterations in mRNA expression of the serotonin transporter were associated with variations in alcohol consumption and could, therefore, be used as a novel biomarker to assess, objectively, variations in alcohol consumption levels in response to ondansetron treatment.
Currently used biomarkers to detect alcohol consumption lack adequate specificity to distinguish moderate from heavy drinking (Anton, 2010; Bearer et al., 2010) and do not provide quantitative assessments of heavy drinking in the recent past. Another shortcoming of the existing biomarkers of heavy drinking, such as percentage of carbohydrate-deficient transferrin and gamma-glutamyltransferase, is that they are not particularly sensitive and do not elevate in significant amounts until heavy drinking has occurred for at least a week (Litten and Fertig, 2003; Niemela, 2007). Also, the direct measurement of alcohol concentration in blood, plasma, and breath can only be detected for 10–12 h after heavy drinking (Jatlow and O’Malley, 2010); thus, it has limited value in quantifying drinking in an outpatient setting. Because self-reported alcohol consumption can be unreliable (Stewart et al., 2009), there is a need to detect biomarkers that are both sensitive and specific. Additionally, the implementation of a novel accurate, quantitative biomarker will enhance pharmacotherapy research by providing a valid measure of alcohol consumption (Lee et al., 2005; Freeman and Vrana, 2010; Jatlow and O’Malley, 2010; Litten et al., 2010).
Materials and Methods
Participants
Forty-one unrelated Caucasian and Hispanic (95 and 5%, respectively) alcohol-dependent subjects were included in the study. All participants were current drinkers recruited from Central Virginia as part of a larger pharmacogenetic clinical trial that tested the efficacy of ondansetron for the treatment of alcohol dependence (Johnson et al., 2011). A detailed description of the inclusion and exclusion criteria of the study and the characteristics of the population has been provided elsewhere (Johnson et al., 2011). Briefly, all participants had no Axis I diagnosis other than nicotine dependence according to the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (American Psychiatric Association, 1994), were aged 26–78 years (mean age, 52.93 years), scored >8 on the Alcohol Use Disorders Identification Test (Bohn et al., 1995), which assessed the severity of alcohol-related problems, reported drinking ≥21 standard drinks/week for women and ≥30 standard drinks/week for men during the 90 days prior to enrollment, and had a negative urine toxicologic screen for narcotics, amphetamines, or sedative–hypnotics at enrollment. Enrollees presented themselves at breath alcohol concentration levels under 0.02% at all clinical assessment and screen visits and were physically healthy, not pregnant, and not using abused drugs at enrollment. All subjects provided informed consent to participate. Demographic characteristics are presented in Table 1.
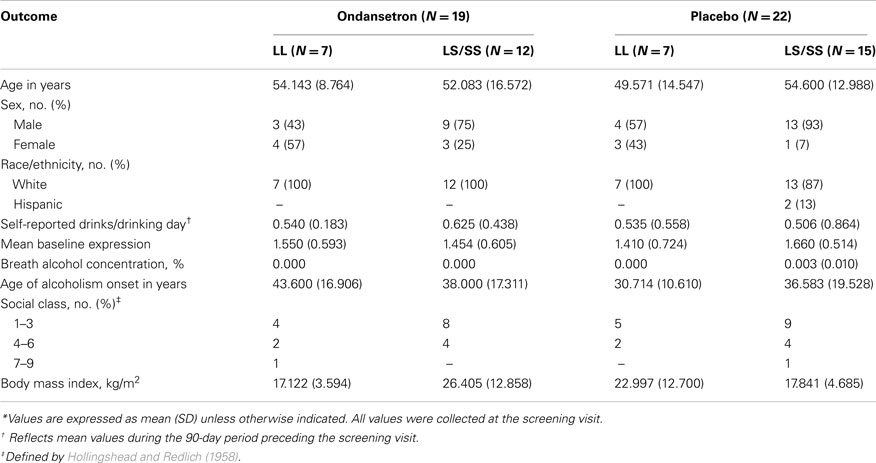
Table 1. Baseline demographic and biological characteristics*.
