1
Laboratorio de Circuitos Corticales, Centro de Tecnología Biomédica, Universidad Politécnica de Madrid, Madrid, Spain
2
Instituto Cajal, Madrid, Spain
3
Institut Neuropatologia, Servei Anatomia Patològica, IDIBELL-Hospital Universitari de Bellvitge, Universitat de Barcelona, Hospitalet de Llobregat, Spain
One of the main pathological hallmarks of Alzheimer’s disease (AD) is the accumulation of plaques in the cerebral cortex, which may appear either in the neuropil or in direct association with neuronal somata. Since different axonal systems innervate the dendritic (mostly glutamatergic) and perisomatic (mostly GABAergic) regions of neurons, the accumulation of plaques in the neuropil or associated with the soma might produce different alterations to synaptic circuits. We have used a variety of conventional light, confocal and electron microscopy techniques to study their relationship with neuronal somata in the cerebral cortex from AD patients and APP/PS1 transgenic mice. The main finding was that the membrane surfaces of neurons (mainly pyramidal cells) in contact with plaques lack GABAergic perisomatic synapses. Since these perisomatic synapses are thought to exert a strong influence on the output of pyramidal cells, their loss may lead to the hyperactivity of the neurons in contact with plaques. These results suggest that plaques modify circuits in a more selective manner than previously thought.
Alzheimer’s disease (AD) is the most common form of senile dementia and it is characterized by a progressive loss of memory, coupled with a deterioration of behavioral and cognitive functions. AD patients display cerebral cortical atrophy, particularly in the temporal and frontal lobes. Among the main pathological hallmarks of this disease in the brain are: the formation of plaques of extracellular fibrillar amyloid beta peptide (Aβ); the appearance of the so-called neuropil threads, intraneuronal neurofibrillary tangles of aggregated hyperphosphorylated tau (in the cell body as well as in the dendritic and axonal processes); and elevated levels of soluble Aβ oligomers (Glenner and Wong, 1984
; Masters et al., 1985
; Grundke-Iqbal et al., 1986
; Ihara et al., 1986
; Kosik et al., 1986
; Goedert et al., 1988
). Plaques and neurofibrillary tangles are mostly found in the cerebral cortex (entorhinal cortex, hippocampal formation and neocortex), where both their number and the proportion of the cortex affected increases progressively as the disease advances (Braak and Braak, 1991
; Dickson, 1997
; Thal et al., 2002
). However, these pathological changes can also be observed in subcortical structures such as the amygdala, nucleus basalis, thalamus, locus coeruleus and raphe nuclei, particularly during the late stages of AD (e.g., Esiri et al., 1997
). Thus, multiple neuronal circuits and neurotransmitter systems may be altered in the brain of AD patients. One hypothesis regarding the pathogenesis of AD is the so-called “amyloid hypothesis”, whereby an accumulation of the neurotoxic Aβ peptide in the cerebral cortex causes the pathological symptoms of AD (e.g., Price and Morris, 2004
). Indeed, it was recently proposed that Aβ oligomers are the main neurotoxins in AD, whose activity is directly linked to tau hyperphosphorylation (De Felice et al., 2008
).
A striking feature of the amyloid plaques associated with AD, particularly during the early stages of the disease, is that there are remarkable differences in their density across the distinct cortical areas and layers (e.g., Terry and Katzman, 1983
; Kemper, 1984
; Kirkpatrick, 1985
; Pearson et al., 1985
; Rogers and Morrison, 1985
; Duyckaerts et al., 1986
; Lewis et al., 1987
; Rafalowska et al., 1988
; Arnold et al., 1991
; Lippa et al., 1992
; Gomez-Isla et al., 1996
; Kuljis and Tikoo, 1997
; Thal et al., 2000
). Plaques have been classified into several types according to their morphological criteria and staining properties (e.g., Wisniewski et al., 1989
; Delaere et al., 1991
; Thal et al., 2000
) and significantly, these different types of plaques are heterogeneously distributed throughout the cerebral cortex (e.g., Kirkpatrick, 1985
; Rogers and Morrison, 1985
; Duyckaerts et al., 1986
; Lewis et al., 1987
; Rafalowska et al., 1988
). Since plaques have traditionally been described in the neuropil, it is generally thought that the neurotoxic effects of Aβ deposits affect axons and dendrites. However, other authors have indicated that plaques come into contact with, or even contain, neuronal cell bodies (Allsop et al., 1989
; Pappolla et al., 1991
; Cummings et al., 1993
; Armstrong, 1995
). Thus, since plaques may appear in the neuropil or in direct association with neuronal cell bodies, the relative abundance of these two types of plaques is likely to influence the alterations to specific synaptic circuits that may occur in AD. For example, pyramidal cells are the most common type of cortical neuron and different regions of the pyramidal cells are innervated by different axonal systems. In general, all axon terminals forming synapses with the perisomatic region of pyramidal cells are GABAergic and they originate from two main types of interneurons: basket cells and chandelier cells. The dendrites in the neuropil establish synapses with a larger variety of axonal systems, including: glutamatergic axons (mainly originating from pyramidal cells and thalamic afferents); GABAergic/peptidergic axons (originating from various types of axo-dendritic interneurons); and noradrenergic, dopaminergic, serotoninergic and cholinergic axons (arising in the brainstem and basal forebrain: DeFelipe, 2002
).
This situation suggests that the plaques in the neuropil will produce different alterations to synaptic circuits than those associated with neuronal somata. However, the relationship between plaques and neuronal somata has not been directly addressed despite the numerous neuropathological studies that have been performed. Finally, different transgenic mice models of AD have been developed that are useful to investigate some aspects of the disease, as well as to evaluate the therapeutic potential of various drugs and treatments that for obvious ethical reasons cannot be tested in human beings (review in McGowan et al., 2006
; Eriksen and Janus, 2007
). In one of these mutants the amyloid pathology displayed is due to the co-expression of a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9) protein, both of which are involved in familial forms of AD. The peculiarity of these double transgenic (APP/PS1) mice is that they do not develop the neurofibrillary changes that are also typical of AD (Irizarry et al., 1997a
,b
). Therefore, this mouse model is useful to examine the effect of Aβ overproduction and deposition on the brain, independently of the neurofibrillary alterations. Here, we have used a variety of conventional light, electron and confocal microscopy techniques to analyze the relationship of plaques with neuronal somata in the cerebral cortex of AD patients and transgenic AD mice. We found that the membrane surface of the neuronal somata in contact with the plaques lacked GABAergic axo-somatic synapses. These results suggest that plaques modify the circuits in a more selective manner than previously thought.
Human Brain Tissue
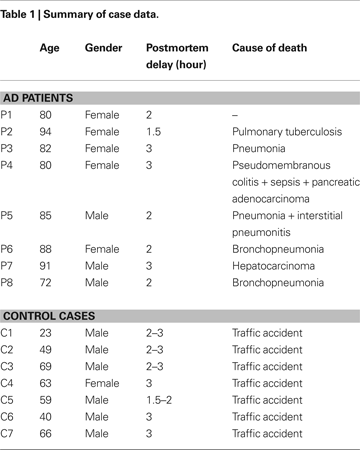
Human brain tissue from eight patients with AD (aged 80–94, average 84) was obtained from the Institut Neuropatologia, Servei Anatomia Patològica, IDIBELL-Hospital Universitari de Bellvitge (Barcelona, Spain). In addition, control human brain tissue was obtained at autopsy from seven individuals (aged 23–69, average 53) that were free of any neurological or psychiatric disease (kindly supplied by Dr. R. Alcaraz, Forensic Pathology Service, Basque Institute of Legal Medicine, Bilbao, Spain: Table 1
).
Following a neuropathological examination, the AD stages were defined according to Braak and Braak (1991)
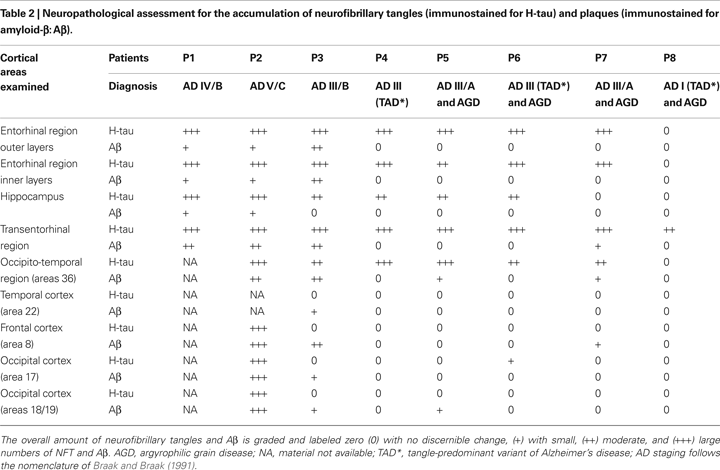
(Table 2
). In all cases, the time between death and tissue processing was between 1.5 and 3 h. Upon removal, the brain tissue was immediately fixed in cold 4% paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4 and after 2 h, the tissue was cut into small blocks and post-fixed in the same fixative for 24–48 h at 4°C. Brain samples were obtained following the guidelines and approval by the Institutional Ethical Committee.
The tissue was obtained from area 4 (motor primary cortex) in the precentral gyrus; area 10 (frontal cortex) in the most rostral portion of the superior frontal gyrus and the middle frontal gyrus; area 17 (striate cortex) located within the calcarine sulcus; area 18 (second visual area) located immediately adjacent to area 17; areas 20 and 21 (the inferior temporal cortex) in the medial and inferior temporal gyri, respectively; area 24 (the cingulate cortex) which occupies most of the anterior cingulate gyrus in an arc around the genu of corpus callosum and that has a poorly defined layer IV. Blocks from the different (Brodmann’s) areas of the cortex were selected from each brain according to their surface anatomy based on the patterns of the gyri and sulci. Furthermore, in this study we included the following regions of the hippocampal formation and adjacent cortex: the dentate gyrus; the hippocampus proper (Cornu Ammonis fields CA1, CA2 and CA3/CA4); the subiculum, the entorhinal cortex; and the perirhinal cortex. In all cases, the identification of each cortical area was later confirmed by analyzing their distinctive cytoarchitectonic features in Nissl-stained sections.
After fixation, all the specimens were immersed in graded sucrose solutions and they were stored in a cryoprotectant solution at −20°C. Serial sections (50 μm) of the cortical tissue were obtained using a vibratome, and the sections from each region and case were batch-processed for immunocytochemical staining. The sections immediately adjacent to those stained immunocytochemically were Nissl-stained in order to identify the cortical areas and the laminar boundaries.
