 Nicola Lopizzo1
Nicola Lopizzo1 Luisella Bocchio Chiavetto1,2
Luisella Bocchio Chiavetto1,2 Nadia Cattane1
Nadia Cattane1 Giona Plazzotta1
Giona Plazzotta1 Frank I. Tarazi3
Frank I. Tarazi3 Carmine M. Pariante4
Carmine M. Pariante4 Marco A. Riva5
Marco A. Riva5 Annamaria Cattaneo1,4*
Annamaria Cattaneo1,4*- 1IRCCS Fatebenefratelli San Giovanni di Dio, Brescia, Italy
- 2Faculty of Psychology, eCampus University, Novedrate, Como, Italy
- 3Department of Psychiatry and Neuroscience Program, McLean Hospital, Harvard Medical School, Belmont, MA, USA
- 4Stress, Psychiatry and Immunology Laboratory, Department of Psychological Medicine, Institute of Psychiatry, King’s College London, London, UK
- 5Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
Major depressive disorder (MDD) is a multifactorial and polygenic disorder, where multiple and partially overlapping sets of susceptibility genes interact each other and with the environment, predisposing individuals to the development of the illness. Thus, MDD results from a complex interplay of vulnerability genes and environmental factors that act cumulatively throughout individual’s lifetime. Among these environmental factors, stressful life experiences, especially those occurring early in life, have been suggested to exert a crucial impact on brain development, leading to permanent functional changes that may contribute to lifelong risk for mental health outcomes. In this review, we will discuss how genetic variants (polymorphisms, SNPs) within genes operating in neurobiological systems that mediate stress response and synaptic plasticity, can impact, by themselves, the vulnerability risk for MDD; we will also consider how this MDD risk can be further modulated when gene × environment interaction is taken into account. Finally, we will discuss the role of epigenetic mechanisms, and in particular of DNA methylation and miRNAs expression changes, in mediating the effect of the stress on the vulnerability risk to develop MDD. Taken together, we aim to underlie the role of genetic and epigenetic processes involved in stress- and neuroplasticity-related biological systems on the development of MDD after exposure to early life stress, thereby building the basis for future research and clinical interventions.
Background
Major depressive disorder (MDD) is a leading cause of disability and disease affecting millions of individuals worldwide (1, 2). MDD includes alterations in mood, neurovegetative functions, cognition, and psychomotor activity, and it represents a major public health concern because of the short- and long-term detrimental effects to the patients and for the co-morbidities to prevalent health outcomes, such as cardiovascular disease and metabolic syndrome. The lifetime prevalence is estimated to be approximately 10–15%, and the World Health Organization has predicted that, by the year 2030, MDD will account for the 13% of the total global burden of disease replacing cardiovascular disorders (3). Due to the heterogeneity of the disorder, the diagnosis requires the presence of several symptoms including mood changes, loss of interest or pleasure, significant loss of appetite and weight, insomnia/hypersomnia, psychomotor agitation/slowing, fatigue/loss of energy, feelings of worthlessness/inappropriate guilt, inability to concentrate/indecisiveness, recurrent thoughts of death/suicide with negative consequences on work, and social relations (4, 5).
Although the etiopathogenesis of the disease is not fully understood yet, a genetic component has been recognized as exerting an important impact (6, 7). Indeed, several genetic vulnerability factors are associated with an increased risk to develop MDD, as indicated by family, twin, and adoption studies (8, 9). In particular, these studies indicate that there is a two to threefold increase in lifetime risk of developing MDD among first-degree relatives, with a heritability risk that is estimated to be around the 0.37 (95% confidence interval 0.31–0.42) based on meta-analysis data (10–12).
However, the genetic vulnerability background is not able to explain by itself the disease development. This is mainly because MDD is a complex disorder where no single gene is sufficient to cause MDD; on the contrary, each susceptibility gene contributes to a small fraction of the total genetic risk. Moreover, to make things even more complicated, MDD is known to be characterized by a complex genetic heterogeneity, meaning that multiple and partially overlapping sets of susceptibility genes interact each other and with the environment, predisposing individuals to mood disorders (7).
Genetic Vulnerability Risk Factors for Major Depression
To date, as part of a concerted effort to understand the genetic contribution to major mental illnesses, many genetic association studies based on candidate genes or genome-wide approaches have been performed with the aim to identify susceptibility loci for MDD. However, a recent meta-analysis of genetic association studies on MDD (13) clearly concluded that candidate genes-based studies have provided only little support for the identification of vulnerability genes. Indeed, although a significant association between MDD and several genes, including apolipoprotein E (APOE) (14–16), piccolo presynaptic cytomatrix protein (PCLO) (17–19), translocase of outer mitochondrial membrane 40 homolog (TOMM40) (20, 21), guanine nucleotide binding protein (G protein) beta polypeptide 3 (GNB3) (22–24), methylenetetrahydrofolate reductase (MTHFR) (25, 26), and solute carrier family 6 (neurotransmitter transporter) member 4 (SLC6A4) (27–29) have been found in several studies, a difficulty to confirm such associations in independent samples has been also reported (30–33).
Over the past years, genome-wide association studies (GWAS), which test simultaneously common SNPs and copy number variations (CNVs), have quickly substituted the candidate genes association studies. However, they have not been able as well to clearly identify gene variants associated with MDD vulnerability (34). One of the first largest GWAS study was performed by Sullivan et al. (17) in 1,738 MDD cases and 1,802 controls, which allowed the identification of 11 signals localized to a 167 kb region overlapping the gene piccolo (PCLO). However, when they undertook validations of these SNPs in five independent samples, they were not able to replicate the findings. Subsequent GWAS and meta-analysis studies of MDD were equally unsuccessful in validating SNPs previously associated with MDD vulnerability (31, 35–37). Recently, also the largest Psychiatric GWAS Consortium study on MDD, where 9,500 cases were taken into account, has not been able to detect any significant genome-wide association (38).
The failure of GWAS analyses to provide robust evidence for loci that exceed genome-wide significance is compatible with a paradigm where MDD results from the combined action of multiple loci of small effect together with a variety of environmental factors. In line with this, when researchers started to look at the interaction between genes of vulnerability for MDD and the environment they found more robust results.
The studies above reported have been summarized in Table 1.
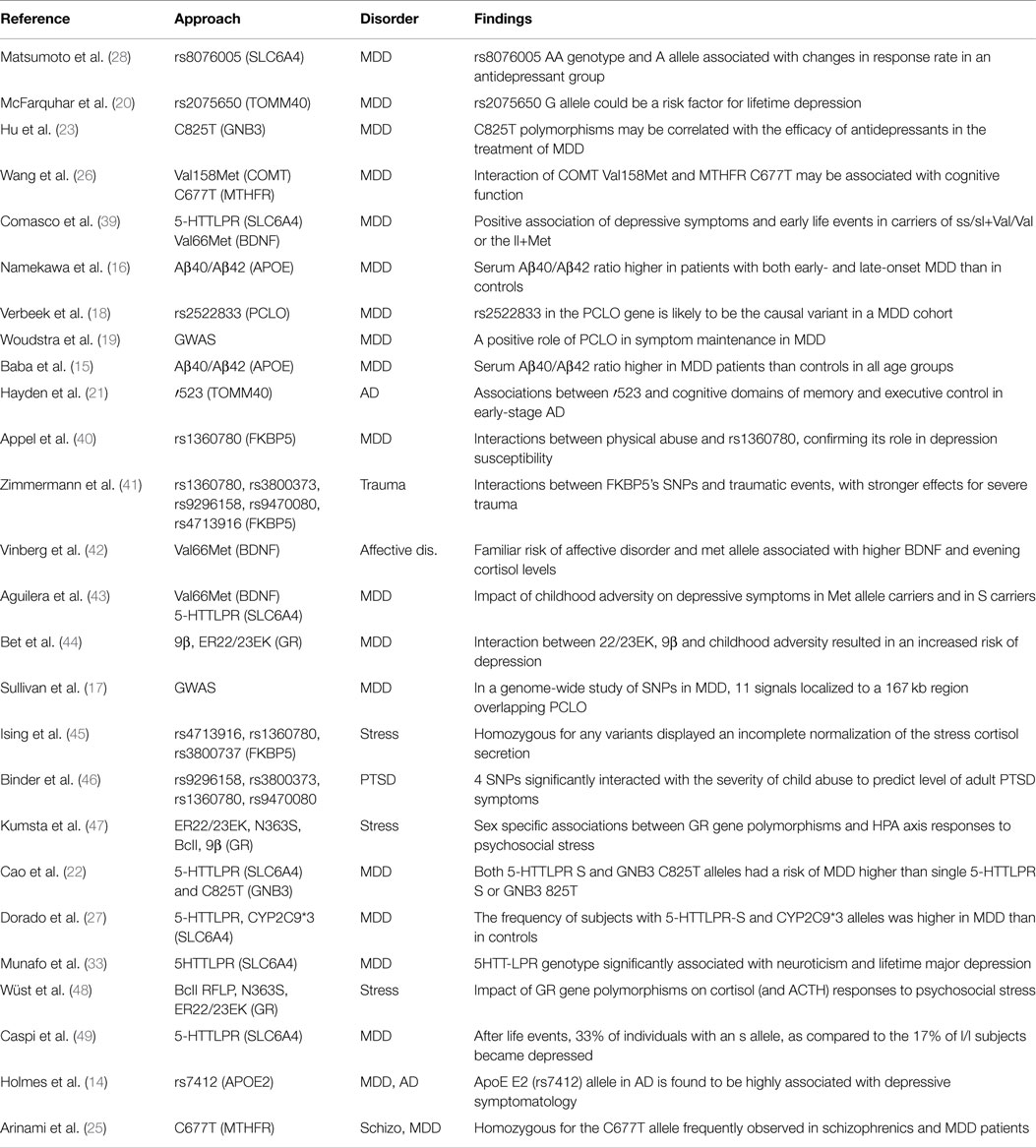
Table 1. Genetic approach for gene–environment interaction.
