Comparison of the efficacy of two anticonvulsants, phenytoin and valproate to improve PCP and D-amphetamine induced deficits in a reversal learning task in the rat
1
The School of Pharmacy, The University of Bradford, Bradford, West Yorkshire, UK
2
Department of Neuropharmacology, GlaxoSmithKline S.p.A., Verona, Italy
Recent studies in our laboratory have shown that PCP (phencyclidine) and D-amphetamine induce a cognitive deficit in rats, in a paradigm of potential relevance for the pathology of schizophrenia. Atypical, but not classical antipsychotics and the anticonvulsant, lamotrigine have been shown to prevent a selective reversal learning deficit induced by PCP. In contrast, only haloperidol reversed the D-amphetamine-induced deficit. The present study aimed to explore the ability of two anticonvulsants with differing mechanism of action, valproate and phenytoin to attenuate the cognitive defi cits induced by PCP and D-amphetamine in the reversal learning paradigm. PCP at 1.5 mg/kg and D-amphetamine at 0.5 mg/kg both produced a selective and significant reduction in performance of the reversal phase with no effect on the initial phase of the task in female-hooded Lister rats. Valproate (25-200 mg/kg) and phenytoin (25-50 mg/kg) had no effect on performance when administered alone. Valproate (100-200 mg/kg), whose principle action is thought to be the enhancement of GABA transmission, was unable to prevent the cognitive deficit induced by either PCP or D-amphetamine. Conversely, phenytoin (50 mg/kg), a use-dependent sodium channel inhibitor, significantly prevented the deficit induced by PCP, but not D-amphetamine. These results add to our earlier work with lamotrigine, and suggest that sodium channel blockade may be a mechanism by which some anticonvulsant drugs can prevent the PCP-induced deficit. These data have implications for the use of anticonvulsant drugs in the treatment of cognitive or psychotic disorders.
The psychotomimetic noncompetitive N-methyl-D-aspartate receptor antagonist phencyclidine (PCP) can mimic many of the positive, negative and cognitive symptoms of schizophrenia in normal humans and precipitate a psychotic episode in schizophrenia patients in remission (Aniline and Pitts, 1982
; Javitt, 1987
; Jentsch and Roth, 1999
; Steinpreis, 1996
). NMDA receptor antagonists also disrupt the behaviour of animals, providing a model to investigate the mechanism by which these agents induce psychotic symptoms and perhaps some insight into the pathophysiology underlying schizophrenia (reviewed recently by Large, 2007
) The study of these models led to the NMDA receptor hypofunction hypothesis of the pathophysiology of schizophrenia, which has stimulated interest in developing new medication strategies. These experimental strategies include potentiation of NMDA receptor-mediated neurotransmission, facilitation of GABAergic neurotransmission, and direct or indirect blockade of excessive release of glutamate. Increasing evidence suggests that agents that inhibit glutamate release may have some application in the treatment of cognitive dysfunction associated with schizophrenia (Muzina et al., 2002
). Previously, we have shown that PCP induces a cognitive deficit in rats in a reversal learning paradigm, which is believed to model aspects of cognitive impairments in schizophrenia (Abdul-Monim et al., 2003
; Idris et al., 2005
). The atypical antipsychotic, clozapine with its high affinity for the 5-HT2A receptor and low affinity for the dopamine D2 receptor prevented the disruption of reversal learning induced by PCP, and the anticonvulsant, lamotrigine, a use-dependent sodium channel blocker (Xie et al., 1995
) that may act in part by inhibiting glutamate release (Cunningham and Jones, 2000
; Lingamaneni and Hemmings, 1999
; Waldmeier et al., 1996
), was also shown to be effective. Both compounds failed to prevent a D-amphetamine induced cognitive deficit, which was attenuated by haloperidol (Idris et al., 2005
).
These findings suggest that different mechanisms underlie the cognitive impairment induced by PCP and D-amphetamine. We hypothesise that PCP induces a deficit in the reversal learning task as a consequence of increased glutamate transmission arising downstream of NMDA receptor inhibition (Adams and Moghaddam, 1998
; Moghaddam et al., 1997
; Olney and Farber, 1995
). Currently used antipsychotic therapies do not target directly glutamate transmission, thus the validity of this hypothesis remains untested. However, several lines of evidence suggest that glutamate is involved in the pathophysiology of schizophrenia (Carlsson et al., 1999
; Deakin et al., 1989
; Dursun et al., 1999
; Hirsch et al., 1997
; Tamminga, 1998, 1999
), and a hyperglutamate hypothesis was first proposed by Deakin et al. (1989)
on the basis of neurochemical evidence of an excess of glutamatergic synapses in the frontal cortex of schizophrenic patients (Deakin and Simpson, 1997
; Deakin et al., 1997
; Reynolds et al., 2001
; Simpson et al., 1998
). Importantly, PCP and ketamine have been shown to increase glutamate levels in the prefrontal cortex (López-Gil et al., 2006
; Moghaddam et al., 1997
). Furthermore, sub-chronic PCP treatment produces a significant reduction in parvalbumin containing GABAergic interneurones in the frontal cortex (Abdul-Monim et al., 2003
; Cochran et al., 2002
, 2003
). This is consistent with human postmortem studies that indicate a deficit in GABAergic neurons in the prefrontal cortex of schizophrenia patients (Beasley and Reynolds, 1997
).