Study Procedures
Subjects were enrolled in an 11-week, placebo-controlled, double-blind, randomized study, between May 13, 2006, and October 20, 2008, at the University of Virginia while they were still drinking (i.e., without a period of abstinence). At enrollment, we collected self-reported drinking amounts over the past 90 days, based on the timeline follow-back method (Sobell and Sobell, 1992), and other screening parameters, including those of physical health, breath alcohol concentration, and additional psychosocial measures associated with drinking behavior, as detailed in our previous trials (Johnson et al., 2000, 2006, 2008b). Subjects were randomized by serotonin transporter genotype (LL vs. LS/SS) and then into the ondansetron and placebo groups. A detailed description of subject randomization into the ondansetron and placebo groups and the clinical trial procedures can be found in Johnson et al. (2011). From a subset of the total study cohort, whole blood samples were drawn from all consenting subjects for mRNA expression studies at the screening visit (baseline) and at weeks 4, 8, and 12 after baseline. Daily drinking amounts for all time periods were also collected using the timeline follow-back method at each weekly visit to the study center.
Dna Extraction and Genotyping
Genomic DNA was extracted using a Gentra Puregene® kit (QIAGEN Inc., Valencia, CA, USA). The 5′-HTTLPR L/S alleles were determined as described earlier (Wendland et al., 2006; Johnson et al., 2008a) with custom-made primers (Forward: TCCTCCGCTTTGGCGCCTCTTCC, and Reverse: TGGGGGTTGCAGGGGAGATCCTG).
Rna Extraction and qRT-PCR
From each participant, 8 ml of whole blood was drawn at screen and then 4, 8, and 12 weeks afterward. Blood collections were performed at the same time of the day to minimize the effects of circadian cycle variations on mRNA expression levels. Following blood collection, whole blood was lysed to isolate white blood cells (WBC) using red blood cell lysis solution (QIAGEN Inc.). Isolated WBC then were washed with phosphate-buffered saline for further purification, suspended in RNAlater® solution to protect the RNA from degradation (Ambion®; Applied Biosystems, Foster City, CA, USA), and stored at −80°C until the total RNA was extracted with a RiboPure™ Kit (Ambion®; Applied Biosystems) according to the manufacturer’s instructions. cDNA synthesis and quantitative real-time polymerase chain reaction (qRT-PCR) assays were carried out as described previously (Seneviratne et al., 2009). Each qRT-PCR experiment was performed in triplicate. To minimize potential technical variations among samples from the four time points within subjects, cDNA synthesis and qRT-PCR reactions from total RNA samples collected at all four time points from a given subject were performed simultaneously in a 384-well PCR plate (Applied Biosystems). The resulting mRNA levels for the serotonin transporter gene and the glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH), which was used as the housekeeping gene, were quantified by an ABI PRISM® 7900HT (Applied Biosystems) sequence detection system using the comparative threshold (Ct) method (Winer et al., 1999). Mean Ct ratios of the serotonin transporter to GAPDH mRNA were calculated for each time point in all subjects at baseline and at weeks 4, 8, and 12 after baseline for statistical analyses of genotype and treatment group differences.
Statistical Analyses
Data quality control
All the samples were tested in triplicate with the reference gene GAPDH, a housekeeping gene for normalization of data, to correct for variations in cDNA and/or RNA quantity. Mean Ct ratios of serotonin transporter to GAPDH gene mRNA calculated from each triplicate for each sample were tested for variations due to sample handling and preparation by comparing the SD with the sample mean. All triplicates included in our analyses varied less than 1.5 SDs from the mean. Additionally, each PCR run included non-template controls that generated low-level signals (Ct values > 40). Quality of all clinical data was assessed by a database coordinator and a statistician.
Statistical method
The dependent variable, drinking severity, was measured as weekly drinks per drinking day (DDD), and then all DDD values for each subject at each time point were normalized by dividing by the subject’s body mass index (BMI); the BMI-adjusted DDD is denoted by DDD′ in Figure 1. BMI strongly correlates with blood alcohol concentrations (Wang et al., 1992; Dettling et al., 2007); therefore, the adjustment of DDD with BMI is particularly important as we utilized a peripheral WBC model for expression analyses. As explained above, for each participant, we quantified serotonin transporter mRNA expression levels at four time points (baseline and weeks 4, 8, and 12); as these measures were collected from the same participant, they were considered as repeated measures that reflected alterations throughout the treatment period. Therefore, to analyze how serotonin transporter mRNA expression levels vary across the treatment period (baseline and weeks 4, 8, and 12) in response to varying drinking amounts (adjusted by BMI; DDD′), we employed a repeated-measures, mixed-effects linear regression model. The fitted model included baseline DDD′, age, gender, and self-defined ethnicity as covariates. Prior to statistical analyses, all ratios of serotonin transporter to GAPDH mRNA were transformed to natural logarithms. The effects of DDD′ on repeated measures of mRNA expression levels were assessed first for the total cohort and then for each of the four “genotype × treatment” groups separately – i.e., LL genotype carriers treated with ondansetron, LL carriers treated with placebo, LS/SS carriers treated with ondansetron, and LS/SS carriers treated with placebo. This approach allowed us to examine both genotype and ondansetron treatment effects on trends of serotonin transporter mRNA expression alterations in response to varying alcohol doses. Statistical significance for the associations between DDD′ and serotonin transporter mRNA expression levels were assessed by the p-values derived from the repeated-measures, mixed-effects linear regression model.
