Engineered G-protein coupled receptors are powerful tools to investigate biological processes and behaviors
- 1 Department of Pharmacology and Therapeutics, Louisiana State University Health Sciences Center, New Orleans, LA, USA
- 2 Department of Pharmacology, University of North Carolina, Chapel Hill, NC, USA
Understanding how discreet tissues and neuronal circuits function in relation to the whole organism to regulate physiological processes and behaviors is a fundamental goal of modern biological science. Powerful and important new tools in this discovery process are modified G-protein coupled receptors (GPCRs) known as ‘Receptors Activated Solely by Synthetic Ligands (RASSLs),’ and ‘Designer Receptors Exclusively Activated by a Designer Drug (DREADDs).’ Collectively, these are GPCRs modified either through rational design or directed molecular evolution, that do not respond to native ligand, but functionally respond only to synthetic ligands. Importantly, the utility of these receptors is not limited to examination of the role of GPCR-coupled effector signal transduction pathways. Due to the near ubiquitous expression of GPCRs throughout an organism, this technology, combined with whole animal transgenics to selectively target expression, has the ability to regulate activity of discreet tissues and neuronal circuits through effector pathway modulation to study function and behavior throughout the organism. Advantages over other systems currently used to modify in vivo function include the ability to rapidly, selectively and reversibly manipulate defined signal transduction pathways both in short term and long term studies, and no need for specialized equipment due to convenient systemic treatment with activating ligand.
Introduction
The perturbation of normal biological processes has been one of the primary strategies employed to investigate the role of specific genes, proteins, tissues, and neural circuits in the intact organism or system. A primary methodology long used to modify neuronal activity and function has been with pharmacological tools. The discovery and utilization of agonist and antagonist ligands selective for specific neurotransmitter receptors and related signaling pathways and processes has allowed researchers to probe the role of select neuronal circuits in CNS function with tremendous power. Even so, there are certain limitations associated with pharmacological treatments, especially in vivo, that include off target affinities and undesired side-effects effects of drugs, and inability to effectively target a drug to act only in a restricted subset of tissues or neuronal circuits normally expressing the target. A more recent method to study neuronal function has involved the creation and use of transgenic animals. Initially it was possible to only knock-out a gene or knock-in a modified gene systemically. Modified genes usually represented hypo- or hyperactive versions, or particular splice isoforms. Subsequent developments both in insect and mammalian models allowed for the creation of expression systems where, through the use of a bipartite genetic system, transgene expression can be induced in defined tissues, and conditionally at defined time points by the administration of a drug, hormone, or temperature shift (Brand and Perrimon, 1993 ; Wells and Carter, 2001 ; McGuire et al., 2004 ; Aiba and Nakao, 2007 ; Gaveriaux-Ruff and Kieffer, 2007 ). Advantages of inducible genetic systems over constitutive ones include more precise control of expression and potential assessment of reversibility of expression. Targeted inactivation and activation of neurotransmitter related genes, as well as those for other neuronally expressed genes, has allowed for more precise elucidation of the function of neurotransmitters and circuits within the CNS of the intact organism. Other approaches that involve selective expression of toxins within defined neural circuits have also been informative (Drago et al., 1998 ; Martin et al., 2002 ; Yu et al., 2004 ; Nakashiba et al., 2008 ). These methods, however, are not without their limitations. Non-inducible systems are often associated with confounding developmental issues, inducible systems are slow to turn on and off, eliminating expression of an endogenous gene or introduction of a constitutively active isoform can have confounding effects on a particular cell’s physiology with respect to its role in neuronal function, and toxins can damage or kill the neurons they are expressed in.
Recently, transgenic technologies have been used to create novel systems able to primarily influence neuronal activity states. These systems rely upon targeted expression of particular receptor proteins that, when activated, can inhibit or enhance neuronal activity. These approaches are more systems-based, allowing for functional analysis of distinct neuronal circuits as a whole, rather than examination of the role of a particular gene/protein within a neuron. One strategy has been to use potassium channels to inhibit neuronal activity. Both targeted expression of modified constitutively open potassium channels in Drosophila (Nitabach et al., 2002 ), and targeted expression of the inward rectifying Kir2.1 potassium channel (Yu et al., 2004 ) in mice, have been found to be effective in silencing neuronal activity. To modulate neuronal activity, a system has been developed to target expression of GABAA chloride channels that respond to the allosteric modulator zolpidem within a transgenic mouse model insensitive to zolpidem (Wulff et al., 2007 ). Difficulties with these approaches include potential developmental effects of constitutively expressed channels, and temporal induction methods of expression can take days or weeks to become effective.
A very interesting approach to perform targeted modification of neuronal activity has involved the use of light to activate ion channels and proteins. One method utilizes targeted expression of a light-activated non-selective cationic channel protein isolated from unicellular green algae, channelrhodopsin, to excite neuronal activity. (Nagel et al., 2003 , 2005 ; Schroll et al., 2006 ; Lin et al., 2009 ). Whereas this is a very powerful method for rapidly activating neurons, channelrhodopsins are useful only for short-term neuronal modulation because they are hindered by rapid desensitization. Another light-based method that is used to promote neuronal inhibition uses lentiviral mediated expression of the light-activated halorhodopsin chloride pump from the microorganism Natronomonas pharaonis to hyperpolarize neurons in the CNS (Tonnesen et al., 2009 ). The most recent of these optical techniques are a collection of chimeras between rhodopsin and β2 and α1 adrenergic G-protein coupled receptors (GPCRs). Lentiviral mediated expression of these optoXR proteins in CNS tissues was found to modify firing rates of neurons in slice culture upon exposure to light consistent with the effects of neuronal Gαs or Gαq signal pathway activation to enhance or decrease firing respectively (Airan et al., 2009 ). Advantages of these optic techniques include very rapid activation, however, limitations include poor penetration of light into whole organisms and the need of specialized equipment including light sources and fiber optics. Furthermore, lentiviral methods involve surgical procedures and the effects are usually transient.
Other studies have employed transgenic expression of certain wild type GPCRs to induce neuronal silencing. For example, selective targeted restoration of 5-HT1A serotonin receptor expression within a 5-HT1A receptor knockout background followed by convenient administration of receptor selective agonist has been found to be effective (Tsetsenis et al., 2007 ) in mice. Similarly, selective targeting of different Drosophila 5-HT receptors to defined tissues has also had some success to modulate tissue function (Kerr et al., 2004 ). Whereas these methods utilizing native GPCRs are a promising and powerful avenue, care must be taken with respect to data interpretation due to potential confounding effects of endogenous neurotransmitters, and some of these models require specialized genetic backgrounds. Another approach involves heterologous targeted expression of the Drosophila melanogaster allostatin peptide GPCR within the mouse CNS followed by application of allostatin, an insect peptide hormone not normally found in mammals, to induce neuronal silencing in the CNS (Tan et al., 2006 ; Wehr et al., 2009 ). Peptides, however, have limited use because of their low solubility and must therefore be directly applied to tissues of interest.
A new approach to study biological function and behaviors that circumvents many of the disadvantages of other technologies has been developed that combines the tissue specificity of transgenics with the rapid and reversible effects of pharmacological agents. The methodology involves targeted expression of GPCRs that have been modified to respond only to non-endogenous chemicals to rapidly and reversibly modulate effector pathway activity in defined tissues and neural circuits. Importantly, these receptors can be used to produce both short-term and long-term modulation of activity, and only require simple and convenient systemic administration of ligand by feeding or peripheral injection. Invasive procedures including stereotactic injection, and specialized equipment like fiber optics and light sources are not required. These modified G-protein receptors receptors have been termed: ‘Receptors Activated Solely by Synthetic Ligands (RASSLs)’ and ‘Designer Receptors Exclusively Activated by a Designer Drug (DREADDs)’ (Table 1 ).