Anticonvulsant drugs are a group of compounds that suppress neuronal firing through a broad array of mechanisms. Phenytoin, like lamotrigine, has been shown to inhibit voltage-gated sodium channels and may also inhibit glutamate release (Kwan and Brodie, 2000
; Macdonald and Greenfield, 1997
). Sodium valproate has been shown to influence levels of GABA (Baldino and Geller, 1981
; Loscher, 1989
), and is unlikely to influence sodium channels at therapeutic concentrations. The present study aimed to explore further the mechanism by which lamotrigine prevents the PCP-induced disruption of reversal learning by examining the efficacy of these additional anticonvulsants that have differing mechanisms of action. For comparison with our previous studies with clozapine, haloperidol, and lamotrigine (Idris et al., 2005
), we also examine the ability of the two drugs to prevent a disruption of reversal learning produced by D-amphetamine.
Subjects and Housing Conditions
Subjects were 40 female-hooded Lister rats (Harlan, UK) obtained as adults and randomly housed in groups of four (cages measured 38 cm × 59 cm × 24 cm). Animals were maintained under standard laboratory conditions at a temperature of 21°C (±2°C) and humidity of 40–50%. They were maintained on a 12-h light/dark cycle (lights on at 07:00 hours) and the experimental procedure was undertaken in the last part of the light phase. Rats were gradually food deprived to approximately 85% of their free-feeding body weight (225–250 g). The reduced body weight was maintained by restricting the amount of food given (Standard laboratory chow, Special Diet Services, Essex, UK) to each rat per day (12 g/day). The availability of water was not restricted.
Experiments were carried out in accordance with the Animals Scientific Procedures Act, 1986 and were approved by the University of Bradford ethical review process.
Apparatus
All rats were tested in one of eight identical operant chambers (constructed in-house). Each chamber (29 cm × 30 cm × 30 cm) consisted of Plexiglas walls and ceiling and metal grid floor over sawdust. A hinged Plexiglas panel (6 cm × 6 cm) provided access to a food hopper containing food pellets (25 mg Noyes pellets, Sandown Scientific, UK). Two retractable levers (4 cm × 2 cm) were positioned either side of the food hopper. A light emitting diode (LED) was positioned centrally above each lever and a house-light was located in the ceiling of each chamber. The chambers were placed individually within ventilated sound-attenuating hardboard boxes (69 cm × 38 cm × 42 cm) containing a Perspex window to allow viewing. A small fan was built into each chamber to mask external noise. Each animal was tested in the same operant chamber throughout the study. All boxes were controlled by Med-PC software (Version 2.0 for DOS, Med Associates Inc. Lafayette, Indiana). Programmes were written using Medstate notation.
Operant Training Sessions
Following habituation to the operant chambers, rats were trained to respond for food on an FR1 (Fixed Ratio 1) schedule of reinforcement with both levers active, as previously described (Abdul-Monim et al., 2003
). When responding stabilised, rats were trained to press either the left or right lever for food delivery. Both levers were present, but only one lever was active. The active lever was varied from day to day using a pseudorandom Gellerman schedule, which randomly assigns either the left or right lever as active, thus avoiding generation of a lever bias. Each session lasted 20 min and counts were recorded on each lever. This first phase of training was complete in approximately 2 weeks.
As described previously (Abdul-Monim et al., 2003
), the rats were then trained to respond for food according to the position of a visual cue (a lit LED). Half were trained to press the lever under the lit LED in order to receive a food reward, the other half were trained on the opposite contingency, in other words, to press the lever under the non-lit LED.
Each training session began with illumination of the house-light. After 3 s both levers, together with the visual cue, were presented. The cue was presented for 1 s. Following a lever press, a correct response resulted in delivery of a food pellet. The levers were then retracted and the house light was extinguished for a 3-s time-out period, during which the pellet was consumed (if a correct response had been made). The house-light was then turned on again and the cycle repeated. The experimental session was terminated following a total of 128 lever presses, which took approximately 30 min. Each rat had one training session per day. Rats were required to make at least 115 responses on the correct lever (90% correct responding) on at least three consecutive days, which they generally achieved within 2 weeks. The contingency (visual cue, i.e. LED on or off, relative to active lever) remained constant during this period, though the position of the active lever varied from day to day according to a pseudo-random Gellerman schedule. Subsequently, rats were trained until they again reached criterion on the opposite contingency (again this generally took 2 weeks). Rats achieved criterion at the same rate irrespective of whether they responded to one rule or the other. Following a total of approximately 6 weeks of habituation and training, the reversal learning task was introduced.
Reversal Learning Task
The day before each reversal task session, a full 30-min operant training session (as described above) was conducted in order to ensure stable responding. The reversal task session then took place as follows. Animals were first exposed to a 5-min period during which the contingency (cue position relative to active lever) was the same as for the operant training session. During this period, responses on both correct and incorrect levers were recorded. This part of the session is termed the initial phase. This was followed by a 2-min time-out period, which was signalled by the house-light being turned off. In the subsequent 5-min period, the contingency was reversed. Responses made on the correct and incorrect levers were again recorded. This second period is termed the reversal phase. Animals undertook several of these reversal learning task sessions before the beginning of the drug studies in order to ensure that they attained stable level of performance in both phases of the task. In general, four to six sessions were required before this was achieved. In total, animals required about 6 weeks of training to reach this point.