Transgenic Mouse Brain Tissue
In this study, we used the B6C3-Tg 85DBo/J transgenic mice (APPswe, PSEN1dE9) expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9) in CNS neurons (from The Jackson Laboratory, Bar Harbor, ME, USA). Six 12-month-old male mice were anaesthetized with sodium pentobarbital and perfused through the heart with physiological saline, followed by 4% paraformaldehyde in PB. The brain was then removed and post-fixed for 24 h at 4°C. Coronal sections (50 μm) were obtained with the aid of a vibratome. In these animals, the analysis was focused on the granular neocortical areas between bregma −1.10 and −3.00 mm (Hof et al., 2000a
), areas including: the dorsal part of the auditory cortex; the primary auditory cortex; the ventral part of the auditory cortex; the anteromedial visceral cortex; the primary visceral cortex; the primary somatosensory cortex; the secondary somatosensory cortex; the anteromedial visceral cortex; the primary visceral cortex; and the visual cortex (rostrolateral and anterolateral area). The experiments were approved by the ethical institutional committee according to National Institutes of Health guidelines.
Immunohistochemistry
Free-floating sections were pre-treated in 1% H2O2 for 30 min to inactivate the endogenous peroxidase. Subsequently, the sections were incubated for 1 h in PB with 0.25% Triton-X and 3% normal horse serum (Vector Laboratories Inc., Burlingame, CA, USA). Thereafter, two methods were used to examine the relationship between neuronal cell somata and the plaques: combined fluorescence immunohistochemistry and histochemical staining; or dual peroxidase immunohistochemistry in the same sections.
Fluorescence immunohistochemistry and histochemistry
Dual labeling. Sections were double stained using a mouse antibody against the neuron specific nuclear protein (NeuN, 1:2000; Chemicon, Temecula, CA, USA) to label the neurons, and Thioflavine-S to label the plaques. The sections were incubated overnight at 4°C in a solution containing anti-NeuN and then for 2 h at room temperature with a biotinylated horse anti-mouse IgG (1:200, Vector Laboratories). After rinsing in PB, the sections were incubated for 2 h at room temperature in Alexa fluor 594 coupled streptavidin (1:1000; Molecular Probes, Eugene, OR, USA). Thereafter, sections were stained for 10 min in a 1% solution of Thioflavine-S and quickly rinsed with water. The sections were then rapidly rinsed with three changes of 100, 70, 50% ethanol, and then in PB. Finally, the sections were washed and mounted with ProLong Gold Antifade Reagent (Invitrogen Corporation, Carlsbad, CA, USA) and examined on a LeicaDMI 600B confocal laser scanning system equipped with an argon/krypton mixed gas laser with excitation peaks at 498 and 594 nm. The Alexa 594 and Thioflavine-S fluorescence was recorded through separate channels. In order to examine the 3D relationship between neuronal cell somata and plaques, we obtained image stacks (physical size 76.88 mm × 76.88 mm, logical size 1024 × 1024 pixels) that consisted of 19–43 image planes. A 63× glycerol-immersion lens was used (NA, 1.3, working distance, 280 mm, refraction index, 1.45) with a calculated optimal zoom factor 3.2 and with z-step 0.24 μm.
Triple labeling. Some sections were triple stained using a mouse antibody against NeuN (1:2000), Thioflavine-S and two different antibodies to visualize GABAergic terminals: a rabbit antiserum raised against the high-affinity GABA plasma membrane transporter GAT1 (1:500; Chemicon International, Temecula, CA, USA) or a rabbit antiserum raised against the vesicular GABA transporter (VGAT) (1:2000; Synaptic Systems, Goettingen, Germany). The sections were incubated overnight at 4°C in a solution containing anti-NeuN and either VGAT or GABA transporter 1 (GAT-1) and then for 2 h at room temperature with a biotinylated goat anti-rabbit IgG (1:200, Vector Laboratories). After rinsing in PB, the sections were incubated for 2 h at room temperature in Alexa fluor goat-anti mouse 594 (1:2000; Molecular Probes) and streptavidin coupled to Alexa fluor 488 (1:1000; Molecular Probes). Thereafter, sections were stained for Thioflavine-S under the same conditions indicated above. After rinsing in PB, the sections were treated with Autofluorescence Eliminator Reagent (Chemicon International) to reduce or eliminate lipofuscin like-autofluorescence without adversely affecting other fluorescent label in the sections. Finally, sections were washed and mounted with ProLong Gold Antifade Reagent and examined on the LeicaDMI 600B confocal laser scanning system. The Alexa 594, Alexa 488 and Thioflavine-S fluorescence was recorded through separate channels. Since thioflavine fluoresces in the blue–green range (380–480 nm), it was recorded in blue to differentiate it from Alexa 488. The 3D relationship between neuronal cell somata, GABAergic terminals and plaques, was analyzed using image stacks (physical size 123 μm × 123 μm and 98.4 μm × 98.4 μm for human and transgenic mice sections, respectively, and logical size 1024 × 1024 pixels) that consisted of 17–55 image planes. A 63× glycerol-immersion lens was used (NA, 1.3, working distance, 280 mm, refraction index, 1.45) with a calculated optimal zoom factor 2.5 and 2.0 (for human and transgenic mice sections, respectively) and with z-step 0.2 μm.
Dual peroxidase immunohistochemistry
Sections from human and mouse tissue were incubated for 20 min in formic acid 55% in PB. Then the sections were incubated overnight at 4°C with a mouse antibody against Aβ (clone 6F/3D diluted 1:50; Dako, Glostrup, Denmark) and a mouse antibody against NeuN under the same conditions indicated above. The following day, the sections were rinsed and incubated for 1 h with a biotinylated horse anti-mouse IgG (1:200; Vector), then for 30 min in a Vectastain ABC immunoperoxidase kit (Vector). Finally, the antibodies were visualized with the 3,3′-diaminobenzidine (DAB) tetrahydrochloride chromogen (Sigma-Aldrich, St Louis, MO, USA) and they were then dehydrated, cleared with xylene and coverslipped.
For all immunocytochemical procedures, the controls involved processing selected sections either after replacing the primary antibody with normal serum, omitting the secondary antibody, or by replacing the secondary antibody with an inappropriate secondary antibody. No significant staining was detected under these control conditions.
Electron Microscopy
Human and mouse sections adjacent to those used for histopathological assessment were processed for electron microscopy. These sections were post-fixed in 2% glutaraldehyde in PB for 1 h, treated with 1% osmium tetroxide, dehydrated and flat embedded in Araldite resin. Plastic-embedded sections were studied using a correlative light and electron microscopy method that was described in detail elsewhere (DeFelipe and Fairen, 1993
). Briefly, sections were photographed under the light microscope and then serially cut into semi-thin (2 μm thick) sections with a Reichert ultramicrotome. The semi-thin sections were stained with 1% toluidine blue in 1% borax, they were examined under the light microscope, and then photographed to locate the region of interest. Selected semi-thin sections were further sectioned into serial ultrathin sections (50–70 nm thick) with a diamond knife using a Reichert ultramicrotome. The ultrathin sections were collected on formvar-coated, single-slot nickel grids, and stained with uranyl acetate and lead citrate. Digital pictures were captured at different magnifications in a Jeol 150 transmission electron microscope (JEOL USA, Inc., Peabody, MA, USA) equipped with a SIS Megaview III CCD digital camera.
Quantitative and Statistical Analyses
A BX51 Olympus microscope equipped with a motorized stage, a DP70 digital camera and the Neurolucida package (MicroBrightField, Williston, VT, USA), was used to count the different types of plaques in sections double stained for Aβ and NeuN. Cortical layers were traced with the 4× objective and using the Neurolucida package, the complete surface of the previously defined counting fields was scanned with a 40× objective in successive and non-overlapping frames of 17250 μm2. The white matter (WM) in the neocortex was divided into the superficial and deep white matter (SWM and DWM, respectively) with an arbitrary thickness of 175 μm for each layer. The SWM extended from just below layer VI to a depth of 175 μm. The upper limit of the DWM was situated at a distance of 225 μm from the SWM (i.e., 400 μm below of layer VI) and the lower limit was situated at a depth 175 μm below its upper limit. All the plaques were recorded and classified as diffuse or neuritic (including non-cored and cored; see Section “Results”). In addition, the plaques were subdivided depending on whether they were in contact or not with neurons. A neuron was considered in contact with a plaque when at least a portion of the soma was observed in close apposition with the plaque in the same focal plane. Furthermore, when the plaques were in contact with neurons we recorded the number of these neurons and whether they were at the periphery or within the plaques.
Statistical comparisons between the different types of plaques in each layer were performed using one-way ANOVA or the Kruskal–Wallis non-parametric test, depending on whether the datasets fitted a normal distribution and passed the test for homogeneity of variances. The data are presented as the mean ± SEM. All statistical studies were performed with the aid of the GraphPad Prism 5.1 statistical package (Prism, San Diego, CA, USA).
To generate the figures, light microscopy images were captured using a digital camera (Olympus DP50) attached to an Olympus light microscope and Adobe Photoshop CS3 software was used to generate the figure plates (Adobe Systems Inc., San Jose, CA, USA).
Plaques were observed in all cortical layers and sub-cortical WM of the neocortex, as well as in the hippocampal formation and adjacent cortex. Detailed quantitative analyses were performed on tissue from three AD cases (Figures 1
–3
) and six transgenic mice (Figure 4
). Despite the variability in the size of the different types of plaques, around 80% were between 200 and 1200 μm2 in humans and between 100 and 700 μm2 in mouse. As described previously (Jaffar et al., 2001
), the appearance of the plaques was similar in humans and mice (compare Figures 1
G–L with Figures 4
C–E).
