
- 1Faculty of Health Sciences, Camilo José Cela University, Madrid, Spain
- 2Department of Pharmacology, Faculty of Medicine, University of Alcalá, Madrid, Spain
Active metabolites of some antipsychotic drugs exhibit pharmacodynamic and pharmacokinetic properties that may be similar to or differ from the original compound and that can be translated by a different profile of responses and interactions to clinical level. Some of these antipsychotics’ active metabolites might participate in mechanisms of antidepressant activity, as m-chlorophenylpiperazine (aripiprazole), 9-OH-risperidone and norquetiapine. Norquetiapine exhibits distinct pharmacological activity from quetiapine and plays a fundamental role in its antidepressant efficacy. In this review, we analyze the differential pharmacological aspects between quetiapine and norquetiapine, both from the pharmacokinetic and pharmacodynamic perspectives (affinity for dopaminergic, noradrenegic, and/or serotonergic receptors, etc.), as well as differential neuroprotective role. The pharmacological differences between the two drugs could explain the differential clinical effect, as well as some differences in tolerability profile and drug interactions. The available data are sufficient to arrive at the conclusion that antidepressant activity of quetiapine is mediated, at least in part, by the active metabolite norquetiapine, which selectively inhibits noradrenaline reuptake, is a partial 5-HT1A receptor agonist, and acts as an antagonist at presynaptic α2, 5-HT2C, and 5-HT7 receptors.
Introduction
The body treats drugs as foreign, or xenobiotic, substances, so it tends to metabolize them to facilitate elimination. Usually, the products of biotransformation are referred to as metabolites and tend to be more water soluble than the original molecule so the kidneys can more easily eliminate them (1, 2).
Usually, biotransformation produces chemically stable metabolites that are neither pharmacologically nor toxicologically active. In some cases, though, chemically reactive metabolites are generated and may be toxic. In addition, the reactions involved in biotransformation may produce chemically stable and pharmacologically active metabolites, termed “active metabolites” (3); those will be the primary focus of this article.
Active metabolites exhibit pharmacodynamic and pharmacokinetic properties that may be similar to or differ from the original compound. In either case, they alter its activities and play a role in its end result. In fact, the presence of active metabolites is akin to polytherapy (2–4). Many medications give way to active metabolites. They are only considered clinically relevant when this occurs in high enough quantities to have a pharmacological impact, are at least as active as the original compound, or exist in specific tissues, like the brain (1, 2).
An active metabolite can be entirely or only partially responsible for a drug’s therapeutic effect. Others may be pharmacologically active in a way not related to the original molecule’s main activity. Sometimes, a metabolite reverses or deactivates the original molecule’s pharmacological activity (3). It is widely believed that many psychoactive drugs’ effects are the result not only of the original molecule, but its active metabolites. Moreover, some drugs are inactive per se, and must be biotransformed in the body to become pharmacologically active; these have been called prodrugs.
At times, when an active metabolite improves the original compound’s pharmacodynamic, pharmacokinetic, or toxicological properties, it gets commercialized. Examples of this in psychopharmacology include desipramine (desmethyl imipramine), derivatives of the antidepressant imipramine, mesoridazine and paliperidone, derivatives of the antipsychotics thioridazine and risperidone, and oxazepam, diazepam’s metabolite (1, 3).
Active Metabolites of Antidepressant Drugs
Antidepressants are a wide and varied group of drugs involving many active metabolites. Some such metabolites have even been commercialized as active agents in their own right. Table 1 presents the major known active metabolites and their corresponding antidepressants.
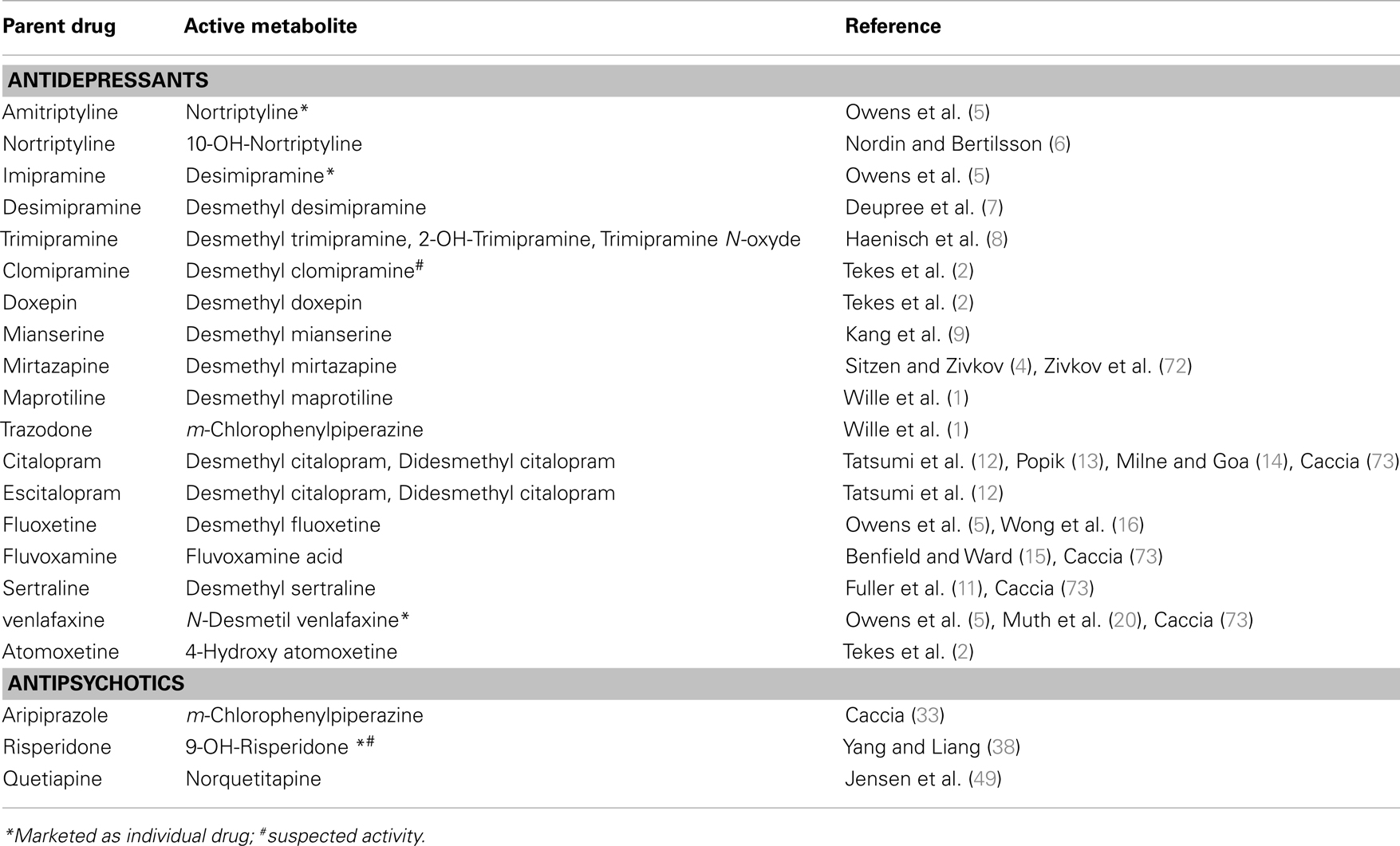
Table 1. Active metabolites of psychotropic drugs with antidepressants properties.