Experimental Design-Drug Studies
An independent design was used in the present study and drug treated groups were compared with their appropriate controls. The experimental design typically consisted of a training day (day 1), followed by a drug test day for which the data are shown (day 2). On a drug test day, 40 animals were used in a complete drug study, e.g. to test effects of phenytoin versus PCP or D-amphetamine. On the following day, animals were trained on the same reversal task reward contingency used on the previous day (day 3) and on the next day, a drug free reversal test was performed to ensure that animals’ performance had returned to baseline levels (day 4). If baseline responding had been restored, this sequence of events would be repeated for the next drug study.
In this way, drug treatments were separated by at least 4 days and a drug-free reversal test was conducted before the next drug study began. The drug treatment given to each rat (and within each home-cage) over the course of these six experiments was randomised. Every effort was made to keep the number of drug and vehicle treatments equivalent between animals and to randomise the order of treatments (treatments for an example animal are shown in the final column of Table 1
). Thus if an animal received vehicle in experiment 1, it received a drug combination in experiment 2 and so on. As all drugs were given acutely, baseline performance was restored following the drug-free training session.
Results are only shown for day 2, i.e. the drug test day. The general design for testing and training is shown below:
Day 1 – training, approximately 30 min.
Day 2 – test day for drug study 1, two 5 min tests (initial and reversal phases). 40 animals tested in a complete drug study.
Day 3 – re-training on the same reward contingency used in the reversal task of the drug test day (day 2), approximately 30 min.
Day 4 – drug free reversal test, two 5 min tests (initial and reversal phases).
Day 5 – re-training on the same reward contingency used in the reversal task of the previous day, approximately 30 min.
Day 6 – re-training, approximately 30 min.
Day 7 – test day for drug study 2, two 5 min tests (initial and reversal phases) and so on….
This regimen was repeated for each drug study carried out, with 40 rats used to test the effects of each drug combination (n = 8–10 rats per group). Forty rats were used for all these drug studies. Baseline performances were restored before the next drug study was conducted, as shown above and control levels of performance in this task had not changed significantly by the end of these experiments (see Results).
Drugs
All drugs were administered in a volume of 1 ml/kg via the intraperitoneal route (i.p.). Drug doses were calculated as the base equivalent weight. PCP and D-amphetamine were administered 30 min prior to testing. In the event of co-administration, sodium valproate or phenytoin was given 30 min prior to administration of PCP or D-amphetamine. Phenytoin (Sigma, Germany), sodium valproate (Sigma, Germany) D-amphetamine (Sigma, UK) and PCP HCl (Sigma, UK) were dissolved in distilled water. The doses for PCP and D-amphetamine were selected on the basis of our previous dose response studies in the reversal learning paradigm, as described in Idris et al., (2005)
. The doses of anticonvulsants, valproate and phenytoin were based on previous studies (Farber et al., 2002a
) and from personal communications (C. Large, GSK).
Statistical Analysis
Data from this study are presented in two ways: mean total number of lever presses (±S.E.M.) and percent correct responses (±S.E.M.); in each case values for the initial and reversal phase are presented for the different drug treatment groups. The raw lever press data were used to determine whether there were significant effects of phase of task or drug(s) on overall responding (e.g. that might reflect changes in motor co-ordination or motivation). The percentage correct response data were used to determine whether there was a significant effect of phase of task or drug(s) on response accuracy (e.g. that might reflect cognitive dysfunction); these data were Arc-Sin transformed prior to analysis. Statistical significance was assumed when P < 0.05 and was determined as follows:
Analysis of Variance for repeated measures was performed in order to detect main effect of drug(s) treatment. Where a significant effect was detected, a post hoc Dunnett’s test was performed in order to compare treatment groups versus the appropriate control.
Baseline Levels of Performance
Rats were regularly tested to determine whether baseline performance was disrupted over the course of the study (approximately 8 weeks). At the beginning of the study the mean ± S.E.M. percentage correct responding in the initial phase was 82.9 ± 3.52 and 79.6 ± 1.84 for the reversal phase. The average values were 89.5 ± 3.22 (initial phase) and 80.38 ± 3.56 (reversal phase) at the end of the drug studies.
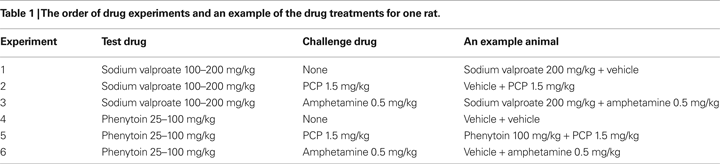
Experiment 1: Phenytoin Dose-Response Evaluation
The vehicle treated group made 22.7 ± 1.8 (mean ± S.E.M., n = 10) correct responses out of a total of 27.3 ± 1 lever presses in the initial phase and 22.4 ± 1.2 out of 28.2 ± 0.9 in the reversal phase of the task (Figure 1
A). Phenytoin (25 and 50 mg/kg) had no effect on the accuracy of responding in the initial or reversal phases of the task, but did cause a significant reduction in the total number of lever presses in the reversal phase alone at 25 mg/kg (P < 0.01) and in both phases at 50 mg/kg (P < 0.001; Figure 1
A). At 100 mg/kg, phenytoin significantly reduced correct responding in both the initial and reversal phases of the task (P < 0.05; Figure 1
B). This impairment was accompanied by a highly significant reduction in the total number of lever presses in both the initial and the reversal phases of the task (P < 0.001; Figure 1
A). Overall analysis of the experiment suggests that there was no significant effect of phenytoin on correct responding in the reversal phase when compared with the initial phase [F (3, 36) = 0.5, NS; Figure 1
B].