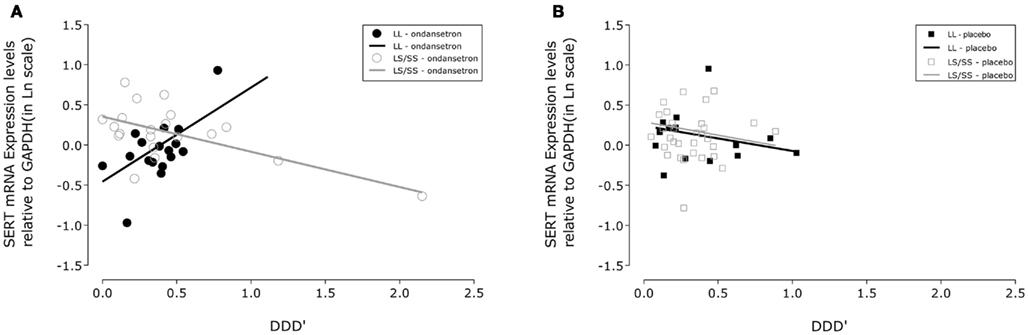
Figure 1. Association between drinking severity and serotonin transporter (SERT) mRNA expression levels during 11 weeks of treatment. SLC6A4 gene expression changes in human white blood cells with the amount of severe drinking (drinks per drinking day; DDD′) and genotypic variation of the serotonin transporter-linked polymorphic region in subjects treated with (A) ondansetron (N = 19) and (B) placebo (N = 22). The closed symbols represent covariate adjusted individual expression levels, and the dark lines represent expression level changes associated with drinking severity in LL subjects; the open symbols and light lines represent the corresponding changes in LS/SS subjects. The intercepts and slopes for expression levels in the LL and LS/SS ondansetron groups were estimated from a linear mixed-effects model: y = −0.4566 + 1.1698 × x and y = 0.3524 − 0.4385 × x, respectively. The corresponding estimates for the LL and LS/SS placebo groups were y = 0.2417 − 0.3150 × x and y = 0.2919 − 0.3363 × x, respectively. GAPDH, glyceraldehyde-3-phosphate dehydrogenase gene.
Results
At baseline, DDD′ and serotonin transporter mRNA expression levels did not differ significantly among the four genotype × treatment groups (LL-ondansetron, LS/SS-ondansetron, LL-placebo, and LS/SS-placebo). Baseline demographic and biological characteristics of the cohort are given in Table 1.
From Table 2, it can be seen that the analyses of the total cohort showed a significant three-way interactive effect of DDD′, 5′-HTTLPR genotype, and treatment on predicting the mRNA expression levels (N = 41, F = 4.64, p = 0.0396). Interestingly, when the two treatment groups were analyzed separately, the interaction between DDD′ and serotonin transporter genotype was significant only within the ondansetron treatment group, but not within the placebo group (N = 19, F = 4.96, p = 0.0385, and N = 22, F = 0.07, p = 0.7938, respectively). The mRNA expression levels in all subjects who received ondansetron and placebo are shown in Figures 1A,B, respectively; the symbols in the two panels represent mRNA expression levels at each time point (weeks 4, 8, and 12) for all the individuals included in the “genotype × treatment” groups. As described in the Figure 1 legend, we then estimated slopes for the associations between DDD′ and mRNA expression throughout the entire study period (baseline to week 12) by utilizing a repeated-measures, mixed-effects model. As illustrated in Figure 1, subjects carrying the LL genotype who were treated with ondansetron showed a trend toward having decreased serotonin transporter mRNA expression levels with decreasing drinking levels (slope = +1.1698) during the 11-week treatment period. In contrast, those with the LS or SS genotype who were treated with ondansetron showed a trend toward having increased serotonin transporter mRNA expression levels with decreasing drinking levels (slope = −0.4385) during the 11-week treatment period. Serotonin transporter mRNA expression levels in both LL and LS/SS subjects who were treated with placebo showed a trend toward increased expression with decreasing drinking levels (slope = −0.3150 for LL, slope = −0.3363 for LS/SS).