Nortriptyline is an active metabolite in amitriptyline and features distinctly potent inhibition of monoamine transporters, noradrenaline (NET), and serotonin (SERT) beyond that of its parent compound. Thus, while amitriptyline inhibits both transporters, its metabolite, nortriptyline, acts primarily as an inhibitor of NET reuptake (5). In turn, 10-OH-nortriptyline, considered an active metabolite of nortriptyline, is a stronger NET inhibitor and is less anticholinergic (6).
Desipramine is an active metabolite in imipramine, which inhibits reuptake of NET and SERT; the metabolite features almost selective NET inhibition. Desipramine and nortriptyline, in fact, are the strongest NET reuptake inhibitors of the tricyclic antidepressants commercially available (5). Desipramine, too, is converted into an active metabolite, desmethyl desipramine, which has more affinity for SERT than for NET. This metabolite reduces desipramine’s selectivity and adds to its overall antidepressant effect (7).
Trimipramine also involves several pharmacologically active metabolites. One is desmethyl trimipramine, which inhibits NET and SERT transporters as strongly as its parent compound. However, 2-OH-trimipramine is weaker and trimipramine-N-oxide preferentially inhibits SERT (8).
Other tricyclic antidepressants have active metabolites, but their differential profiles relative to their parent compounds have yet to be clearly elucidated. However, clomipramine is known to give way to desmethyl clomipramine, and doxepin to the metabolite desmethyl doxepin (2). Mianserin is demethylated to produce an active metabolite, N-desmethyl mianserin, whose longer half-life increases the effect of mianserin (9).
Mirtazapine undergoes extensive metabolism during its first pass through the liver, the major metabolic pathways in humans being N-demethylation, N-oxidation, and 8-hydroxylation, followed by conjugation. The demethylated metabolite, normirtazapine, remains pharmacologically active, but is three to four times less active than mirtazapine (4, 10).
Among selective serotonin reuptake inhibitors (SSRIs), citalopram, fluoxetine, and sertraline are initially demethylated, generating desmethyl citalopram, norfluoxetine, and norsertraline, which also inhibit SERT (11). Norcitalopram’s steady state plasma concentration approaches nearly 40% of citalopram’s, and it actively inhibits SERT three to eight times less than citalopram (12, 13). What is more, norcitalopram is converted into desmethyl citalopram, which acts as a weak SERT reuptake inhibitor (4, 14). Fluvoxamine is metabolized into multiple metabolites and the carboxylic acid derived from it has been shown to inhibit SERT weakly compared to the parent compound (4, 15).
Fluoxetine is metabolized through N-demethylation into its major active metabolite, norfluoxetine, which is comparable to fluoxetine, from pharmacological perspective (5, 16). However, norfluoxetine easily permeates the blood-brain barrier, reaches higher plasma concentrations, and has a higher half-life than fluoxetine (17, 18). In patients suffering from depression, norfluoxetine reaches concentrations comparable or even higher than fluoxetine (19), so it likely contributes considerably to the drug’s major SERT inhibition, therapeutic effectiveness, and adverse effects (4).
Sertraline gives way to a demethylated metabolite called desmethyl sertraline, which is a weaker SERT reuptake inhibitor than sertraline in vivo. However, it may contribute to the prolonged SERT transporter inhibition achieved by administering sertraline; laboratory research has shown this to be more the case for mice than for rats. Nevertheless, it remains unclear how much norsertraline boosts the original compound’s therapeutic activity in humans (4, 11).
Venlafaxine is a dual uptake inhibitor of SERT and NET. In humans, most venlafaxine metabolism, almost 60%, takes place through O-demethylation. Other minor metabolic routes include N-demethylation, which produces N-desmethyl venlafaxine. O-desmethyl venlafaxine exhibits similar pharmacological activity to that of its parent compound, both in vitro and in vivo. The derivative N-desmethyl is also active, but has less capacity for SERT and NET reuptake inhibition than venlafaxine does (4, 5, 20).
Atomoxetine is a selective NET reuptake inhibitor employed in the treatment of attention deficit hyperactivity disorder (ADHD). It generates a series of oxidative metabolites. Of those, 4-hydroxy-atomoxetine stands out because it blocks NET with demonstrably similar ability to atomoxetine. Atomoxetine’s remaining oxidative metabolites, including N-desmethyl atomoxetine, all seem to be pharmacologically inactive (21).
Active Metabolites of Atypical Antipsychotic Drugs with Antidepressant Activity
Antipsychotics have a long history of effectiveness at treating depressive profiles. In 1954, Kielhoz suggested chlorpromazine was effective at treating depressive and manic episodes. Then Nahunek et al. (22) reported that the first atypical antipsychotic, clozapine, effectively treated endogenous depression. Today, atypical antipsychotics are recognized as effective at treating bipolar depression (23, 24), but in unipolar depression profiles, its efficacy has been mainly shown as a combined, or adjunctive, treatment in resistant profiles, an indication for which certain antipsychotics have received official approval (25, 26). The question this article aims to address is whether any of these antipsychotics’ active metabolites (see Tables 1 and 2) might participate in mechanisms of antidepressant activity.