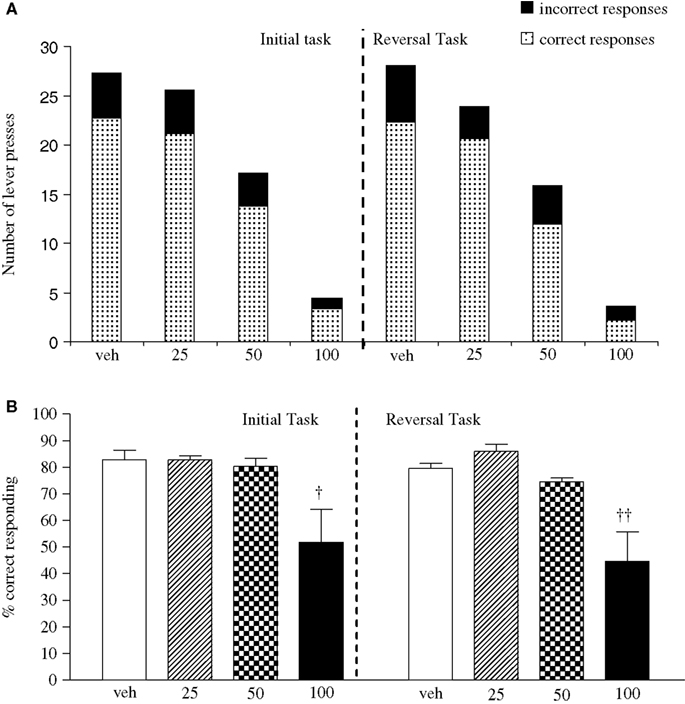
Figure 1. (A) The influence of phenytoin (25–100 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as the mean total number of lever presses, correct responses are shown as clear bars and incorrect responses are shown as shaded bars (n = 10 per group). (B) The influence of phenytoin (25–100 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as mean ± S.E.M. percentage correct responding (n = 10 per group). Significant reduction in performance of the initial and reversal phase compared with the respective vehicle group; †P < 0.05, ††P < 0.01; ANOVA followed by Dunnett’s t-test.
Experiment 2: Effect of Phenytoin on PCP-Induced Impairment
The vehicle treated group made 24.8 ± 0.6 (mean ± S.E.M., n = 8) correct responses out of a total of 27.5 ± 0.4 in the initial phase and 24.1 ± 0.7 out of 27.6 ± 0.7 in the reversal phase of the task (Figure 2
A). Overall analysis showed that there was a significant effect of phase of task on correct responding [F (1, 35) = 6.9, P < 0.05] and a significant effect of drug treatment on correct responding [F (4, 35) = 3.4, P < 0.05] such that animals made fewer correct responses in the reversal phase compared to the initial phase of the task (Figure 2
B). Post hoc analysis showed that PCP (1.5 mg/kg) significantly reduced correct responding in the reversal, but not initial phase of the task (P < 0.01). This reduction was dose-dependently and significantly attenuated by phenytoin (25–50 mg/kg, P < 0.05; Figure 2
B). However, the highest dose of phenytoin (100 mg/kg) significantly reduced correct responding in both the initial and reversal phases of the task (P < 0.01), as observed in the first experiment. Phenytoin significantly reduced total lever pressing in both phases of the task at 50 mg/kg (P < 0.01) and 100 mg/kg (P < 0.001; Figure 2
A).
