National Centre for Biological Sciences, Bangalore, India
Python is emerging as a common scripting language for simulators. This opens up many possibilities for interoperability in the form of analysis, interfaces, and communications between simulators. We report the integration of Python scripting with the Multi-scale Object Oriented Simulation Environment (MOOSE). MOOSE is a general-purpose simulation system for compartmental neuronal models and for models of signaling pathways based on chemical kinetics. We show how the Python-scripting version of MOOSE, PyMOOSE, combines the power of a compiled simulator with the versatility and ease of use of Python. We illustrate this by using Python numerical libraries to analyze MOOSE output online, and by developing a GUI in Python/Qt for a MOOSE simulation. Finally, we build and run a composite neuronal/signaling model that uses both the NEURON and MOOSE numerical engines, and Python as a bridge between the two. Thus PyMOOSE has a high degree of interoperability with analysis routines, with graphical toolkits, and with other simulators.
In computational biology there are two approaches to developing a simulation. First, write your custom program to do a specific simulation, and second, write a model and run it in a general-purpose simulator. While the first approach is very common, it requires the scientist to be a good programmer (or have one at her/his disposal) and moves the focus towards programming rather than science. Furthermore, it is very difficult for others to read such a program and understand how it relates to the targeted biological system. In this context, a model is a well-defined set of equations and parameters that is meant to represent and predict the behavior of a biological system. Ideally, a general-purpose simulator allows the model to be separated from the low-level data-structures and control. The scientist is no longer concerned with minutiae of software engineering and can concentrate on the biological system of interest. The model can be shared by other people and understood relatively easily using intermediate-level descriptions of the model with a more obvious mapping to the real biological system. General simulators also lend themselves to declarative, high-level model descriptions that have now become important part of scientific interchange in the computational neuroscience and systems biology communities (Beeman and Bower, 2004
; Cannon et al., 2007
; Goddard et al., 2001
; Hucka et al., 2002
; http://www.morphml.org/
; http://neuroml.org
, http://sbml.org
). The goal of this paper is to show how the simulator Multi-scale Object Oriented Simulation Environment (MOOSE; http://moose.ncbs.res.in/
, mirrored at http://moose.sourceforge.net/
) uses Python to address these issues of interoperability with analysis software, graphical interfaces, and other simulators.
General-purpose simulators have been in use since the venerable circuit simulator SPICE was utilized to solve compartmental models (Bunow et al., 1985
; Segev et al., 1985
). While this level of generality ran into limitations of computing power, more specialized neuronal simulators such as GENESIS and NEURON (Bower and Beeman, 1998
; Carnevale and Hines, 2006
; Hines, 1993
) included optimized custom code that would allow the simulation to be run in affordable time and memory. This process of building domain-specific general simulators has continued with several simulators devoted to different aspects of computational and systems biology (e.g., VCell, Smoldyn, COPASI). This proliferation of simulators brings back the problems of model exchange and interoperability, albeit at a higher-level than raw Fortran or C code. While these simulators now have a common set of shared higher-level concepts (e.g., compartments, channels, synapses), they use entirely different vocabularies and languages for set up and control.
MOOSE is a new simulator project that supports simulations across a wide range of scales in computational biology, including computational neuroscience and systems biology. In order to improve interoperability, MOOSE uses two existing languages: the GENESIS scripting language, and Python. The Neurospaces (Cornelis and De Schutter, 2003
; http://neurospaces.sourceforge.net/
) project takes a distinct approach to supporting some GENESIS capabilities using backward-compatible scripting, and it too can utilize Python.
Most established simulators have their own scripting languages. For example, NEURON uses hoc along with modl files to set up simulations. GENESIS has its own custom scripting language. MOOSE avoids introducing a new language, and instead inherits the GENESIS parser. To increase compatibility, MOOSE has equivalents for most objects in GENESIS, and many old scripts can be run on MOOSE with little or no modification. Given these existing capabilities, why add Python scripting? Despite its flexibility, the GENESIS scripting language has several limitations:
1. Domain specificity: It is not used outside GENESIS. This forces the user to learn a special-purpose scripting language.
2. Problem with extensibility: While it is easy to write a script to define functions that can be included in other scripts, these interpreted functions are much slower than compiled code. The GENESIS scripting language itself provides for some degree of extensibility, but this is difficult to implement. Adding a single command requires implementation in C, as well as definition of the command in a configuration file that must be pre-processed to include into the interpreter. The addition of a new class is still more involved.
3. Lack of existing libraries: The GENESIS scripting language is a special-purpose language and has no additional features other than those written into the language.
4. Syntax: The syntax is complex and inconsistent as a result of accretion of features by many developers and users. For example, arrays are implemented in three inconsistent ways in the GENESIS scripting language: as arrays of elements, entries within tables and extended fields.
To harness the capabilities provided by a modern widely used scripting language, we chose a Python interface. Among the plethora of programming languages, Python has some special advantages:
1. Interactive: We need a scripting language that comes with a command line interpreter. Python is suited for this. User interaction is as important as running standalone scripts. Simulations are built incrementally, and it is important that users can try out bits and pieces of code and get quick feedback from the system. Moreover, this practice helps in identifying errors early in the development process, which saves considerable time and computational resources.
2. It is easy to interface with other programming languages: Python itself is written in C. It has a standard developers’ API for creating extension libraries. This simplifies creating Python interface for C/C++ code. Moreover, tools like Simplified Wrapper and Interface Generator (SWIG), Qt sip, boost-Python can automate the task of creating a Python interface from existing C/C++ code.
3. It is portable: Python runs on Linux, Solaris, Macintosh and Windows operating systems and many other platforms (http://www.python.org/about/
).
4. Free: Python is free and open-source.
5. Widely used: Python is widely used in scientific community. There is a large repertoire of third-party libraries for Python. Many of these libraries are free, open source and mature.
In this study we show how PyMOOSE harnesses each of these capabilities.
There are two common approaches to create a Python interface to a C/C++ library: (1) statically link it with the Python interpreter – which involves compiling the Python interpreter source-code, (2) create a dynamic link library and provide it as a Python module. We took the second approach as it provides more flexibility on the choice of the Python interpreter and reduces the burden on the maintainer.