Gene–Environment Interaction in Major Depression
Gene–environment interactions reflect a causal mechanism where one or more genetic variants and one or more environmental factors contribute to the causation of a condition in the same individual with genetic factors influencing the sensitivity to environmental exposures (50). A number of environmental factors have been found to contribute to depression vulnerability, including in utero exposure to infection, lack of nutrients, maternal stress, perinatal complications, social disadvantage, urban upbringing, ethnic minority status, childhood maltreatment, bullying, traumatic events, cannabis use, and exposure to stress (51–55). Among these environmental factors, stressful life experiences, especially those occurring early in life, have been suggested to exert a crucial impact on brain development leading to permanent functional changes that may contribute to lifelong risk for mental health outcomes (56–58). Indeed, during periods of heightened neural plasticity throughout development, brain regions involved in the regulation of emotion and in the mediation of the stress response appear to be particularly sensitive to the effects of stressful events. Such experience-dependent plasticity may produce altered neural circuits and maladaptive responsiveness to the environment that, ultimately, lead to an enhanced risk for depression (59).
This also suggests that genes and environment start to interact early during the development and that higher liability to psychopathology may originate when environmental challenges do occur in the pre-perinatal period. This situation may result in a variety of outcomes based on the severity of both genetic and environmental profiles for a particular disorder, and also on the presence or absence of other protective factors, which may modulate the risk for subsequent psychopathology (60). Stressful experiences during early childhood can thus significantly undermine the development of adaptive mechanisms required to deal with challenges in the adulthood and may also contribute for unhealthy lifestyles, negative interpersonal relationship, and poor health outcomes (61–63).
The mechanisms that mediate the impact of early life adversities on depression risk have been object of studies over decades. Chapman et al. (64) reported a dose–response relationship between the severity of experienced childhood adversities and the presence of a depressive episode or lifetime chronic depression. The experience of any childhood adversity has been indeed associated with an increased risk of suicide attempts in different period of life including childhood, adolescence, and also adulthood (65). In addition to maltreatment, parental loss due to death or separation or an adverse family environment characterized by poor paternal relationships or maternal overprotection has been also associated with increased risk for depressive disorders (66–69). However, it is important to mention that, although early life stressful experiences have an impact on MDD vulnerability, they do not lead to psychiatric disorders in all the exposed individuals, since the outcome is highly dependent on the individual genetic background, which, in turn, can regulate/influence the mechanisms of coping to stressful stimuli (70, 71).
On these bases, a large body of research has thus focused on identifying genetic variations that interact with early life adversities in predicting current or lifetime MDD diagnosis, or symptom severity. Here below, we discuss how genes involved in stress response and neuroplasticity interact with the environment, in particular with stressful experiences, modulating the risk for MDD.
Caspi et al. (49) were the first showing the effect of gene and environment interaction in modulating the risk for depression development. They focused on the role of a common functional 43 bp insertion/deletion polymorphism (5-HTTLPR) in the promoter region of the serotonin transporter gene (SLC6A4 or SERT). This polymorphism is known to involve either short (s) or long (l) alleles, with the short allele variant being associated with lower promoter transcriptional efficiency (59). The authors, studying the Dunedin Birth Cohort of New Zealand, showed that 5-HTTLPR interacts with stressful life events, in particular childhood maltreatment, to predict current and lifetime diagnoses of MDD as well as suicide attempts. They reported that, in the context of exposure to stressful life events, subjects’ homozygotes (s/s) or heterozygotes (s/l) for the short allele exhibited more depressive symptoms, diagnosable depression, and increased suicide behavior compared to individuals homozygous for the long allele (l). In particular, if exposed to stressful experiences during life, the 33% of individuals with an s allele, as compared to the 17% of l/l homozygotes subjects became depressed (49).
Other similar studies have been then performed to investigate the role of 5-HTTLPR short variant in increasing the vulnerability for depression development upon exposure to environmental adversity, providing both replications and failures of the original finding (72–76). Recent meta-analysis indicated, however, an absence of a clear association: indeed, although 17 positive replication studies were reported, also 8 partial replications (interaction only in females or only with one of several types of adversity) and 9 non-replications (no interaction or an interaction in the opposite direction) were indicated (74, 77, 78). The possible causes of such discrepancies could be due to potential sources of this heterogeneity, mainly stemming from methodological differences between studies (77). For example, the method of assessing early life stressful experiences, as well as the type and timing of ELS, appear to be important modifiers (75). In addition, the definition of the outcome variable seems to exert a particular influence. Furthermore, it has now been reported that the interaction between 5-HTTLPR and ELS in predicting depression is moderated by other polymorphisms in other genes (39, 79).
Another gene that may modulate the impact of stress on depression vulnerability is the brain-derived neurotrophic factor (BDNF). BDNF is a neurotrophin widely expressed in the brain where it is implicated in neuronal growth, synaptic plasticity, and neuronal survival, and it plays important roles in structural brain abnormalities observed in depressed individuals, such as reduced hippocampal volume or cognitive deficits (80–83).
A functional SNP (rs6265) within the BDNF gene promoter, which causes a valine to methionine substitution at codon 66 in the BDNF gene (Val66Met), has been shown to influence the activity of the BDNF protein (84–86). Moreover, the Met allele of the BDNF Val66Met polymorphism has been found associated with a reduced BDNF activity (87), memory impairment (84), harm avoidance (88), brain volume reduction, and has been also shown to affect intracellular trafficking (89). Moreover, individuals, which are heterozygotes or homozygotes for the Met allele, have elevated evening cortisol levels, suggesting an altered HPA axis functionality (42). Interestingly, still in the context of stress, interaction between this polymorphism and childhood sexual abuse in the prediction of adult depression has been demonstrated (43). Several studies have further provided evidence that the BDNF Val66Met polymorphism interact with ELS in predicting depression (59, 90) and a recent meta-analyses, which combined the results from 22 different studies, supported the idea that BDNF Val66Met polymorphism significantly moderates the relationship between life stress and depression (91).
The regulating effects of stress on brain and behavior are mediated by the binding of the stress hormone, cortisol, to specific receptors: the high-affinity mineralocorticoid receptors (MR), which exert tonic inhibitory effects on basal HPA axis activity, and the high-affinity glucocorticoid receptors (GR), which are critical in regulating stress responses characterized by elevated cortisol levels. The GR is kept at cytosolic level in an inactive state by the binding with FK506 binding protein 51 (FKBP5), a co-chaperone of hsp90, which prevents its translocation to the nucleus. When cortisol binds the GR-hsp90 complex, FKBP-5 acquires a reduced affinity for the complex and it is substituted by another co-chaperone named FKBP4 that, on the contrary, facilitates nuclear GR translocation leading to transcription of GR-dependent genes. An alteration in GR functionality, also known as GR resistance, is a well-characterized feature of depression and it has been associated with HPA axis hyperactivity. Given the important role of GR in regulating stress responses and the evidence for GR resistance in depression, polymorphisms within GR or FKBP5 genes have been studied as possible genetic factors for depression vulnerability. To this regard, Wüst et al. (48) have shown an impact of polymorphisms within GR gene on cortisol and ACTH responses to psychosocial stress. Specifically, 112 healthy males were studied to estimate the impact of three GR gene polymorphisms (BclI RFLP, N363S, ER22/23EK) on cortisol and ACTH responses to psychosocial stress (Trier social stress test) with the conclusion of a strong association (48).