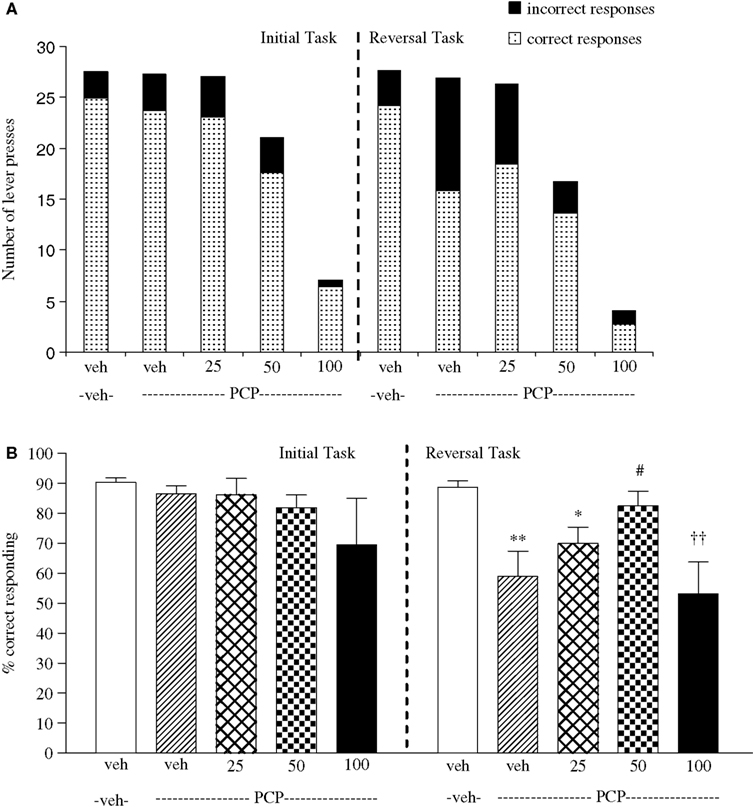
Figure 2. (A) The influnce of phenytoin pretreatment (25–100 mg/kg, i.p.) on the effect of PCP (1.5 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as the mean total number of lever presses, correct responses are shown as clear bars and incorrect responses are shown as shaded bars (n = 8 per group). (B) The effect of phenytoin pretreatment (25–100 mg/kg, i.p.) on the deficit produced by PCP (1.5 mg/kg, i.p.) on performance of the reversal phase in a reversal learning task. Data are shown mean ± S.E.M. percentage correct responding (n = 8 per group). Significant reduction in performance of the reversal phase compared with the initial phase; *P < 0.05, **P < 0.01. Significant reduction in performance of the reversal phase compared with the vehicle, vehicle controls; ††P < 0.01. Significant improvement in responding compared to PCP alone in the reversal phase; #P < 0.05, ANOVA followed by Dunnett’s t-test.
Experiment 3: Effect of Phenytoin on Amphetamine-Induced Impairment
The vehicle treated group in this experiment made 24.5 ± 1.3 (mean ± S.E.M., n = 10) correct responses out of a total of 28 ± 0.9 in the initial phase, and 22.6 ± 0.9 out of 26.5 ± 0.7 in the reversal phase (Figure 3
A). Overall analysis of the experiment suggests that there was a significant effect of phase of the task on correct responding [F (1, 35) = 5.0, P < 0.01] and also a significant effect of drug treatment on correct responding [F (4, 35) = 4.5, P < 0.01] such that animals made fewer correct responses during the reversal phase than the initial phase of the task. Post hoc analysis showed that D-amphetamine (0.5 mg/kg) significantly reduced correct responding in the reversal phase (P < 0.001), with no effect on the initial phase of the task. Phenytoin (25–100 mg/kg) did not prevent the reduction in correct responding induced by D-amphetamine. Furthermore, correct responding was significantly reduced by phenytoin at 100 mg/kg in the presence of D-amphetamine in both initial and reversal phases of the task when compared with the respective vehicle groups (P < 0.01; Figure 3
B). D-amphetamine alone produced a small, but significant reduction in total lever pressing in the reversal phase when compared with the appropriate vehicle group. In the presence of D-amphetamine, phenytoin (25 mg/kg) significantly reduced lever pressing in the initial phase alone (P < 0.05), whereas at 50 and 100 mg/kg phenytoin significantly reduced total lever pressing in both the initial and reversal phases (P < 0.05, P < 0.01, respectively, Figure 3
A).
Figure 3. (A) The effect of phenytoin pretreatment (25–100 mg/kg, i.p.) on the deficit produced by D-amphetamine (0.5 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as the mean total number of lever presses, correct responses are shown as clear bars and incorrect responses are shown as shaded bars (n = 8 per group). (B) The effect of phenytoin pretreatment (25–100 mg/kg, i.p.) on the deficit produced by D-amphetamine (0.5 mg/kg, i.p.) on performance of the reversal phase in a reversal learning task. Data are shown as mean ± S.E.M. percentage correct responding (n = 8 per group). Significant reduction in performance of the reversal phase compared with the initial phase; *P < 0.05, **P < 0.01, ***P < 0.001. Significant reduction in performance of the initial and reversal phase compared with the respective vehicle group; ††P < 0.01, ANOVA followed by Dunnett’s t-test.
Experiment 4: Valproate Dose-Response Evaluation
The vehicle treated group made 24.3 ± 1.4 (mean ± S.E.M., n = 10) correct responses out of a total of 27.8 ± 0.3 lever presses in the initial phase and 20.9 ± 1.8 out of 28.3 ± 0.3 in the reversal phase (Figure 4
A). Overall analysis suggests that there was no significant effect of phase of the task on correct responding [F (1, 36) = 0.58, NS]. There was no significant effect of valproate (100–200 mg/kg i.p.) on correct responding [F (3, 36) = 0.92, NS] or on the total number of lever presses [F (3, 36) = 1.9, NS; Figures 4
A,B].