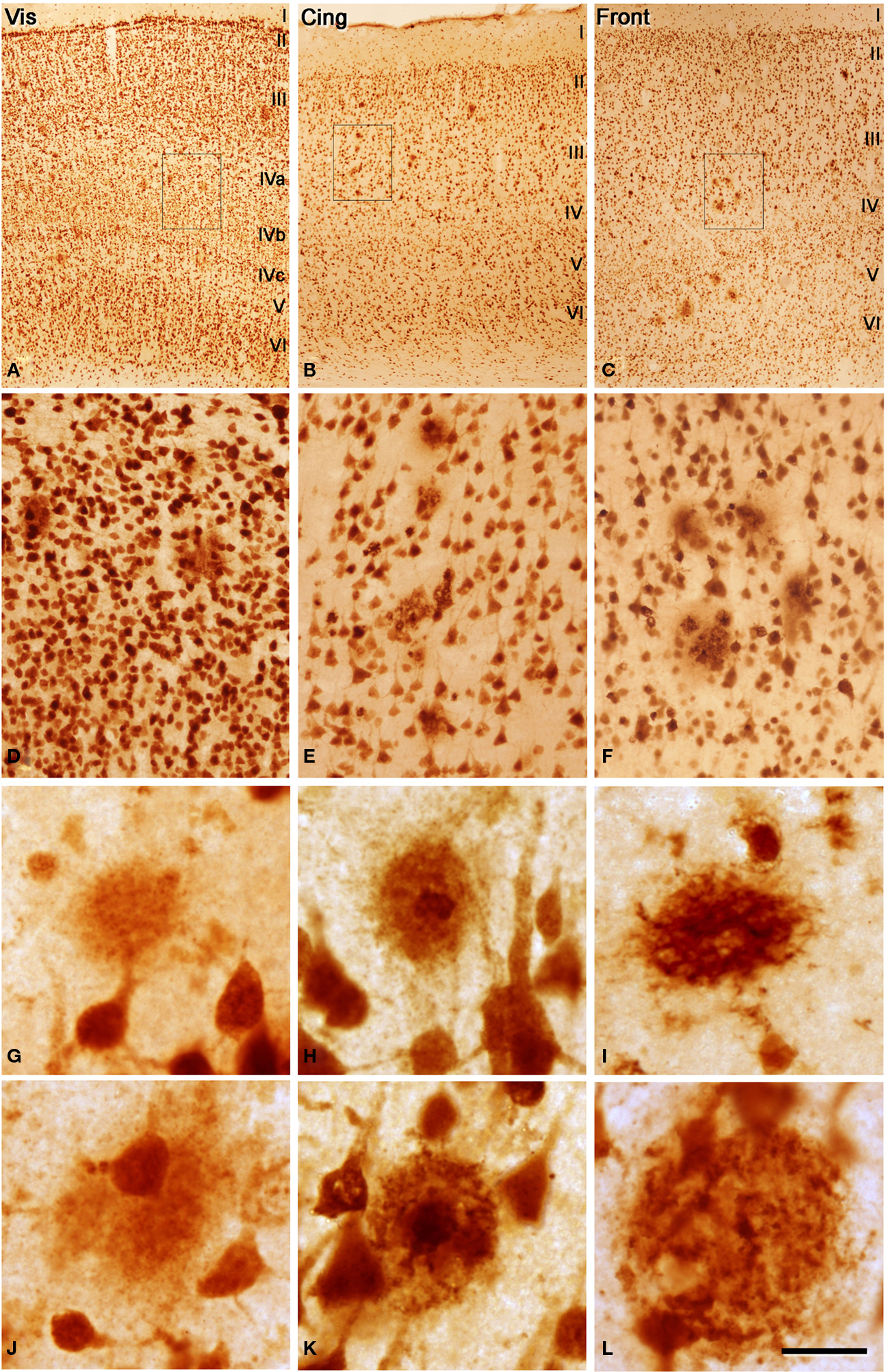
Figure 1. (A–C) Low power photomicrographs from sections double stained for anti-Aβ and NeuN, showing the pattern of immunostaining in layers I–VI of different cortical areas from patient P3. (A) Visual cortex, (B) cingulate cortex, (C) frontal cortex. (D–F) Higher magnification of boxed areas in (A–C), respectively. (G–L) High-power photomicrographs showing different types of plaques and their relationship with neurons: (G–I) diffuse, cored neuritic and non-cored neuritic plaques, respectively, in the neuropil. (J–L) diffuse, cored neuritic and non-cored neuritic plaques, respectively, in contact with neurons. Scale bar, 100 μm in (A–C); 20 μm in (D–F), 120 μm; 20 μm in (G–L).
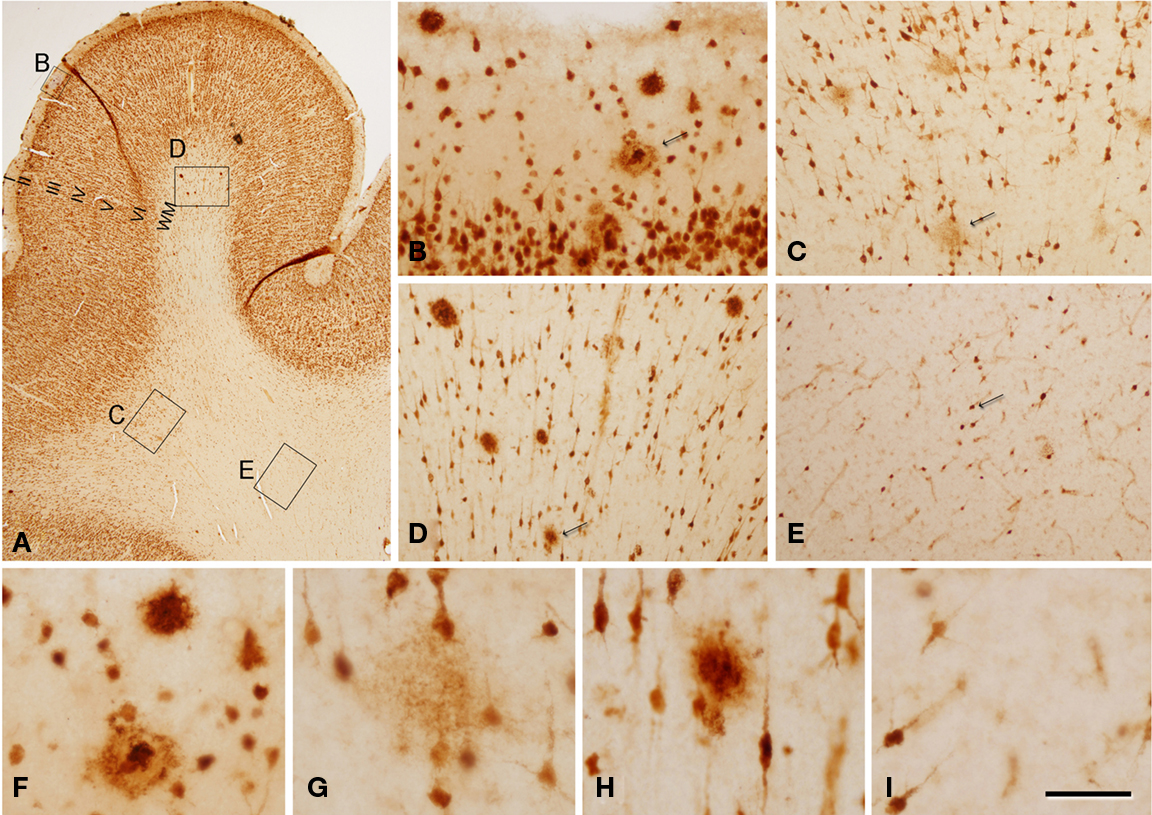
Figure 2. Photomicrographs from a section double stained for anti-Aβ and NeuN, showing the pattern of immunostaining in layer I and in the white matter of the temporal cortex of patient P2. (A) Panoramic view showing the cortical layers and white matter. (B,F) Low- and high-magnification of layer I, illustrating a non-cored neuritic and a cored neuritic plaque. (C,G), (D,H), and (E,I) are low- and high-magnifications of the boxed areas in the white matter in (A), respectively. Arrows in (B–E) point out plaques (B–D) or a neuron (E) also shown at higher magnification in (F–I), respectively. Note that in the superficial white matter (C,D) there are numerous plaques, whereas in the deep white matter (E) no or very few plaques are present. (F–H) Cored neuritic, diffuse and non-cored neuritic plaques, respectively. Scale bar, 100 μm in (A); 50 μm in (B–E); 25 μm in (F–I).
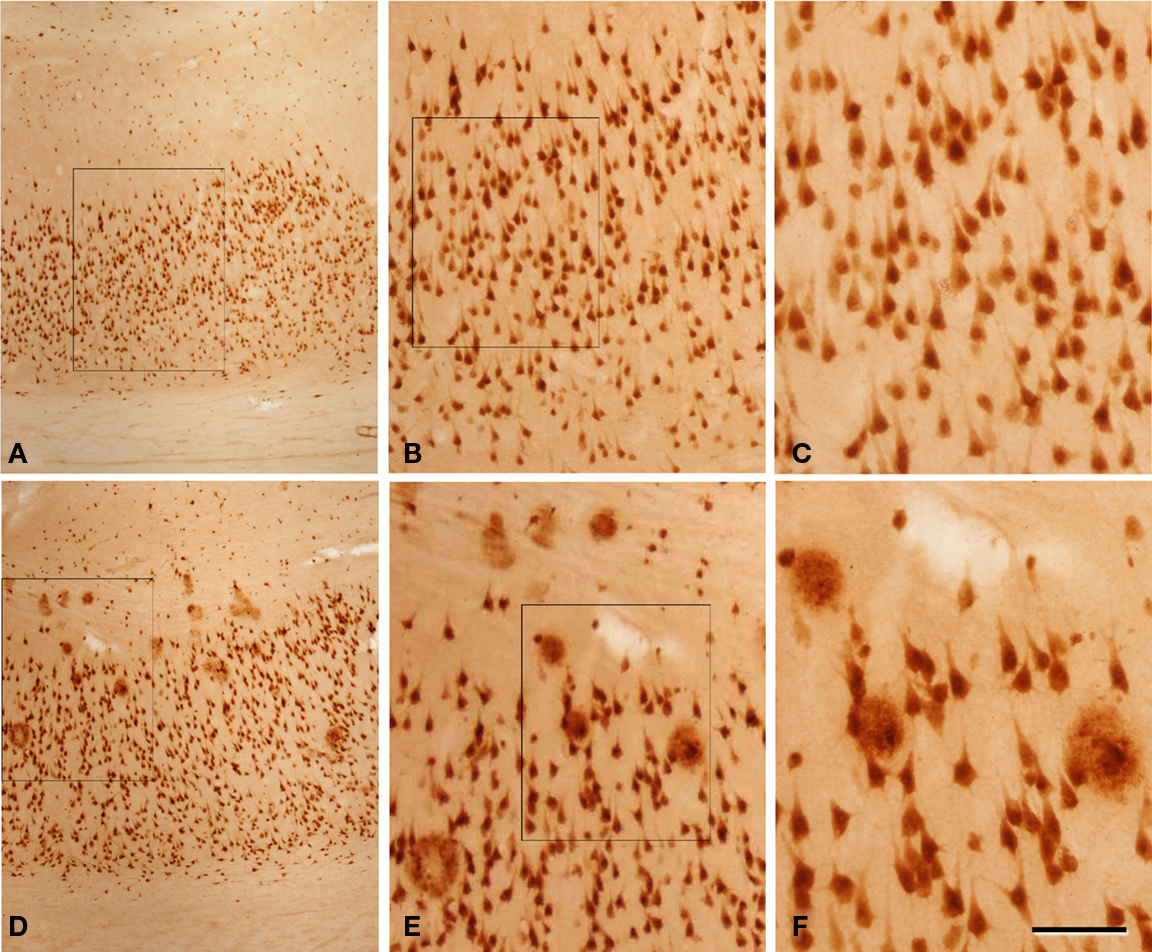
Figure 3. Photomicrographs from sections double stained for anti-Aβ and NeuN showing the pattern of immunostaining in the CA1 of patient P3 (A–C) and patient P1 (D–F). (B,C) and (E,F) are successive higher magnifications of the boxed areas in (A,B) and (D,E), respectively. In these microscopic fields there are no plaques in patient P3, whereas in patient P1 a large number of plaques are observed. Scale bar, 100 μm in (A,D); 50 μm in (B,E); 20 μm in (C,F).