Moreover, a strong gene × environment interaction involving functional polymorphisms within GR gene has also been shown by Bet et al. (44). In particular, the authors investigated the GR 22/23EK and 9β polymorphisms showing how they do predict the development of clinically relevant depression by interacting with environmental adversities during adolescence and childhood including war experiences, sexual abuse, parental loss, or physical illness (44). This evidence has been supported by Kumsta and colleagues, which showed a significant sex specific association between GR gene polymorphisms in 9β and BclI and HPA axis responses to psychosocial stress as well as GC sensitivity (47). These findings support the relevance of GR gene polymorphisms in HPA axis regulation and suggest that they might represent a risk factor in the development of stress-related disorders like MDD.
Also, variants within the FKBP5 gene have been shown to modulate the risk of developing MDD in relation to stressful experiences (92). For example, Appel et al. (40) reported a significant gene × environment interaction by investigating the effect of a functional SNP within FKBP5 gene (rs1360780) on depression development in more than 2,000 German people. In particular, they found that TT subjects with a history of physical abuse had an enhanced risk for depression development when compared to CC/CT individuals. This interaction between the rs1360780 and traumatic life events in predicting the onset of MDD was confirmed by another study performed in 884 adolescent and young adult individuals characterized for traumatic life events (UK environmental risk longitudinal twin study) (41). Subsequently, the same SNPs within the FKBP5 gene have been associated also with peritraumatic dissociation among injured children (93), with recovery from psychosocial stress in normal controls (45), and also with the association between childhood abuse and the development of post-traumatic stress disorder (46). Another evidence that supports the role of FKBP5 as mediator of stress exposure comes from a study where a particular FKBP5 haplotype of four SNPs (rs3800373, rs9296158, rs1360780, rs9470080) was found associated with an increased risk of suicide attempts but only in individuals with a history of childhood trauma (94).
These results, taken together, strongly support a role of the FKBP5 gene in the pathogenesis of stress-related depression, likely mediated through the influence of individual level of GR resistance and, consequently, glucocorticoid signaling.
Epigenetic Mechanisms as Mediators of the Effect of the Environment on the Genome: Focus on DNA Methylation and miRNAs
The term “epigenetics” refers to the potentially heritable, but environmentally modifiable, regulation of genetic function and expression (95). Among epigenetic processes, DNA methylation is one of the major epigenetic processes studied in the context of early life adversities as a potential mechanism to explain the long-term effects on gene transcription (96–98). DNA methylation is a covalent modification of the cytosine residues that are located primarily at CpG dinucleotide sequences in mammals; methylation changes within promoter and enhancer regions of the gene are particularly important as they reduce the access of transcription factors to regulatory elements and promote silencing of gene expression (99). The contribution of DNA methylation has been extensively investigated especially in the context of pathologies related to exposure to stressful life events (especially those occurring early in life, ELS) including depression or post-traumatic stress disorder (73, 95, 98, 100, 101).
Studies conducted both in animal models and in humans have shown that ELS can leave persistent epigenetic marks on the genome, which can influence neurobiological substrates until adulthood (56). Indeed, early-life exposures can disrupt epigenetic programing in the brain, with long-lasting consequences for gene expression and behavior (102).
Altered DNA methylation profiles in response to ELS have been observed not only in specific candidate genes, both at central or peripheral level (103, 104), but also on a genome-wide level (105, 106). Recently, Nagy and colleagues found differentially methylated regions of DNA in astrocytes, which are glial cells specific to the CNS, of human prefrontal cortex between cases (depressed patients) and control subjects, revealing reduced methylation levels in cases (107). Indeed, emerging evidence has suggested that ELS can induce structural brain changes through epigenetic mechanisms (108). These alterations include a loss of dendritic spines and synapses, a reduced dendritic arborization together with diminished glial cells and have been widely found in the hippocampus of MDD subjects (109). Interestingly, antidepressant treatment can reverse stress-induced structural changes augmenting dendritic arborization and synaptogenesis (109).
Another quite new area in epigenetic research is represented by small RNAs, in particular microRNAs (miRNAs), which are small non-coding RNAs (20–22 nt) that play a major role in post-transcriptional regulation of gene expression. miRNAs are predicted to influence the expression of more than 60% of all the protein-coding genes through a post-transcriptional mechanism by base-pairing to target mRNAs. Generally, miRNAs inhibit protein synthesis either by repressing translation or by inducing deadenylation and degradation of target mRNAs, but were also reported to activate translation (110, 111). Individual miRNAs have the potential to target hundreds of different mRNAs, and a single gene could be modulated by several different miRNAs, thus implying a coordinate and fine-tuned expression of proteins in a cell and even, in particular, cell compartments (112, 113).
It is also known that environmental factors may modify gene expression through the regulation of miRNA synthesis (114), and there is evidence for a bilateral interaction between miRNA expression and other biological processes modulated by environment including DNA methylation or histone modifications (95, 115–117).
Examples of the effect of environmental factors on miRNAs come from preclinical data, where rats exposed to chronic stress showed an increased expression of miR-186 and miR-381 and a down-regulation of miR-709; interestingly, such alterations persisted over time as miR-709 alterations were still present also after 2 weeks of recovery from stress, suggesting that miRNAs changes can persist over time and can mediate altered stress-induced behaviors (118). Moreover, a maternal separation paradigm was able to up-regulate the expression of miR-132 and miR-124 in the prefrontal cortex of 14-day-old pups, an effect that, again, can be observed also in adult rats (119). These long-lasting modifications of miRNAs may also result from epigenetic changes since a recent human study identified miRNAs possible target of genome methylation after childhood abuse exposure (120). In this study, the authors were able to demonstrate an abuse-associated hypermethylation in 31 miRNAs in a sample of adult males exposed to childhood abuse and, at least for 6 of these miRNAs, the hypermethylated state was consistent with the hypomethylation of their gene targets (120).
Here below, we now report some examples of epigenetic processes (DNA methylation and miRNAs changes) as modulators of target genes involved in neuroplasticity, serotoninergic transmission, and GR functionality and, in particular, we will focus on the same genes we have already discussed in previous paragraphs as involved in gene × environment interaction.
The neurotrophin BDNF is a molecule highly sensible to stress, and its expression is reduced in key brain regions of animal models of depression (121–123) and also in the blood of depressed patients (124–126). In order to better understand the possible mechanisms underlying these changes, researchers investigated the role of DNA methylation within BDNF promoter regions. For example, adult rats exposed to early maltreatment showed reduced BDNF mRNA levels in the prefrontal cortex, as a result of an increase in the methylation of CpG sites within the promoter region of the BDNF exon IV. These changes persisted through adolescence and were maintained up to adulthood. Moreover, alterations in the methylation pattern at BDNF IV exon was also found in the offspring of females that had previously experienced the maltreatment regimen, indicating that these epigenetic processes can be perpetuated from one generation to the next (127).
These data have been supported by human studies. Fuchikami and collaborators examined the methylation profile of two CpG islands localized within two promoter regions (I and IV) of the BDNF gene in peripheral blood from depressed patients and controls reporting a significant difference at the promoter of exon I between the two groups (128). In another study, Kang and colleagues (129) found a significant increase of BDNF gene methylation in depressed patients with suicidal attempt history, (129). Moreover, an increased methylation of BDNF exon IV promoter was associated with decreased BDNF mRNA levels in brain Wernicke’s area from suicide victims who were affected by MDD (130).
Alterations of miRNAs expression may also contribute to BDNF dysregulation. Interestingly, Li et al. (131) showed that reduced serum BDNF levels in depressed patients was accompanied by an up-regulation of two miRNAs (miR-132, miR-182), which have been previously described to regulate BDNF (132). Moreover, a reduction of BDNF expression in the brain of animals exposed to maternal deprivation was significantly associated with an up-regulation of miR-16 levels (86).
Also, the SLC6A4 (or SERT) gene has been widely studied for changes in its epigenetic status, especially in relation to stressful life events exposure or in relation to MDD. Several in vivo and in vitro studies found site-specific methylation changes within SLC6A4 gene, which were associated with decreased levels of the SERT mRNA. Moreover, Philibert and colleagues showed, by using lymphoblast cell lines, that this effect was shown only when the 5-HTTLPR genotype was taken into account (133). Higher SERT promoter methylation status was significantly associated with childhood adversities, family history of depression, higher perceived stress, and more severe psychopathology (129). There is also evidence for reduced SERT mRNA levels in individuals exposed to maternal prenatal stress or childhood maltreatment, observed by an inverse correlation between SERT mRNA levels and the magnitude of prenatal/early adversity (56). Maternal separation has been associated with lower SERT mRNA levels in rodents and decreased SERT availability in non-human primates (134). Rhesus macaques exposed to maternal aggression displayed a reduced SERT mRNA levels in peripheral blood cells, indicating that stress-induced changes of SERT expression may not be limited to the brain (135).