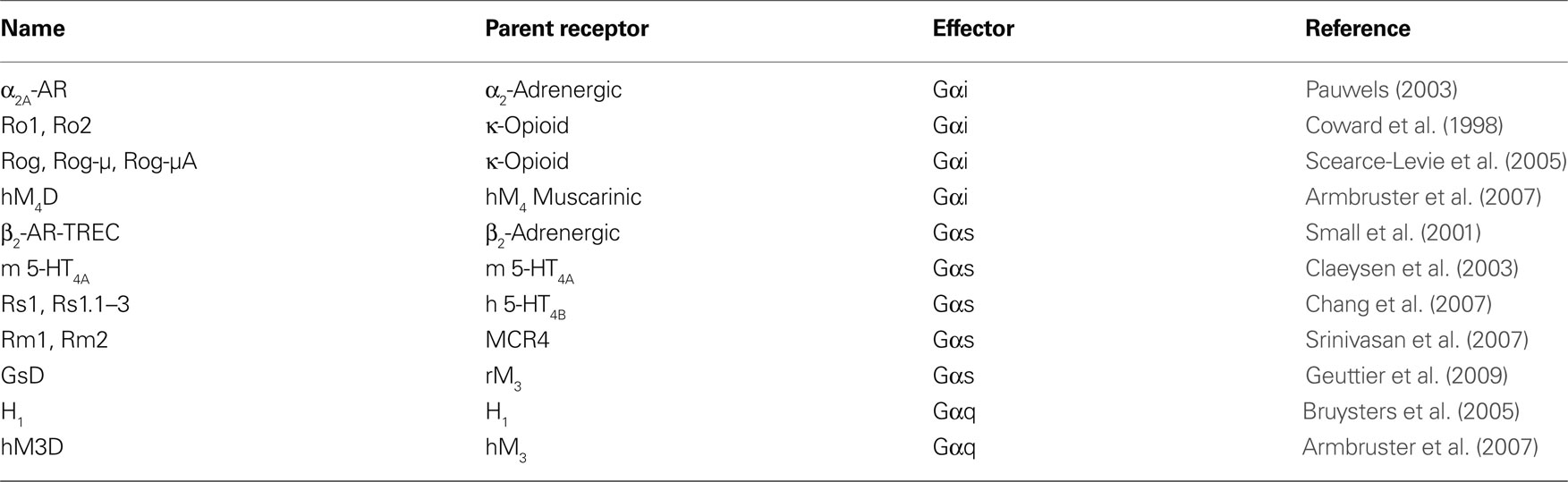
Table 1. The major RASSL and DREADD receptors grouped according to the primary G-protein activated. The parent receptor of each is listed.
GPCRs
G-protein coupled receptors are 7-α-helical transmembrane proteins that transduce and amplify extracellular signals to multiple pathways inside the cell. GPCRs are the most widespread receptor class throughout the organism, and modulate not only cellular processes directly, but also the function of other receptor families, and are the primary mechanism of communication between cells (Kroeze et al., 2003 ; Armbruster and Roth, 2005 ). The physiological processes that GPCRs modulate are diverse and include neurotransmission, development, cardiovascular function, gut motility, and odor detection, among others. About 80% of hormones and neurotransmitters involved in signal transduction are thought to act through GPCRs. Significantly, >50% of current drugs on the market target GPCR function as a major mechanism of therapeutics (Roth et al., 2004 ; Strachan et al., 2006 ). Thus, GPCRs remain the most popular family of targets used in drug discovery.
G-protein coupled receptors are functionally coupled to heterotrimeric G-proteins at the intracellular loops and C-terminus of the GPCR. Heterotrimeric G proteins are comprised of a Gα subunit and a dimeric Gβγ subunit, and are grouped into four classes: Gαs, Gαi/o, Gαq/11, and Gα12/13 (Gilman, 1987 ). Whereas Gαs stimulates the production of cAMP through activation of adenylate cyclase, Gαi/o produces the opposite effect, and reduces levels of intracellular cAMP through inhibition of adenylate cyclase. The Gαq subunit stimulates phospholipase-Cβ, which catalyzes the production of phosphoinositides and the release of intracellular calcium, among other processes. Gα12/13 interacts with a number of effectors including RhoGEFs to modulate cell growth and cytoskeleton structure (Kelly et al., 2007 ).
Because of the widespread expression of GPCRs (Regard et al., 2008 ), and the variety of effector pathways that they can couple to (Urban et al., 2007 ), targeted expression of modified GPCRs is an ideal tool to utilize to probe not only the role of how specific signal transduction pathways influence cellular function, but to also probe the role of specific tissues and neuronal circuits underlying physiological and behavioral processes. The primary RASSL/DREADD strategy is to engineer a collection of modified GPCRs, each coupling to different primary effector pathways, that respond to synthetic or non-endogenous compounds.
RASSLs
The first published report on the creation of a modified GPCR to respond to a non-endogenous ligand was by Strader et al. (1991) . In the process of exploring the nature of ligand-receptor interactions for the β2-adrenergic receptor, a single point mutant (S113A) was found to eliminate binding of endogenous ligands while simultaneously conferring activity for a class of compounds with little affinity for the wild type receptor (Strader et al., 1991 ). Whereas the potencies of these synthetic compounds was relatively low at the modified β2AR, this work nevertheless demonstrated that GPCRs could be modified to lose native ligand recognition while maintaining affinity for synthetic ligands. The Strader et al. (1991) study was thus an important proof-of-concept study for this approach.
The next major development was the creation of modified human kappa-opioid receptors with dramatically reduced affinity for the natural peptide ligands (1000-fold reduction), but retained affinity for small molecule drugs like bremazocine and spiradoline (Coward et al., 1998 ). Two receptor variants were created: Ro1 and Ro2. Ro1 is a chimeric receptor of the κ-opioid receptor containing the second extracellular loop of the delta opioid receptor, and Ro2 is essentially the Ro1 receptor with an additional mutation at the top of the sixth transmembrane helix. The overall design of the modified receptors arose from previous studies of chimeras between κ and δ receptors indicating that the second extracellular loop contains a major determinant for binding of a κ selective ligand, dynorphin. What was achieved by this manipulation was a Gαi-coupled κ-opioid receptor with significantly reduced binding to κ-specific peptides, but retained affinity for κ-selective small molecule synthetic ligands whose binding regions were not determined by the second extracellular loop region (Coward et al., 1998 ). These receptors were termed RASSLs, and when expressed in rat fibroblast cells found to be functional for inducing proliferation, a process known to be stimulated by Gαi signaling, with the synthetic ligand spiradoline (Coward et al., 1998 ).
Many studies using the Ro1 RASSLs were subsequently published utilizing this receptor as a tool to explore the effects of activating the Gαi effector pathway in various tissues as defined by specific transgene targeting (Conklin et al., 2008 ; Pei et al., 2008 ). The first transgenic mouse study detailed the conditional expression and activation of the Ro1 receptor in mouse heart, liver, and brain (Redfern et al., 1999 ). Activation of Gαi signaling in the heart with the κ-opioid receptor agonist spiradoline produced significant and dose dependent bradycardia with a ∼50% reduction in heart rate in less than a minute after drug administration (Redfern et al., 1999 ). In a subsequent study, it was found that the induction of expression of Ro1 in the heart induces cardiomyopathy in the absence of spiradoline (Redfern et al., 2000 ). Partially inhibiting expression of Ro1, treatment with the κOR antagonist nor-binaltomorphine, and treatment with pertussis toxin, restored normal function demonstrating that the heart defects were indeed due to excessive Gαi signaling from the Ro1 receptor in the absence of ligand stimulation (Redfern et al., 2000 ). Significantly, these experiments indicate that there is a level of basal constitutive activity associated with the Ro1 receptor that is able to influence physiological processes in the absence of pharmacological activation. Nevertheless, this system has proven valuable as a new model for the study of dilated cardiomyopathy (Baker et al., 2001 ; McCloskey et al., 2008 ). The inducible doxycycline-responding Ro1 mouse strain was also utilized by Sweger et al. (2007) to investigate the role of astrocytes in mouse brain. They created a transgenic κOR knockout mouse with conditional expression of Ro1 in astrocytes induced by discontinuation of feeding doxycycline and found that even in the absence of the κOR agonist spiradoline the mice developed severe hydrocephalus (Sweger et al., 2007 ) through mechanisms regulating CSF production, defining a new role for Gαi in astrocytes. Importantly, these results independently confirmed that the Ro1 receptor is associated with a certain physiologically relevant level of constitutive activity.