Mapping MOOSE Classes into Python
MOOSE has a set of built-in classes for representing simulation entities. These classes provide a mapping from the concept space to the computational space. Physical or chemical properties and other relevant parameters are accessible as member fields of the classes and the time-evolution of these parameters is calculated by a special process method of each class. These classes add another layer over ordinary C++ classes to provide messaging and scheduling as well as customized access to the member fields. MOOSE provides introspection (Maes, 1987
; Smith, 1982
), so that full field information for each class is accessible to the programmer. This class information is statically initialized for each class at startup time. We utilized this class information and SWIG (Beazley, 1996
; http://www.swig.org
) to build the Python interface.
SWIG is a mature software with good support for Python and C/C++ interfacing as well as many other languages. While it is rather simple to create an interface for ordinary C++ class using SWIG, our task was complicated because MOOSE classes have another layer over ordinary C++ classes. For this reason we created a framework for Python interface with additional C++ classes to wrap MOOSE classes and a few classes to manage the system.
Simulator Control Through Python
All operations on MOOSE objects are carried out via a special class, Shell, of which there is a single instance on each processor node that is running MOOSE. In PyMOOSE we implemented a singleton context object to communicate with the Shell. The context object provides a set of functions that can be called to pass appropriate messages to the Shell. The user can call global MOOSE functions by calling the corresponding methods of the context object. Operations like creation of objects, setting integration time step, running the simulation are all done through the context object.
We created a one-to-one mapping of MOOSE classes to Python classes by means of light-weight C++ wrapper classes. All the wrapper classes were derived from one common base class. Each MOOSE object is identified by an Identifier (ID) field. The main data content of a wrapper class instance is the ID of the corresponding object in MOOSE. Additionally, the wrapper classes have a static pointer to the single instance of the context object. Wrapper classes provide accessor methods that can be used to access the fields in the corresponding MOOSE object.
These C++ wrapper classes were input to SWIG to create the Python module. After translation to Python, the user sees the member fields in the Python classes in place of the accessor methods in the C++ wrapper classes. Behind the scene the Python interpreter calls these accessor methods whenever the user script tries to access MOOSE object fields (Figure 1
A).
Manually developing C++ wrapper classes for all MOOSE classes was a tedious but repetitive task. We therefore embedded stub code in the MOOSE initialization code to generate most of the wrapper code programmatically using Run-Time Type Information in C++. This auto-generated code was used with a few modifications to generate a Python module using SWIG. SWIG takes an interface file with SWIG-specific directives and generates a single C++ file for the library and a Python source-code file that contains support code. We completed the PyMOOSE code generation by compiling and linking the SWIG-generated C++ source-code as a dynamic library. This dynamic library can be imported in any Python program.
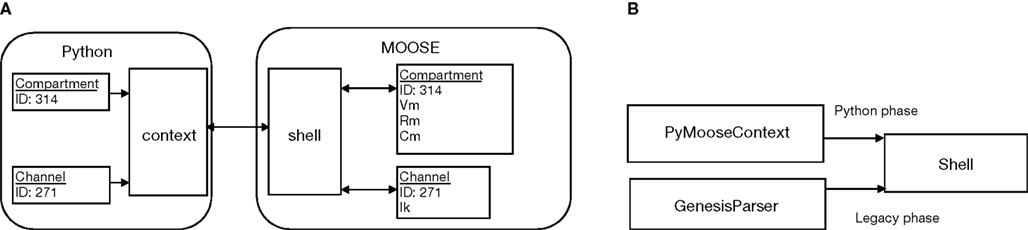
Figure 1. PyMOOSE interface. (A) Communication between Python and MOOSE. MOOSE represents concepts through objects and manipulates them using the singleton Shell object. PyMOOSE provides a light-weight mirror representation of each MOOSE object. Operations on PyMOOSE objects are communicated to MOOSE via the context and the Shell object. (B) Accessing legacy scripts through PyMOOSE. The Shell object is usually controlled through the PyMooseContext. When loading a GENESIS script, control is temporarily passed to the legacy GENESIS script language parser, and then returned to the PyMooseContext.
Legacy Models and PyMOOSE
The PyMOOSE context object keeps a single instance of the GenesisParser class in order to run legacy GENESIS scripts. Whenever the user asks for executing a GENESIS statement, the context object disconnects itself from the Shell and connects the GenesisParser object instead. The GENESIS statement string is passed to the GenesisParser object, which executes it as if the user typed it in at the MOOSE command prompt. After execution of the statement (or script) the GenesisParser object is disconnected from the Shell and the context object is reconnected (Figure 1
B).
While it is valuable to run GENESIS scripts within PyMOOSE, this feature is intended only to support legacy code and is better avoided in new model development. The use of GENESIS scripting language inside Python defeats the whole purpose of moving to a general-purpose programming language. It reduces readability and the user needs to know both languages in order to understand the code.
We used the Python interface of MOOSE to achieve three key targets: (1) Interfacing with standard libraries in a mature scientific computing language, (2) giving access to a portable GUI library for developing user interface and (3) enabling MOOSE to work together with other simulators.
Interfacing Simulations with Python Libraries
We used Python scientific and graphing libraries to analyze and display the output of a PyMOOSE simulation. The interface with Python gives the user freedom to choose from a wide variety of scientific and numerical libraries available from third parties. We demonstrate the use of two libraries along with PyMOOSE for developing simulations with plotting and data analysis within Python. The first of these, NumPy, is a library that provides data structures and algorithms for fast matrix manipulation (http://numpy.scipy.org/
). Even though Python is interpreted, with attendant slow execution, NumPy library provides access to compiled code and hence the functions from the library are as fast as compiled code. The second library, matplotlib, provides a rich set of functions for plotting 2D data both in hardcopy formats and interactively (http://matplotlib.sourceforge.net/
). It can use NumPy for fast matrix operations in Python and several portable GUI toolkits (GTK/Qt/Tk/wxWidgets) as graphical back-end.
We implemented a simulation of the squid giant axon using Hodgkin–Huxley Na+ and K+ channels and parameters (script attached in Appendix). We applied an injection current with random amplitude uniformly distributed between 0 and 100 nA. We recorded the time-series for the membrane potential during the simulation in a MOOSE table object, which can accumulate a time-series of simulation output (Figure 2
A). The interface to Python was done using the MOOSE table class. This class is exposed to Python with methods to emulate iterable type (Martelli et al., 2005
). The array constructor in NumPy accepts an iterable object and creates a NumPy array with a copy of the contents of the object. Thus the user is relieved of explicitly iterating over the table entries and copying them to a NumPy array. This completes the interface from the MOOSE simulation output to NumPy (Figure 2
B). We used the fast Fourier transform operation available in NumPy to compute the discrete Fourier transform of the time-series of the simulated membrane potential. We used matplotlib to plot the original time-series, as well as the output of the FFT (Figure 2
C).