SERT expression can also be influenced by specific miRNAs. Interestingly, it has been shown that miR-16, which targets BDNF, is also involved in the modulation of SERT both in human and rat tissues (136), suggesting that the interaction between BDNF and the serotonergic system may be due to the action of common regulatory miRNAs (137). Interestingly, Baudry and colleagues reported that a chronic treatment with the selective serotonin reuptake inhibitors (SSRI) fluoxetine increases miR-16 levels in serotonergic raphe nuclei, and that this increase is accompanied by a reduction of SERT expression and in BDNF modulation, suggesting a role for miR-16 in the therapeutic action of SSRI antidepressants (138).
Epigenetic mechanisms have been also proposed as linking bridges between environmental factors and genes involved in the HPA axis functionality (139). In rodents, increased maternal care, such as pup licking and grooming (LG) and arched-back nursing (ABN), has been associated with long-lasting changes in DNA methylation within the exon 17 in the promoter region of the glucocorticoid receptor GR gene. In particular, the NGFI-A binding site in the GR promoter 17 was highly methylated in the offspring of low caring mothers, resulting in reduced expression of GR exon 17.
Similar studies have been performed in humans in order to investigate alterations in the GR 1F promoter, the human homolog of the rat exon 17 NR3C1 promoter, in relationship with early life adverse events. To this regard, McGowan et al. (103) have found that the methylation levels of the GR 1F promoter were significantly increased in the hippocampus from suicide victims with a history of childhood abuse, as compared with those from suicide victims with no childhood abuse or with control samples, suggesting an effect of child abuse, independent from suicide, on GR methylation status and gene expression.
Epigenetic changes on human GR promoter as consequence of early environmental factors have also been reported by Oberlander et al. (140), which showed that prenatal exposure to maternal depressed/anxious mood led to increased methylation levels of the NR3C1 promoter in cord blood of newborns and that this methylation pattern was also associated with increased salivary cortisol in response to stress (140). Childhood maltreatment was also associated with increased methylation of NR3C1 promoter in the blood of patients affected by psychiatric disorders including MDD (139). Furthermore, the severity, the number, and the type of maltreatments positively correlated with the level of methylation (139). Interestingly, GR methylation marks might be even transmitted to subsequent generations, suggesting that the vulnerability risk of the offspring toward stress-related disorders may be related to the induction of specific epigenetic signatures (141–146).
For example, Radtke et al. (147) investigated whether intrauterine exposure to maternal stress could affect the epigenetic patterns beyond infancy. In particular, they analyzed the methylation status of the NR3C1 gene both in mothers, subjected to intimate partner violence during pregnancy, and also in their adolescent children. The authors found an increased methylation of NR3C1 promoter in children exposed to prenatal stress, confirming that epigenetic modifications could be transmitted from one generation to another and could be established already in utero.
Also, the GR co-chaperonine, FKBP5, has been widely investigated in term of epigenetic changes induced by ELS, because of its role in modulating the stress response and its relationship to stress-related neuropsychiatric disorders, such as MDD. For example, a transient decreased methylation status accompanied by an increased expression of FKBP5 has been shown in the hippocampus, hypothalamus, and in the blood of mice after prolonged exposure to glucocorticoids (148). Reduced methylation levels of FKBP5 gene within regions containing functional glucocorticoid responsive elements were observed also in the blood of individuals exposed to childhood abuse when compared to subjects without any childhood trauma (104). The authors suggested that changes in FKBP5 methylation might increase the differential responsiveness of FKBP5 to GR activation that, if installed during developmentally critical periods, then can remain stable over time.
Recent evidences suggest that, above to DNA methylation, also miRNAs can persistently influence HPA axis responsiveness. For example, an increase of miR-18a levels, which targets the GR, was found in the hypothalamus of a stress-sensitive rat strain, while miR-34c was found markedly up-regulated after acute and chronic stress in mice (149). Differential modifications in miRNAs expression were demonstrated in relation to normal (non-learned helplessness) versus aberrant (learned helplessness) response to a repeated shock paradigm in rats (150). It was also suggested that stress-inducible cognitive impairments could be attributable to cholinergic-mediated induction of miR-132 in hippocampus. Interestingly, excess in glucocorticoids was shown to interfere with the BDNF/miR-132 cascade (151). Another GR-targeting miRNA is miR-124a, which has been suggested to be critical for maintaining GR expression at permissive levels to neurogenesis (152).
We summarized the above-mentioned studies on epigenetics in Table 2.
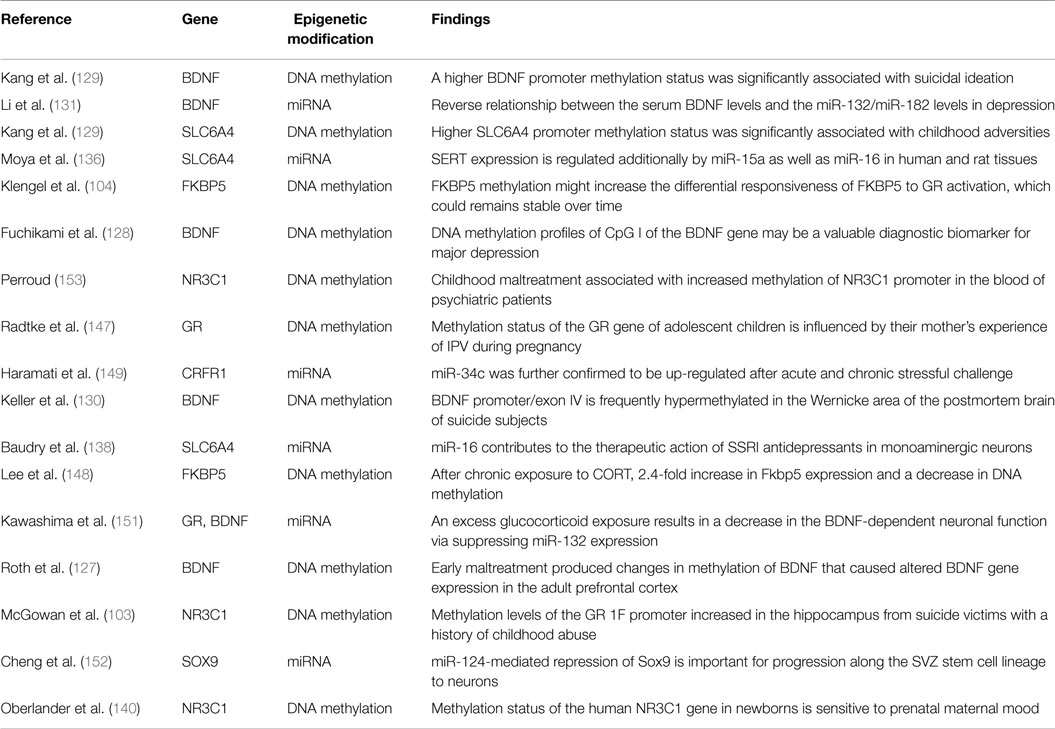
Table 2. Correlation between epigenetic modification, stress, and genetics.
Overall, these evidences suggest that a GR functionality deficit due to changes in DNA methylation or miRNAs expression could lead to long-term changes in stress hormone system regulation, to alterations of neuronal circuits and other glucocorticoid receptor responsive systems, resulting in a higher risk for the development of stress-related psychiatric disorders.
Conclusion
Despite the relative importance of genetic risk factors in the pathogenesis of MDD, gene association studies have identified, to date, only a very small number of candidate vulnerability genes that, however, can explain little of the variance. This is known also as “missing heritability” and it could be accounted by several factors including the evidence that each susceptibility gene contributes to a small fraction of the total genetic risk for MDD, and that multiple and partially overlapping sets of susceptibility genes interact each other and with the environment, modulating the individual risk to MDD.
Among environmental factors, a prominent role has been recognized for ELS. However, not all the individuals exposed to ELS develop MDD or other psychiatric illnesses, and this is mainly because the mental outcome is dependent, once again, on the individual genetic background. Thus, the best paradigm to predict the MDD risk and MDD onset is explained by gene–environment interactions, which reflect causal mechanisms, where one or more genetic variants and one or more environmental factors contribute to the causation of a condition in the same individual with the genetic factors influencing the sensitivity to environmental exposures.
Epigenetic mechanisms, and in particular DNA methylation and miRNAs expression changes, have been widely identified as processes that could play an important role in the pathogenesis of MDD as they represent the main mediators of the effect of the environment in enhancing the vulnerability risk to develop MDD. Interestingly, epigenetic changes can be transmitted from one generation to another and this is an interesting aspect, as it could also explain the transmission of MDD vulnerability across families. In addition, ELS has been also found to act, still through epigenetic mechanism, directly on brain connections and structure by reducing the number of glial cells in the brain.