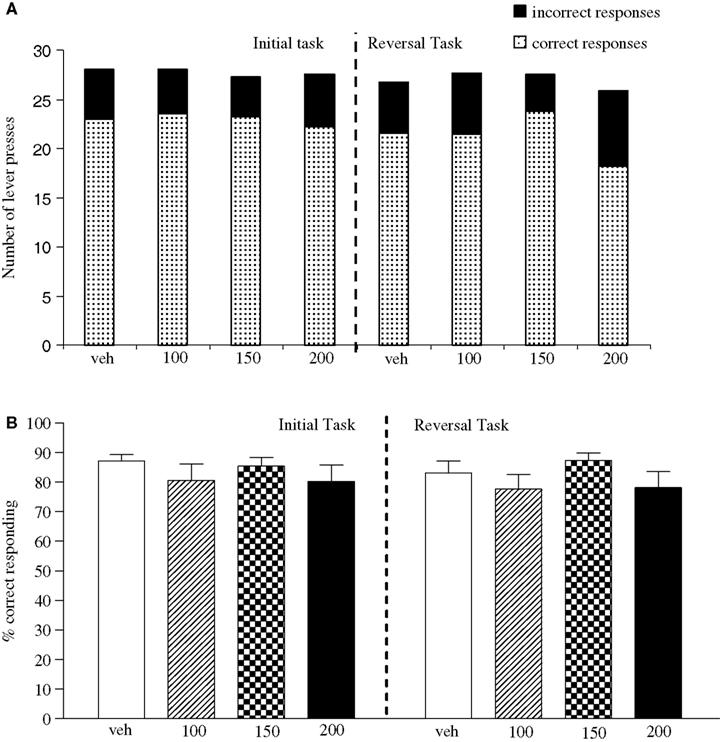
Figure 4. (A) The influence of sodium valproate (100–200 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as the mean total number of lever presses, correct responses are shown as clear bars and incorrect responses are shown as shaded bars (n = 10 per group). (B) The influence of sodium valproate (100–200 mg/kg, i.p.) on performance of the initial and reversal phase in a reversal learning task. Data are shown as mean ± S.E.M. percentage correct responding (n = 10 per group).
Experiment 5: Effect of Valproate on PCP-Induced Impairment
The vehicle treated group in this experiment made 24.3 ± 1.4 (mean ± S.E.M., n = 8) correct responses out of a total of 27.8 ± 0.3 in the initial phase and 20.9 ± 1.9 out of 28.3 ± 0.3 in the reversal phase of the task (Figure 5
A). Overall analysis showed that there was a significant effect of phase of task on correct responding [F (1, 35) = 33.4, P < 0.001] and a significant effect of drug treatment on correct response [F (4, 35) = 15.4, P < 0.01] such that animals made fewer correct responses in the reversal phase compared to the initial phase of the task (Figure 5
B). Post hoc analysis showed that PCP (1.5 mg/kg) significantly reduced correct responding in the reversal, but not in the initial phase of the task (P < 0.01; Figure 5
B). Valproate (100–200 mg/kg) did not prevent the reduction in correct responding induced by PCP, since in the presence of valproate there was still a significant reduction in correct responding in the reversal phase when compared with the initial phase (P < 0.001, P < 0.01 respectively; Figure 5
B). PCP alone had no significant effect in the total number of lever presses, but valproate at 150–200 mg/kg plus PCP produced a small, but significant reduction in the total number of lever presses in both the initial and the reversal phases of the task when compared with the vehicle group (P < 0.05; Figure 5
A).
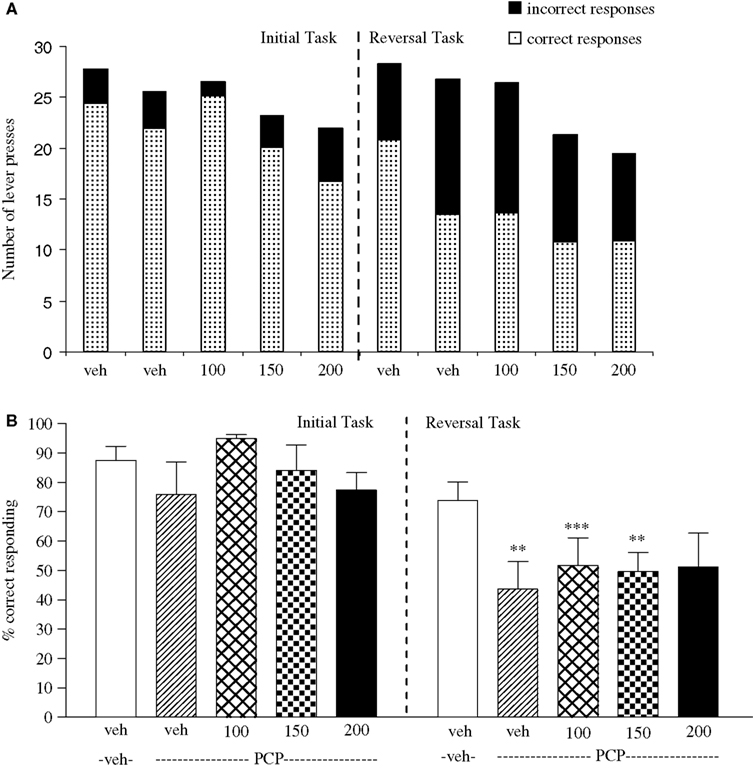
Figure 5. (A) The effect of sodium valproate pretreatment (100–200 mg/kg, i.p.) on the deficit produced by PCP (1.5 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as the mean total number of lever presses, correct responses are shown as clear bars and incorrect responses are shown as shaded bars (n = 8 per group). (B) The effect of sodium valproate pretreatment (100–200 mg/kg, i.p.) on the deficit produced by PCP (1.5 mg/kg, i.p.) on performance of the reversal phase in a reversal learning task. Data are shown as mean ± S.E.M. percentage correct responding (n = 8 per group). Significant reduction in performance of the reversal phase compared with the initial phase; **P < 0.01, ***P < 0.001, ANOVA followed by Dunnett’s t-test.