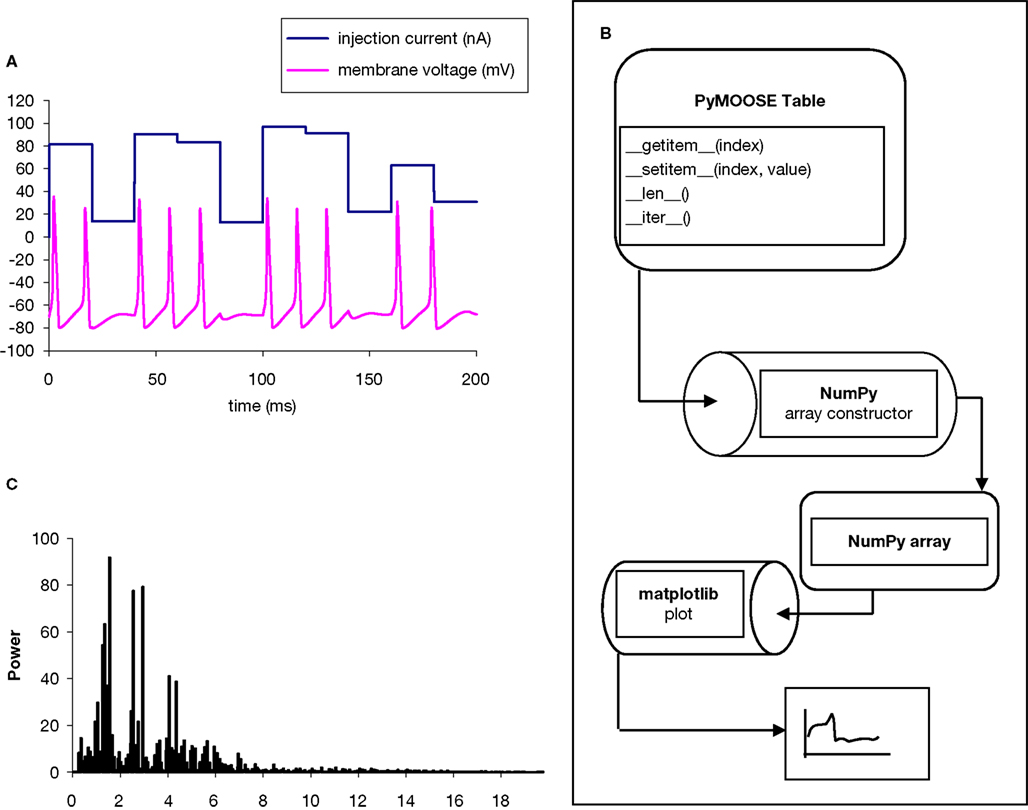
Figure 2. Analysis and graphing of a PyMOOSE simulation. (A) Simulation input (random input current) and output (membrane potential). (B) Data flow. The simulation time-series is recorded in the MOOSE table object, which is visible to Python as a sequence object. This is accessed as an array in NumPy. The fast Fourier transform is applied to this array, and the result plotted in Matplotlib. (C) Output of FFT analysis (with the fundamental frequency removed).
Overall, this example simulation illustrates how PyMOOSE facilitates interoperability of Python numerical and graphing libraries with MOOSE.
Portable GUI Through Python
The use of Python separates the problem of GUI development from simulator development. Moreover, it gives one the freedom to choose from a number of free GUI toolkits. The major platform independent GUI toolkits with Python interfaces are Qt(TM) available as PyQt, wxWidgets (wxPython), Tk and GTK (http://wiki.python.org/moin/GuiProgramming
; http://www.python.org/doc/faq/gui/
). We used PyQt4 to develop a simple user interface for a clone of the GENESIS squid tutorial in MOOSE. We selected Qt4 as it is a mature and clean toolkit that is freely distributed and runs well on all the major operating systems.
The program was divided into three modules – (1) the squid axon compartment with Hodgkin–Huxley channels, (2) a model object which combined a few tables with the squid compartment to record various parameters through the time of the simulation, and (3) the GUI to take user inputs and to plot data. We implemented the squid axon model as described in the previous section, using PyMOOSE to set up and parameterize the model. As before, the model was interfaced with table objects to monitor time-series output of the simulation. Finally, we implemented the GUI by loading in the PyQt4 libraries, and using Python calls to set up the interface (Figure 3
). While there are Qt IDEs available (http://trolltech.com/products/qt/
), we constructed the interface through explicit Python calls to create widgets, assign actions, and manage output data. Qt uses a signal-slot mechanism for passing event information. PyQt allows the use of arbitrary Python methods to be used as slots. Hence we could connect the GUI widgets to methods in the PyMOOSE model class and thus provided simulation control through the GUI in a clean manner. We used PyQwt, a Python interface of the Qt-based plotting library Qwt, for creating output graphs. Since PyQwt can take NumPy arrays as data, we converted the tables in MOOSE to NumPy arrays and used PyQwt plotting widgets to display them.