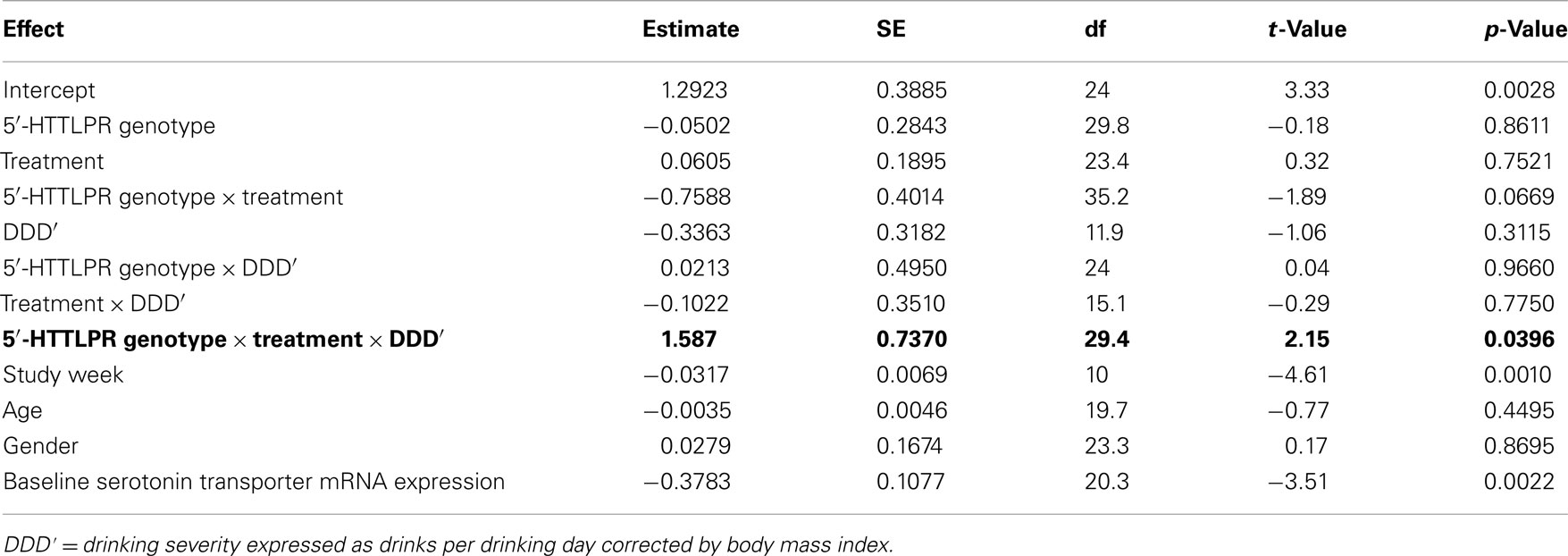
Table 2. Effects of variables included in the mixed-effects linear regression model on serotonin mRNA expression levels.
Discussion
The primary goal of this study was to examine for the possibility of utilizing the combination of mRNA expression levels and the LL genotype of the serotonin transporter gene as a genomic biomarker to assess, objectively and quantitatively, changes in drinking severity in response to ondansetron treatment. During the 11-week treatment period, serotonin transporter mRNA expression levels within LL subjects treated with ondansetron decreased at a rate of ln(1.164) with decreasing drinking severity, implying approximately a threefold reduction of serotonin transporter mRNA expression levels for an increase in one standard drink per drinking day.