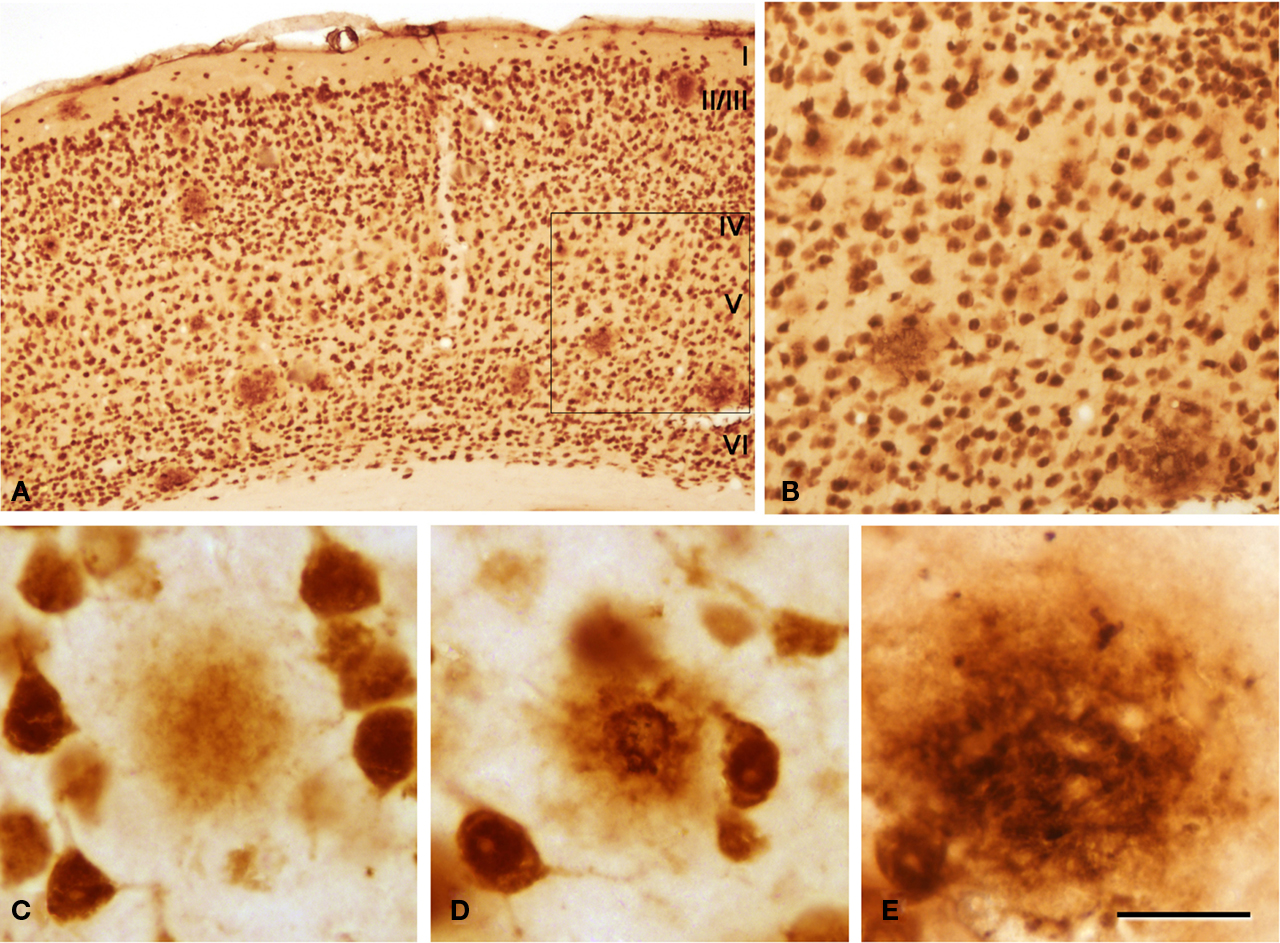
Figure 4. (A) Low-power photomicrograph from a section double stained for anti-Aβ and NeuN, showing the pattern of immunostaining in layers I–VI of the neocortex of an APP/PS1 transgenic mouse. (B) Higher magnification of the boxed area in (A). (C–E) Examples of diffuse, cored neuritic and non-cored neuritic plaques, respectively. Scale bar, 200 μm in (A); 100 μm in (B); 50 μm in (C–E).
Types of Plaques
We could distinguish two main types of plaques in sections immuocytochemically stained for Aβ using DAB as the chromogen: Type 1 plaques that contain irregular extracellular deposits of lightly stained Aβ with no large punctate structures (Figures 1
G and 4
C); and Type 2 plaques that are characterized by Aβ deposits and large punctate structures (Figures 1
I and 4
E). When analyzed by correlative light and electron microscopy (see below), these large punctate structures represent dystrophic neurites. Thus, on the basis of previous studies (e.g., Bancher et al., 1989
; Delaere et al., 1991
; Thal et al., 2000
, 2006
) we called the Type 1 and Type 2 plaques, diffuse and neuritic plaques, respectively. Furthermore, some neuritic plaques contained a round, dense and darkly stained central region, or a nucleus of Aβ that was frequently surrounded by a lightly stained halo. This halo consisted of amorphous material with some large and irregular punctate elements (Figures 1
H and 4
D). Hence, neuritic plaques were sub-divided into “cored” (classic plaques) and “non-cored” neuritic plaques. Plaques were evident in the neuropil (Figures 1
G–I and 4
C) or associated with neurons (in contact with or contained within neurons, Figures 1
J–L and 4
D,E).
Thus, in the present study the main objective was to examine the spatial relationship of the plaques with neurons, their perisomatic GABAergic innervation and the ultrastructural characteristics of this relationship.
The Relationship between Plaques and Neurons
In order to study a relatively large number of plaques and their relationship with neurons, we examined at the light microscopy level sections that were processed for dual peroxidase immunohistochemistry for anti-Aβ and NeuN in the neocortex. Here, we shall first describe the results from the human brain tissue before addressing the data obtained from the transgenic mice.
Conventional light microscopy studies
A total of 2742 plaques (diffuse, 25%; non-cored neuritic, 41%; cored neuritic, 34%) were examined in the neocortex of three AD cases. In all cases, plaques were frequently associated with neurons in layers II–VI since 36% were localized in the neuropil and the remaining 64% were observed in contact with neurons (Figure 5
A). However, in layer I and in the SWM the percentage of plaques observed in the neuropil was greater than those found in layers II–VI (86 and 65%, respectively). Moreover, in layers II–VI, 70% of cored neuritic plaques and 73% of diffuse plaques were in contact with neurons, whereas neurons were only in contact with 51% of non-cored neuritic plaques. In layer I and SWM, the percentage of diffuse, non-cored neuritic and core neuritic plaques in contact with neurons was 28 and 31%, 13 and 22%, and 8 and 20%, respectively (Figure 5
A). Significant differences were found in the representation of each plaque type in the distinct cortical layers (Table 3
).
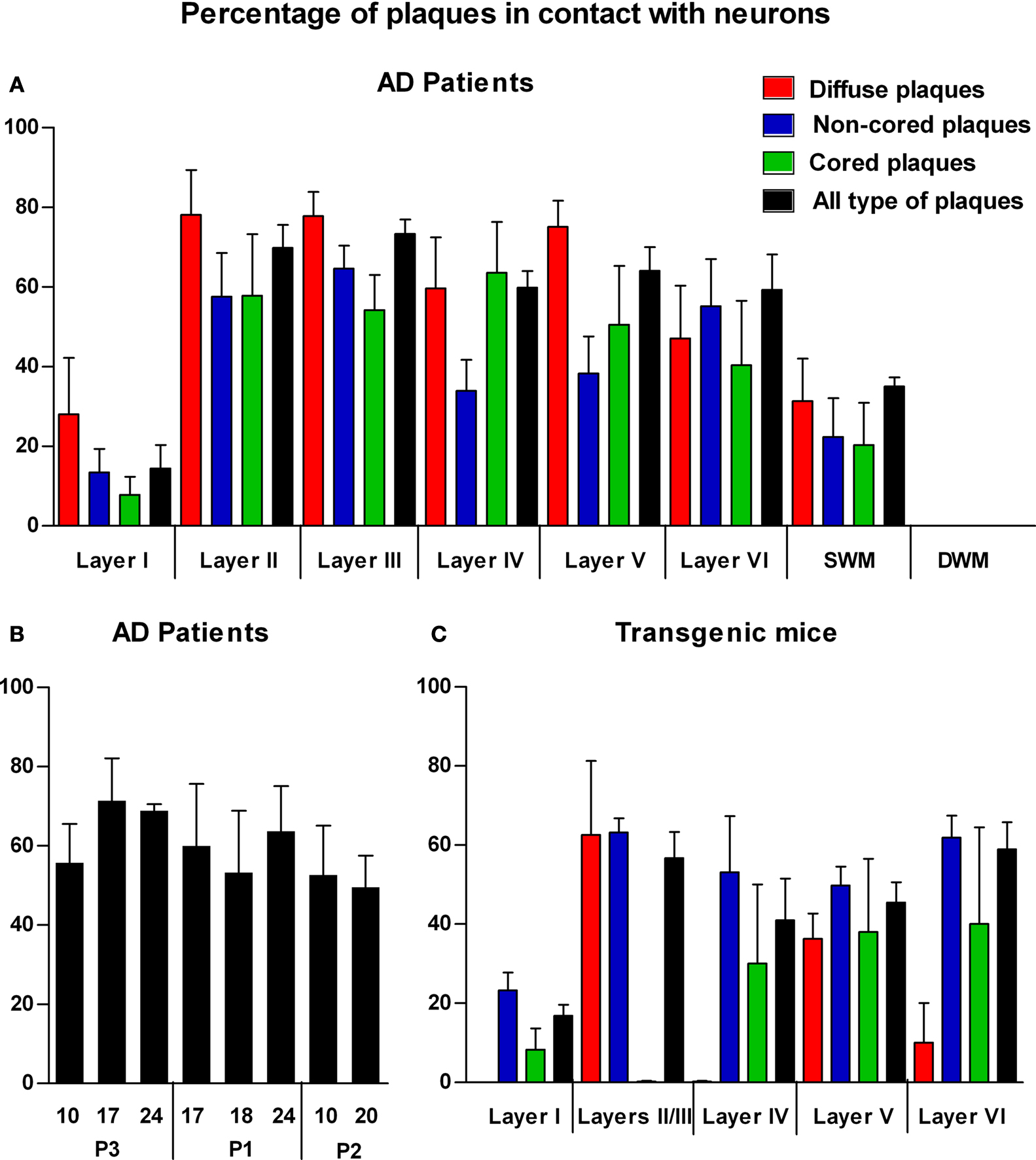
Figure 5. (A) Bar diagram illustrating the percentage of the different types of plaques (diffuse, non-cored neuritic, and cored neuritic) in contact with neurons by cortical layers in the neocortex of AD patients. (B) Bar diagram illustrating the percentage of plaques (all types) in contact with neurons in different cortical areas (areas 10, 17, 24 and 20) of patients P1, P2 and P3. (C) Bar diagram illustrating the percentage of the different types of plaques in contact with neurons by cortical layers in the neocortex of transgenic mice (see Tables 3 and 4
for statistical comparisons).