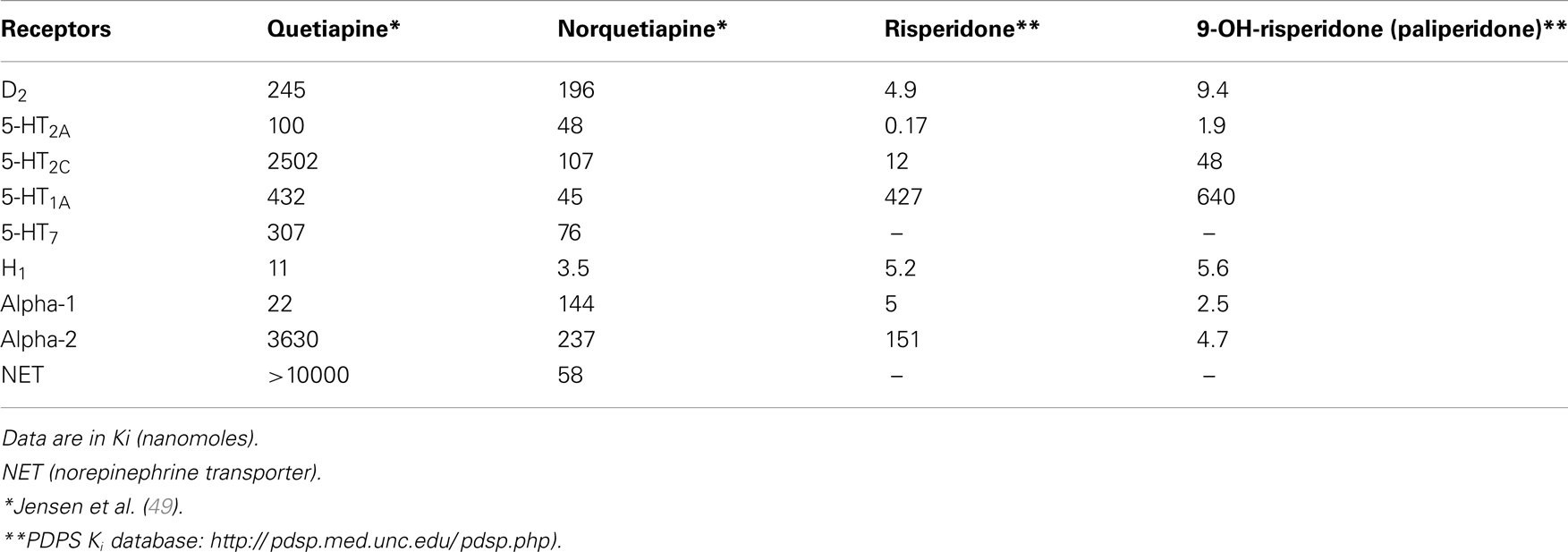
Table 2. Pharmacodynamic properties of quetiapina, risperidone, and their active metabolites (norquetiapine and paliperidone).
Olanzapine, coupled with fluoxetine, was the first case of an atypical antipsychotic drug showing greater efficacy than each active principle separately (26, 27). Olanzapine produces various metabolites, of which conjugated 10-N-glucuronide has noteworthy applications in humans. Other minor metabolites include N-desmethyl olanzapine, 4-N-oxide-olanzapine, 2-hydroxymethyl olanzapine, and 4-N-glucuronide. It is noteworthy that both in vitro and in vivo, its primary metabolite, 10-N-glucuronide, is inactive. Of the three remaining, minor metabolites, N-desmethyl olanzapine and 2-hydroxymethylolanzapine display similar activity in vitro as their parent compound, while the rest are inactive or nearly so. These data indicate that all of olanzapine’s metabolites are less active than it, so their activity probably does not contribute to its pharmacological profile (28).
Of the available atypical antipsychotics, aripiprazole has proven effective as adjunctive treatment for major depression when it is resistant to treatment with antidepressants (29, 30). FDA has officially approved this indication. Aripiprazole’s antidepressant action may be due, first, to its role as a partial agonist of D2, D3, and 5-HT1A receptors, and second, to its ability to block 5-HT2 receptors (29). It is a piperazine derivative and may undergo an array of metabolic processes, depending on the species. Dehydro-aripiprazole is aripiprazole’s major metabolite in humans and exhibits a similar pattern of activity at the D2 receptor (31). Dehydro-aripiprazole has a longer half-life than the original compound (32), suggesting it may contribute to its pharmacological efficacy. One of its metabolites, 2-3-dichlorophenylpiperazine, is an analog of m-Chlorophenylpiperazine (mCPP), an active metabolite of trazodone and nefazodone. This metabolite reaches a concentration in the brain 7% that of aripiprazole, so it could contribute only partially to that particular atypical antipsychotic’s antidepressant effect (33).
Risperidone has shown its effectiveness as adjunctive therapy, especially in conjunction with SSRIs, in patients with major depression (26). It is metabolized mainly by CYP450-2D6 (CYP2D6), generating 9-OH-risperidone, or paliperidone, through hydroxylation. When risperidone is administered, its pharmacological effects are due to the original molecule as well as its active metabolite. Considering that many patients who take risperidone orally exhibit plasma levels of 9-OH-risperidone 5–10 times higher than risperidone (34), we consider the metabolite to play an important role in the antipsychotic’s antidepressant effect (Table 2) (35). In fact, extended-release paliperidone, whether taken orally (36) or as an injection (37), has proven effective in treating schizoaffective disorder, a profile marked by a set of schizophrenia symptoms in addition to major depressive disorder. According to these clinical data, paliperidone seems to behave like a metabolite of risperidone and to have antidepressant activity. Nevertheless, to date, paliperidone has never been studied in the treatment of major depression, as monotherapy or adjunctive therapy. Yang and Liang (38) did, however, report one case where a patient with treatment-resistant depression was successfully treated with paliperidone as adjunctive therapy.
With regard to quetiapine, this particular atypical antipsychotic drug is extensively metabolized in the liver, where many of its metabolites are produced. Its most important metabolite is N-desalkyl quetiapine, also known as norquetiapine, which exhibits distinct pharmacological activity from quetiapine and plays a fundamental role in its antidepressant efficacy. This article will explore norquetiapine in depth in a special section below.
Norquetiapine: Active Metabolite of Quetiapine with Antidepressant Activity
The efficacy of quetiapine in treating psychotic disorders, both schizophrenia and mania, as well as mood disorders and anxiety disorders, leads us to view it as a multifunctional psychoactive drug. Its broad spectrum of efficacy is likely due to its ability to modify systems of dopaminergic, noradrenergic, and serotonergic neurotransmission. Its effects appear to be mediated by the actions of both quetiapine and norquetiapine.
Quetiapine is a dibenzothiazepine derivative available in two different pharmaceutical forms: immediate-release (IR) and extended-release (XR) (39). Both forms are metabolized extensively by the liver into various metabolites, with only 1% eliminated unaltered, in this case in urine (40).