This constitutive activity of Ro1 was exploited to investigate the role of Gαi signaling in bone development by Peng et al. (2008) . It had been previously known that inactivation of Gαs signaling, the main effector pathway coupled to the PTH/PTHrP (PPR) receptors in osteoblasts, resulted in a reduction of bone formation and turnover (Sakamoto et al., 2005 ). Furthermore, that various means of indirectly inhibiting Gαi pathways, increased bone turnover (Peng et al., 2008 ). To directly test the role of Gαi signaling, the Ro1 receptor was conditionally expressed in mouse osteoblasts at various developmental time points from embryogenesis to weaning. At each time point, expression of the Ro1 receptor resulted in decreased bone formation and turnover (Peng et al., 2008 ), mimicking the effects of Gαs inactivation. Together, these results indicate that proper bone formation results from a balance between Gαi and Gαs signaling activity in osteoblasts (Peng et al., 2008 ).
An interesting study using the doxycycline-responding Ro1 transgenic mice created by Redfen and colleagues was to examine the nature of the sweet and umami taste. Here, Ro1 was expressed in taste buds under the control of the T1R2 receptor promoter in T1R2 null mice. These mice responded to the κ-selective agonist spiradoline as control mice did to sweet taste, indicating that Gαi signaling through the T1R2 receptor is responsible for perception of sweet taste (Zhao et al., 2003 ).
Following the success of the initial Ro1 receptor, attempts were made to modify it. First, the Rog (RASSL opioid green) receptor was created by fusing GFP to the N-terminus of Ro1 to visualize receptor localization in the living cell (Scearce-Levie et al., 2005 ). After validating its ability to internalize upon agonist stimulation, signaling, and RASSL function, subsequent modifications included mutation of the four C-terminal phosphorylation sites to alanine (Rog-A), replacement of the entire C-terminus with the μ-opioid receptor (Rog-μ), and mutation of five C-terminal serine and glutamic acid residues of the Rog-μ receptor to alanine (Rog-μA) (Scearce-Levie et al., 2005 ). The loss of C-terminal phosphorylation sites in the Rog-A resulted in a significant reduction in agonist induced internalization and a resistance to desensitization, however maximal cAMP inhibition was unaffected (Scearce-Levie et al., 2005 ). The Rog-μ receptor demonstrated increased agonist induced receptor internalization, as was predicted based upon the ability of the μ receptor to more readily internalize than the δ receptor (Scearce-Levie et al., 2005 ). Whereas the Rog-μA receptor was predicted to be resistant to agonist induced internalization as the Rog-A receptor was, it surprisingly demonstrated constitutive internalization (Scearce-Levie et al., 2005 ). Addition of an antagonist, nor-BNI, was found to rescue cell surface expression (Scearce-Levie et al., 2005 ). Another physiological effect, adenylate cyclase superactivation, was examined in these receptors. Long-term activation of Gαi signaling pathways can lead to an increase in adenylate cyclase activity and an enhanced response to forskolin stimulation over baseline conditions (Watts and Neve, 2005 ). Neither the Rog-μ nor-Rog-μA receptors showed this effect after overnight treatment with spiradoline followed by forskolin stimulation (Scearce-Levie et al., 2005 ). The authors speculated that it is not constitutive activity of Gαi from the Ro1 receptors that is responsible for cardiomyopathy observed in their previous studies, but rather an increase in Gαs signaling through adenylate cyclase superactivation that produces the phenotype (Scearce-Levie et al., 2005 ). It is possible that these newly modified Ro1-related RASSLs will address this issue as well as others.
Through a series of structure/function studies, another modified Gαi coupled receptor, the α2A-adrenergic receptor, was demonstrated to have RASSL-like properties. Based upon previous work predicting the importance of two conserved serine residues within the putative transmembrane binding site the serine at position 200, and the serine at position 204 were each mutated to alanine and the resulting variants pharmacologically characterized. The resulting receptors had reduced (S200A), to negligent (S204A) affinity and activity for the native ligand, but high affinity and activity for certain classes of drug including synthetic imidazoline derivatives (Pauwels and Colpaert, 2000 ). Interestingly, several α2A-adrenergic receptor antagonists, including atipamezole and SKF86466, retained antagonist properties at the S200A variant but demonstrated significant partial agonist activity at the S204A receptor (Pauwels and Colpaert, 2000 ). Significantly, these studies demonstrated that aspects of functional selectivity can be potentially engineered in to a RASSL where the response of a receptor to a particular ligand is not only lost or retained in mutant RASSL variants, but can be fundamentally altered to a different response.
There have been three independent RASSL families developed to probe Gαs signaling. The first was an attempt to modify the β2-adrenergic receptor as a tool for use in gene therapies and create a ‘modified therapeutic receptor–effector complex’. In the human β2-adrenergic receptor, glutamine at position 27 was mutated to glutamate to reduce ligand-induced receptor internalization, aspartate at position 113 was mutated to serine to alter ligand binding properties, 15 potential phosphorylation sites were changed to alanine, and the entire open reading frame of the rat Gαs was fused to the C-terminus coding region (Small et al., 2001 ). This modified receptor complex was found to be unresponsive to catacholamines, but responsive to a single tested ligand L158870 at micromolar concentrations (Small et al., 2001 ). Due to the low potency of the modified receptor complex to the synthetic ligand its utility in vivo is likely limited.
The second approach at developing a Gαs RASSL involved modifying the serotonin 5-HT4A receptor. Key amino acids in the native ligand binding pocket were deduced based upon the published crystal structure of the β-adrenergic receptor to identify Asp100 in TM3, which was predicted to interact with the amine in the indole ring of serotonin. Mutation of this Asp to Ala in the mouse 5-HT4A receptor significantly reduced binding as well as activation of the receptor by serotonin and other tryptamines with respect to adenylate cyclase activation, while maintaining affinity and activity for other ligands without a protonated amine in the core structure (Claeysen et al., 2003 ). Interestingly, a number of antagonists at the native receptor were found to have agonist activity with respect to adenylate cyclase at the modified receptor (Rs1) (Claeysen et al., 2003 ). This may allow for selective activation of the modified receptor in animal models, while keeping native receptors inactive, something not possible with the Ro1 receptors. In a subsequent study, conditional expression of the Rs1 receptor in transgenic mouse osteoblasts resulted in increased bone mass in the absence of activating ligand, presumably by constitutive Gαs signaling from the Rs1 receptor (Hsiao et al., 2008 ). These bone-enhancing effects were observed to be different from a mouse expressing a constitutively activated form of the Gαs-coupled PPR receptor, indicating that additional factors are differentially recruited to the receptors to mediate physiological effects.
To refine the RASSL characteristics of a receptor like Rs1, a series of modifications of the human 5-HT4B D100A receptor, which has high constitutive adenylate cyclase activity, were created (Chang et al., 2007 ). To reduce constitutive activity two separate mutations were made, based upon previous reported studies examining structure and activity, changing Asp at position 66 to Asn (Rs1.2), and Trp at position 272 to Ala. Although each resulting RASSL had significantly reduced constitutive adenylate cyclase activity, there was also a large reduction in the levels of activation by the ligand zacopride, limiting their utility (Chang et al., 2007 ). Nevertheless, the RASSLs were significantly stimulated by agonist. An interesting feature of the Rs1 receptor includes functional selectivity where certain classes of ligand predominantly activate Gαs, while other classes, represented by zacopride, also induce coupling to Gαq. Surprisingly, the ability of zacopride to induce coupling to Gαq was lost in the two mutant 5-HT4B RASSLs (Chang et al., 2007 ).