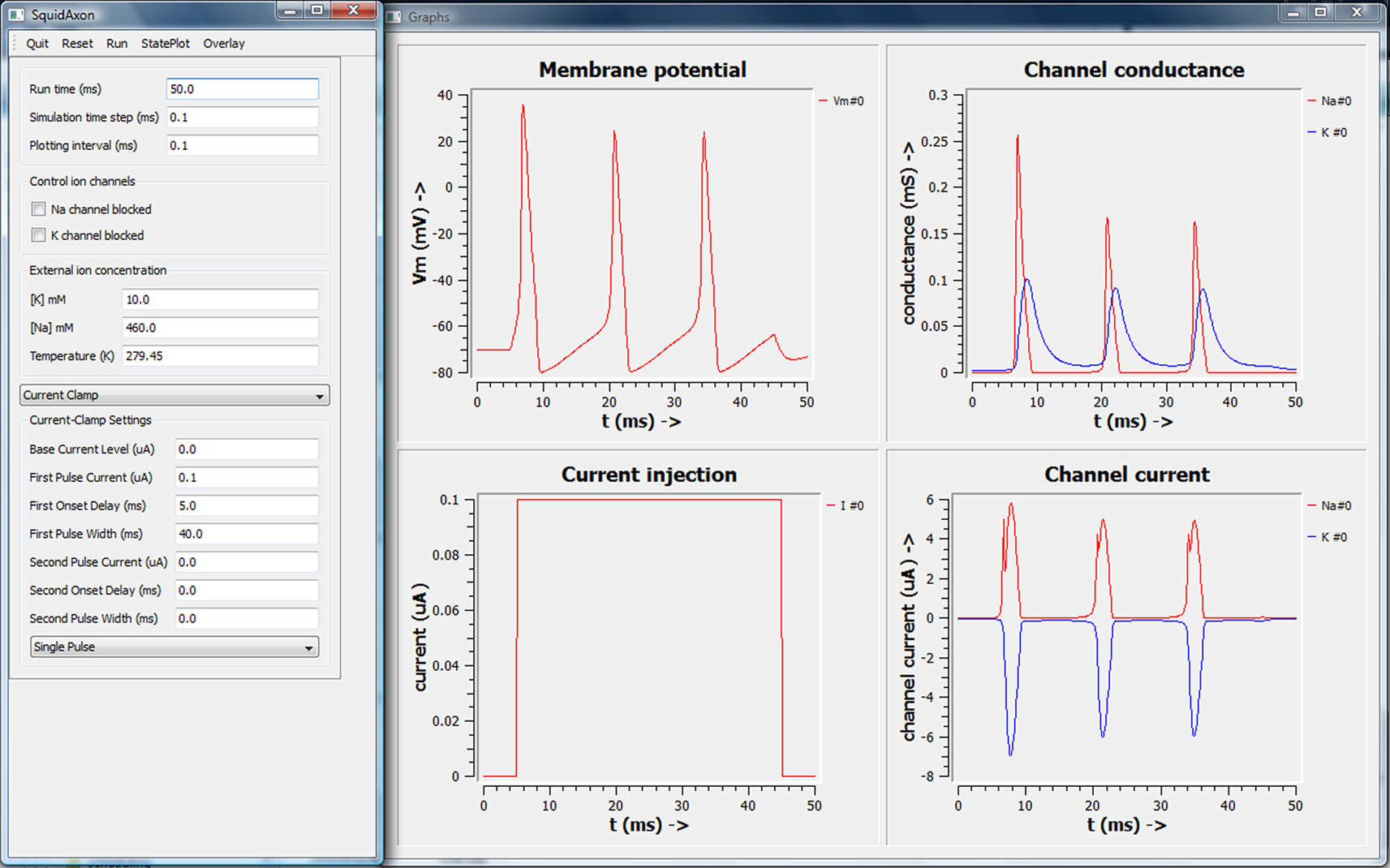
Figure 3. Screen shot of PyMOOSE/Qt interface for the Hodgkin–Huxley model. The layout is closely modeled on the Squid demo from GENESIS.
We based the layout of the simulation on the widely used GENESIS Squid tutorial program. To confirm portability of the system, we ran the model on Linux as well as the Windows operating system.
This exercise demonstrated the capability of PyMOOSE to draw upon existing graphical libraries for its graphical requirements. This is an important departure from GENESIS. The GENESIS graphical libraries (XODUS) were an integral part of the C code-base and XODUS objects were visible as, and manipulated in the same way as other GENESIS objects. In contrast, PyMOOSE did not need to implement any graphical objects within the MOOSE C++ code, but instead reused extant third-party graphical libraries available for Python. Furthermore the existing libraries are professionally designed and have a much more consistent look-and-feel than did the original GENESIS graphical library, XODUS (Bhalla, 1998
).
Simulator Interoperability
With Python becoming a popular language for developing platform independent scripts, several neuronal simulators have implemented Python interfaces. This raises the possibility of using Python as a glue language to run simulations that span different simulators. As a final demonstration of interoperability, we used PyMOOSE with PyNEURON to build a multi-scale, multi-simulator model that incorporates neuronal electrical activity as well as biochemical signaling (Figure 4
A).
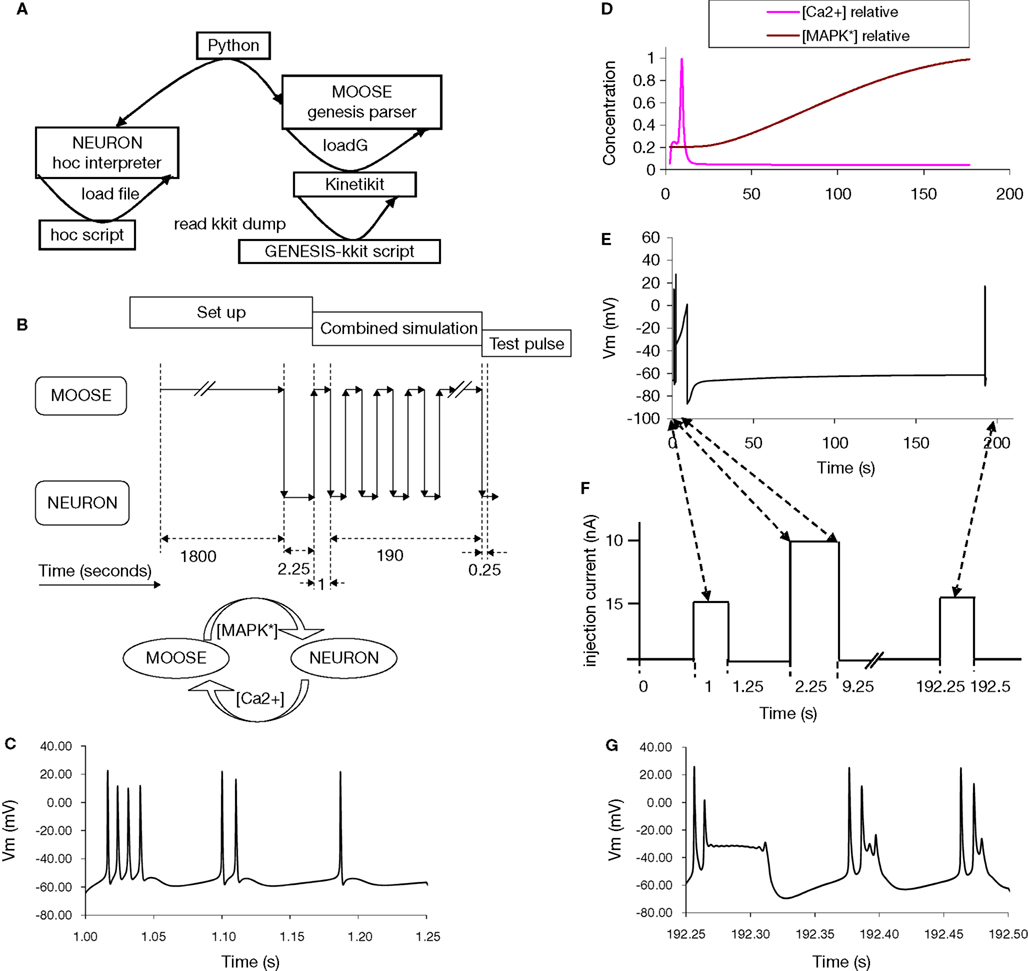
Figure 4. A combined, multi-scale NEURON and GENESIS model. (A) Setup of combined model, using NEURON and GENESIS model definition files. (B) Information flow during simulation. The two models were run independently for an initial settling period and for the test pulse to the NEURON model. During the combined simulation phase, each model was advanced for 1 s and then data was transferred via Python to the other model. Finally a second test pulse was delivered. (C) Response of NEURON model to first test pulse. (D) Calcium and MAPK levels in the signaling model. (E) Voltage responses from the NEURON model. (F) Experiment design and input to NEURON model. A test current pulse of 0.15 nA was delivered for 0.25 s to the NEURON model in the initialization phase. At the start of the combined simulation, a stimulus of 10 nA was delivered for 7 s. After 180 s of combined simulation a second test pulse (0.15 nA, 0.25 s) was applied. (G) Response of NEURON model to second test pulse. The difference is due to modulation of KCa by the elevated MAPK activity.