Experiment 6: Effect of Valproate on Amphetamine-Induced Impairment
The vehicle treated group in this experiment made 24.9 ± 0.9 (mean ± S.E.M., n = 8) correct responses out of a total of 27.8 ± 0.3 in the initial phase and 22.5 ± 1.2 out of 28.1 ± 0.5 in the reversal phase (Figure 6
A). Overall analysis suggests that there was a significant effect of phase of the task on correct responding [F (1, 35) = 41.6, P < 0.001] and also a significant effect of drug treatment on correct responding [F (4, 35) = 5.5, P < 0.001] such that animals made fewer correct responses in the reversal phase compared to the initial phase of the task (Figure 6
B). Post hoc analysis showed that D-amphetamine (0.5 mg/kg) significantly reduced correct responding in the reversal phase [F (1, 35) = 10.5, P < 0.001], with no effect on the initial phase of the task (Figure 6
B). Valproate 100–200 mg/kg) did not prevent the reduction in correct responding induced by D-amphetamine, since a significant reduction in correct responding in the reversal phase was still observed with the addition of valproate (100–200 mg/kg) in the presence of D-amphetamine when compared with initial phase (P < 0.05 to P < 0.001, n = 8 per group; Figures 6
A,B). Neither D-amphetamine alone nor valproate at 100–200 mg/kg plus 0.5 mg/kg D-amphetamine had a significant effect in the total number of lever presses (Figure 6
A).
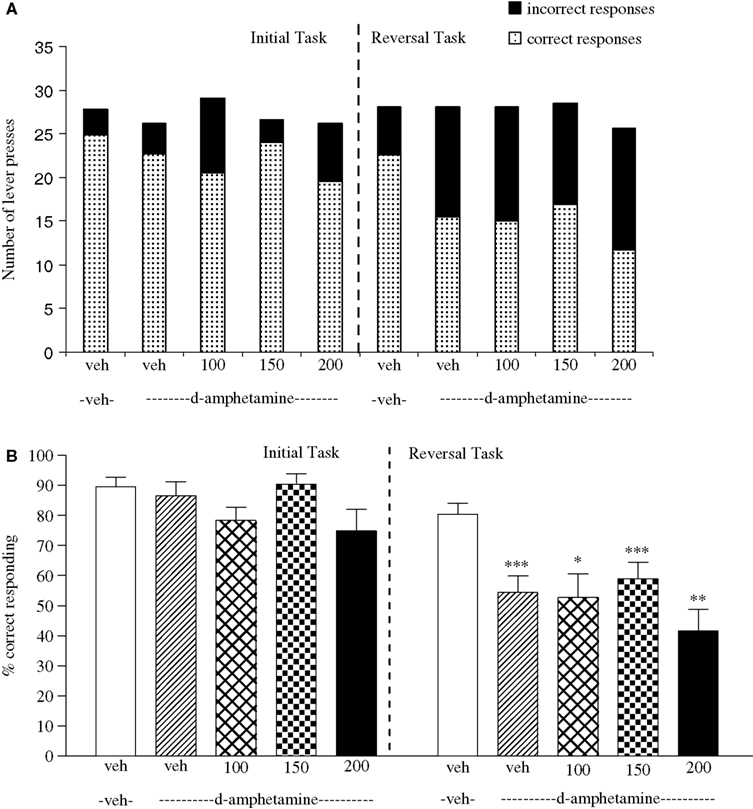
Figure 6. (A) The effect of sodium valproate pretreatment (100–200 mg/kg, i.p.) on the effect of D-amphetamine (0.5 mg/kg, i.p.) on performance of the initial and reversal phase of the reversal learning task. Data are shown as the mean total number of lever presses, correct responses are shown as clear bars and incorrect responses are shown as shaded bars (n = 8 per group). (B) The effect of sodium valproate pretreatment (100–200 mg/kg, i.p.) on the deficit produced by D-amphetamine (0.5 mg/kg, i.p.) on performance of the reversal phase in a reversal learning task. Data are shown as mean ± S.E.M. percentage correct responding (n = 8 per group). Significant reduction in performance of the reversal phase compared with the initial phase; *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA followed by Dunnett’s t-test.
The Reversal Learning Task
The current study confirms previous work showing that doses of 1.5 mg/kg of PCP and 0.5 mg/kg of amphetamine produce a selective deficit in the reversal phase of the reversal leaning task. In the presence of the two psychotomimetic drugs, animals were able to maintain performance of the initial task requiring memory for the learned rule. However, in the reversal phase, where animals are required to stop responding according to the original rule, and respond in the opposite manner, both drugs significantly affected performance.. Thus, PCP and D-amphetamine significantly impaired performance of a complex cognitive task without disrupting simple rule directed responding. Such patterns of cognitive deficit have been clearly demonstrated in schizophrenia, for example the inability of patients to perform the Wisconsin card-sorting test (Lidde, 2000
). PCP or D-amphetamine alone did not affect the total number of lever presses, irrespective of the phase of the task, which would suggest no sedation, behavioural or motivational impairment induced by these compounds at the doses used here. Thus, the deficits observed were due to incorrect responding rather than a loss in ability or motivation to lever press.