In an attempt to create a predominantly Gαq-coupled RASSL, the intracellular loops of the human Rs1 were replaced with those from the Gαq-coupled human 5-HT2C receptor to create Rs1-5-HT2C (Rs1.1). In this process, 12 different chimeras were created and tested that each had different splicing junctions and combinations of intracellular loops. It was found that replacement of the second or third intracellular loops eliminated both Gαs and Gαq activity, and only replacement of the C-terminus was able to increase coupling to Gαq (Chang et al., 2007 ). This new receptor, Rs1-C-5-HT2C, however, still had significant constitutive and ligand-induced Gαs activity. Following similar techniques utilizing intracellular domains from the human 5-HT1A receptor, attempts were made to engineer a Gαi-couple RASSL from Rs1. Replacement of the third intracellular loop was found to abolish Gαs and Gαq activity and to confer Gαi activity to the Rs1-i3-5-HT1A (Rs1.3) receptor (Chang et al., 2007 ). The potency of this effect, however, was weak thus limiting the utility of this receptor. Importantly, these studies with the Rs1 series of receptors demonstrated that it was possible to continue to modify a RASSL to alter and refine physiological properties to something more desirable as a tool for in vivo use.
The third approach to develop a Gαs coupled RASSL involved modifying the melanocortin-4 receptor (MCR4). An extensive review of previous work examining the location and functional result of individual mutations in the MCR4 led to the identification of five candidate mutations for further study as potential RASSLs (Srinivasan et al., 2007 ). Of these, two were designated RASSLs: L106P-MCR4 (Rm1), and D122A-MCR4 (Rm2). The native RCM4 has significant constitutive basal activity associated with Gαs signaling, and although Rm1 has a 30% reduction in activity, Rm2 has a nearly twofold increase in basal activity over the native receptor (Srinivasan et al., 2007 ). Importantly, neither Rm1 nor-Rm2 respond to native melanocortin peptides, but do respond to nanomolar concentrations of the synthetic MCR4 selective ligand THIQ with respect to measured cAMP release (Srinivasan et al., 2007 ).
The third major class of G-protein effector is Gαq, and one attempt has been made to generate a RASSL coupled to this pathway. Based upon previous structure function studies examining ligand binding properties, the phenylalanine at position 435 of the human H1 receptor was mutated to an alanine and the resulting receptor characterized (Bruysters et al., 2005 ). Rather than assess Gαq coupling by measuring PI turnover or calcium mobilization, a luciferase reporter assay for NFkB activity was used to measure activity. This was based upon previous work that demonstrated direct functional coupling of Gαq/11 to NFkB activation (Bakker et al., 2001 ). Therefore, RASSL activity from this receptor can only be reliably extended to activation of the NFkB signaling pathway. Interestingly, whereas the F453A H1 receptor variant maintained affinity for histamine, affinity and potency at NFkB activation increased up to 1000-fold for certain synthetic ligands (Bruysters et al., 2005 ). In the case of ClPheHA, potency was increased from an EC50 of ∼1 mM at the native receptor to ∼1 nM at the F453A variant. Rather than engineering loss of affinity and potency for native ligand while retaining those properties for a synthetic ligand, greatly enhancing affinity and potency for a synthetic ligand through receptor modification was demonstrated here to be a new strategy to develop a RASSL. The F453A H1 receptor has negligent basal constitutive activity with respect to NFkB signaling, however, additional pathways coupled to Gαq have yet to be investigated, as well as the utility of this receptor to study in vivo processes.
Of significant concern with nearly all RASSLs created thus far is the presence of physiologically relevant basal constitutive activity with the primary Gα effector protein. Whereas this may effectively allow functional analysis of an effector pathway under certain conditions, constitutive activity present from embryogenesis has the potential to seriously confound results examining acute function. Another considerable shortcoming of the conventional RASSLs is that they by and large utilize synthetic ligands that have significant affinity and potency for the endogenous wild type receptor as well as potential off-target actions. Therefore, ligands used to stimulate RASSLs in vivo could induce behaviors and physiological processes from native receptors and confound results. Nonetheless, based on the large number of important papers, conventional RASSLs continue to be widely used with great success.
DREADDs
Ideally, a modified receptor would have no basal constitutive activity, and respond to a synthetic ligand that had no natural targets within the organism. To develop such a receptor system we followed a novel directed molecular evolution approach to generate a collection of modified GPCRs termed DREADDs (Armbruster et al., 2007 ). This strategy involved choosing a biologically inert metabolite of clozapine, clozapine-N-oxide (CNO) as the synthetic ligand, and the human M3 receptor (M3R) for initial mutagenesis. Whereas the parent compound clozapine has affinity at multiple GPCRs, including high affinity and weak partial agonist activity for the M3R, the metabolite CNO was shown to lack appreciable affinity for any receptor by screening of nearly the entire relevant recepterome (Armbruster et al., 2007 ). A yeast model system where activation of the heterologously expressed modified rat M3R was required for cell growth (Erlenbach et al., 2001 ) was used as a screening platform for random mutagenesis to generate a set of mutants that were each activated by clozapine. A subset of these clones were selected for a second round of mutagenesis and selection for activation by 1 μM CNO, followed by a third round of mutagenesis and selection for growth on 5 nM CNO to generate a final collection of two modified M3 receptors with high affinity for CNO (Armbruster et al., 2007 ) (Figure 1 ). Fortuitously, loss of affinity for the native ligand, acetylcholine, as well as carbachol was lost during this process. Whereas these modified rat M3 receptors demonstrated functional activation by CNO and insensitivity to acetylchole, they had high basal activity with respect to Gαq signaling and PI turnover in human HEK T cells. Based upon the results of the yeast screening experiments, a focused library of mutant full length human M3 receptors was created and screened in HEK T cells to identify a receptor demonstrating high affinity for CNO, extremely low affinity for acetylcholine, and minimal constitutive activity associated with Gαq signaling and PI turnover (Armbruster et al., 2007 ). The final receptor, hM3D, contained two point mutations with tyrosine at position 149 mutated to cystine, and alanine at position 239 mutated to glycine (Armbruster et al., 2007 ). Importantly, not only was CNO able to activate Gαq signaling and PI turnover, but CNO was also shown to activate MAPK signaling through interactions with β-arrestin, indicating that coupling to multiple effector pathways was preserved in the DREADD receptor. Mutation of the conserved tyrosine and glycine residues in the other human muscarinic receptors resulted in the creation of hM2D and hM4D DREADD receptors coupled to Gαi/o, and hM1D coupled to Gαq. Additional functionality of the hM4D was determined by demonstrating that stimulation of the receptor with CNO, but not with ACh, activated inwardly rectifying potassium channels (GIRKs) in both transfected HEK cells and transfected hippocampal neurons (Armbruster et al., 2007 ). These results indicate that the hM4D receptor has potential as a tool for in vivo neuronal silencing.