We used NEURON to model a multicompartmental electrical model of a Type A neuron from the CA3b region of the rat hippocampus (Migliore et al., 1995
; http://senselab.med.yale.edu/ModelDB/ShowModel.asp?model=3263
). This is a morphologically detailed model with experimentally constrained distribution of membrane ion channels. It reproduces experimental observations of firing behavior and intracellular Ca2+ dynamics. We modified the hoc script for the model, to run it for arbitrary time intervals. We directed the output data to Vector objects in NEURON. The Python wrapper class for this model provided a handle for the simulation parameters and functions defined in the hoc script. As described in the PyNEURON documentation (http://www.neuron.yale.edu/neuron/docs/help/neuron/neuron/classes/python.html
), Python commands were directed to the NEURON engine by constructing hoc statement strings and executing them through the hoc interpreter instance provided by the neuron module. Moreover, hoc object references are directly available in Python as attributes of the hoc interpreter object. Thus accessing hoc objects was quite clean in Python (Figure 4
A).
We used MOOSE to model calcium-triggered biochemical signaling events at the synapse. We used a model of a bistable MAPK-PKC-PLA2 feedback loop that was originally implemented in GENESIS/Kinetikit (Ajay and Bhalla, 2004
; Bhalla and Iyengar, 1999
; Bhalla et al., 2002
) and uploaded to the DOQCS database (http://doqcs.ncbs.res.in/template.php?&y=accessiondetails&an=79
). The model was defined in the GENESIS scripting language. We used the legacy scripting mode of PyMOOSE to load the GENESIS/kinetikit model. The simulation objects thus instantiated were standard MOOSE objects, and were accessible using Unix-like path strings. The PyMOOSE interface exposed these objects as regular Python objects. Thus access to the MOOSE objects, representing GENESIS data concepts, was also straightforward in Python (Figure 4
A).
We used the Python interface to accomplish three critical operations to combine the two simulations: (1) Initialization, (2) run-time control and synchronization, and (3) variable communication and rescaling.
1. To initialize the models, we used PyNEURON command load_file to load the hoc script. Once the script is loaded, variables and functions defined in the script become available as members of the hoc interpreter instance inside Python. In this case we defined a setup function to initialize the NEURON simulation. This function is called in the constructor (__init__) of the Python wrapper class over the NEURON simulation. At this stage we applied a test pulse of 1 nA for 250 ms to measure the firing properties of the neuron before potentiation. We then ran the NEURON model for 1 s to allow the model to settle. Similarly we loaded the GENESIS/Kinetikit model using the loadG command, and ran this simulation for 1800 s to settle.
2. In the Python wrapper class for each model, we defined a run method to advance the simulation in time. That for the NEURON model uses a run function we defined in the custom hoc script. This run function calls NEURON’s fadvance command to advance the simulation. In the wrapper class for the GENESIS/Kinetikit model the run method calls the step command to advance the simulation (Figure 4
B).
3. We used the Python interface to read out somatic calcium levels from the NEURON model and insert them into the MOOSE model, and to feed back MAPK activity changes from the MOOSE model to modulate KCa conductances.
We wrote another higher-level function run to advance the coupled simulations using the two wrapper classes (not to be confused with the member method run of these classes). This function (1) creates instances of both wrappers, which involves initializing the models, (2) runs the NEURON simulation for 1 s, (3) reads out the calcium level, performs rescaling and updates the kinetic model with this value, (4) advances the kinetic simulation for 1 s to catch up with the electrical model, (5) reads out the activity level of MAPK from the GENESIS/Kinetikit model and modifies the [Ca2+] dependent K+ channel conductances in the NEURON model in inverse proportion to this (Figures 4
E,F).
Our simulated experiment is illustrated in Figure 4
F. We loaded the models and allowed them to settle. We measured baseline neuronal responses at this stage using a 250-ms, 0.15 nA current pulse. Following this we used the run function for the further time-evolution of the system. We applied a strong LTP-inducing stimulus to the neuronal model for 7 s, and then allowed the simulation to continue for 183 s. Finally we repeated the 250 ms, 0.15 nA test for neuronal responses.
The time-evolution of membrane potential, Ca2+ levels, and MAPK activity are shown in Figures 4
D,E. The initial and final burst waveforms of the neuron are shown in Figures 4
C,G. We observe that the coupled model shows how electrical stimulation can lead to signaling events, with feedback effects on the electrical properties of the neuron. We should point out that this simulation is only a demonstration and the relationship between the chemical system and the biophysical properties of the neuron is over-simplified, although the two component models we used are realistic within their respective domains.
This example also illustrates the efficiency of using Python for data transfer when traffic volumes are small compared to the computational times. The neuronal calculations in NEURON took about 91% of the simulation run-time, the signaling calculations in MOOSE took ∼8.5%, and the data transfer through Python accounted for only around 0.5%. As we discuss below, there may be other interface contexts where more efficient, low-level data transfer protocols may be needed, and the relatively facile Python interface may not be appropriate.