Taken together, we underlie the role of genetic and epigenetic processes involved in stress and neuroplasticity-related biological systems on development of MDD after exposure to ELS, thereby building the basis for future research and clinical interventions.
Conflict of Interest Statement
All the authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Cattaneo and Prof. Pariante have been funded through research obtained from the Medical Research Council (UK) (MR/J002739/1) and from the Psychiatry Research Trust, UK (McGregor 97). Prof. Pariante has been also funded by the Commission of European Communities Seventh Framework Programme (Collaborative Project Grant Agreement no. 22963, Mood Inflame); by the National Institute for Health Research Biomedical Research Centre in Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London. Dr. Cattaneo has additionally been supported by the Italian Ministry of Health (Ricerca Corrente) and by ERANET Neuron Grant. Prof. M. A. Riva has been funded by grants from the Italian Ministry of University and Research (PRIN 20107MSMA4), from Fondazione CARIPLO (grant no. 2012-0503) and from European Union/ERANET. Dr. Bocchio-Chiavetto was supported by grants from Cariplo Foundation (MICROMOOD Project 2009-2701) and from the Italian Ministry of Health (Ricerca Corrente).
References
1. Shenal BV, Harrison DW, Demaree HA. The neuropsychology of depression: a literature review and preliminary model. Neuropsychol Rev (2003) 13:33–42. doi:10.1023/A:1022300622902
2. Reddy MS. Depression: the disorder and the burden. Indian J Psychol Med (2010) 32:1–2. doi:10.4103/0253-7176.70510
3. World Health Organization. The World Health Report 2001 – Mental Health: New Understanding. New Hope (2001).
4. Kessler D, Sharp D, Lewis G. Screening for depression in primary care. Br J Gen Pract (2005) 55:659–60.
5. Sibille E, French B. Biological substrates underpinning diagnosis of major depression. Int J Neuropsychopharmacol (2013) 16(8):1893–909. doi:10.1017/S1461145713000436
6. Lotan A, Fenckova M, Bralten J, Alttoa A, Dixson L, Williams RW, et al. Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Front Neurosci (2014) 8:331. doi:10.3389/fnins.2014.00331
7. Lohoff FW. Overview of the genetics of major depressive disorder. Curr Psychiatry Rep (2010) 12:539–46. doi:10.1007/s11920-010-0150-6
8. Lesch KP. Gene-environment interaction and the genetics of depression. J Psychiatry Neurosci (2004) 29:174–84.
9. Züchner S, Roberts ST, Speer MC, Beckham JC. Update on psychiatric genetics. Genet Med (2007) 9:332–40. doi:10.1097/GIM.0b013e318065a9fa
10. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry (2000) 157:1552–62. doi:10.1176/appi.ajp.157.10.1552
11. Menke A, Domschke K, Czamara D, Klengel T, Hennings J, Lucae S, et al. Genome-wide association study of antidepressant treatment-emergent suicidal ideation. Neuropsychopharmacology (2012) 37:797–807. doi:10.1038/npp.2011.257
12. Bogdan R, Nikolova YS, Pizzagalli DA. Neurogenetics of depression: a focus on reward processing and stress sensitivity. Neurobiol Dis (2013) 52:12–23. doi:10.1016/j.nbd.2012.05.007
13. Lopez-Leon S, Janssens AC, Gonzalez-Zuloeta Ladd AM, Del-Favero J, Claes SJ, Oostra BA, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry (2008) 13:772–85. doi:10.1038/sj.mp.4002088
14. Holmes C, Russ C, Kirov G, Aitchison KJ, Powell JF, Collier DA, et al. Apolipoprotein E: depressive illness, depressive symptoms, and Alzheimer’s disease. Biol Psychiatry (1998) 43:159–64. doi:10.1016/S0006-3223(97)00326-0
15. Baba H, Nakano Y, Maeshima H, Satomura E, Kita Y, Suzuki T, et al. Metabolism of amyloid-beta protein may be affected in depression. J Clin Psychiatry (2012) 73:115–20. doi:10.4088/JCP.10m06766
16. Namekawa Y, Baba H, Maeshima H, Nakano Y, Satomura E, Takebayashi N, et al. Heterogeneity of elderly depression: increased risk of Alzheimer’s disease and Abeta protein metabolism. Prog Neuropsychopharmacol Biol Psychiatry (2013) 43:203–8. doi:10.1016/j.pnpbp.2012.12.016
17. Sullivan PF, De Geus EJ, Willemsen G, James MR, Smit JH, Zandbelt T, et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry (2009) 14:359–75. doi:10.1038/mp.2008.125
18. Verbeek EC, Bevova MR, Bochdanovits Z, Rizzu P, Bakker IM, Uithuisje T, et al. Resequencing three candidate genes for major depressive disorder in a Dutch cohort. PLoS One (2013) 8:e79921. doi:10.1371/journal.pone.0079921
19. Woudstra S, Van Tol MJ, Bochdanovits Z, Van Der Wee NJ, Zitman FG, Van Buchem MA, et al. Modulatory effects of the piccolo genotype on emotional memory in health and depression. PLoS One (2013) 8:e61494. doi:10.1371/journal.pone.0061494
20. McFarquhar M, Elliott R, Mckie S, Thomas E, Downey D, Mekli K, et al. TOMM40 rs2075650 may represent a new candidate gene for vulnerability to major depressive disorder. Neuropsychopharmacology (2014) 39:1743–53. doi:10.1038/npp.2014.22
21. Hayden KM, McEvoy JM, Linnertz C, Attix D, Kuchibhatla M, Saunders AM, et al. A homopolymer polymorphism in the TOMM40 gene contributes to cognitive performance in aging. Alzheimers Dement (2012) 8:381–8. doi:10.1016/j.jalz.2011.10.005
22. Cao MQ, Hu SY, Zhang CH, Xia DS. Study on the interrelationship between 5-HTTLPR/G-protein beta3 subunit (C825T) polymorphisms and depressive disorder. Psychiatr Genet (2007) 17:233–8. doi:10.1097/YPG.0b013e3280c1e5de
23. Hu Q, Zhang SY, Liu F, Zhang XJ, Cui GC, Yu EQ, et al. Influence of GNB3 C825T polymorphism on the efficacy of antidepressants in the treatment of major depressive disorder: a meta-analysis. J Affect Disord (2014) 172C:103–9. doi:10.1016/j.jad.2014.09.039
24. Klenke S, Kussmann M, Siffert W. The GNB3 C825T polymorphism as a pharmacogenetic marker in the treatment of hypertension, obesity, and depression. Pharmacogenet Genomics (2011) 21:594–606. doi:10.1097/FPC.0b013e3283491153
25. Arinami T, Yamada N, Yamakawa-Kobayashi K, Hamaguchi H, Toru M. Methylenetetrahydrofolate reductase variant and schizophrenia/depression. Am J Med Genet (1997) 74:526–8. doi:10.1002/(SICI)1096-8628(19970919)74:5<526::AID-AJMG14>3.0.CO;2-E
26. Wang X, Wang Z, Wu Y, Yuan Y, Hou Z, Hou G. Association analysis of the catechol-O-methyltransferase/methylenetetrahydrofolate reductase genes and cognition in late-onset depression. Psychiatry Clin Neurosci (2014) 68:344–52. doi:10.1111/pcn.12133
27. Dorado P, Penas-Lledo EM, Gonzalez AP, Caceres MC, Cobaleda J, Llerena A. Increased risk for major depression associated with the short allele of the serotonin transporter promoter region (5-HTTLPR-S) and the CYP2C9*3 allele. Fundam Clin Pharmacol (2007) 21:451–3. doi:10.1111/j.1472-8206.2007.00501.x
28. Matsumoto Y, Fabbri C, Pellegrini S, Porcelli S, Politi P, Bellino S, et al. Serotonin transporter gene: a new polymorphism may affect response to antidepressant treatments in major depressive disorder. Mol Diagn Ther (2014) 18:567–77. doi:10.1007/s40291-014-0110-7
29. Gatt JM, Burton KL, Williams LM, Schofield PR. Specific and common genes implicated across major mental disorders: a review of meta-analysis studies. J Psychiatr Res (2015) 60C:1–13. doi:10.1016/j.jpsychires.2014.09.014
30. Bosker FJ, Hartman CA, Nolte IM, Prins BP, Terpstra P, Posthuma D, et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol Psychiatry (2011) 16:516–32. doi:10.1038/mp.2010.38
31. Muglia P, Tozzi F, Galwey NW, Francks C, Upmanyu R, Kong XQ, et al. Genome-wide association study of recurrent major depressive disorder in two European case-control cohorts. Mol Psychiatry (2010) 15:589–601. doi:10.1038/mp.2008.131
32. Hirschhorn N. Creativity and affective illness. Am J Psychiatry (2002) 159:677. doi:10.1176/appi.ajp.159.4.677
33. Munafo MR, Clark TG, Roberts KH, Johnstone EC. Neuroticism mediates the association of the serotonin transporter gene with lifetime major depression. Neuropsychobiology (2006) 53:1–8. doi:10.1159/000089915