Of the plaques associated with neurons, 68% were in contact with neurons but not inside the plaques while at least one neuron was found inside the plaque in the remaining 32% of plaques. Of the plaques with no neurons inside them, most displayed one to three neurons (92%) in contact with the plaques and less frequently (8%), there were up to nine neurons associated. With regards the plaques containing neurons, most of them had one to three neurons within the plaques (90%) while the remainder contained four neurons (10%).
In order to investigate possible variations in the percentage of plaques in contact with neurons in different cortical areas in patients with different stages of the disease, we compared patients P1, P2 and P3 that were classified as IV/B, V/C and III/B, respectively, according to Braak and Braak (1991
) (Table 2
). As shown in Figure 5
B, no significant differences were observed in the proportion of the plaques that were in contact with neurons in different cortical regions and patients.
We also quantified the relationship between plaques and the neuronal cell bodies in each cortical layer in six transgenic mice (Figures 4 and 5
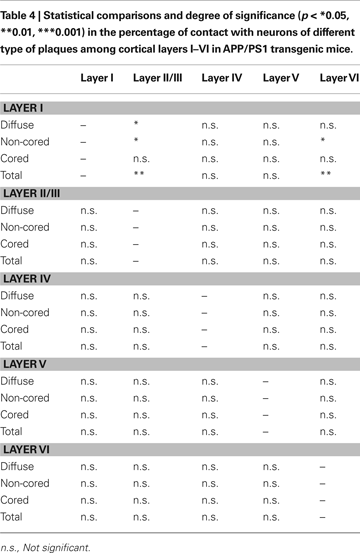
C). A total of 538 plaques were examined (18% diffuse, 72% non-cored neuritic, 11% cored neuritic). In layers II–VI, 52% of plaques were in contact with neurons whereas in layer I this percentage was 17%. When the relationship between the different types of plaques and neuronal cell bodies was examined, the non-cored plaques were preferentially associated with neurons (57%) while only 41% of cored neuritic plaques and 34% of diffuse plaques were in contact with neurons. Indeed, there were significant differences between the different types of plaques in the distinct layers (Table 4
). Of the plaques in contact with neurons, 81% had one to three neurons at the periphery of the plaque, whereas the remaining 19% of plaques had neurons both at the periphery and within (one to three neurons) the plaque.
Diminished Perisomatic GABAergic Terminals on Neurons Adjacent to Plaques
Confocal microscopy studies
When the spatial association of neurons and plaques was studied across confocal z-series, staining the same sections for both NeuN to visualize neurons and for Thioflavine-S to visualize plaques, it was clear that some neuronal somata and/or their proximal processes were in intimate contact with the plaques (Figure 6
), suggesting that no perisomatic boutons were present. Since virtually all cell somata and proximal processes in layers II–VI are outlined by GABAergic terminals (Ribak, 1978
; see Merchan-Perez et al., 2009
for a recent study), we examined whether the perisomatic innervation of neurons in contact with plaques was altered in the human and mouse cerebral cortex. We used triple fluorescence confocal microscopy to analyze neurons immunohistochemically labeled with NeuN that were in contact with plaques labeled with Thioflavine-S, and which in turn were innervated by GABAergic terminals recognized by antibodies against the GAT-1 or the VGAT (Minelli et al., 1995
, 2003
; Yan et al., 1997
). As previously described, GAT-1 and VGAT immunoreactivity was evident in puncta around the cell bodies and in the neuropil of the normal human and mouse cerebral cortex, either that derived from control material or from cortical regions without plaques. However, the part of the neuron’s membrane (mainly pyramidal cells) that is in contact with the plaques lacks GABAergic perisomatic terminals. Indeed, of the several hundred neurons that we found to come into contact with plaques, in no cases were perisomatic terminals found at the surface of the neuron that was directly touching the plaque. We verified this observation by analyzing the z-projections of optical sections of neurons in contact with plaques from the human (n = 17) and mouse (n = 6) cerebral cortex. In these experiments, these neurons were partially or completely “disconnected” at the perisomatic level (Figure 7
).
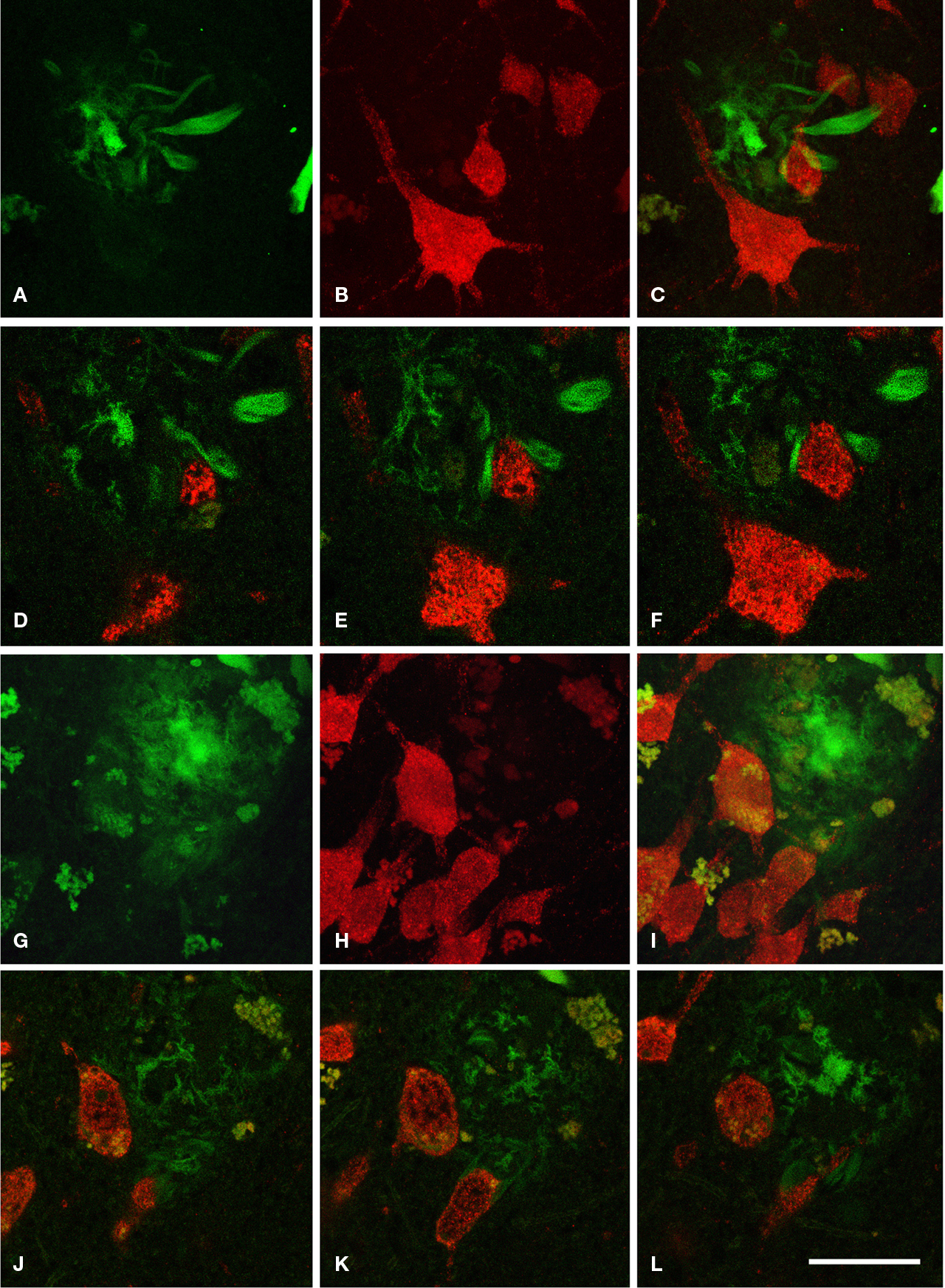
Figure 6. Series of confocal images from patient P2 of two plaques (A–F) and (G–L) in contact with neurons doubled labeled for NeuN (red) and Thioflavine-S (green). (A–C) Represent stacks of 11 optical sections separated by 1 μm in the z-axis (total 9 μm). (D–F) Single optical sections of the plaque shown in (A–C) at different levels (6, 8, and 10 steps, respectively). (G–I) Represent stacks of 43 optical images separated by 0.50 μm in the z-axis (total 21 μm). (J–L) Single optical sections at different levels (10, 14, and 21 steps, respectively) of the plaque shown in (G–I). Scale bar, 25 μm in (A–L).