We have used PyMOOSE, the Python interface to MOOSE, to achieve interoperability at three levels. First, we used standard mathematical packages in Python to analyze MOOSE output. Second, we used the QT graphical toolkit from within Python to build a GUI for a MOOSE simulation. Third, we used Python as a glue language to run a cross-simulator model combining an electrophysiological model set up in NEURON with a biochemical signaling model set up in GENESIS/Kinetikit.
Issues with Python Interoperability
The strengths of the Python language make it perhaps too easy to repeat well-known mistakes in simulation development. We consider two such issues. First, Python is an interpreted language in most implementations. In the context of simulations, it is not meant for number crunching. Well-designed libraries like NumPy can hide some of these limitations from the user, and fast hardware can conceal other inefficiencies. However, given the same specialized algorithms, a compiled language will perform better than an interpreted one. Therefore, for large simulations, we need to combine the best possible algorithms with optimized and compiled languages. MOOSE has as one of its goals the capability of managing the low-level, high-traffic flow of data between different numerical engines incorporated into MOOSE. We do not consider Python appropriate for such operations. Second, many aspects of model specification should be done using declarative rather than procedural approaches (Cannon et al., 2007
; Crook et al., 2005
, 2007
). However, Python makes procedural model definition very easy, and may even provide a certain level of interoperability if several simulators provide equivalent calls for model setup. For example, there are some impressive recent efforts to develop a standard vocabulary for network definitions across simulators (http://neuralensemble.org/trac/PyNN/
; this issue). While the presence of Python as a common link language may temporarily address the interoperability issues of this approach, we feel that it would be a cleaner design to use a separate, declarative definition for networks such as NeuroML (http://neuroml.org
). Nevertheless, we completely agree that a standard vocabulary for model definitions is an important first step toward this goal.
Model Specification vs. Simulator Control
Model specification and exchange issues have been ably addressed by the communities developing model specification languages (Le Novère et al., 2005
; Qi and Crook, 2004
; http://neuroml.org
; http://sbml.org
). The current paper focuses on the second problem, that of making it easier for researchers to control and set up these diverse simulation tools. We have shown how this can be done with the simulator MOOSE, using Python as a glue language. Run-time communication between simulators has previously been achieved using the NEOSIM framework, which uses Java (Goddard et al., 2001
; Howell et al., 2002
). More recently, the MUSIC framework specifies an API for simulators to use to communicate with each other (Ekeberg and Djurfeldt, 2008
). Our study is novel in two respects. First, we use the built-in Python capabilities of two simulators to achieve run-time communication, without the need to modify either simulator or to build an additional framework for communication. Second, we carry out bidirectional communications across scales (biophysical to biochemical models) and involving continuous data types (channel conductance and calcium concentrations) rather than spike events.
The evolution of neuronal simulator technology has seen a gradual separation of different aspects of modeling, with a corresponding improvement in interoperability. The first step was to develop higher-level simulation tools (e.g., NEURON and GENESIS) to separate the numerical and housekeeping code from the model-specific code. This let people share models, provided they were written for the same simulator. The second was the development of declarative model specifications that were separate from the simulator. This initially took the form of semi-declarative cell morphology files (NEURON ‘.geom’ files and GENESIS ‘.p’ files), which required additional files for channel specification. This process of separation of model definition from simulator control has continued. The Neuroconstruct suite refines the declarative definition of models, with NeuroML and ChannelML as declarative definitions sufficient for most single-neuron models. Importantly, at this level quite different simulators can use the same original model definition to run simulations. A third stage is the convergence of different simulators to use the same link language, in this case Python. This makes it possible to explicitly separate model definition from simulator control. In the current paper, we have illustrated this with a composite signaling-neuronal model drawing on NEURON and MOOSE. We have utilized two legacy models, one written for NEURON, and one written for GENESIS. Even though the legacy models themselves were not entirely set up in a declarative manner, we used the original model definitions only to load in the model specifications. We used Python as the procedural language to control these operations, and to mediate communication between the models at run-time.
Sustainability of Python Interoperability
Simulator interoperability has long been regarded as important (Crook et al., 2005
, 2007
; Goddard et al., 2001
). Such projects have been difficult to execute, and still harder to maintain, because they depend on multiple underlying simulator projects, each with different APIs, directions and life-cycles. Python is a potential way out of this problem. First, Python itself is a well-established language with a strong community and support. Second, the issues of interfacing to Python are now being undertaken by individual simulator development teams. Interoperability emerges from these independent efforts rather than requiring a separate project to achieve coordination. Third, PyMOOSE itself will be maintained for the long-term, since Python will be the default scripting language for MOOSE. We suggest that long-term improvements in interoperability will be driven both by widespread simulator support for declarative model specifications, and by a richer ecosystem of simulators fluent in Python.
Program listing: ca3_db.hoc provides the functions to load and initialize the NEURON CA3 cell model as well as for advancing the simulation for a specified interval and for updating parameters.