N-desalkyl quetiapine, also known as norquetiapine, is quetiapine’s key metabolite and is produced by the action of isoenzyme CYP3A4 in cytochrome P450. No genetic polymorphisms affect CYP3A4, so variations in quetiapine metabolism based on race or genetics are unlikely. However, there may be interaction at the level of this isoenzyme with some inductors (carbamazepine, phenytoin) that increase the proportion of norquetiapine, or with potent enzyme inhibitors (ketoconazole, itraconazole, erythromycin, and fluvoxamine) that slow its production (41, 42). On another note, there is more pharmacokinetic variability among older adults and patients with concomitant medications in the case of quetiapine than norquetiapine; norquetiapine levels are more stable (43). CYP2D6 also contributes to quetiapine metabolism, though to a lesser extent. This creates small quantities of 7-hydroxy quetiapine (44), whose activity has not yet been specifically determined. In addition, this isoenzyme metabolizes norquetiapine into 7-hydroxy-desalkyl-quetiapine, which is pharmacologically active (45).
According to Mauri et al. (46), quetiapine’s plasma concentrations do not seem sufficiently high to explain its action at receptors or its clinical effects, suggesting that active metabolites participate in its pharmacodynamics. However, the plasma concentrations of CNS drugs is a poor predictor of central activity because numerous other factors influence target engagement, including blood/brain barrier penetration, CNS accumulation, and receptor association/dissociation kinetics.
Pharmacodynamic Profile of Norquetiapine vs. Quetiapine
Norquetiapine and quetiapine interact with the dopaminergic, noradrenergic, and serotonergic systems, but differ qualitatively and quantitatively in their respective abilities to alter those systems’ functioning. As with other atypical antipsychotics, quetiapine can block 5-HT2A receptors with greater affinity than D2 receptors, which may explain its antipsychotic properties and lower propensity to cause extrapyramidal symptoms and hyperprolactinaemia (40). Furthermore, quetiapine shows affinity for different serotonergic and noradrenergic receptors, which may contribute to its antidepressant and anxiolytic effects (47). Also, its active metabolite, norquetiapine, is highly important in that in addition to inhibiting NET reuptake, it shows affinity for several SERT receptors (41, 42).
An overall picture of the receptor profiles of quetiapine and norquetiapine appears in the Figure 1.
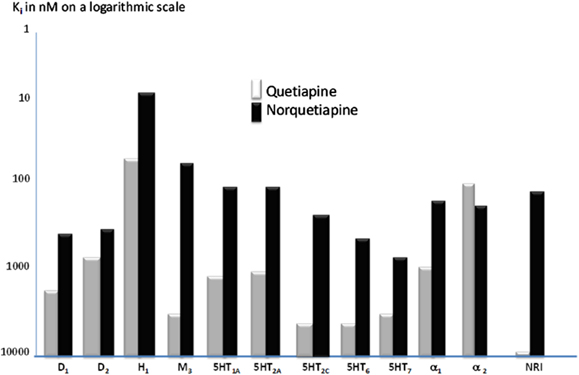
Figure 1. Binding affinities of quetiapine (white bars) and its metabolite N-Desalkylquetiapine (dark bars) at the tested panel of brain receptors neurotransmitter transporters (Ki in nanomoles on a logarithmic scale). Modified to Jensen et al. (49).
Dopaminergic Functioning
D2 receptor occupancy in the limbic system is the primary mechanism for bringing about antipsychotic effects. Quetiapine and norquetiapine both show moderate affinity for D1 and D2 dopamine receptor binding (Ki < 1 μM). Additionally, quetiapine dissociates rapidly from D2 receptors, so it must be administered at high dosages to exert an antipsychotic effect. It also exhibits good extrapyramidial tolerability and a low capacity for up-regulation at these receptors, which explains the low incidence of tardive dyskinesia associated with it (40, 48).
At the level of the nigrostriatal and tuberoinfundibular dopamine pathways, SERT seems to act as an inhibitory modulator by means of 5-HT2A receptor stimulation. Quetiapine and norquetiapine are 5-HT2A receptor antagonists (Ki 100 and 48 nM, respectively) (49), facilitating dopamine release along those pathways. PET studies have revealed higher 5-HT2A than D2 antagonism along said pathways (50), which explains why quetiapine is not linked to hyperprolactinaemia and is well tolerated from an extrapyramidal standpoint. Given that norquetiapine is more adept at blocking 5-HT2A than D2 receptors, its tolerability is similar or even better than quetiapine’s.
Moreover, greater dopamine release by quetiapine and norquetiapine not only improves its tolerability. Through various mechanisms, dopamine release in the mesocortical pathway could explain improved functioning in the prefrontal cortex (PFC) and hippocampus, which, in addition to relieving schizophrenia’s negative and cognitive symptoms, contributes to the drug’s antidepressant impact. Effectively, dopamine deficit in the PFC could explain some of the central symptoms of depression, such as anhedonia, social withdrawal, loss of motivation, and psychomotor retardation (51).
Dopamine release in the PFC is mediated by a complicated balance of several neurotransmitters and their receptors. As mentioned above, 5-HT2A receptor blockage produces a spike in nigrostriatal and tuberoinfundibular dopamine levels, but also facilitates mesocortical dopamine release.
Moreover, 5-HT2C receptor blockage eases cortical dopamine release (52), but that effect is secondary to inhibition of GABAergic interneurons in the brainstem (42). Thus, norquetiapine’s affinity for this receptor is low (Ki = 107 nM), but higher than its affinity for many dopamine receptors. Still, quetiapine would need concentrations 10 times higher (Ki = 2450 nM) than norquetiapine to block D2 receptors (49). Therefore, mesocortical dopamine release, mediated by 5-HT2A blockage, is essentially due to norquetiapine.
The NET transporter is responsible for dopamine reuptake in the PFC, and norquetiapine inhibits that transporter, not quetiapine, increasing dopamine levels in the frontal cortex and adding to the antidepressant effect (49, 52). Since norquetiapine is not formed in rodents, concomitant administration of quetiapine and reboxetine, a norepinephrine reuptake inhibitor, resulted in a synergistic increase in cortical dopamine output (53).
Serotonergic Functioning
In addition to the ability of quetiapine and norquetiapine to antagonize different SERT receptors, thereby easing dopamine transmission, those agents also facilitate SERT transmission. In effect, quetiapine, and to a greater extent its metabolite norquetiapine, facilitate serotonergic transmission by behaving as partial agonists at 5-HT1A receptors. 5-HT1A receptors are associated with antidepressant and anxiolytic effects in humans. Presynaptic 5-HT1A receptors in the raphe nuclei control serotonergic neurons’ release, while those located in postsynaptic limbic and cortical regions modulate serotonergic functioning. In fact, azapirones, like buspirone and gepirone, are partial agonists at 5-HT1A receptors whose anxiolytic and antidepressant properties have been demonstrated in controlled clinical trials (54).