34. Levinson DF, Mostafavi S, Milaneschi Y, Rivera M, Ripke S, Wray NR, et al. Genetic studies of major depressive disorder: why are there no genome-wide association study findings and what can we do about it? Biol Psychiatry (2014) 76:510–2. doi:10.1016/j.biopsych.2014.07.029
35. Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry (2012) 17:36–48. doi:10.1038/mp.2010.109
36. Shi J, Potash JB, Knowles JA, Weissman MM, Coryell W, Scheftner WA, et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry (2011) 16:193–201. doi:10.1038/mp.2009.124
37. Kloiber S, Czamara D, Karbalai N, Muller-Myhsok B, Hennings J, Holsboer F, et al. ANK3 and CACNA1C – missing genetic link for bipolar disorder and major depressive disorder in two German case-control samples. J Psychiatr Res (2012) 46:973–9. doi:10.1016/j.jpsychires.2012.04.017
38. Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry (2013) 18:497–511. doi:10.1038/mp.2012.21
39. Comasco E, Åslund C, Oreland L, Nilsson KW. Three-way interaction effect of 5-HTTLPR, BDNF Val66Met, and childhood adversity on depression: a replication study. Eur Neuropsychopharmacol (2013) 23:1300–6. doi:10.1016/j.euroneuro.2013.01.010
40. Appel K, Schwahn C, Mahler J, Schulz A, Spitzer C, Fenske K, et al. Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology (2011) 36:1982–91. doi:10.1038/npp.2011.81
41. Zimmermann P, Bruckl T, Nocon A, Pfister H, Binder EB, Uhr M, et al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry (2011) 168:1107–16. doi:10.1176/appi.ajp.2011.10111577
42. Vinberg M, Trajkovska V, Bennike B, Knorr U, Knudsen GM, Kessing LV. The BDNF Val66Met polymorphism: relation to familiar risk of affective disorder, BDNF levels and salivary cortisol. Psychoneuroendocrinology (2009) 34:1380–9. doi:10.1016/j.psyneuen.2009.04.014
43. Aguilera M, Arias B, Wichers M, Barrantes-Vidal N, Moya J, Villa H, et al. Early adversity and 5-HTT/BDNF genes: new evidence of gene-environment interactions on depressive symptoms in a general population. Psychol Med (2009) 39:1425–32. doi:10.1017/S0033291709005248
44. Bet PM, Penninx BW, Bochdanovits Z, Uitterlinden AG, Beekman AT, Van Schoor NM, et al. Glucocorticoid receptor gene polymorphisms and childhood adversity are associated with depression: new evidence for a gene-environment interaction. Am J Med Genet B Neuropsychiatr Genet (2009) 150B:660–9. doi:10.1002/ajmg.b.30886
45. Ising M, Depping AM, Siebertz A, Lucae S, Unschuld PG, Kloiber S, et al. Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur J Neurosci (2008) 28:389–98. doi:10.1111/j.1460-9568.2008.06332.x
46. Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA (2008) 299:1291–305. doi:10.1001/jama.299.11.1291
47. Kumsta R, Entringer S, Koper JW, Van Rossum EF, Hellhammer DH, Wust S. Sex specific associations between common glucocorticoid receptor gene variants and hypothalamus-pituitary-adrenal axis responses to psychosocial stress. Biol Psychiatry (2007) 62:863–9. doi:10.1016/j.biopsych.2007.04.013
48. Wüst S, Van Rossum EF, Federenko IS, Koper JW, Kumsta R, Hellhammer DH. Common polymorphisms in the glucocorticoid receptor gene are associated with adrenocortical responses to psychosocial stress. J Clin Endocrinol Metab (2004) 89:565–73. doi:10.1210/jc.2003-031148
49. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science (2003) 301:386–9. doi:10.1126/science.1083968
50. Uher R. Gene-environment interactions in common mental disorders: an update and strategy for a genome-wide search. Soc Psychiatry Psychiatr Epidemiol (2014) 49:3–14. doi:10.1007/s00127-013-0801-0
51. Lin YL, Wang S. Prenatal lipopolysaccharide exposure increases depression-like behaviors and reduces hippocampal neurogenesis in adult rats. Behav Brain Res (2014) 1(259):24–34. doi:10.1016/j.bbr.2013.10.034
52. Gámez-Guadix M, Orue I, Smith PK, Calvete E. Longitudinal and reciprocal relations of cyberbullying with depression, substance use, and problematic internet use among adolescents. J Adolesc Health (2013) 53(4):446–52. doi:10.1016/j.jadohealth.2013.03.030
53. Gullander M, Hogh A, Hansen ÅM, Persson R, Rugulies R, Kolstad HA, et al. Exposure to workplace bullying and risk of depression. J Occup Environ Med (2014) 56(12):1258–65. doi:10.1097/JOM.0000000000000339
54. Cummings CM, Caporino NE, Kendall PC. Comorbidity of anxiety and depression in children and adolescents: 20 years after. Psychol Bull (2014) 140(3):816–45. doi:10.1037/a0034733
55. Feingold D, Weiser M, Rehm J, Lev-Ran S. The association between cannabis use and mood disorders: A longitudinal study. J Affect Disord (2014) 172C:211–8. doi:10.1016/j.jad.2014.10.006
56. Heim C, Binder EB. Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol (2012) 233:102–11. doi:10.1016/j.expneurol.2011.10.032
57. Booij SH, Bos EH, De Jonge P, Oldehinkel AJ. Markers of stress and inflammation as potential mediators of the relationship between exercise and depressive symptoms: findings from the TRAILS study. Psychophysiology (2014) 52(3):352–8. doi:10.1111/psyp.12329
58. Davis DA, Bortolato M, Godar SC, Sander TK, Iwata N, Pakbin P, et al. Prenatal exposure to urban air nanoparticles in mice causes altered neuronal differentiation and depression-like responses. PLoS One (2013) 8:e64128. doi:10.1371/journal.pone.0064128
59. Hornung OP, Heim CM. Gene-environment interactions and intermediate phenotypes: early trauma and depression. Front Endocrinol (2014) 5:14. doi:10.3389/fendo.2014.00014
60. Ming X, Julu PO, Wark J, Apartopoulos F, Hansen S. Discordant mental and physical efforts in an autistic patient. Brain Dev (2004) 26(8):519–24.
61. Garner AS, Shonkoff JP; Committee on Psychosocial Aspects of Child and Family Health, Committee on Early Childhood, Adoption, and Dependent Care, Section on Developmental and Behavioral Pediatrics. Early childhood adversity, toxic stress, and the role of the pediatrician: translating developmental science into lifelong health. Pediatrics (2012) 129:e224–31.
62. Danese A, McEwen BS. Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav (2012) 106:29–39. doi:10.1016/j.physbeh.2011.08.019
63. Shonkoff JP, Garner AS; Committee on Psychosocial Aspects of Child and Family Health, Committee on Early Childhood, Adoption, and Dependent Care, Section on Developmental and Behavioral Pediatrics. The lifelong effects of early childhood adversity and toxic stress. Pediatrics (2012) 129:e232–46. doi:10.1542/peds.2011-2663
64. Chapman DP, Whitfield CL, Felitti VJ, Dube SR, Edwards VJ, Anda RF. Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord (2004) 82:217–25. doi:10.1016/j.jad.2003.12.013
65. Dube SR, Anda RF, Felitti VJ, Chapman DP, Williamson DF, Giles WH. Childhood abuse, household dysfunction, and the risk of attempted suicide throughout the life span: findings from the adverse childhood experiences study. JAMA (2001) 286:3089–96. doi:10.1001/jama.286.24.3089
66. Agid O, Shapira B, Zislin J, Ritsner M, Hanin B, Murad H, et al. Environment and vulnerability to major psychiatric illness: a case control study of early parental loss in major depression, bipolar disorder and schizophrenia. Mol Psychiatry (1999) 4:163–72. doi:10.1038/sj.mp.4000473
67. Tyrka AR, Wier L, Price LH, Ross NS, Carpenter LL. Childhood parental loss and adult psychopathology: effects of loss characteristics and contextual factors. Int J Psychiatry Med (2008) 38:329–44. doi:10.2190/PM.38.3.h
68. Lizardi H, Klein DN, Ouimette PC, Riso LP, Anderson RL, Donaldson SK. Reports of the childhood home environment in early-onset dysthymia and episodic major depression. J Abnorm Psychol (1995) 104:132–9. doi:10.1037/0021-843X.104.1.132
69. Brook CA, Schmidt LA. Social anxiety disorder: a review of environmental risk factors. Neuropsychiatr Dis Treat (2008) 4:123–43.