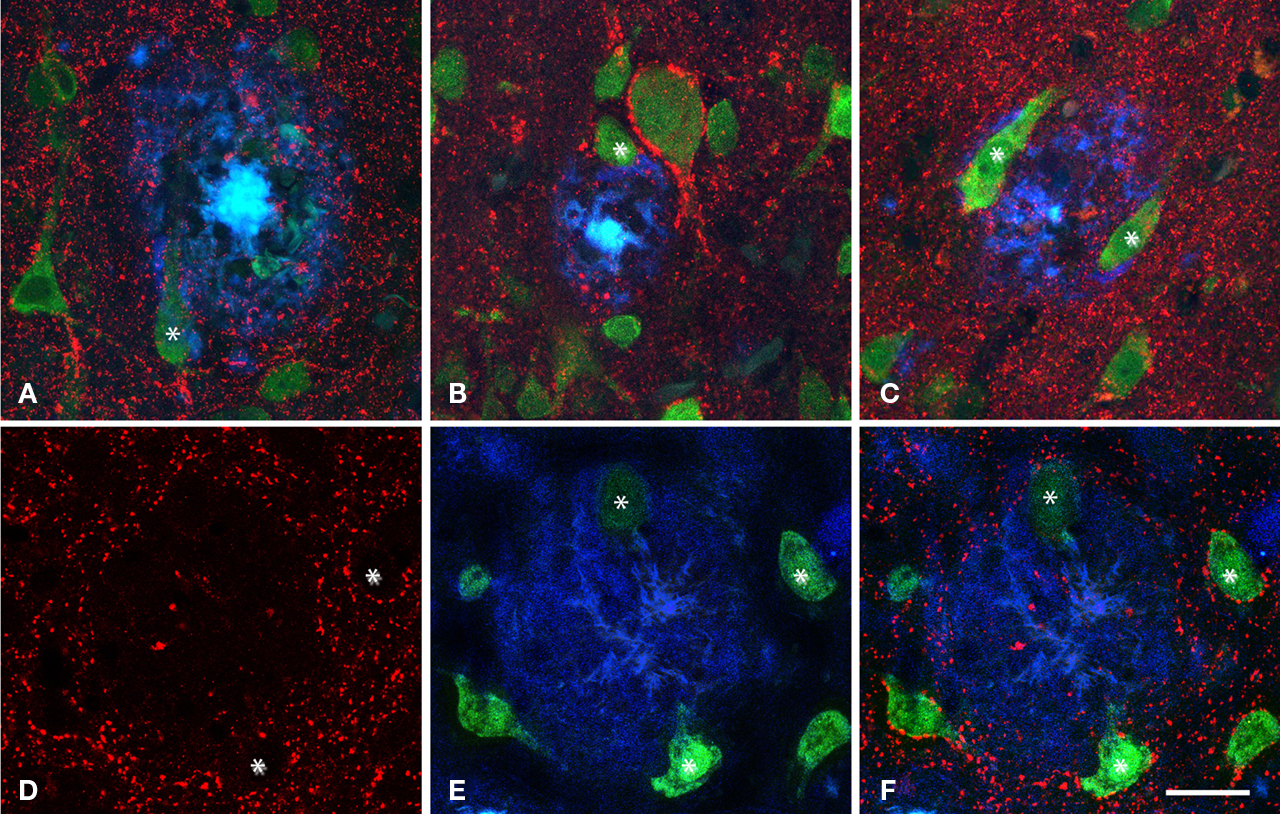
Figure 7. Triple fluorescence immunohistochemistry to analyze neurons visualized with NeuN (green) in contact (some of them indicated with an asterisk) with plaques labeled with Thioflavine-S (blue) that were innervated by GABAergic terminals visualized with an antibody against the vesicular GABA transporter (VGAT, red, B and D–F) or GABA transporter 1 (GAT-1, red, A and C). (A–C), represent stacks of two optical images [10, 15 and 14, 18 steps; (A,B), respectively] and three optical images [33, 43, 49 steps; (C)] each step separated by 0.2 μm in the z-axis for three different plaques from patient P2. (D–F) Single optical sections from the same plane to illustrate the GABAergic innervation of neurons in contact with a plaque in a transgenic mouse. Note the lack of perisomatic GABA-positive terminals on the surface of the neurons in direct contact with the plaques. Scale bar, 30 μm in (A–C); 25 μm in (D–F).
Electron microscopy
The relationship between neurons and plaques was further examined using a correlative light and electron microscopy protocol (DeFelipe and Fairen, 1993
), which was applied to semithin (2 μm) plastic sections with neurons in contact with plaques (n = 13, Figures 8 and 9
). We examined the cerebral cortex from both human and transgenic mice from which similar results were obtained. However, due to the better preservation of the tissue we focused on the ultrastructural analysis of the mouse cerebral cortex. As described previously (Kidd, 1964
; Luse and Smith, 1964
; Terry et al., 1964
), cored neuritic plaques consist of three more-or-less spherical areas. There is a central area that contained a core of electron dense fibrillar Aβ components, and this was surrounded by a second area made up of a light ring of glial processes. No synapses were observed in these two areas. Surrounding this second domain was a region with more diffuse limits that contained dystrophic neurites that were intermingled with the apparently normal neuropil where synapses could be observed. In general, non-cored neuritic and diffuse plaques had similar ultrastructural characteristics to cored neuritic plaques, although the Aβ components did not form a dense core and the light ring of glial processes was also absent. Within these plaques no or very few synapses were observed, whereas synapses were present in the periphery. Importantly, although only GABAergic terminals establish axo-somatic synapses (DeFelipe and Fariñas, 1992
), both the glutamatergic and GABAergic terminals typically found in the perisomatic region (see Merchan-Perez et al., 2009
) were excluded from the regions of the neuronal somata and proximal processes in contact with the plaques. Indeed, the direct apposition of dystrophic or fibrillar Aβ plaque components precluded the establishment of these synapses (Figures 8
E, 9
B and 10
A). However, the neuronal somata in contact with the plaques looked normal, with a normal complement of cytoplasmic organelles (Figures 9
C and 10
A). Furthermore, both glutamatergic and GABAergic perisomatic terminals were present on the parts of the somata or proximal processes of these neurons that were not in contact with the plaques, and the GABAergic terminals established normal looking symmetric synapses (Figure 10
B).
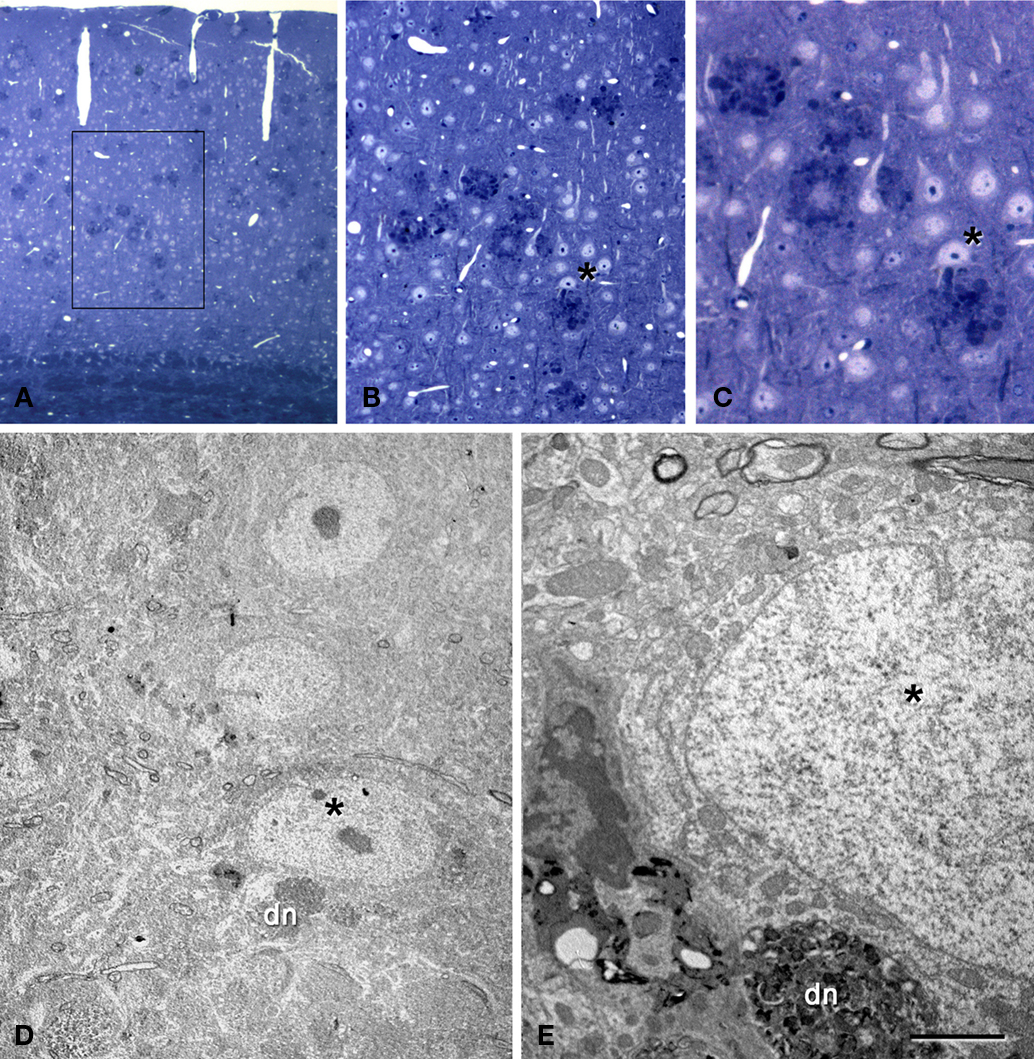
Figure 8. Correlative light and electron microscopy photomicrographs of plaques from the neocortex of an APP/PS1 transgenic mouse. (A–C) Photomicrographs of a 2-μm thick semithin plastic section stained with 1% toluidine blue showing plaques in the neocortex of an APP/PS1 transgenic mouse. (B,C) are successive higher magnifications of the boxed area in (A). (D) Low-power electron micrographs taken after resection of the semithin section shown in (C), illustrating a neuron (asterisk) in contact with a plaque (dn, indicates a dystrophic neurite). The same neuron is indicated in (B,C). (E) Higher magnification of (D) to illustrate the neuron in contact (asterisk) with the dystrophic neurite (dn). Scale bar, 150 μm in (A); 50 μm in (B); 12 μm in (C); 10 μm in (D); 2 μm and (E).