Norquetiapine has a high affinity for 5-HT1A receptors (Ki = 45 nM), similar to that of conventional 5-HT1A agonists buspirone and gepirone (55) and 10 times greater than quetiapine’s (Ki = 430 nM). Moreover, in functional studies of GTPg-35s, the metabolite was also more efficacious than quetiapine itself (75 vs. 47% of maximum SERT response) (49).
Hence, norquetiapine can bring about activation at postsynaptic 5-HT1A receptors in the hippocampus (56), an area of the brain altered by depression. This could spur neuron regeneration in that area by increasing the release of nerve growth factors like brain-derived neurotrophic factor (BDNF) (57). Also, 5-HT1A receptor stimulation increases acetylcholine release in the PFC, promoting synaptic plasticity linked to learning and memory (58). Norquetiapine’s effect as a partial agonist of 5-HT1A receptors may contribute, at least in part, to quetiapine’s clinically demonstrated antidepressant and anxiolytic action (59).
Norquetiapine also has a higher affinity for the 5-HT7 receptor (Ki = 76 nM) than quetiapine (Ki = 307 nM). This receptor’s involvement in depression and sleep-related, circadian rhythm disorders has been experimentally documented (60). While no data are available about 5-HT7 antagonists’ effects in humans, it is possible that norquetiapine’s 5-HT7 receptor antagonism contributes to quetiapine’s antidepressant action (49).
As discussed above, quetiapine, and to a greater extent norquetiapine, have antagonistic properties at 5-HT2A receptors. Use of 5-HT2A antagonists has been proposed to treat insomnia, so perhaps norquetiapine, which is also a potent H1 receptor antagonist, contributes to the highly sedative effects observed during quetiapine treatment (49).
Norquetiapine has a high affinity (Ki = 14 nM) for 5-HT2B receptors (49). It is well-known that drugs that stimulate that particular receptor are closely tied to acute heart valve disease (61). However, norquetiapine binds to said receptor purely as an antagonist, not as an agonist, ruling out the possibility that it would cause valvular disease (49).
Some researchers have postulated that 5-HT2C receptor blockage, paired with H1 histamine receptor blockage, is linked to weight gain caused by treatment with antipsychotics (62, 63). Since norquetiapine binds to 5-HT2C receptors (Ki = 107 nM) with much more potency than quetiapine (Ki = 2502 nM), while also blocking histamine receptors (Ki = 3.5 nM), weight gain may be primarily the fault of this metabolite (49).
Noradrenergic Functioning
Norquetiapine acts as a potent NET transporter inhibitor (Ki = 23 nM), while quetiapine does not actively inhibit said transporter. Norquetiapine’s affinity for the NE transporter (Ki = 58 nM) is similar to that of certain other antidepressants like nortriptyline, amitriptyline, and duloxetine (49). Quetiapine, in contrast, cannot bind to the NET transporter. This differential feature of norquetiapine versus quetiapine may have an impact on antidepressant ability.
Also, norquetiapine increases noradrenergic functioning by blocking presynaptic α2 receptors (Ki = 237 nM) considerably more than quetiapine (Ki = 3630 nM) (49). 5-HT2A and 5-HT2C antagonism promotes this increase in NET, which in turn raises dopamine and NET levels in the PFC, as mentioned above (42, 49, 64, 65). By inhibiting the NET transporter, norquetiapine increases dopamine levels in the PFC, because dopamine also employs the NET transporter for reuptake (49, 52).
According to data collected in vivo, norquetiapine at dosages as low as 0.1 mg/Kg displayed antidepressant activity in the tail suspension test in mice (49).
Other Mechanisms Involved in the Antidepressant Effect of Quetiapine
Other alternative, much less thoroughly studied mechanisms than those involving monoamines may be at work in quetiapine’s antidepressant action. For example, stress and depression are known to increase neurodegeneration, and antidepressants may counteract that effect or undo it altogether. In experimental studies, quetiapine has been shown to prevent BDNF reduction as well as chronic stress-related cellular degeneration in the hippocampus. It has been postulate that these effects could improve some of the symptoms and cognitive deficits implicated in depression (66).
Additionally, quetiapine has been observed to modulate the activity of glutamate receptors to hypothetically restore them to normal functioning. This would decrease the neurotoxicity caused by an excess of the neurotransmitter glutamate (67).
Other authors have postulated that quetiapine may exert antidepressant effects by controlling inflammatory cytokines, cytoprotective molecules, and the antioxidant effect, all of which are mechanisms associated with the onset of depressive symptomatology (68), though there is scarcely any experimental or clinical support for that hypothesis. Norquetiapine’s role in those mechanisms has not yet been studied.
Norquetiapine’s Receptor Binding to Blame for Adverse Effects
The receptor profiles of quetiapine and norquetiapine may explain the adverse effects that often arise in treating depression. The ability of quetiapine (Ki = 11 nM) and norquetiapine (Ki = 3.5 nM) to bind to H1 histamine receptors is associated with sedation, hypnotic effects, increased appetite, and weight gain.
Blockage of α1 adrenergic receptors, which both quetiapine and norquetiapine exhibit, is linked to orthostatic hypertension in some patients. This is less true of quetiapine XR, which has fewer plasmatic “peaks.”
Norquetiapine shows affinity for muscarine receptors, always acting as an antagonist, especially at subtypes M1, M3, and M5. This antagonism is 20–80 times stronger than what was observed with quetiapine, so it is believed that adverse atropinic effects like dry mouth, urine retention, mydriasis, increased intraocular pressure, and hypothermia result from quetiapine’s active metabolite. On the other hand, some researchers postulate that M3 receptor antagonism is responsible, at least in part, for the onset of hyperglycemia and diabetes caused by antipsychotic treatment (69). It would seem that most of these adverse effects following quetiapine overdose (70) are due primarily to its active metabolite, norquetiapine (49).
Conclusion
Quetiapine is a multifunctional drug endowed with antidepressant properties through various mechanisms: noradrenergic, serotonergic, and dopaminergic. Quetiapine displays clinically significant efficacy in treating unipolar depression, both as a monotherapy (off-label use; only in Australia and Canada is this an officially approved indication) and as adjunctive therapy with antidepressants in patients who respond unsatisfactorily or not at all to antidepressants alone. Furthermore, quetiapine is the only drug demonstrated to be effective at treating unipolar and bipolar depression with no associated risk of veering into mania (68, 71).
The available data are sufficient to arrive at the conclusion that quetiapine’s antidepressant activity is mediated, at least in part, by the active metabolite norquetiapine, which selectively inhibits NET reuptake, is a partial 5-HT1A receptor agonist, and acts as an antagonist at presynaptic α2, 5-HT2C, and 5-HT7 receptors. Furthermore, its active metabolite contributes to quetiapine’s profile of undesirable effects because norquetiapine binds at H1 histamine, α1 adrenergic, and muscarine receptors. Norquetiapine’s activity profile is shared by no other known antipsychotic or antidepressant drug (49).