/******************************************************************************
* Derived from Hippocampal CA3 pyramidal neuron model from the paper * M. Migliore, E. Cook, D.B. Jaffe, D.A. Turner and D. Johnston, Computer* simulations of morphologically reconstructed CA3 hippocampal neurons, J.* Neurophysiol. 73, 1157-1168 (1995). * The original model is available in modeldb: accession no: 3263** Modified by: Subhasis Ray , 2008 ******************************************************************************/
objref cvode, vecCai, vecT, vecV, outFile, stim1, stim2, stim3, fihvecV = new Vector()vecCai = new Vector()vecT = new Vector()outFile = new File()cvode = new CVode(0)cvode.active(1)cvode.atol(1e-3)START = 2AMP = 1.0// ************* NEURON A **********FARADAY=96520PI=3.14159secondorder=2dt=0.025celsius=30flagl=0xopen("ca3a.geo")proc conductances() { forall { insert pas e_pas=-65 g_pas=1/60000 Ra=200 insert cadifus insert cal gcalbar_cal=0.0025 insert can gcanbar_can=0.0025 insert cat gcatbar_cat=0.00025 insert kahp gkahpbar_kahp=0.0004 insert cagk gkbar_cagk=0.00055} soma { insert nahh gnabar_nahh=gna insert borgkdr gkdrbar_borgkdr=gkdr insert borgka gkabar_borgka=gka insert borgkm gkmbar_borgkm=gkm} for i=0,1 dend2[i] { insert nahh gnabar_nahh=gna insert borgkdr gkdrbar_borgkdr=gkdr insert borgka gkabar_borgka=gkainsert borgkm gkmbar_borgkm=gkm }for i=0,2 dend3[i] { insert nahh gnabar_nahh=gna insert borgkdr gkdrbar_borgkdr=gkdr insert borgka gkabar_borgka=gka insert borgkm gkmbar_borgkm=gkm}for i=37,38 dend3[i] { insert nahh gnabar_nahh=gna insert borgkdr gkdrbar_borgkdr=gkdr insert borgka gkabar_borgka=gka insert borgkm gkmbar_borgkm=gkm }}proc init() { t=0 coord_cadifus() forall { cao=2 cai=50.e-6 ek=-91 v=-65 if (ismembrane("nahh")) {ena=50} } vecV.record(&soma.v(0.5)) vecCai.record(&soma.cai(0.5)) vecT.record(&t) finitialize(v) fcurrent() forall {if (ismembrane("nahh")) {e_pas=v+(ina+ik+ica)/g_pas} else {e_pas=v+(ik+ica)/g_pas} } cvode.re_init()}proc setup(){ strength = 1.0 /*namps*/ tstim = 50tstop=500 gna=0.015 gkdr=0.03 gka=0.001gkm=0.0001 conductances() /* The schedule of experiment is as follows:
The 1800 s runs with 1 s intervals interspersed with 1 s of kinetic simulation and update of gkbar for all ca dependent k channels. The genesis model needs over 1 uM [Ca2+] for 10 s. */ soma { // first test pulsestim1 = new IClamp(0.5)stim1.amp = 0.15stim1.del = 1000.0stim1.dur = 250// tetanus pulsestim2 = new IClamp(0.5)stim2.amp = 1.0stim2.del = 2250stim2.dur = 7e3// final test pulsestim3 = new IClamp(0.5)stim3.amp = 0.15stim3.del = 192.25e3stim3.dur = 250 } init()}proc update_gkbar(){/* multiply all Ca2+ dependent K+ conductance by $1 */forall { gkahpbar_kahp = gkahpbar_kahp * $1 } soma { print "soma gkdrbar before:", gkdrbar_borgkdr gkdrbar_borgkdr = gkdrbar_borgkdr * $1 gkmbar_borgkm = gkmbar_borgkm * $1 print "soma gkdrbar after", gkdrbar_borgkdr }for i=0,1 dend2[i] { gkdrbar_borgkdr = gkdrbar_borgkdr * $1 gkmbar_borgkm = gkmbar_borgkm * $1}for i=0,2 dend3[i] { gkdrbar_borgkdr = gkdrbar_borgkdr * $1 gkmbar_borgkm = gkmbar_borgkm * $1}for i=37,38 dend3[i] { gkdrbar_borgkdr = gkdrbar_borgkdr * $1 gkmbar_borgkm = gkmbar_borgkm * $1} fcurrent()}access somadistance()/* run for interval specified as argument# 1 */proc run(){ t_start = t while (t < (t_start + $1)){ // print "run() - @t=", tfadvance() }// print "run(): t_start =", t_start, " current time =", t, "run interval =", $1}proc do_run(){ setup() print "setup done. running 7.25s" run(12250) print "t = ", t, "ms. done running. dumping data in test_neuron1.dat" outFile.wopen("test_neuron1.dat") for ii = 0, vecT.size() - 1 { outFile.printf("%g %g %g\n", vecT.x(ii), (vecCai.x(ii) - 50e-6)*2e6,
vecV.x(ii)) // the original GUI plots this function of cai instead of absolute
value - unit is nM*2 } outFile.close() print "done dumping. running for 5s with 0.5nA" run(5000) print "t =", t, "ms. soma.Cai = ", soma.cai(0.5), ". now updating gkbar" update_gkbar(10.0) print "done updating. writing to file" outFile.wopen("test_neuron2.dat") for ii = 0, vecT.size() - 1 { outFile.printf("%g %g %g\n", vecT.x(ii), (vecCai.x(ii) - 50e-6)*2e6,
vecV.x(ii)) // the original GUI plots this function of cai instead of absolute
value - unit is nM*2 } outFile.close() print "done dumping. now running the rest" run(1800300) print "t = ", t, "ms. done running. writing to file" outFile.wopen("test_neuron3.dat") for ii = 0, vecT.size() - 1 { outFile.printf("%g %g %\n", vecT.x(ii), (vecCai.x(ii) - 50e-6)*2e6,
vecV.x(ii)) // the original GUI plots this function of cai instead of absolute
value - unit is nM*2 } outFile.close()}Program listing 2: moosenrn.py – this program wraps the GENESIS model and the NEURON model and provides simulation control and data exchange between the two simulators.#!/usr/bin/env python# Author: Subhasis Rayimport syssys.path.append("/home/subha/lib/python2.5/site-packages")sys.path.append("/home/subha/lib/python2.5/site-packages/neuron")import pylabimport numpyimport neuronimport mooseclass NeuronSim: """Wrapper class for the neuron simulation""" def __init__(self, fileName="ca3_db.hoc"): """Load the file specified by fileName""" self.hoc = neuron.h self.hoc.load_file(fileName) self.hoc.setup() def run(self, interval): """Simulate for interval time in second""" self.hoc.run(interval * 1e3) # neuron keeps time in milli second def cai(self): """Returns cai of in nM""" return self.hoc.soma(0.5).cai def cai_record(self): """Returns a tuple containing the array of time points and the arrayof cai values at the corresponding points""" timeVec = numpy.array(neuron.h.vecT) caiVec = numpy.array(neuron.h.vecCai) return (timeVec, caiVec) def v_record(self): """Returns a tuple containing the array of time points and the arrayof membrane potential