Interestingly, the rates of serotonin transporter mRNA alterations were more than 50% less in the remaining three “genotype × treatment” groups, compared with serotonin transporter mRNA expression level alterations per unit of standard drink per drinking day in alcohol-dependent individuals with the LL genotype who were treated with ondansetron. Furthermore, unlike in the LL-ondansetron group, in all three of these groups (S-carriers treated with ondansetron and LL and S-carrier subjects who received a placebo), associations between serotonin transporter mRNA expression and drinking severity showed negative trends. Additionally, the negative trends within the placebo treatment group did not differ significantly between the two serotonin transporter genotype groups. One possibility for this observation is the lack of significant drinking severity reductions in these groups, or perhaps an unknown direct regulatory effect of ondansetron on serotonin transporter gene expression. To our knowledge, the exact mechanism of serotonin transporter regulation under the condition of severe drinking is not yet characterized. Nevertheless, there is growing evidence that serotonin transporter gene expression differences among alcohol-dependent individuals with the LL genotype, compared with S-carriers, may be regulated by epigenetic mechanisms, such as methylation in alcohol-dependent individuals (Philibert et al., 2007, 2008) and transcription factor binding in subjects with obsessive–compulsive disorder (Hu et al., 2006).
There are at least three promising aspects to quantifying serotonin transporter mRNA levels within LL genotype carriers as a serotonin transporter genomic biomarker for predicting drinking severity. First, unlike any of the currently existing biomarkers of alcohol consumption, the serotonin transporter genomic biomarker targets a pathophysiological mechanism in the brain associated with heavy drinking (Johnson, 2005; Johnson et al., 2011). For this reason, in addition to serving as a marker for treatment outcome, it also may be a surrogate endpoint marker for the severity of the disease itself. Second, because the marker is tested in populations carrying a specific genetic variant (the LL genotype), the individual variability on serotonin transporter mRNA expression levels will be less. Third, the serotonin transporter genomic biomarker can be tested in research or clinical facilities with access to a genetic laboratory.
The main limitation of our study is the small sample size in each of the genotype × treatment groups. We did, however, have a minimum of seven subjects in each group and, therefore, six degrees of freedom (df), which is greater than the minimum of five df recommended by Churchill (2002) for each group in gene expression analyses with the microarray technique. Nevertheless, additional studies with a larger sample size will be needed to replicate our findings. Another notable limitation was that the self-reported drinking data could not be verified independently. Thus, the development of serotonin transporter mRNA levels as a potential novel biomarker would require studies that characterize directly its sensitivity and specificity, in both healthy and alcohol-dependent individuals, under conditions where known amounts of alcohol are administered under either a fixed or varying schedule. Another notable drawback of our study is the lack of consensus among researchers on the association between 5′-HTTLPR genotypes and alcoholism. Although an association with alcoholism would be a preferable aspect of a genetic marker for drinking severity, it should also be noted that the pharmacological response to alcohol may involve genes or genetic variants within those genes that are different from the genetic factors governing disease susceptibility. To our knowledge, there are no genome-wide association studies that have examined genetic associations with drinking severity. In this light, based on our previous biochemical and molecular studies that led us to develop the hypothesis for the present study, we believe that the 5′-HTTLPR genotypes are a reasonable genetic marker to predict serotonin transporter expression level alterations in response to severe drinking. Furthermore, if successfully validated, our biomarker development strategy may provide a model for screening and validation of other potential genomic markers for drinking severity in the future.
In sum, our findings provide the preliminary evidence for the possibility of utilizing the combination of the serotonin transporter LL genotype and mRNA levels as a novel biomarker to assess, quantitatively and objectively, alcohol consumption levels in alcohol-dependent individuals treated with ondansetron.
Conflict of Interest Statement
Professor Bankole A. Johnson has served as a consultant to Johnson & Johnson (Ortho-McNeil Janssen Scientific Affairs, LLC), Transcept Pharmaceuticals, Inc., D&A Pharma, Organon, ADial Pharmaceuticals, LLC (with which he also serves as Chairman), Psychological Education Publishing Company (PEPCo), LLC, and Eli Lilly and Company. Dr. Chamindi Seneviratne reports no financial relationships with commercial interests.
Acknowledgments
We are grateful to the National Institute on Alcohol Abuse and Alcoholism for its generous support through grants 7 U10 AA011776-10, 7 R01 AA010522-12, 2 R01 AA010522-16, 5 R01 AA013964-05, 5 R01 AA014628-04, and 5 R01 AA012964-06 awarded to Professor Bankole A. Johnson. We also thank Robert H. Cormier, Jr., B.A., for his assistance with manuscript preparation.
References
American Psychiatric Association. (1994). Diagnostic and Statistical Manual of Mental Disorders, 4th Edn. Washington, DC: American Psychiatric Association.