Norquetiapine clearly exemplifies the case of an active metabolite that, through metabolism, can make a drug originally introduced as an antipsychotic, in this case quetiapine, become a useful antidepressant agent.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wille SMR, Maudens KE, Van Peteghem CH, Lambert WEE. Development of a solid phase extraction for 13 ‘new’ generation antidepressants and their active metabolites for gas chromatographic–mass spectrometric analysis. J Chromatogr A (2005) 1098:19–29. doi:10.1016/j.chroma.2005.08.059
2. Tekes K, Hashemi F, Szegi P, Sotonyi P, Laufer R, Kalasz H. Prodrugs and active metabolites among antidepressive compounds. Neuropsychopharmacol Hung (2011) 13:103–10.
3. Fura A. Role of pharmacologically active metabolites in drug discovery and development. Drug Discov Today (2006) 11:133–42. doi:10.1016/S1359-6446(05)03681-0
4. Caccia S. Metabolism of the newer antidepressants an overview of the pharmacological and pharmacokinetic implications. Clin Pharmacokinet (1998) 34:281–302. doi:10.2165/00003088-199834040-00002
5. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther (1997) 283:1305–22.
6. Nordin C, Bertilsson L. Active hydroxymetabolites of antidepressants. Emphasis on E-10-hydroxy-nortriptyline. Clin Pharmacokinet (1995) 28:26–40. doi:10.2165/00003088-199528010-00004
7. Deupree JD, Montgomery MD, Bylund DB. Pharmacological properties of the active metabolites of the antidepressants desipramine and citalopram. Eur J Pharmacol (2007) 576:55–60. doi:10.1016/j.ejphar.2007.08.017
8. Haenisch B, Hiemke C, Bönisch H. Inhibitory potencies of trimipramine and its main metabolites at human monoamine and organic cation transporters. Psychopharmacology (Berl) (2011) 217:289–95. doi:10.1007/s00213-011-2281-9
9. Kang MJ, Song WH, Shim BH, Oh SY, Lee HY, Chung EY, et al. Pharmacologically active metabolites of currently marketed drugs: potential resources for new drug discovery and development. Yakugaku Zasshi (2010) 130:1325–37. doi:10.1248/yakushi.130.1325
10. Sitsen JMA, Zivkov M. Mirtazapine: clinical profile. CNS Drugs (1995) 4(Suppl 1):39–48. doi:10.2165/00023210-199500041-00007
11. Fuller RW, Hemrick-Luecke SK, Littlefield ES, Audia JE. Comparison of desmethylsertraline with sertraline as a monoamine uptake inhibitor in vivo. Prog Neuropsychopharmacol Biol Psychiatry (1995) 19:135–49. doi:10.1016/0278-5846(94)00110-4
12. Tatsumi M, Groshan K, Blakely RD, Richelson E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol (1997) 340:249–58. doi:10.1016/S0014-2999(97)01393-9
13. Popik P. Preclinical pharmacology of citalopram. J Clin Psychopharmacol (1999) 19(Suppl 1):4S–22S. doi:10.1097/00004714-199910001-00002
14. Milne CJ, Goa KL. Citalopram: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs (1991) 41:450–77. doi:10.2165/00003495-199141030-00008
15. Benfield P, Ward A. Fluvoxamine: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in depressive illness. Drugs (1986) 32:313–34. doi:10.2165/00003495-198632040-00002
16. Wong DT, Bymaster FP, Engleman EA. Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci (1995) 57:411–41. doi:10.1016/0024-3205(95)00209-O
17. Hirano K, Kimura R, Sugimoto Y, Yamada J, Uchida S, Kato Y, et al. Relationship between brain serotonin transporter binding, plasma concentration and behavioural effect of selective serotonin reuptake inhibitors. Br J Pharmacol (2005) 144:695–702. doi:10.1038/sj.bjp.0706108
18. Qu Y, Aluisio L, Lord B, Boggs J, Hoey K, Mazur C, et al. Pharmacokinetics and pharmacodynamics of norfluoxetine in rats: increasing extracellular level in the frontal cortex. Pharmacol Biochem Behav (2009) 92:469–73. doi:10.1016/j.pbb.2009.01.023
19. Renshaw PF, Guimaraes AR, Fava M, Rosenbaum JF, Pearlman JD, Flood JG, et al. Accumulation of fluoxetine and norfluoxetine in human brain during therapeutic administration. Am J Psychiatry (1992) 149:1592–4.
20. Muth EA, Moyer JA, Haskins JT, Andree TH, Husbands GEM. Biochemical, neurophysiological, and behavioral effects of Wy-45,233 and other identified metabolites of the antidepressant venlafaxine. Drug Dev Res (1991) 23:191–9. doi:10.1002/ddr.430230210
21. Sauer JM, Ponsler GD, Mattiuz EL, Long AL, Witcher JW, Thomasson HR, et al. Disposition and metabolic fate of atomoxetine hydrochloride: the role of CYP2D6 in human disposition and metabolism. Drug Metab Disp (2003) 31:98–107. doi:10.1124/dmd.31.1.98
22. Náhunek K, Rodová A, Svestka J, Kamenická V, Cesková E, Misurec J. Clinical experience with clozapine in endogenous depression. Act Nerv Super (Praha) (1973) 15:111.