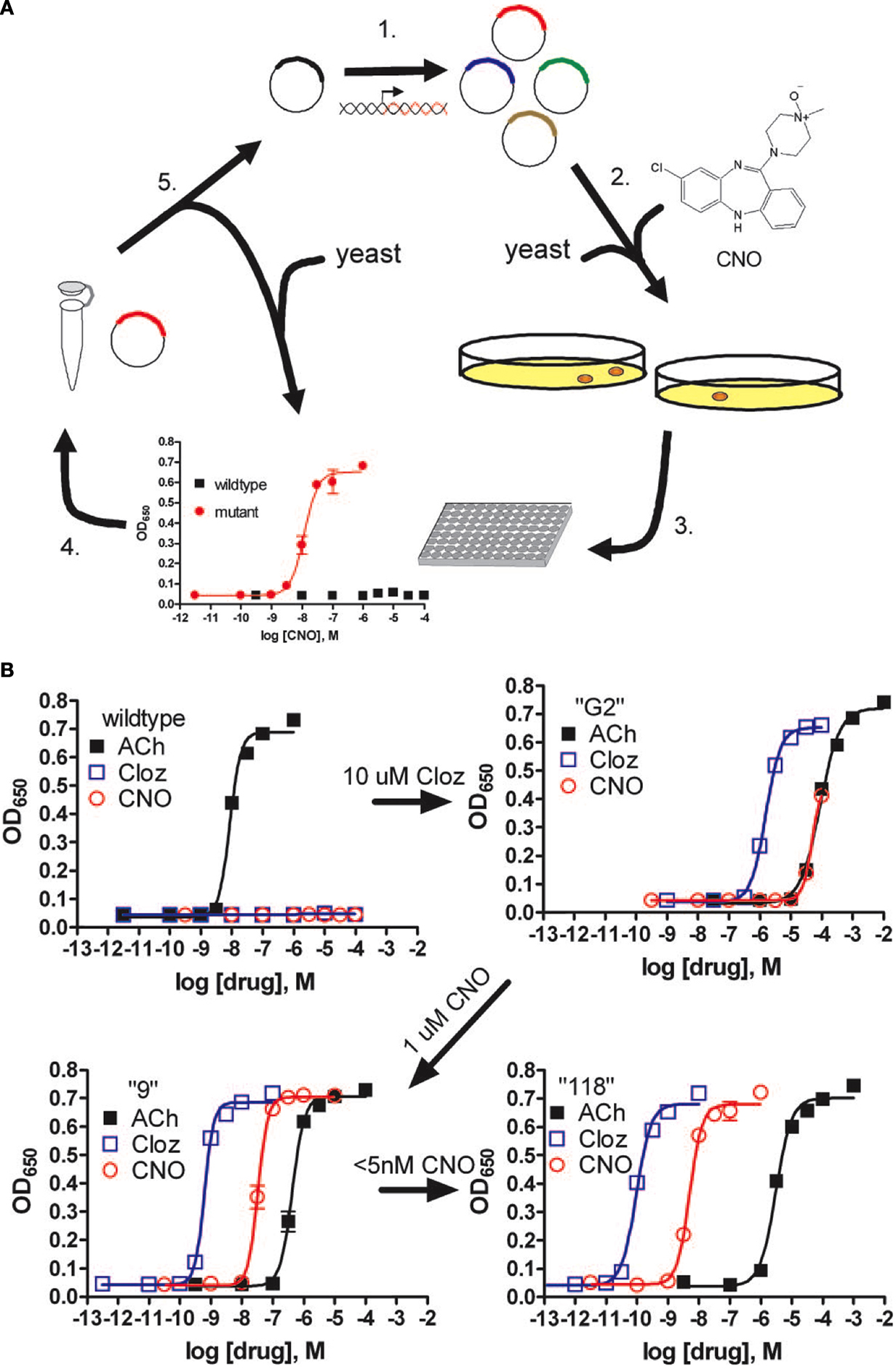
Figure 1. Pharmacological profiles of an rM3Δi3 receptor mutant selected during directed molecular evolution for CNO responsiveness. (A) Experimental design for directed evolution of mammalian GPCRs in yeast to create DREADDs. (1) Libraries of randomly mutated rM3Δi3 receptors were produced by mutagenic PCR; (2) yeast-expressing mutant receptors activated by synthetic ligands (e.g., CNO) were selected for by growth on nutrient deficient medium; (3) mutants were verified by secondary liquid growth assays in 96-well plates; (4) plasmid DNA was isolated from yeast; (5) clones were retransformed into yeast to pharmacologically profile mutants by liquid growth assays, and those with desirable properties were sequenced and remutagenized for subsequent rounds of selection to yield receptors with higher potency. (B) Optical density at 650 nm of liquid cultures of yeast transformed with either wild type, clone “G2” (first library, 10 μM clozapine screen), clone “9” (second library, 1 μM CNO screen), or clone “118” (third library, 5-nM CNO screen) rM3Δi3 receptors incubated with ACh (■), clozapine (□), or CNO (E). Data shown are mean ± SEM values of a representative experiment performed with two independent yeast transformants grown for each clone. Figure used with permission: Copyright (2007) National Academy of Sciences, USA Armbruster et al. (2007) .
To develop the final Gαs coupled DREADD (GsD), we modified the rat M3D receptor by replacing the second and third intracellular loops with the corresponding loops from the Gαs-coupled turkey β1-adrenergic receptor (Geuttier et al., 2009 ). Transgenic mouse lines expressing the hM3D and GsD receptors in pancreatic beta-cells have been made that show stimulation of either receptor has significant effects on beta-cell function including aspects of glucose tolerance and insulin release (Geuttier et al., 2009 ). Additional transgenic mouse studies with all three DREADD receptors are currently being performed to analyze the role of G-protein signaling in a variety of tissues (Figure 2 ). We have demonstrated remote control of neuronal activity in mice expressing the hM3D receptor in hippocampus (Alexander et al., 2009 ). Administration of CNO to these transgenic mice led to both increases in hippocampal neuronal activity as well as behavioral modifications including increased locomotor activity and seizures in a dose dependent fashion (Alexander et al., 2009 ). In slice cultures, the increases in neuronal activity from a single pulse of CNO returned to baseline levels in about 60 min. The locomotor effects of systemic CNO, however, did not return to baseline levels for about 9 h (Alexander et al., 2009 ). Importantly, these data indicate that this system is reversible with in vitro cellular effects of CNO extinguishing more rapidly than the in vivo behavioral effects.
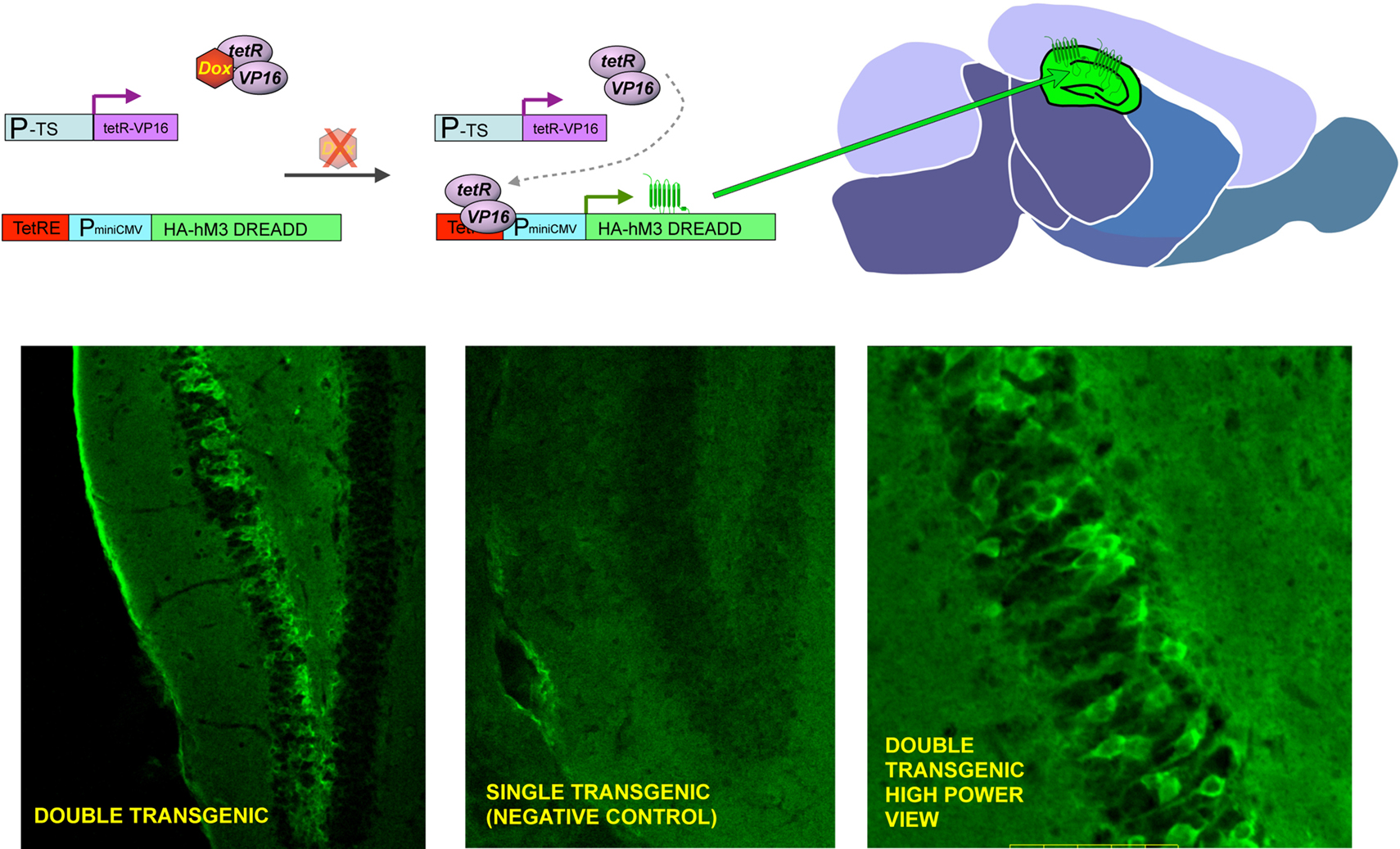
Figure 2. General strategy to achieve inducible, tissue-specific expression of DREADDs. The upper panel shows the basic strategy to generate tissue specific expression of DREADDs in mouse brain. Double transgenic animals are made where one element contains a tissue specific promotor (P-TS) driving expression of the tetR-VP16 fusion protein, and the other contains the tetracycline response element (TetRE), followed by a minimal CMV promoter driving expression of HA-tagged DREADDs. Whereas both transgenic elements are present in every cell throughout the body, expression of tetR-VP19 is only in promoter defined areas (e.g. PET-1 for 5-HT neurons, GFAP for neuronal precursors and DARPP-32 for medium spiny neurons) and transgenic receptor expression is limited to the defined tissue (upper center and right panel). When doxycycline is present (DOX), it binds to the tetR factor and blocks it from interacting with the TetRE, preventing transcription of DREADDs (upper left panel). The lower panels show preliminary success with CAMKII-tet, which achieves forebrain-specific expression; in this set of founders, high levels of expression were achieved in hippocampus.