Anton, R. F. (2010). Editorial commentary: alcohol biomarker papers. Alcohol. Clin. Exp. Res. 34, 939–940.
Barr, C. S., and Goldman, D. (2006). Non-human primate models of inheritance vulnerability to alcohol use disorders. Addict. Biol. 11, 374–385.
Barr, C. S., Newman, T. K., Becker, M. L., Champoux, M., Lesch, K. P., Suomi, S. J., Goldman, D., and Higley, J. D. (2003). Serotonin transporter gene variation is associated with alcohol sensitivity in rhesus macaques exposed to early-life stress. Alcohol. Clin. Exp. Res. 27, 812–817.
Barr, C. S., Newman, T. K., Lindell, S., Shannon, C., Champoux, M., Lesch, K. P., Suomi, S. J., Goldman, D., and Higley, J. D. (2004). Interaction between serotonin transporter gene variation and rearing condition in alcohol preference and consumption in female primates. Arch. Gen. Psychiatry 61, 1146–1152.
Bearer, C. F., Bailey, S. M., and Hoek, J. B. (2010). Advancing alcohol biomarkers research. Alcohol. Clin. Exp. Res. 34, 941–945.
Bierut, L. J., Agrawal, A., Bucholz, K. K., Doheny, K. F., Laurie, C., Pugh, E., Fisher, S., Fox, L., Howells, W., Bertelsen, S., Hinrichs, A. L., Almasy, L., Breslau, N., Culverhouse, R. C., Dick, D. M., Edenberg, H. J., Foroud, T., Grucza, R. A., Hatsukami, D., Hesselbrock, V., Johnson, E. O., Kramer, J., Krueger, R. F., Kuperman, S., Lynskey, M., Mann, K., Neuman, R. J., Nothen, M. M., Nurnberger, J. I. Jr., Porjesz, B., Ridinger, M., Saccone, N. L., Saccone, S. F., Schuckit, M. A., Tischfield, J. A., Wang, J. C., Rietschel, M., Goate, A. M., and Rice, J. P. (2010). A genome-wide association study of alcohol dependence. Proc. Natl. Acad. Sci. U.S.A. 107, 5082–5087.
Bohn, M. J., Babor, T. F., and Kranzler, H. R. (1995). The Alcohol Use Disorders Identification Test (AUDIT): validation of a screening instrument for use in medical settings. J. Stud. Alcohol 56, 423–432.
Buchmann, A. F., Schmid, B., Blomeyer, D., Becker, K., Treutlein, J., Zimmermann, U. S., Jennen-Steinmetz, C., Schmidt, M. H., Esser, G., Banaschewski, T., Rietschel, M., Schumann, G., and Laucht, M. (2009). Impact of age at first drink on vulnerability to alcohol-related problems: testing the marker hypothesis in a prospective study of young adults. J. Psychiatr. Res. 43, 1205–1212.
Churchill, G. A. (2002). Fundamentals of experimental design for cDNA microarrays. Nat. Genet. 32(Suppl.), 490–495.
Dettling, A., Fischer, F., Bohler, S., Ulrichs, F., Skopp, G., Graw, M., and Haffner, H. T. (2007). Ethanol elimination rates in men and women in consideration of the calculated liver weight. Alcohol 41, 415–420.
Freeman, W. M., and Vrana, K. E. (2010). Future prospects for biomarkers of alcohol consumption and alcohol-induced disorders. Alcohol. Clin. Exp. Res. 34, 946–954.
Gorwood, P., Batel, P., Ades, J., Hamon, M., and Boni, C. (2000). Serotonin transporter gene polymorphisms, alcoholism, and suicidal behavior. Biol. Psychiatry 48, 259–264.
Heinz, A., Higley, J. D., Gorey, J. G., Saunders, R. C., Jones, D. W., Hommer, D., Zajicek, K., Suomi, S. J., Lesch, K. P., Weinberger, D. R., and Linnoila, M. (1998). In vivo association between alcohol intoxication, aggression, and serotonin transporter availability in nonhuman primates. Am. J. Psychiatry 155, 1023–1028.
Heinz, A., Jones, D. W., Gorey, J. G., Bennet, A., Suomi, S. J., Weinberger, D. R., and Higley, J. D. (2003). Serotonin transporter availability correlates with alcohol intake in non-human primates. Mol. Psychiatry 8, 231–234.