23. Cruz N, Sánchez-Moreno J, Torres F, Goikolea JM, Valentí M, Vieta E. Efficacy of modern antipsychotics in placebo-controlled trials in bipolar depression: a meta-analysis. Int J Neuropsychopharmacol (2010) 13:5–14. doi:10.1017/S1461145709990344
24. Vieta E, Günther O, Locklear J, Ekman M, Miltenburger C, Chatterton ML, et al. Effectiveness of psychotropic medications in the maintenance phase of bipolar disorder: a meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol (2011) 14:1029–49. doi:10.1017/S1461145711000885
25. Nelson JC, Papakostas GI. Atypical antipsychotic augmentation in major depressive disorder: a meta-analysis of placebo-controlled randomized trials. Am J Psychiatry (2009) 166:980–91. doi:10.1176/appi.ajp.2009.09030312
26. Chen J, Gao K, Kemp DE. Second-generation antipsychotics in major depressive disorder: update and clinical perspective. Curr Opin Psychiatry (2011) 24:10–7. doi:10.1097/YCO.0b013e3283413505
27. Shelton RC, Tollefson GD, Tohen M, Stahl S, Gannon KS, Jacobs TG, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry (2001) 158:131–4. doi:10.1176/appi.ajp.158.1.131
28. Calligaro DO, Fairhurst J, Hotten TM, Moore NA, Tupper DE. The synthesis and biological activity of some known and Putative metabolites of the atypical antipsychotic agent Olanzapine (ly170053). Bioorg Med Chem Lett (1997) 7:25–30. doi:10.1016/S0960-894X(96)00567-7
29. Casey DE, Laubmeie KK, Marler SV, Forbes RA, Baker RA. Efficacy of adjunctive aripiprazole in major depressive disorder: a pooled response quartile analysis and the predictive value of week 2 early response. Prim Care Comp CNS Disord (2012) 14(3). doi:10.4088/PCC.11m01251
30. Nelson JC, Thase ME, Bellocchio EE, Rollin LM, Eudicone JM, McQuade RD, et al. Efficacy of adjunctive aripiprazole in patients with major depressive disorder who showed minimal response to initial antidepressant therapy. Int Clin Psychopharmacol (2012) 27:125–33. doi:10.1097/YIC.0b013e3283502791
31. Wood MD, Scott C, Clarke K, Westaway J, Davies CH, Reavill C, et al. Aripiprazole and its human metabolite are partial agonists at the human dopamine D2 receptor, but the rodent metabolite displays antagonist properties. Eur J Pharmacol (2006) 546:88–94. doi:10.1016/j.ejphar.2006.07.008
32. Swainston HT, Perry CM. Aripiprazole: a review of its use in schizophrenia and schizoaffective disorder. Drugs (2004) 64:1715–36. doi:10.2165/00003495-200464150-00010
33. Caccia S. Pharmacokinetics and metabolism update for some recent antipsychotics. Exp Opin Drug Metab Toxicol (2011) 7:829–46. doi:10.1517/17425255.2011.575061
34. De León J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics (2010) 51:80–8. doi:10.1176/appi.psy.51.1.80
35. Álamo C, López-Muñoz F. The pharmacological role and clinical applications of antipsychotics’ active metabolites: risperidone and paliperidone (9-OH risperidone). Clin Exp Pharmacol (2013) 3:117. doi:10.4172/2161-1459.1000117
36. Yang LP. Oral paliperidone: a review of its use in the management of schizoaffective disorder. CNS Drugs (2011) 25:523–38. doi:10.2165/11207440-000000000-00000
37. Canuso CM, Turkoz I, Fu DJ, Bossie CA. Role of paliperidone extended-release in treatment of schizoaffective disorder. Neuropsychiatr Dis Treat (2010) 6:667–79. doi:10.2147/NDT.S12612
38. Yang FW, Liang CS. Paliperidone augmentation for treatment-resistant depression: a case report. J Clin Psychopharmacol (2011) 31:245–6. doi:10.1097/JCP.0b013e31821083fb
39. Riedel M, Musil R, Spellmann I, Seemüller F, Möller HJ. Quetiapine XR – a new retard formulation in the treatment of schizophrenia. Eur Psychiatr Rev (2008) 1:70–5.
40. Peuskens J. The management of schizophrenia: focus on extended-release quetiapine fumarate. Neuropsychiatr Dis Treat (2011) 7:549–64. doi:10.2147/NDT.S3380
41. Winter HR, Earley WR, Hamer-Maanson JE, Davis PC, Smith MA. Steady-state pharmacokinetic, safety, and tolerability profiles of quetiapine, norquetiapine, and other quetiapine metabolites in pediatric and adult patients with psychotic disorders. J Child Adol Psychopharmacol (2008) 18:81–98. doi:10.1089/cap.2007.0084
42. Prieto E, Micó JA, Meana JJ, Majadas S. Neurobiological bases of quetiapina antidepresant effect in the bipolar disorder. Actas Esp Psiquiatr (2010) 38:22–32.
43. Bakken GV, Rudberg I, Molden E, Refsum H, Hermann M. Pharmacokinetic variability of quetiapine and the active metabolite N-desalkylquetiapine in psychiatric patients. Ther Drug Monit (2001) 33:222–6. doi:10.1097/FTD.0b013e31821160c4
44. Fisher DS, Handley SA, Taylor D, Flanagan RJ. Measurement of quetiapine and four quetiapine metabolites in human plasma by LC-MS/MS. Biomed Chromatogr (2012) 26:1125–32. doi:10.1002/bmc.2672
45. Bakken GV, Molden E, Knutsen K, Lunder N, Hermann M. Metabolism of the active metabolite of quetiapine, N-desalkylquetiapine in vitro. Drug Metab Dispos (2012) 40:1778–84. doi:10.1124/dmd.112.045237
46. Mauri MC, Volonteri LS, Colasanti A, Fiorentini A, De Gaspari IF, Bareggi SR. Clinical pharmacokinetics of atypical antipsychotics: a critical review of the relationship between plasma concentrations and clinical response. Clin Pharmacokinet (2007) 46:359–88. doi:10.2165/00003088-200746050-00001
47. Sanford M, Keating GM. Quetiapine: a review of its use in the management of bipolar depression. CNS Drugs (2012) 26:435–60. doi:10.2165/11203840-000000000-00000
48. Seeman P, Tallerico T. Rapid release of antipsychotic drugs from dopamine D2 receptors: an explanation for low receptor occupancy and early clinical relapse upon withdrawal of clozapine or quetiapine. Am J Psychiatry (1999) 156:876–84.