We have recently created transgenic Drosophila melanogaster expressing the Gαs-coupled hM4D, Gαi-coupled rM3D-βar, and Gαq-coupled hM1D receptors under the control of the bipartite GAL4/UAS system. The GAL4/UAS system is a genetic method to express transgenes selectively in defined tissues (Brand and Perrimon, 1993 ). In preliminary studies we have targeted DREADD expression to distinct neural circuits and have found that behaviors are dramatically altered when adult flies expressing DREADDS are fed CNO (unpublished results). Importantly, wild type and parental strains maintained on food containing CNO (at least to 10 mM) exhibit no overt abnormal developmental or behavioral effects, indicating that the effects of CNO on the transgenic flies expressing DREADDs are indeed due to DREADD activation. Given the conservation of biological processes between mammalian systems and the power of the fly as a genetic model (Nichols, 2006 ), studies performed in the fly are likely to be informative to both insect and mammalian models with respect to elucidation of signal transduction pathways and molecular processes underlying neuronal function as they relate to behaviors. Furthermore, this system is anticipated to be useful in the fly to probe the role of signal transduction pathways and discreet tissues in developmental processes.
Aside from using DREADDs to directly probe the role of individual signal transduction pathways in physiological processes and the role of specific tissues and circuits in behaviors, the hM4D receptor has recently proven to be a valuable tool in understanding mechanisms of allosteric modulation in GPCRs. A known allosteric modulator of the M4 receptor, LY2033298, was shown to act cooperativity with the orthosteric binding site to restore affinity and functionality of acetylcholine to the orthosteric site of the hM4D receptor (Nawaratne et al., 2008 ). These muscarinic DREADD receptors, and potentially other similarly modified receptors, may represent novel tools to screen for allosteric modulators that act solely through interactions at distinct allosteric sites and have cooperativity with the orthosteric site. Compounds that would be able to restore affinity and activity to the native ligand would therefore potentially represent novel therapeutics that enhance the ability of native ligand to activate its receptor.
Summary
An exciting and powerful method of probing the role of G-protein effector signaling, as well as the function of discreet tissues and neural circuits in mediating physiological processes and behaviors has recently been developed and refined. The first generation of engineered GPCRs, RASSLs, were primarily rationally designed based upon structure/function studies to eliminate native ligand binding, while maintaining affinity for synthetic ligands. These receptors have been used to generate conditionally expressing transgenic mice, where they have been used to study how G-protein effector pathways affect various processes including heart function, bone growth, and brain development. They have also been used to define the function of other GPCRs as well as particular tissues, as they were used to elucidate the receptor and effector pathway underlying the perception of sweet taste. Advantages that this system offers are the ability to very rapidly turn on and off signaling pathways by simply administering a synthetic ligand. Importantly, because GPCRs and their associated effector pathways are ubiquitous throughout an organism, studies are not limited to examining the native role of the particular receptor a RASSL is derived from, but can be used to define the function of entire tissues and neural circuits in a more systems-based approach. These first generation RASSLs, unfortunately, are frequently associated with physiologically relevant basal constitutive activity, as well as response to synthetic ligands that also target the endogenous wild type receptors. Whereas these properties may be advantageous for some studies, they likely present certain limitations and challenges for widespread use.
The next generation of engineered GPCRs, DREADDs, created through a process of directed molecular evolution overcome many of the limitations present in the first generation RASSLs. DREADDS have negligent basal constitutive activity associated with them, and are activated by synthetic ligands with no appreciable affinity for any of the known receptorome. Additionally, CNO has minimal activity at a variety of relevant kinases (Figure 3 ). Transgenic expression models in both mouse and fly have validated their functionality to probe both physiological processes and complex behaviors. Given the advantages of the DREADD receptors over the RASSLs, there are certain caveats that must be considered for them, as well as for the RASSLs, involving expression levels.
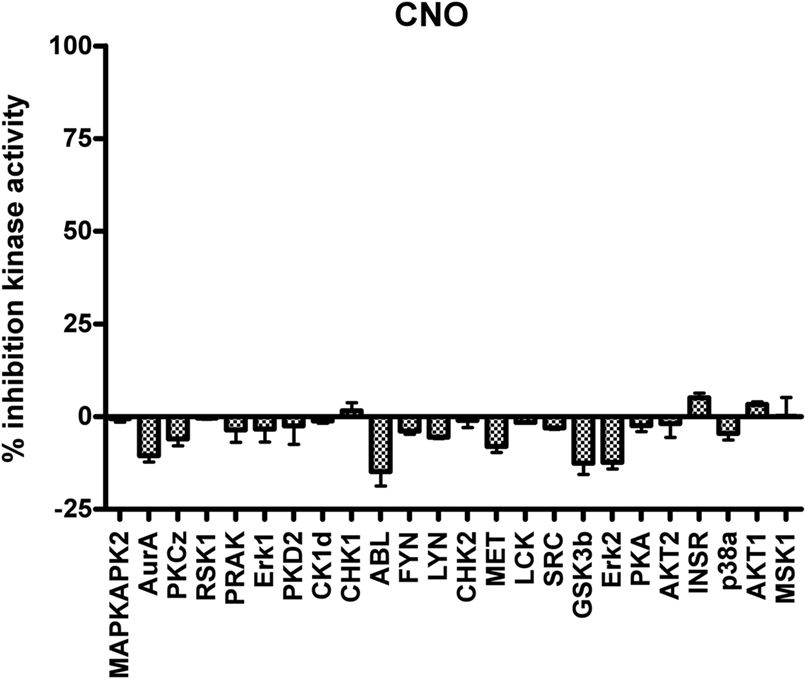
Figure 3. Clozapine-N-oxide is devoid of activity at essential neuronal protein kinases. As is shown, CNO at a screening concentration of 10 μM is devoid of appreciable agonist or antagonist activity at a large number of the neuronal kinases implicated in signaling (V. Setola and B. L. Roth, unpublished observations).