Heinz, A., Jones, D. W., Mazzanti, C., Goldman, D., Ragan, P., Hommer, D., Linnoila, M., and Weinberger, D. R. (2000). A relationship between serotonin transporter genotype and in vivo protein expression and alcohol neurotoxicity. Biol. Psychiatry 47, 643–649.
Hinckers, A. S., Laucht, M., Schmidt, M. H., Mann, K. F., Schumann, G., Schuckit, M. A., and Heinz, A. (2006). Low level of response to alcohol as associated with serotonin transporter genotype and high alcohol intake in adolescents. Biol. Psychiatry 60, 282–287.
Ho, P. S., Shih, M. C., Ma, K. H., Huang, W. S., Ho, K. K., Yen, C. H., Lu, R. B., and Huang, S. Y. (2011). Availability of the serotonin transporter in patients with alcohol dependence. World J. Biol. Psychiatry 12, 134–142.
Hollingshead, A. B., and Redlich, F. C. (1958). Social Class and Mental Illness: A Community Study. New York: John Wiley & Sons.
Hu, X. Z., Lipsky, R. H., Zhu, G., Akhtar, L. A., Taubman, J., Greenberg, B. D., Xu, K., Arnold, P. D., Richter, M. A., Kennedy, J. L., Murphy, D. L., and Goldman, D. (2006). Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am. J. Hum. Genet. 78, 815–826.
Jatlow, P., and O’Malley, S. S. (2010). Clinical (nonforensic) application of ethyl glucuronide measurement: are we ready? Alcohol. Clin. Exp. Res. 34, 968–975.
Johnson, B. A. (2000). Serotonergic agents and alcoholism treatment: rebirth of the subtype concept – an hypothesis. Alcohol. Clin. Exp. Res. 24, 1597–1601.
Johnson, B. A. (2005). Recent advances in the development of treatments for alcohol and cocaine dependence: focus on topiramate and other modulators of GABA or glutamate function. CNS Drugs 19, 873–896.
Johnson, B. A., Ait-Daoud, N., Seneviratne, C., Roache, J. D., Javors, M. A., Wang, X.-Q., Liu, L., Penberthy, J. K., DiClemente, C. C., and Li, M. D. (2011). Pharmacogenetic approach at the serotonin transporter gene as a method of reducing the severity of alcohol drinking. Am. J. Psychiatry 168, 265–275.
Johnson, B. A., Javors, M. A., Roache, J. D., Seneviratne, C., Bergeson, S. E., Ait-Daoud, N., Dawes, M. A., and Ma, J. Z. (2008a). Can serotonin transporter genotype predict serotonergic function, chronicity, and severity of drinking? Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 209–216.
Johnson, B. A., Rosenthal, N., Capece, J. A., Wiegand, F., Mao, L., Beyers, K., McKay, A., Ait-Daoud, N., Addolorato, G., Anton, R. F., Ciraulo, D. A., Kranzler, H. R., Mann, K., O’Malley, S. S., Swift, R. M., and Topiramate for Alcoholism Advisory Board and Topiramate for Alcoholism Study Group. (2008b). Improvement of physical health and quality of life of alcohol-dependent individuals with topiramate treatment: US multisite randomized controlled trial. Arch. Intern. Med. 168, 1188–1199.
Johnson, B. A., Roache, J. D., Ait-Daoud, N., Javors, M. A., Harrison, J. M., Elkashef, A., Mojsiak, J., Li, S. H., and Bloch, D. A. (2006). A preliminary randomized, double-blind, placebo-controlled study of the safety and efficacy of ondansetron in the treatment of cocaine dependence. Drug Alcohol Depend. 84, 256–263.
Johnson, B. A., Roache, J. D., Javors, M. A., DiClemente, C. C., Cloninger, C. R., Prihoda, T. J., Bordnick, P. S., Ait-Daoud, N., and Hensler, J. (2000). Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: a randomized controlled trial. JAMA 284, 963–971.
Laucht, M., Treutlein, J., Schmid, B., Blomeyer, D., Becker, K., Buchmann, A. F., Schmidt, M. H., Esser, G., Jennen-Steinmetz, C., Rietschel, M., Zimmermann, U. S., and Banaschewski, T. (2009). Impact of psychosocial adversity on alcohol intake in young adults: moderation by the LL genotype of the serotonin transporter polymorphism. Biol. Psychiatry 66, 102–109.