Phenytoin
Phenytoin significantly impaired responding in the reversal learning paradigm in a manner that was not related to the phase of the task; however, despite this, the drug did appear to preserve the accuracy of responses (% correct responding), and also significantly prevented the reduction in response accuracy induced by PCP in the reversal phase of the task. Previously, we have shown that lamotrigine could also reduce the effect of PCP on response accuracy in the reversal phase, but lamotrigine had the advantage that it did not affect the number of lever presses (Idris et al., 2005
). Furthermore, both lamotrigine and phenytoin can prevent the neurotoxic effects of the potent non-competitive NMDA antagonist MK-801 (Farber et al., 2002b
). These data suggest that the mechanism of action of these agents to improve the reversal learning deficit induced by PCP (i.e. an inhibition of voltage-gated sodium channels inducing decreased release of glutamate) probably underlies their ability to prevent the PCP induced disruptive effect including NRHypo neurotoxicity (Farber, 2003
; Farber et al., 2002b
). To begin to understand the mechanism(s) underlying the consequence of the NRHypo state in humans, several research groups have begun examining the consequence of PCP a non-competitive NMDA receptor antagonist induced NRHypo in adult rodents. One typical consequence is excessive release of glutamate (Moghaddam et al., 1997
; Noguchi et al., 1998
) in the prefrontal cortex. It has been proposed that this excessive release of excitatory neurotransmitters and consequent overstimulation of postsynaptic neurons might explain the cognitive deficits associated with NMDA receptor antagonism (Moghaddam et al., 1997
; Olney et al., 1989
). These data indicate that inhibition of voltage-gated sodium channels is a likely mechanism through which these agents prevent the cognitive deficit induced by PCP.
In the case of phenytoin, the strong effect of the drug to reduce lever pressing makes it difficult to draw the same conclusion, despite the positive statistical analysis. However, consistent with previous results with lamotrigine (Idris et al., 2005
), phenytoin was unable to prevent the disruption of reversal phase performance by D-amphetamine despite significantly reducing lever pressing. A comparison between the PCP and D-amphetamine experiments thus provides confidence that the prevention by phenytoin of the reduction in correct responding by PCP was not a statistical artifact. In conclusion, we suggest that the results with phenytoin add support to the hypothesis that sodium channel blockers have a beneficial effect on the disruption by PCP of a cognitive task; however, overall responding was significantly impaired by phenytoin, and this might preclude the use of this drug in a therapeutic setting.
Sodium Valproate
Sodium valproate had little or no effect on performance of either phase of the reversal learning task. A minor reduction in lever pressing was observed only at the highest dose tested during the reversal phase of the task. However, valproate failed to reduce the disruption by PCP or D-amphetamine of correct responding in the reversal phase of the reversal learning task. This result is consistent with previous studies that found that valproate was unable to prevent ketamine- or D-amphetamine-induced disruption of prepulse inhibition in mice (Ong et al., 2005
). Furthermore, valproate was unable to prevent an increase in locomotor activity induced by D-amphetamine (Arban et al., 2005
). The doses of valproate used in the present study were considered sufficient, since doses of 75 and 150 mg/kg, given orally, were found to be effective at preventing increases in locomotor activity induced by a mixture of D-amphetamine and chlordiazepoxide (Arban et al., 2005
), and doses in the range of 50–200 mg/kg, given by the intraperitoneal route are anticonvulsant in rodent models of seizure (C. Large, unpublished observations). Furthermore, in the present study, valproate at all doses did produce a reduction in lever pressing when in combination with PCP. These data indicate a pharmacodynamic interaction between valproate and PCP. A similar interaction was observed in the study by Arban et al. (2005)
, which found that valproate enhanced the sedating effects of chlordiazepoxide, consistent with the ability of valproate to enhance GABAergic transmission (Baldino and Geller, 1981
; Loscher, 1989
). In contrast, no reduction in lever pressing was observed in the present study with the combination of valproate and D-amphetamine, suggesting that there was no pharmacodynamic interaction in this case.
The present study adds to previous results with the reversal learning paradigm and supports the efficacy of sodium channel blocking drugs to prevent disruption of the reversal phase of the task by NMDA receptor antagonists. These results are also consistent with the hypothesis that the behavioural disruption induced by PCP is linked to an increase in glutamate transmission, since phenytoin, like lamotrigine, has been shown to reduce glutamate release under some circumstances (Cunningham and Jones, 2000
). In addition, this study adds to data from other models suggesting that enhancing GABAergic transmission does not reduce the effects of PCP, but instead can exacerbate effects. This finding apparently contradicts the proposed mechanism by which low doses of NMDA receptor antagonists cause a reduction in GABAergic feedback, which then leads to a disinhibition of principal neurons in cortex and hippocampus (Olney and Farber, 1995
). However, the majority of the data linking valproate to increased GABA transmission is derived from chronic dosing studies (see also Schechter et al., 1978
), so it remains to be demonstrated that the drug does increase GABA in an acute setting.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The study was supported by grants from the GlaxoSmithKline.
Cochran, S. M., Kennedy, M., McKerchar, C. E., Steward, L. J., Pratt, J. A., and Morris, B. J. (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28, 265–275.