49. Jensen NH, Rodriguiz RM, Caron MG, Wetsel WC, Rothman RB, Roth BL. N-desalkylquetiapine, a potent norepinephrine reuptake inhibitor and partial 5-HT1A agonist, as a putative mediator of quetiapine’s antidepressant activity. Neuropsychopharmacology (2008) 33:2303–12. doi:10.1038/sj.npp.1301646
50. Gefvert O, Lundberg T, Wieselgren IM, Bergström M, Langstrom B, Wiesel F, et al. D(2) and 5HT(2A) receptor occupancy of different doses of quetiapina in schizophrenia: a PET study. Eur Neuropsychopharmacol (2001) 11:105–10. doi:10.1016/S0924-977X(00)00133-4
51. Montgomery SA. The under-recognize role of dopamine in the treatment of major depressive disorder. Int Clin Psychopharmacol (2008) 23:63–9. doi:10.1097/YIC.0b013e3282f2b3cb
52. Möller HJ. Antipsychotic and antidepressive effects of second generation antipsychotics. Two different pharmacological mechanisms? Eur Arch Psychiatry Clin Neurosci (2005) 255:190–201. doi:10.1007/s00406-005-0587-5
53. Björkholm C, Jardemark K, Marcus MM, Malmerfelt A, Nyberg S, Schilström B, et al. Role of concomitant inhibition of the norepinephrine transporter for the antipsychotic effect of quetiapine. Eur Neuropsychopharmacol (2012). doi:10.1016/j.euroneuro.2012.05.012 Available at: http://dx.doi.org/10.1016/j.euroneuro.2012.05.012,
54. El-Khalili N, Joyce M, Atkinson S, Buynak RJ, Datto C, Lindgren P, et al. Extended-release quetiapina fumarate (quetiapine XR) as adjunctive therapy in major depressive disorder (MDD) in patients with an inadequate response to ongoing antidepressant treatment: a multicentre, randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol (2010) 13:917–32. doi:10.1017/S1461145710000015
55. Becker OM, Dhanoa DS, Marantz Y, Chen D, Shacham S, Cheruku S, et al. An integrated in silico 3D model-driven discovery of a novel, potent, and selective amidosulfonamide 5-HT1A agonist (PRX-00023) for the treatment of anxiety and depression. J Med Chem (2006) 49:3116–35. doi:10.1021/jm0508641
56. McIntyre RS, Muzina DJ, Adams A, Lourenco MT, Law CW, Soczynska JK, et al. Quetiapine XR efficacy and tolerability as monotherapy and as adjunctive treatment to conventional antidepressants in the acute and maintenance treatment of major depressive disorder: a review of registration trials. Exp Opin Pharmacother (2009) 10:3061–75. doi:10.1517/14656560903448837
57. Fumagalli F, Molteni R, Bedogni F, Gennarelli M, Pérez J, Racagni G, et al. Quetiapine regulates FGF-2 and BDNF expression in the hippocampus of animals treated with MK-801. Neuroreport (2004) 15:2109–12. doi:10.1097/00001756-200409150-00022
58. Ichikawa J, Li Z, Dai L, Meltzer HY. Atypical antipsychotic drugs, quetiapine, iloperidone, and melperone, preferentially increase dopamine and acetylcholine release in rat medial prefrontal cortex: role of 5-HT1A receptor agonism. Brain Res (2002) 956:349–57. doi:10.1016/S0006-8993(02)03570-9
59. Figueroa C, Brecher M, Hamer-Maansson JE, Winter H. Pharmacokinetic profiles of extended release quetiapine fumarate compared with quetiapina immediate release. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33:199–204. doi:10.1016/j.pnpbp.2008.09.026
60. Guscott M, Bristow LJ, Hadingham K, Rosahl TW, Beer MS, Stanton JA, et al. Genetic knockout and pharmacological blockade studies of the 5-HT7 receptor suggest therapeutic potential in depression. Neuropharmacology (2005) 48:492–502. doi:10.1016/j.neuropharm.2004.11.015
62. Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet (2005) 20:368–78. doi:10.2133/dmpk.20.368
63. Kirk SL, Glazebrook J, Grayson B, Neill JC, Reynolds GP. Olanzapine-induced weight gain in the rat: role of 5-HT2C and histamine H1 receptors. Psychopharmacology (Berl) (2009) 207:119–25. doi:10.1007/s00213-009-1639-8
64. Nyberg S, Takano A, Grimm S, Gulyas B, McCarthy D, Lee C, et al. PET-measured D2, 5-HT2, and norepinephrine transporter (NET) occupancy by quetiapina and N-desalkyl-quetiapine in non-human primates. Eur Neuropsychopharmacol (2007) 17:S254–5. doi:10.1016/S0924-977X(07)70346-2
65. Möller H-J, Johnson S, Mateva T, Brecher M, Svensson O, Miller F, et al. Evaluation of the feasibility of switching from quetiapine immediate release to quetiapine extended release in stable outpatients with schizophrenia. Int Clin Psychopharmacol (2008) 23:95–105. doi:10.1097/YIC.0b013e3282f2d42c
66. Xu H, Chen Z, He J. Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stress-induced decrease in cell proliferation and BDNF expression in rat hippocampus. Hippocampus (2006) 16:551–9. doi:10.1002/hipo.20184
67. Ichikawa J, Ishii H, Bonaccorso S, Fowler WL, O’Laughlin IA, Meltzer HY. 5-HT(2A) and D(2) receptor blockade increases cortical DA release via 5-HT(1A) receptor activation: a possible mechanism of atypical antipsychotic-induced cortical dopamine release. J Neurochem (2001) 76:1521–31. doi:10.1046/j.1471-4159.2001.00154.x
68. Shelton RC, Papakostas GI. Augmentation of antidepressants with atypical antipsychotics for treatment-resistant major depressive disorder. Acta Psychiatr Scand (2008) 117:253–9. doi:10.1111/j.1600-0447.2007.01130.x
69. Johnson DE, Yamazaki H, Ward KM, Schmidt AW, Lebel WS, Treadway JL, et al. Inhibitory effects of antipsychotics on carbachol-enhanced insulin secretion from perifused rat islets: role of muscarinic antagonism in antipsychotic-induced diabetes and hyperglycemia. Diabetes (2005) 54:1552–8. doi:10.2337/diabetes.54.5.1552
70. Balit CR, Isbister GK, Hackett LP, Whyte IM. Quetiapine poisoning: a case series. Ann Emerg Med (2003) 42:751–8. doi:10.1016/S0196-0644(03)00600-0
71. Bauer M, Pretorius HW, Constant EL, Earley WR, Szamosi J, Brecher M. Extended-release quetiapina as adjunct to an antidepressant in patients with major depressive disorder: results of a randomized, placebo-controlled, double-blind study. J Clin Psychiatry (2009) 70:540–9. doi:10.4088/JCP.08m04629
Keywords: active metabolites, antipsychotic drugs, norquetiapine, quetiapine, mood disorders
Citation: López-Muñoz F and Álamo C (2013) Active metabolites as antidepressant drugs: the role of norquetiapine in the mechanism of action of quetiapine in the treatment of mood disorders. Front. Psychiatry 4:102. doi: 10.3389/fpsyt.2013.00102
Received: 28 February 2013; Accepted: 27 August 2013;
Published online: 12 September 2013.
Edited by:
Andrew C. McCreary, Brains On-Line, NetherlandsReviewed by:
Adrian Newman-Tancredi, Neurolixis Inc., USABart Ellenbroek, Victoria University Wellington, New Zealand
Copyright: © 2013 López-Muñoz and Álamo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco López-Muñoz, Faculty of Health Sciences, Camilo José Cela University, C/Castillo de Alarcón, 49, Urb. Villafranca del Castillo, Villanueva de la Cañada, 28692 Madrid, Spain e-mail: francisco.lopez.munoz@gmail.com