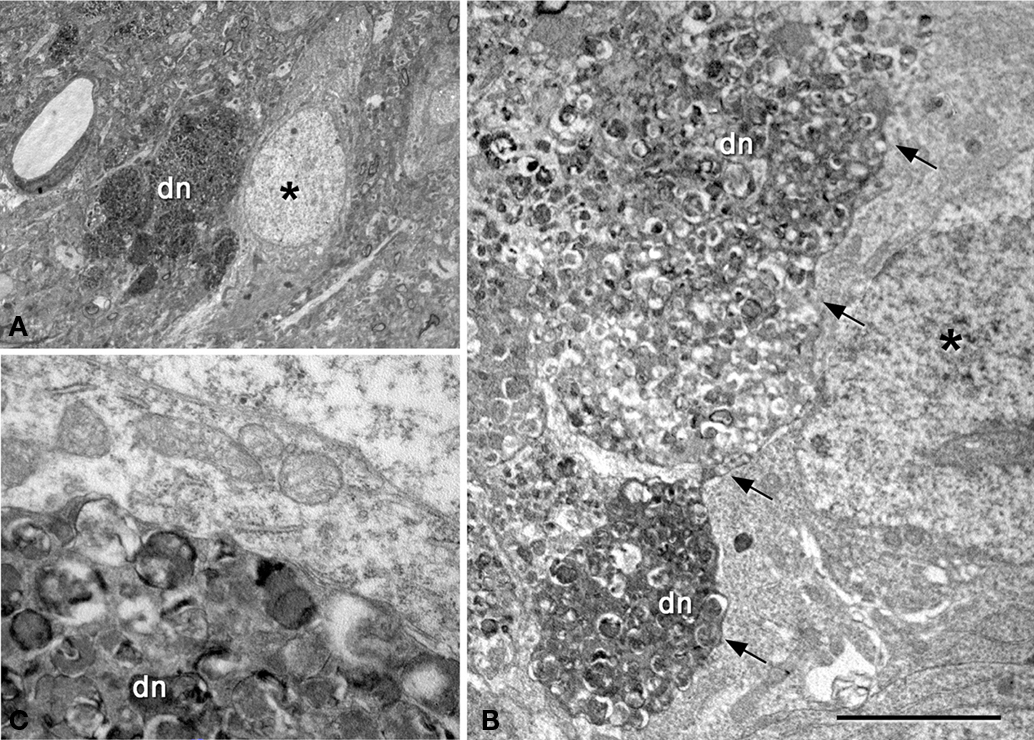
Figure 9. (A) Low-power electron micrograph illustrating a neuron (asterisk) in contact with a plaque (dn, indicates dystrophic giant neurites filled with degenerating organelles). (B) Higher magnification of (A) showing that a large portion of the surface of the soma (arrows) is in contact with dystrophic neurites (dn). (C) Higher magnification of the dystrophic neurites in contact with the membrane of the soma of a normal looking neuron. Scale bar, 10 μm in (A); 3.7 μm in (B); 1 μm in (C).
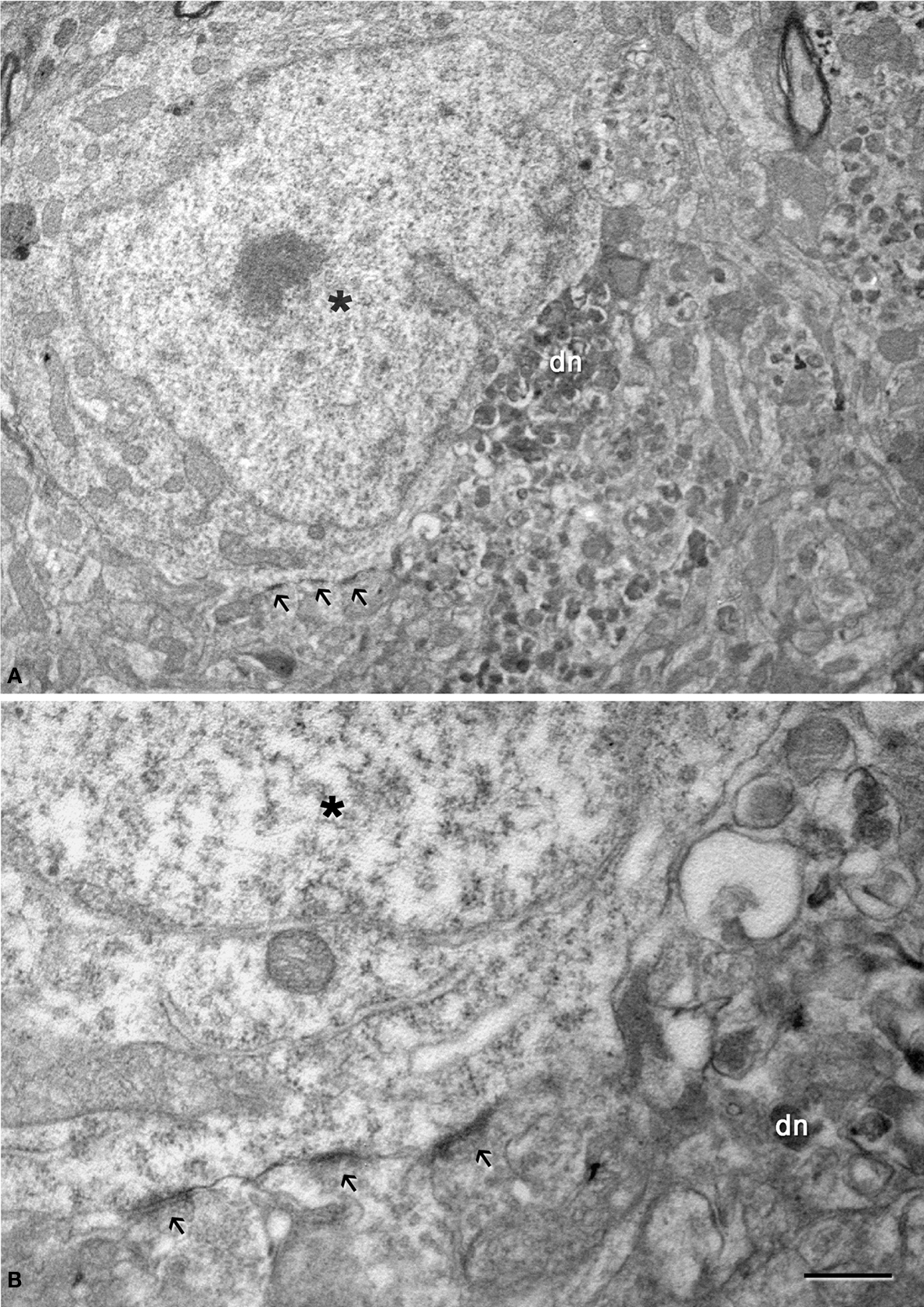
Figure 10. (A) Low magnification electron micrograph illustrating a neuron (asterisk) in contact with a dystrophic giant neurite (dn). Note that the portion of the soma in contact with the dystrophic neurite lacks axo-somatic synapses, whereas synapses are established (arrows) in the portion of the soma that is not in contact with the dystrophic neurite. (B) Higher magnification of the axo-somatic synapses that are typically symmetric (GABAergic) synapses (see text for further details). Scale bar, 4 μm in (A); 1 μm in (B).
We can draw two main conclusions from the data generated in this study. Firstly, a high proportion of plaques are in contact with the somata and proximal processes of neurons. Secondly, the membrane of the neuronal somata in contact with the plaque lacked perisomatic terminals.
Association between Different Types of Plaques with Neurons
The relationship between different types of plaques and neuronal cell bodies has been addressed in relatively few studies. For example, in one study over 50% of non-cored neuritic plaques present in the hippocampal formation and the parahippocampal gyri of five AD cases were associated with neurons (Pappolla et al., 1991
). When diffuse plaques were examined in the superior temporal gyrus of four patients with Down’s syndrome, the vast majority of plaques had at least one neuronal cell body within or immediately adjacent to the plaque (Allsop et al., 1989
). Finally, in a study of four patients with Down’s syndrome that focused on the parahippocampal gyrus and the lateral occipitotemporal gyrus, diffuse deposits were more frequently in contact with neurons than non-cored neuritic plaques (Armstrong, 1995
).
In the present study, we found that the plaques in AD patients were very frequently associated with neurons, particularly in the case of diffuse and cored neuritic plaques and less so for non-cored neuritic plaques. Furthermore, we observed little variation in the proportion of plaques that were in contact with neurons in different cortical regions and patients. In transgenic mice, plaques were also frequently associated with neurons and this phenomenon was also relatively homogeneous in all the mice analyzed. In all cases, the proportion of non-cored neuritic plaques in contact with neurons was always slightly higher or equivalent to those observed in the neuropil, whereas cored neuritic and diffuse plaques were more commonly observed in the neuropil than in contact with neurons.
We wondered whether the relationship between the deposits of Aβ and the somata of neurons might be associated with the pathogenesis of AD. A large proportion of plaques are in contact with neurons and indeed, it has been proposed that Aβ could be released from the somata of neurons (Allsop et al., 1989
; Gouras et al., 2000
; D’Andrea et al., 2001
; Takahashi et al., 2002
). The fact that plaques are also frequently observed in the neuropil could indicate that those plaques in contact with neurons would eventually kill these neurons. Thus, plaques in the neuropil could represent a final pathogenic stage. In turn, this might help to explain the loss of neurons reported in AD patients. However, although the percentage of plaques associated to neurons was also very high in the transgenic mice, no neuronal loss has been found in these animals (Irizarry et al., 1997a
,b
; Knafo et al., 2009a
). Thus, this relationship would seem to be non-specific and that the large proportion of plaques associated with neurons might be explained by the relatively large size of plaques and the high density of neurons. This is in line with the observation that the percentage of plaques associated with neurons is low in layer I or in the WM adjacent to layer VI, where neurons are relatively sparse. However, if this were the case then there would be an equal chance for all types of plaques to come into contact with neurons in any given cortical area. We found that non-cored neuritic plaques are less frequently associated with neurons in AD patients, as was the case for diffuse and cored neuritic plaques in transgenic mice. Thus, whether there is a direct relationship between the soma of neurons and plaques remains to be elucidated, although it certainly does not seem to be the case for all types of plaques.