Under most normal transgenic conditions, expression levels are likely high, such that even under non-activated conditions the stoichiometric balance between receptor and precoupled effector may be perturbed such that the normal function of the tissue and circuit may be affected. Furthermore, the presence of high levels of activated receptor may induce activation of effector pathways not normally functionally coupled to the particular GPCR, confounding results. To achieve the most relevant data for a particular system, expression levels should be determined, and manipulated if possible, with those having expression closest to naturally occurring GPCRs used for experimentation. Nevertheless, both RASSLs and DREADDs present a very effective tool to elucidate biological function and behaviors. Advantages over other current systems include the specificity of transgenic targeting, the convenience of systemic administration of small molecule drugs with no other appreciable biological targets within the organism, rapidity and reversibility of effect, and no need for specialized equipment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aiba, A., and Nakao, H. (2007). Conditional mutant mice using tetracycline-controlled gene expression system in the brain. Neurosci. Res. 58, 113–117.
Airan, R. D., Thompson, K. R., Fenno, L. E., Bernstein, H., and Deisseroth, K. (2009). Temporally precise in vivo control of intracellular signalling. Nature 458, 1025–1029.
Alexander, G. M., Rogan, S. C., Abbas, A. I., Armbruster, B. N., Pei, Y., Allen, J. A., Nonneman, R. J., Hartmann, J., Moy, S. S., Nicolelis, M. A., McNamara, J. O., and Roth, B. L. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39.
Armbruster, B. N., Li, X., Pausch, M. H., Herlitze, S., and Roth, B. L. (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U.S.A. 104, 5163–5168.
Baker, A. J., Redfern, C. H., Harwood, M. D., Simpson, P. C., and Conklin, B. R. (2001). Abnormal contraction caused by expression of G(i)-coupled receptor in transgenic model of dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 280, H1653–H1659.
Bakker, R. A., Schoonus, S. B., Smit, M. J., Timmerman, H., and Leurs, R. (2001). Histamine H(1)-receptor activation of nuclear factor-kappa B: roles for G beta gamma- and G alpha(q/11)-subunits in constitutive and agonist-mediated signaling. Mol. Pharmacol. 60, 1133–1142.
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415.
Bruysters, M., Jongejan, A., Akdemir, A., Bakker, R. A., and Leurs, R. (2005). A G(q/11)-coupled mutant histamine H(1) receptor F435A activated solely by synthetic ligands (RASSL). J. Biol. Chem. 280, 34741–34746.
Chang, W. C., Ng, J. K., Nguyen, T., Pellissier, L., Claeysen, S., Hsiao, E. C., and Conklin, B. R. (2007). Modifying ligand-induced and constitutive signaling of the human 5-HT4 receptor. PLoS ONE 2, e1317. doi: 10.1371/journal.pone.0001317.
Claeysen, S., Joubert, L., Sebben, M., Bockaert, J., and Dumuis, A. (2003). A single mutation in the 5-HT4 receptor (5-HT4-R D100(3.32)A) generates a Gs-coupled receptor activated exclusively by synthetic ligands (RASSL). J. Biol. Chem. 278, 699–702.
Conklin, B. R., Hsiao, E. C., Claeysen, S., Dumuis, A., Srinivasan, S., Forsayeth, J. R., Guettier, J. M., Chang, W. C., Pei, Y., McCarthy, K. D., Nissenson, R. A., Wess, J., Bockaert, J., and Roth, B. L. (2008). Engineering GPCR signaling pathways with RASSLs. Nat. Methods 5, 673–678.
Coward, P., Wada, H. G., Falk, M. S., Chan, S. D., Meng, F., Akil, H., and Conklin, B. R. (1998). Controlling signaling with a specifically designed Gi-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 352–357.
Drago, J., Padungchaichot, P., Wong, J. Y., Lawrence, A. J., McManus, J. F., Sumarsono, S. H., Natoli, A. L., Lakso, M., Wreford, N., Westphal, H., Kola, I., and Finkelstein, D. I. (1998). Targeted expression of a toxin gene to D1 dopamine receptor neurons by cre-mediated site-specific recombination. J. Neurosci. 18, 9845–9857.
Erlenbach, I., Kostenis, E., Schmidt, C., Hamdan, F. F., Pausch, M. H., and Wess, J. (2001). Functional expression of M(1), M(3) and M(5) muscarinic acetylcholine receptors in yeast. J. Neurochem. 77, 1327–1337.
Gaveriaux-Ruff, C., and Kieffer, B. L. (2007). Conditional gene targeting in the mouse nervous system: insights into brain function and diseases. Pharmacol. Ther. 113, 619–634.
Geuttier, J. M., Gautam, D., Scarselli, M., Ruiz de Azula, I., Li, J. H., Rosemond, E., Ma, X., Gonzales, F., Armbruster, B., Lu, H., Roth, B. L., and Wess, J. (2009). A chemical-genetic approach to study G protein regulation of β cell function in vivo. Proc. Natl. Acad. Sci. U.S.A. (in press).
Gilman, A. G. (1987). G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649.
Hsiao, E. C., Boudignon, B. M., Chang, W. C., Bencsik, M., Peng, J., Nguyen, T. D., Manalac, C., Halloran, B. P., Conklin, B. R., and Nissenson, R. A. (2008). Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc. Natl. Acad. Sci. U.S.A. 105, 1209–1214.
Kelly, P., Casey, P. J., and Meigs, T. E. (2007). Biologic functions of the G12 subfamily of heterotrimeric g proteins: growth, migration, and metastasis. Biochemistry 46, 6677–6687.
Kerr, M., Davies, S. A., and Dow, J. A. (2004). Cell-specific manipulation of second messengers; a toolbox for integrative physiology in Drosophila. Curr. Biol. 14, 1468–1474.
Kroeze, W. K., Sheffler, D. J., and Roth, B. L. (2003). G-protein-coupled receptors at a glance. J. Cell. Sci. 116, 4867–4869.
Lin, J. Y., Lin, M. Z., Steinbach, P., and Tsien, R. Y. (2009). Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys. J. 96, 1803–1814.
Martin, J. R., Keller, A., and Sweeney, S. T. (2002). Targeted expression of tetanus toxin: a new tool to study the neurobiology of behavior. Adv. Genet. 47, 1–47.
McCloskey, D. T., Turcato, S., Wang, G. Y., Turnbull, L., Zhu, B. Q., Bambino, T., Nguyen, A. P., Lovett, D. H., Nissenson, R. A., Karliner, J. S., and Baker, A. J. (2008). Expression of a Gi-coupled receptor in the heart causes impaired Ca2+ handling, myofilament injury, and dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 294, H205–H212.
McGuire, S. E., Roman, G., and Davis, R. L. (2004). Gene expression systems in Drosophila: a synthesis of time and space. Trends Genet. 20, 384–391.
Nagel, G., Szellas, T., Huhn, W., Kateriya, S., Adeishvili, N., Berthold, P., Ollig, D., Hegemann, P., and Bamberg, E. (2003). Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. U.S.A. 100, 13940–13945.
Nagel, G., Szellas, T., Kateriya, S., Adeishvili, N., Hegemann, P., and Bamberg, E. (2005). Channelrhodopsins: directly light-gated cation channels. Biochem. Soc. Trans. 33, 863–866.
Nakashiba, T., Young, J. Z., McHugh, T. J., Buhl, D. L., and Tonegawa, S. (2008). Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science 319, 1260–1264.
Nawaratne, V., Leach, K., Suratman, N., Loiacono, R. E., Felder, C. C., Armbruster, B. N., Roth, B. L., Sexton, P. M., and Christopoulos, A. (2008). New insights into the function of M4 muscarinic acetylcholine receptors gained using a novel allosteric modulator and a DREADD (designer receptor exclusively activated by a designer drug). Mol. Pharmacol. 74, 1119–1131.
Nichols, C. D. (2006). Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol. Ther. 112, 677–700.