70. Mirescu C, Peters JD, Gould E. Early life experience alters response of adult neurogenesis to stress. Nat Neurosci (2004) 7:841–6. doi:10.1038/nn1290
71. Mann JJ, Currier DM. Stress, genetics and epigenetic effects on the neurobiology of suicidal behavior and depression. Eur Psychiatry (2010) 25:268–71. doi:10.1016/j.eurpsy.2010.01.009
72. Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA (2009) 301:2462–71. doi:10.1001/jama.2009.878
73. Dalton ED, Hammen CL, Najman JM, Brennan PA. Genetic susceptibility to family environment: BDNF Val66met and 5-HTTLPR influence depressive symptoms. J Fam Psychol (2014) 28:947–56. doi:10.1037/fam0000032
74. Sharpley CF, Palanisamy SK, Glyde NS, Dillingham PW, Agnew LL. An update on the interaction between the serotonin transporter promoter variant (5-HTTLPR), stress and depression, plus an exploration of non-confirming findings. Behav Brain Res (2014) 273:89–105. doi:10.1016/j.bbr.2014.07.030
75. Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry (2011) 68:444–54. doi:10.1001/archgenpsychiatry.2010.189
76. Blakely RD, Veenstra-Vanderweele J. Genetic indeterminism, the 5-HTTLPR, and the paths forward in neuropsychiatric genetics. Arch Gen Psychiatry (2011) 68:457–8. doi:10.1001/archgenpsychiatry.2011.34
77. Wankerl M, Wust S, Otte C. Current developments and controversies: does the serotonin transporter gene-linked polymorphic region (5-HTTLPR) modulate the association between stress and depression? Curr Opin Psychiatry (2010) 23:582–7. doi:10.1097/YCO.0b013e32833f0e3a
78. Gao Z, Yuan H, Sun M, Wang Z, He Y, Liu D. The association of serotonin transporter gene polymorphism and geriatric depression: a meta-analysis. Neurosci Lett (2014) 578:148–52. doi:10.1016/j.neulet.2014.06.046
79. Zannas AS, McQuoid DR, Steffens DC, Chrousos GP, Taylor WD. Stressful life events, perceived stress, and 12-month course of geriatric depression: direct effects and moderation by the 5-HTTLPR and COMT Val158Met polymorphisms. Stress (2012) 15:425–34. doi:10.3109/10253890.2011.634263
80. Nestler EJ, Barrot M, Dileone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron (2002) 34:13–25. doi:10.1016/S0896-6273(02)00653-0
81. Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci (2007) 10:1089–93. doi:10.1038/nn1971
82. Oral E, Canpolat S, Yildirim S, Gulec M, Aliyev E, Aydin N. Cognitive functions and serum levels of brain-derived neurotrophic factor in patients with major depressive disorder. Brain Res Bull (2012) 88:454–9. doi:10.1016/j.brainresbull.2012.03.005
83. Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrinology (2009) 34(Suppl 1):S208–16. doi:10.1016/j.psyneuen.2009.05.014
84. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell (2003) 112:257–69. doi:10.1016/S0092-8674(03)00035-7
85. Rybakowski JK. BDNF gene: functional Val66Met polymorphism in mood disorders and schizophrenia. Pharmacogenomics (2008) 9:1589–93. doi:10.2217/14622416.9.11.1589
86. Baj G, Carlino D, Gardossi L, Tongiorgi E. Toward a unified biological hypothesis for the BDNF Val66Met-associated memory deficits in humans: a model of impaired dendritic mRNA trafficking. Front Neurosci (2013) 7:188. doi:10.3389/fnins.2013.00188
87. Post RM. Role of BDNF in bipolar and unipolar disorder: clinical and theoretical implications. J Psychiatr Res (2007) 41:979–90. doi:10.1016/j.jpsychires.2006.09.009
88. Jiang X, Xu K, Hoberman J, Tian F, Marko AJ, Waheed JF, et al. BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects. Neuropsychopharmacology (2005) 30:1353–61.
89. Ribeiro L, Busnello JV, Cantor RM, Whelan F, Whittaker P, Deloukas P, et al. The brain-derived neurotrophic factor rs6265 (Val66Met) polymorphism and depression in Mexican-Americans. Neuroreport (2007) 18:1291–3. doi:10.1097/WNR.0b013e328273bcb0
90. Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry (2009) 14:681–95. doi:10.1038/mp.2008.143
91. Hosang GM, Shiles C, Tansey KE, McGuffin P, Uher R. Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta-analysis. BMC Med (2014) 12:7. doi:10.1186/1741-7015-12-7
92. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology (2009) 34(Suppl 1):S186–95. doi:10.1016/j.psyneuen.2009.05.021
93. Koenen KC, Saxe G, Purcell S, Smoller JW, Bartholomew D, Miller A, et al. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry (2005) 10:1058–9. doi:10.1038/sj.mp.4001727
94. Koenen KC, Uddin M. FKBP5 polymorphisms modify the effects of childhood trauma. Neuropsychopharmacology (2010) 35:1623–4. doi:10.1038/npp.2010.60
95. Sun H, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology (2013) 38:124–37. doi:10.1038/npp.2012.73
96. Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci (2005) 6:108–18. doi:10.1038/nrn1604
97. Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem (2008) 89:599–603. doi:10.1016/j.nlm.2007.07.016
98. Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci (2007) 8:355–67. doi:10.1038/nrn2132
99. Szyf M. Early life, the epigenome and human health. Acta Paediatr (2009) 98:1082–4. doi:10.1111/j.1651-2227.2009.01382.x
100. Lutz PE, Turecki G. DNA methylation and childhood maltreatment: from animal models to human studies. Neuroscience (2014) 264:142–56. doi:10.1016/j.neuroscience.2013.07.069
101. Meaney MJ, Ferguson-Smith AC. Epigenetic regulation of the neural transcriptome: the meaning of the marks. Nat Neurosci (2010) 13:1313–8. doi:10.1038/nn1110-1313
102. Kundakovic M. Postnatal risk environments, epigenetics, and psychosis: putting the pieces together. Soc Psychiatry Psychiatr Epidemiol (2014) 49:1535–6. doi:10.1007/s00127-014-0927-8
103. McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci (2009) 12:342–8. doi:10.1038/nn.2270
104. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci (2013) 16:33–41. doi:10.1038/nn.3275
105. Provencal N, Suderman MJ, Guillemin C, Massart R, Ruggiero A, Wang D, et al. The signature of maternal rearing in the methylome in rhesus macaque prefrontal cortex and T cells. J Neurosci (2012) 32:15626–42. doi:10.1523/JNEUROSCI.1470-12.2012
106. Nieratschker V, Massart R, Gilles M, Luoni A, Suderman MJ, Krumm B, et al. MORC1 exhibits cross-species differential methylation in association with early life stress as well as genome-wide association with MDD. Transl Psychiatry (2014) 4:e429. doi:10.1038/tp.2014.75
107. Nagy C, Suderman M, Yang J, Szyf M, Mechawar N, Ernst C, et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry (2014) 20(3):320–8. doi:10.1038/mp.2014.21
108. Lucassen PJ, Naninck EFG, van Goudoever JB, Fitzsimons C, Joels M, Korosi A. Perinatal programming of adult hippocampal structure and function; emerging roles of stress, nutrition and epigenetics. Trends Neurosci (2013) 36:621–31. doi:10.1016/j.tins.2013.08.002
109. Serafini G, Hayley S, Pompili M, Dwivedi Y, Brahmachari G, Girardi P, et al. Hippocampal neurogenesis, neurotrophic factors and depression: possible therapeutic targets? CNS Neurol Disord Drug Targets (2014) 13:1708–21. doi:10.2174/1871527313666141130223723
110. Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol (2009) 21:452–60. doi:10.1016/j.ceb.2009.04.009
111. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet (2011) 12:99–110. doi:10.1038/nrg2936
112. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet (2010) 11:597–610. doi:10.1038/nrg2843
113. O’Carroll D, Schaefer A. General principals of miRNA biogenesis and regulation in the brain. Neuropsychopharmacology (2013) 38:39–54. doi:10.1038/npp.2012.87
114. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi:10.1016/j.cell.2009.01.035
115. Watanabe K, Takai D. Disruption of the expression and function of microRNAs in lung cancer as a result of epigenetic changes. Front Genet (2013) 4:275. doi:10.3389/fgene.2013.00275
116. Fabbri M, Calore F, Paone A, Galli R, Calin GA. Epigenetic regulation of miRNAs in cancer. Adv Exp Med Biol (2013) 754:137–48. doi:10.1007/978-1-4419-9967-2_6