Local Toxic Effects of Aβ
The neurons we found in contact with plaques in the mouse cerebral cortex do not seem to have any sign of abnormalities at the level of the cell body. In addition, the portions of the soma of these neurons that were not in contact with the plaques established typical synapses, whereas the region in direct contact with the plaques lacks synapses. Thus, the direct contact of the plaque with the cell body does not induce perisomatic disconnection further away from the contact domain. This is interesting since plaques induce clear morphological alterations of those dendrites that are in contact with Aβ (Tsai et al., 2004
; Spires et al., 2005
). Indeed, in the same transgenic mice as those used in the present study we have recently shown that dendrites in contact or passing through Aβ plaques suffer alterations that include the sprouting of spines on dendrites contacting Aβ plaques, the loss of dendritic spines and the thinning of dendritic shafts passing through Aβ plaques (Knafo et al., 2009a
,b
). Since dendritic spines represent the main postsynaptic elements of excitatory glutamatergic synapses in the cerebral cortex (Gray, 1959
; Colonnier, 1968
), the loss or morphological alteration of spines provoked by plaques in the neuropil may be related to local dampening or alterations to the glutamatergic system. However, plaques in contact with neurons suffer alterations to the GABAergic system (see below). Thus, our results suggest that dendrites are more susceptible to the toxic effect of Aβ than the cell body and that this may have important consequences in the function of cortical circuits.
Possible Functional Significance of the Relationship of Plaques with the Perisomatic Regions of Neurons
It has been proposed that the Aβ pathology progresses in a neurotransmitter-specific manner, such that cholinergic terminals are the most vulnerable, followed by the glutamatergic terminals and finally, the GABAergic terminals (Bell et al., 2003
). Indeed, it has been suggested that GABAergic circuits in the cerebral cortex of AD are relatively well preserved when compared with circuits that use other neurotransmitters systems (Young, 1987
; Lowe et al., 1988
; Reinikainen et al., 1988
; Ferrer et al., 1991
; Hof et al., 1991
; Nägga et al., 1999
; Rissman et al., 2007
). However, alterations of neurons labeled for parvalbumin were identified in the cerebral cortex of AD patients in several studies (Fonseca et al., 1993
; Solodkin et al., 1996
; Brady and Mufson, 1997
; Mikkonen et al., 1999
), which are responsible for most of the GABAergic pericellular innervation of pyramidal cells (basket and chandeliers cells). Irrespective of this possible pathological course, we find that the perisomatic region of the neurons (mostly pyramidal neurons since they represent the vast majority of cortical neurons) that is in contact with plaques is free of glutamatergic and GABAergic perisomatic terminals. We don’t know whether this is simply due to the interposition of dystrophic neurites or other pathological components of the plaques between the axon terminals and their targets, or whether axon terminals are not present because they have degenerated or they have been eliminated by glial phagocytosis induced by the proximity of the plaques (“synaptic stripping”: see Blinzinger and Kreutzberg, 1968
; Moran and Graeber, 2004
). Whatever the cause the final result would be local synaptic disconnection. In general, the soma, proximal dendrites and the axon initial segment only establish symmetric synapses with axon terminals from GABAergic interneurons (mostly basket and chandelier cells; DeFelipe and Fariñas, 1992
). These perisomatic interneurons are presumed to exert a strong influence on the output of pyramidal cells (Miles et al., 1996
; DeFelipe, 1999
). For example, chandelier cells that exclusively innervate the axon initial segment are considered to control the generation and back propagation of action potentials, as well as participating in complex activities such as the synchronization of firing patterns of large populations of pyramidal cells in different states of consciousness (see Cobb et al., 1995
; Miles et al., 1996
; DeFelipe, 1999
; Klausberger et al., 2004
; Howard et al., 2005
).
In experimental animals small reductions in the efficacy of GABA-mediated inhibition can lead to synchronized epileptiform activity (Chagnac-Amitai and Connors, 1989
) and hence, a loss of perisomatic GABA axon terminals may induce epilepsy (see also Fonseca et al., 1993
). Indeed, in vivo calcium-imaging experiments have shown a decrease in activity in about one-third of cortical neurons in layer 2/3 of transgenic APP/PS1 mice, yet an increase in the activity of 21% of neurons when spontaneous calcium bursts are measured (Busche et al., 2008
). Since numerous studies have identified widespread synaptic dysfunction or loss, even in early stages of the disease (Coleman et al., 2004
), a decrease in activity would not be unexpected. Importantly, hyperactive neurons were found directly in the vicinity of Aβ plaques, consistent with the loss of perisomatic GABAergic innervation of neurons in contact with plaques observed here.
Since our findings were consistent and they were widespread observations in both AD patients and transgenic mice, it is likely that similar hyperactive neurons may exist near plaques in AD patients. In this regard, it is important to emphasize that AD patients display a higher incidence of epileptic seizures (Amatniek et al., 2006
). Nevertheless, the physiological significance of hyperactive neurons near plaques and its possible relationship with epilepsy is not clear. Why do some of AD patients manifest seizures whereas others do not? or why do seizures only appear several years after the onset of the AD? In a study performed on eight AD patients bearing the PS1[E280A] mutation, five developed epilepsy some years after the onset of dementia whereas the remaining three remained seizure-free (Velez-Pardo et al., 2004
). In these AD PS1[E280A] patients, two distinct patterns of neuronal loss in the CA1 field were found depending on whether they developed seizures: diffuse–patchy neuronal loss in the non-epileptic AD patients as opposed to a sclerotic-like neuronal loss in the epileptic patients similar to that found typically in the CA1 field of temporal lobe epilepsy patients with hippocampal sclerosis (that is, a large segment of CA1 was practically devoid of neurons). We would like to emphasize that atrophy of the hippocampus is typically considered an early sign of AD but this is also typical of patients with temporal lobe epilepsy. Thus, it is possible that seizures in AD patients arise as a consequence of a particular pattern of neuronal loss similar to that observed in epileptic patients. The present results suggest that the progressive appearance of plaques throughout the cerebral cortex would steadily increase the susceptibility to develop and propagate seizures by increasing the numbers of hyperactive neurons near plaques. Furthermore, the tight spatial association of excitatory axo-dendritic synapses with axo-somatic inhibitory terminals around the perisomatic regions of pyramidal cells has recently been proposed to be responsible for the non-synaptic glutamatergic and GABAergic interactions observed in electrophysiological and pharmacological studies (Ren et al., 2007
), probably due to neurotransmitter spillover (Merchan-Perez et al., 2009
). Since both types of terminals disappear from the perisomatic region in contact with the plaques, not only will GABAergic transmission in the perisomatic region be affected but also, other neurotransmission phenomena that occur in this region. Finally, it should also be considered that glial cells display increased astrocyte activity in APP/PS1 transgenic mice (Kuchibhotla et al., 2009
), which may also affect the activity of synaptic circuits.
Caveats
Animal models are no doubt very useful to examine the different morphological, physiological, molecular aspects of AD in depth, and to develop anti-Aβ immunotherapies (Brendza et al., 2003
; Oddo et al., 2003
; Brendza and Holtzman, 2006
; Eriksen and Janus, 2007
; Götz et al., 2007
; Busche et al., 2008
; Meyer-Luehmann et al., 2008
; Radde et al., 2008
). However, the data obtained in animal models should be treated with caution when trying to extrapolate to the human brain, particularly if we consider that human cortical circuits show remarkable differences when compared to those of mice (Glezer et al., 1993
; Hof et al., 2000b
; DeFelipe, 2002
; Preuss, 2006
; DeFelipe et al., 2007
). Furthermore, neurofibrillar tangles do not develop in the mouse model of AD used but rather, only deposits of Aβ are evident. Thus, there is a clear difference in the pathogenesis between human patients and this mouse model. Nevertheless, the relationship of the plaques of Aβ deposits and neurons was similar, suggesting that the model is useful to study this particular aspect of the disease. Finally, the primary quantitative data on the relationship between the neurons and plaques was obtained as the number of plaque profiles and neurons per single sections (2-D analysis). As a result, no correction was introduced for the possibility that plaques and neurons of different sizes appear with different probabilities in the sections (i.e., larger plaques or neurons will have a greater chance of appearing in more than one section). Thus, it is likely that the number of neurons in contact with the plaques was underestimated in the 2-D analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that should be construed as a potential conflict of interest.
We thank to A.I. García for technical assistance and C. Hernandez and B. García for assistance with the confocal microscopy. This work was supported by grants from the following entities: CIBERNED CB06/05/0066, Fundación CIEN (Financiación de Proyectos de Investigación de Enfermedad de Alzheimer y enfermedades relacionadas 2008), Fundación Caixa (BM05-47-0), the EU 6th Framework Program (PROMEMORIA LSHM-CT-2005-512012), the Spanish Ministerio de Educación Ciencia e Innovación (grants BFU2006-13395 and SAF2009-09394 to J.D.), and L.B-L was awarded a research fellowship from the Spanish Ministry of Science and Technology (grant FPU AP2005-0690). We also thank Ana Martínez and Gabriel Santpere for genotyping the mice.
Brendza, R. P., O’Brien, C., Simmons, K., McKeel, D. W., Bales, K. R., Paul, S. M., Olney, J. W., Sanes, J. R., and Holtzman, D. M. (2003). PDAPP; YFP double transgenic mice: a tool to study amyloid-beta associated changes in axonal, dendritic, and synaptic structures. J. Comp. Neurol. 456, 375–383.
De Felice, F. G., Wu, D., Lambert, M. P., Fernandez, S. J., Velasco, P. T., Lacor, P. N., Bigio, E. H., Jerecic, J., Acton, P. J., Shughrue, P. J., Chen-Dodson, E., Kinney, G. G., and Klein, W. L. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29, 1334–1347.
DeFelipe, J., Alonso-Nanclares, L., Arellano, J. I., Ballesteros-Yáñez, I., Benavides-Piccione, R., and Munoz, A. (2007). Specializations of the cortical microstructure of humans. In Evolution of the Nervous Systems: A Comprehensive Reference, Vol. 4. Primates, J. H. Kaas and T. M. Preuss, eds (Amsterdam, Elsevier), pp. 167–190.
Meyer-Luehmann, M., Spires-Jones, T. L., Prada, C., Garcia-Alloza, M., de Calignon, A., Rozkalne, A., Koenigsknecht-Talboo, J., Holtzman, D. M., Bacskai, B. J., and Hyman, B. T. (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724.
Spires, T. L., Meyer-Luehmann, M., Stern, E. A., McLean, P. J., Skoch, J., Nguyen, P. T., Bacskai, B. J., and Hyman, B. T. (2005). Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 25, 7278–7287.