Nitabach, M. N., Blau, J., and Holmes, T. C. (2002). Electrical silencing of Drosophila pacemaker neurons stops the free-running circadian clock. Cell 109, 485–495.
Pauwels, P. J., and Colpaert, F. C. (2000). Disparate ligand-mediated Ca(2) responses by wild-type, mutant Ser(200)Ala and Ser(204)Ala alpha(2A)-adrenoceptor: G(alpha15) fusion proteins: evidence for multiple ligand-activation binding sites. Br. J. Pharmacol. 130, 1505–1512.
Pauwels, P. J. (2003). Unravelling multiple ligand-activation binding sites using RASSL receptors. Trends Pharmacol. Sci. 24, 504–507.
Pei, Y., Rogan, S. C., Yan, F., and Roth, B. L. (2008). Engineered GPCRs as tools to modulate signal transduction. Physiology (Bethesda) 23, 313–321.
Peng, J., Bencsik, M., Louie, A., Lu, W., Millard, S., Nguyen, P., Burghardt, A., Majumdar, S., Wronski, T. J., Halloran, B., Conklin, B. R., and Nissenson, R. A. (2008). Conditional expression of a Gi-coupled receptor in osteoblasts results in trabecular osteopenia. Endocrinology 149, 1329–1337.
Redfern, C. H., Coward, P., Degtyarev, M. Y., Lee, E. K., Kwa, A. T., Hennighausen, L., Bujard, H., Fishman, G. I., and Conklin, B. R. (1999). Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat. Biotechnol. 17, 165–169.
Redfern, C. H., Degtyarev, M. Y., Kwa, A. T., Salomonis, N., Cotte, N., Nanevicz, T., Fidelman, N., Desai, K., Vranizan, K., Lee, E. K., Coward, P., Shah, N., Warrington, J. A., Fishman, G. I., Bernstein, D., Baker, A. J., and Conklin, B. R. (2000). Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 97, 4826–4831.
Regard, J. B., Sato, I. T., and Coughlin, S. R. (2008). Anatomical profiling of G protein-coupled receptor expression. Cell 135, 561–571.
Roth, B. L., Sheffler, D. J., and Kroeze, W. K. (2004). Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 3, 353–359.
Sakamoto, A., Chen, M., Nakamura, T., Xie, T., Karsenty, G., and Weinstein, L. S. (2005). Deficiency of the G-protein alpha-subunit G(s)alpha in osteoblasts leads to differential effects on trabecular and cortical bone. J. Biol. Chem. 280, 21369–21375.
Scearce-Levie, K., Lieberman, M. D., Elliott, H. H., and Conklin, B. R. (2005). Engineered G protein coupled receptors reveal independent regulation of internalization, desensitization and acute signaling. BMC Biol. 3, 3.
Schroll, C., Riemensperger, T., Bucher, D., Ehmer, J., Voller, T., Erbguth, K., Gerber, B., Hendel, T., Nagel, G., Buchner, E., and Fiala, A. (2006). Light-induced activation of distinct modulatory neurons triggers appetitive or aversive learning in Drosophila larvae. Curr. Biol. 16, 1741–1747.
Small, K. M., Brown, K. M., Forbes, S. L., and Liggett, S. B. (2001). Modification of the beta 2-adrenergic receptor to engineer a receptor–effector complex for gene therapy. J. Biol. Chem. 276, 31596–31601.
Srinivasan, S., Santiago, P., Lubrano, C., Vaisse, C., and Conklin, B. R. (2007). Engineering the melanocortin-4 receptor to control constitutive and ligand-mediated GS signaling in vivo. PLoS ONE 2, e668. doi: 10.1371/journal.pone.0000668.
Strachan, R. T., Ferrara, G., and Roth, B. L. (2006). Screening the receptorome: an efficient approach for drug discovery and target validation. Drug Discov. Today 11, 708–716.
Strader, C. D., Gaffney, T., Sugg, E. E., Candelore, M. R., Keys, R., Patchett, A. A., and Dixon, R. A. (1991). Allele-specific activation of genetically engineered receptors. J. Biol. Chem. 266, 5–8.
Sweger, E. J., Casper, K. B., Scearce-Levie, K., Conklin, B. R., and McCarthy, K. D. (2007). Development of hydrocephalus in mice expressing the G(i)-coupled GPCR Ro1 RASSL receptor in astrocytes. J. Neurosci. 27, 2309–2317.
Tan, E. M., Yamaguchi, Y., Horwitz, G D., Gosgnach, S., Lein, E. S., Goulding, M., Albright, T. D., and Callaway, E. M. (2006). Selective and quickly reversible inactivation of mammalian neurons in vivo using the Drosophila allatostatin receptor. Neuron 51, 157–170.
Tonnesen, J., Sorensen, A. T., Deisseroth, K., Lundberg, C., and Kokaia, M. (2009). Optogenetic control of epileptiform activity. Proc. Natl. Acad. Sci. U.S.A. 106, 12162–12167.
Tsetsenis, T., Ma, X. H., Lo Iacono, L., Beck, S. G., and Gross, C. (2007). Suppression of conditioning to ambiguous cues by pharmacogenetic inhibition of the dentate gyrus. Nat. Neurosci. 10, 896–902.
Urban, J. D., Clarke, W. P., von Zastrow, M., Nichols, D. E., Kobilka, B., Weinstein, H., Javitch, J. A., Roth, B. L., Christopoulos, A., Sexton, P. M., Miller, K. J., Spedding, M., and Mailman, R. B. (2007). Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1.
Watts, V. J., and Neve, K. A. (2005). Sensitization of adenylate cyclase by Galpha i/o-coupled receptors. Pharmacol. Ther. 106, 405–421.
Wehr, M., Hostick, U., Kyweriga, M., Tan, A., Weible, A. P., Wu, H., Wu, W., Callaway, E. M., and Kentros, C. (2009). Transgenic silencing of neurons in the mammalian brain by expression of the allatostatin receptor (AlstR). J. Neurophysiol. 2009; 0: 00480.2009v1 [Epub ahead of print].
Wells, T., and Carter, D. A. (2001). Genetic engineering of neural function in transgenic rodents: towards a comprehensive strategy? J. Neurosci. Methods 108, 111–130.
Wulff, P., Goetz, T., Leppa, E., Linden, A. M., Renzi, M., Swinny, J. D., Vekovischeva, O. Y., Sieghart, W., Somogyi, P., Korpi, E. R., Farrant, M., and Wisden, W. (2007). From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat. Neurosci. 10, 923–929.
Yu, C. R., Power, J., Barnea, G., O’Donnell, S., Brown, H. E., Osborne, J., Axel, R., and Gogos, J. A. (2004). Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron 42, 553–566.
Keywords: G-protein coupled receptors, Receptors Activated Solely by Synthetic Ligands, Designer Receptors Exclusively Activated by a Designer Drug, signal transduction, muscarinic receptor, opioid receptor, serotonin receptor, transgenic
Citation: Nichols CD and Roth BL (2009) Engineered G-protein coupled receptors are powerful tools to investigate biological processes and behaviors. Front. Mol. Neurosci. 2:16. doi: 10.3389/neuro.02.016.2009
Received: 29 May 2009;
Paper pending published: 08 July 2009;
Accepted: 12 September 2009;
Published online: 23 October 2009
Edited by:
William Wisden, Imperial College, UKReviewed by:
Cornelius Gross, European Molecular Biology Laboratory, ItalyWilliam Wisden, Imperial College, UK
Copyright: © 2009 Nichols and Roth. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence:
Charles D. Nichols, Department of Pharmacology and Experimental Therapeutics, LSU Health Sciences Center, 5258 MEB, 1901 Perdido St., New Orleans, LA 70112, USA. e-mail: cnich1@lsuhsc.edu;
Bryan L. Roth, Department of Pharmacology, University of North Carolina, 4072 Genetic Medicine, CB # 7365, Chapel Hill, NC 27599-7365, USA. e-mail: bryan_roth@med.unc.edu