Lee, J. W., Weiner, R. S., Sailstad, J. M., Bowsher, R. R., Knuth, D. W., O’Brien, P. J., Fourcroy, J. L., Dixit, R., Pandite, L., Pietrusko, R. G., Soares, H. D., Quarmby, V., Vesterqvist, O. L., Potter, D. M., Witliff, J. L., Fritche, H. A., O’Leary, T., Perlee, L., Kadam, S., and Wagner, J. A. (2005). Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: a conference report. Pharm. Res. 22, 499–511.
Litten, R. Z., Bradley, A. M., and Moss, H. B. (2010). Alcohol biomarkers in applied settings: recent advances and future research opportunities. Alcohol. Clin. Exp. Res. 34, 955–967.
Litten, R. Z., and Fertig, J. (2003). Self-report and biochemical measures of alcohol consumption. Addiction 98(Suppl. 2), iii–iv.
Matsushita, S., Yoshino, A., Murayama, M., Kimura, M., Muramatsu, T., and Higuchi, S. (2001). Association study of serotonin transporter gene regulatory region polymorphism and alcoholism. Am. J. Med. Genet. 105, 446–450.
Paula, H., Asrani, S. K., Boetticher, N. C., Pedersen, R., Shah, V. H., and Kim, W. R. (2010). Alcoholic liver disease-related mortality in the United States: 1980-2003. Am. J. Gastroenterol. 105, 1782–1787.
Philibert, R. A., Madan, A., Andersen, A., Cadoret, R., Packer, H., and Sandhu, H. (2007). Serotonin transporter mRNA levels are associated with the methylation of an upstream CpG island. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 101–105.
Philibert, R. A., Sandhu, H., Hollenbeck, N., Gunter, T., Adams, W., and Madan, A. (2008). The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 543–549.
Schuckit, M. A., and Smith, T. L. (1996). An 8-year follow-up of 450 sons of alcoholic and control subjects. Arch. Gen. Psychiatry 53, 202–210.
Seneviratne, C., Huang, W., Ait-Daoud, N., Li, M. D., and Johnson, B. A. (2009). Characterization of a functional polymorphism in the 3′ UTR of SLC6A4 and its association with drinking intensity. Alcohol. Clin. Exp. Res. 33, 332–339.
Sobell, L. C., and Sobell, M. B. (1992). “Timeline follow-back: a technique for assessing self-reported alcohol consumption,” in Measuring Alcohol Consumption: Psychosocial and Biochemical Methods, eds R. Z. Litten, and J. P. Allen (Totowa, NJ: Humana Press Inc.), 41–72.
Stewart, S. H., Reuben, A., Brzezinski, W. A., Koch, D. G., Basile, J., Randall, P. K., and Miller, P. M. (2009). Preliminary evaluation of phosphatidylethanol and alcohol consumption in patients with liver disease and hypertension. Alcohol Alcohol. 44, 464–467.
Wang, M. Q., Nicholson, M. E., Jones, C. S., Fitzhugh, E. C., and Westerfield, C. R. (1992). Acute alcohol intoxication, body composition, and pharmacokinetics. Pharmacol. Biochem. Behav. 43, 641–643.
Wendland, J. R., Martin, B. J., Kruse, M. R., Lesch, K. P., and Murphy, D. L. (2006). Simultaneous genotyping of four functional loci of human SLC6A4, with a reappraisal of 5-HTTLPR and rs25531. Mol. Psychiatry 11, 224–226.
Keywords: 5′-HTTLPR, genotype, mRNA, biomarker, alcohol, ondansetron, pharmacogenetic, serotonin
Citation: Seneviratne C and Johnson BA (2012) Serotonin transporter genomic biomarker for quantitative assessment of ondansetron treatment response in alcoholics. Front. Psychiatry 3:23. doi: 10.3389/fpsyt.2012.00023
Received: 13 August 2011; Accepted: 04 March 2012;
Published online: 28 March 2012.
Edited by:
Margit Burmeister, University of Michigan, USAReviewed by:
Herb Lachman, Albert Einstein College of Medicine of Yeshiva University, USAMichael E. Zwick, Emory University, USA
Copyright: © 2012 Seneviratne and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Bankole A. Johnson, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, P.O. Box 800623, Charlottesville, VA 22908-0623, USA. e-mail: bankolejohnson@virginia.edu