117. Hu W, Wang T, Xu J, Li H. MicroRNA mediates DNA methylation of target genes. Biochem Biophys Res Commun (2014) 444(4):676–81. doi:10.1016/j.bbrc.2014.01.171
118. Babenko O, Golubov A, Ilnytskyy Y, Kovalchuk I, Metz GA. Genomic and epigenomic responses to chronic stress involve miRNA-mediated programming. PLoS One (2012) 7:e29441. doi:10.1371/journal.pone.0029441
119. Uchida S, Hara K, Kobayashi A, Funato H, Hobara T, Otsuki K, et al. Early life stress enhances behavioral vulnerability to stress through the activation of REST4-mediated gene transcription in the medial prefrontal cortex of rodents. J Neurosci (2010) 30:15007–18. doi:10.1523/JNEUROSCI.1436-10.2010
120. Suderman M, Borghol N, Pappas JJ, Pinto Pereira SM, Pembrey M, Hertzman C, et al. Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med Genomics (2014) 7:13. doi:10.1186/1755-8794-7-13
121. Elfving B, Plougmann PH, Muller HK, Mathe AA, Rosenberg R, Wegener G. Inverse correlation of brain and blood BDNF levels in a genetic rat model of depression. Int J Neuropsychopharmacol (2010) 13:563–72. doi:10.1017/S1461145709990721
122. Molteni R, Cattaneo A, Calabrese F, Macchi F, Olivier JD, Racagni G, et al. Reduced function of the serotonin transporter is associated with decreased expression of BDNF in rodents as well as in humans. Neurobiol Dis (2010) 37:747–55. doi:10.1016/j.nbd.2009.12.014
123. Qiao H, An SC, Ren W, Ma XM. Progressive alterations of hippocampal CA3-CA1 synapses in an animal model of depression. Behav Brain Res (2014) 275:191–200. doi:10.1016/j.bbr.2014.08.040
124. Vinberg M, Bukh JD, Bennike B, Kessing LV. Are variations in whole blood BDNF level associated with the BDNF Val66Met polymorphism in patients with first episode depression? Psychiatry Res (2013) 210:102–8. doi:10.1016/j.psychres.2013.04.023
125. Cattaneo A, Gennarelli M, Uher R, Breen G, Farmer A, Aitchison KJ, et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology (2013) 38(3):377–85. doi:10.1038/npp.2012.191
126. Molendijk ML, Spinhoven P, Polak M, Bus BA, Penninx BW, Elzinga BM. Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol Psychiatry (2014) 19:791–800. doi:10.1038/mp.2013.105
127. Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry (2009) 65:760–9. doi:10.1016/j.biopsych.2008.11.028
128. Fuchikami M, Morinobu S, Segawa M, Okamoto Y, Yamawaki S, Ozaki N, et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One (2011) 6:e23881. doi:10.1371/journal.pone.0023881
129. Kang HJ, Kim JM, Lee JY, Kim SY, Bae KY, Kim SW, et al. BDNF promoter methylation and suicidal behavior in depressive patients. J Affect Disord (2013) 151:679–85. doi:10.1016/j.jad.2013.08.001
130. Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, et al. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch Gen Psychiatry (2010) 67:258–67. doi:10.1001/archgenpsychiatry.2010.9
131. Li YJ, Xu M, Gao ZH, Wang YQ, Yue Z, Zhang YX, et al. Alterations of serum levels of BDNF-related miRNAs in patients with depression. PLoS One (2013) 8:e63648. doi:10.1371/journal.pone.0063648
132. Villanueva R. Neurobiology of major depressive disorder. Neural Plast (2013) 2013:873278. doi:10.1155/2013/873278
133. Philibert R. Merging genetic and environmental effects in the Iowa adoption studies: focus on depression. Ann Clin Psychiatry (2006) 18:219–22. doi:10.1080/10401230600948399
134. Ichise M, Vines DC, Gura T, Anderson GM, Suomi SJ, Higley JD, et al. Effects of early life stress on [11C]DASB positron emission tomography imaging of serotonin transporters in adolescent peer- and mother-reared rhesus monkeys. J Neurosci (2006) 26:4638–43. doi:10.1523/JNEUROSCI.5199-05.2006
135. Kinnally EL, Tarara ER, Mason WA, Mendoza SP, Abel K, Lyons LA, et al. Serotonin transporter expression is predicted by early life stress and is associated with disinhibited behavior in infant rhesus macaques. Genes Brain Behav (2010) 9:45–52. doi:10.1111/j.1601-183X.2009.00533.x
136. Moya PR, Wendland JR, Salemme J, Fried RL, Murphy DL. miR-15a and miR-16 regulate serotonin transporter expression in human placental and rat brain raphe cells. Int J Neuropsychopharmacol (2013) 16:621–9. doi:10.1017/S1461145712000454
137. Hansen KF, Obrietan K. MicroRNA as therapeutic targets for treatment of depression. Neuropsychiatr Dis Treat (2013) 9:1011–21. doi:10.2147/NDT.S34811
138. Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science (2010) 329:1537–41. doi:10.1126/science.1193692
139. Perroud N, Paoloni-Giacobino A, Prada P, Olie E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry (2011) 1:e59. doi:10.1038/tp.2011.60
140. Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics (2008) 3:97–106. doi:10.4161/epi.3.2.6034
141. Champagne FA. Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol (2008) 29:386–97. doi:10.1016/j.yfrne.2008.03.003
142. Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet (2007) 8:253–62. doi:10.1038/nrg2045
143. Skinner MK, Anway MD, Savenkova MI, Gore AC, Crews D. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One (2008) 3:e3745. doi:10.1371/journal.pone.0003745
144. Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab (2010) 21:214–22. doi:10.1016/j.tem.2009.12.007
145. Whitelaw NC, Whitelaw E. Transgenerational epigenetic inheritance in health and disease. Curr Opin Genet Dev (2008) 18:273–9. doi:10.1016/j.gde.2008.07.001
146. Youngson NA, Whitelaw E. Transgenerational epigenetic effects. Annu Rev Genomics Hum Genet (2008) 9:233–57. doi:10.1146/annurev.genom.9.081307.164445
147. Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, et al. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry (2011) 1:e21. doi:10.1038/tp.2011.21
148. Lee RS, Tamashiro KL, Yang X, Purcell RH, Harvey A, Willour VL, et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology (2010) 151:4332–43. doi:10.1210/en.2010-0225
149. Haramati S, Navon I, Issler O, Ezra-Nevo G, Gil S, Zwang R, et al. MicroRNA as repressors of stress-induced anxiety: the case of amygdalar miR-34. J Neurosci (2011) 31:14191–203. doi:10.1523/JNEUROSCI.1673-11.2011
150. Smalheiser NR, Lugli G, Rizavi HS, Zhang H, Torvik VI, Pandey GN, et al. MicroRNA expression in rat brain exposed to repeated inescapable shock: differential alterations in learned helplessness vs. non-learned helplessness. Int J Neuropsychopharmacol (2011) 14(10):1315–25. doi:10.1017/S1461145710001628
151. Kawashima H, Numakawa T, Kumamaru E, Adachi N, Mizuno H, Ninomiya M, et al. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience (2010) 165:1301–11. doi:10.1016/j.neuroscience.2009.11.057
152. Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci (2009) 12:399–408. doi:10.1038/nn.2294
Keywords: vulnerability genes, stressful life events, DNA methylation, miRNAs, depression
Citation: Lopizzo N, Bocchio Chiavetto L, Cattane N, Plazzotta G, Tarazi FI, Pariante CM, Riva MA and Cattaneo A (2015) Gene–environment interaction in major depression: focus on experience-dependent biological systems. Front. Psychiatry 6:68. doi: 10.3389/fpsyt.2015.00068
Received: 29 December 2014; Paper pending published: 16 March 2015;
Accepted: 21 April 2015; Published: 08 May 2015
Edited by:
Bruno Etain, University Paris East, FranceReviewed by:
Vaibhav A. Diwadkar, Wayne State University School of Medicine, USABernhard J. Mitterauer, Volitronics-Institute for Basic Research Psychopathology and Brain Philosophy, Austria
Copyright: © 2015 Lopizzo, Bocchio Chiavetto, Cattane, Plazzotta, Tarazi, Pariante, Riva and Cattaneo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annamaria Cattaneo, Stress, Psychiatry and Immunology Laboratory, Department of Psychological Medicine, Institute of Psychiatry, King’s College London, Coldharbour Lane, London SE5 9NU, UK, annamaria.cattaneo@kcl.ac.uk; acattaneo@fatebenefratelli.it