values at the corresponding points""" timeVec = numpy.array(neuron.h.vecT) vmVec = numpy.array(neuron.h.vecV) return (timeVec, vmVec) def update_kconductance(self, factor): """Modify the k hcannel conductances in inverse proportion of mapk_star_conc""" self.hoc.update_gkbar(factor) self.hoc.fcurrent() def saveplots(self, suffix): cai = "nrn_cai_" + str(suffix) + ".plot" vm = "nrn_vm_" + str(suffix) + ".plot" t_series, vm_series, = self.v_record() t_series, cai_series, = self.cai_record() numpy.savetxt(cai, cai_series) numpy.savetxt(vm, vm_series) numpy.savetxt("nrn_t_" + str(suffix) + ".plot", t_series)class MooseSim: """Wrapper class for moose simulation""" volume_scale = 6e20 * 1.257e-16 def __init__(self, fileName="acc79.g"): self._settle_time = 1800.0 self._ctx = moose.PyMooseBase.getContext() self._t_table = [] self._t = 0.0 self._ctx.loadG(fileName) self.ca_input = moose.Molecule("/kinetics/Ca_input") self.mapk_star = moose.Molecule("/kinetics/MAPK*") self.pkc_active = moose.Molecule("/kinetics/PKC-active") self.pkc_active_table = moose.Table("/graphs/conc2/PKC-active.Co") self.pkc_ca_table = moose.Table("/graphs/conc1/PKC-Ca.Co") self.mapk_star_table = moose.Table("/moregraphs/conc3/MAPK*.Co") self.mapk_star_table.stepMode = 3 self.mapk_star_table.connect("inputRequest", self.mapk_star, "conc") self.mapk_star_table.useClock(2) self.ca_input_table = moose.Table("/moregraphs/conc4/Ca_input.Co") self.ca_input_table.stepMode = 3 self.ca_input_table.connect("inputRequest", self.ca_input, "conc") self.ca_input_table.useClock(2) self._ctx.reset() self._ctx.reset() def set_ca_input(self, ca_input): """Sets the conc. of Ca_input molecule""" print "set_ca_input: BEFORE: nInit =", self.ca_input.nInit, ", n =",
self.ca_input.n, ", setting to: ", ca_input* MooseSim.volume_scale self.ca_input.nInit = ca_input * MooseSim.volume_scale self.ca_input.n = ca_input * MooseSim.volume_scale print "set_ca_input: AFTER: nInit =", self.ca_input.nInit, ", n =",
self.ca_input.n def ca_input(self): """Returns scaled value of Ca_input conc.""" return self.ca_input.conc def run(self, interval): """Run the simulation for interval time.""" self._ctx.step(float(interval)) # Now expand the list of time points to be plotted points = len(self.pkc_ca_table) - len(self._t_table) delta = interval * 1.0 / points for ii in range(points): self._t_table.append(self._t) self._t += delta def pkc_ca_record(self): """Returns the time series for pkc_ca conc.""" return (self._t_table, self.pkc_ca_table) def pkc_active_record(self): """Returns time series for pkc_active conc.""" return (self._t_table, self.pkc_active_table) def mapk_star_conc(self): """Returns MAPK* conc. in uM""" return self.mapk_star.n / MooseSim.volume_scale def mapk_star_record(self): """Returns time series for [MAPK*]""" return (self._t_table, self.mapk_star_table) def saveplots(self, suffix): pkc_a = "mus_pkc_act_" + str(suffix) + ".plot" pkc_ca = "mus_pkc_ca_" + str(suffix) + ".plot" mapk_star = "mus_mapk_star_" + str(suffix) + ".plot" ca_input = "mus_ca_input_" + str(suffix) + ".plot" numpy.savetxt("mus_t_" + str(suffix) + ".plot", self._t_table) self.mapk_star_table.dumpFile(mapk_star) self.pkc_ca_table.dumpFile(pkc_ca) self.pkc_active_table.dumpFile(pkc_a) self.ca_input_table.dumpFile(ca_input) def test_run(self): self.run(500) print "After 500 steps of uninited run: [MAPK*] =", self.mapk_star_conc() self.ca_input.nInit = 10 * MooseSim.volume_scale self.ca_input.n = 10 * MooseSim.volume_scale self.run(5) print "After another 5 s with 10uM ca input: [MAPK*] =", self.mapk_star_conc() self.ca_input.nInit = 0.08 * MooseSim.volume_scale self.ca_input.n = 0.08 * MooseSim.volume_scale self.run(500) print "finished run. going to plot" print "After another 500 s with 0.08 uM ca input: [MAPK*] =",
self.mapk_star_conc() pylab.plot(pylab.array(self._t_table), pylab.array(self.pkc_active_table), pylab.array(self._t_table), pylab.array(self.pkc_ca_table))pylab.show()if __name__ == "__main__": mus = MooseSim() mus.set_ca_input(0.08) mus.run(1800.0) mus.saveplots("1") start_mapk = mus.mapk_star_conc() nrn = NeuronSim() nrn.run(2.25) nrn.saveplots("1") file_ = open("cai_setings.txt", "w") # Interleaved execution of MOOSE and NEURON model # Synchronizing after every 1 s of simulation while nrn.hoc.t < 192.25e3 scaled_cai = scale_nrncai(nrn.cai()) mus.set_ca_input(scaled_cai) print "scaled_cai =",scaled_cai file_.write(str(nrn.cai()) + " " + str(scaled_cai)+"\n") mus.run(1.0) gkbar_scale = start_mapk / mus.mapk_star_conc() start_mapk = mus.mapk_star_conc() print "[mapk*] = ", start_mapk nrn.update_kconductance(gkbar_scale) nrn.run(1.0) print "time is ", nrn.hoc.t * le-3, "s" file_.close() nrn.saveplots("2") mus.saveplots("2") # final test pulse run nrn.run(0.3) nrn.saveplots("3") t_series, vm_series, = nrn.v_record() t_series, cai_series, = nrn.cai_record() pylab.subplot(121) pylab.plot(t_series, numpy.array(vm_series), t_series, numpy.array(cai_series) * 1e6) t_series, pkc_act, = mus.pkc_active_record() t_series, pkc_ca, = mus.pkc_ca_record() t_series, mapk_star, = mus.mapk_star_record() pylab.subplot(122) pylab.plot(numpy.array(t_series), numpy.array(pkc_act), numpy.array(t_series), numpy.array(pkc_ca), numpy.array(t_series), numpy.array(mapk_star)) pylab.show()The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The development of MOOSE is supported by grants from the Department of Biotechnology, India, and the NIGMS/Systems Biology Center of New York. We acknowledge support from FACETS to S. Ray to attend the FACETS/CodeJam meeting at CNRS, Gif-sur-Yvette, which further stimulated PyMOOSE development.
Crook, S., Beeman, D., Gleeson, P., and Howell, F. (2005). XML for model specification in neuroscience. In Special Issue on Realistic Neuro Modeling – Wam-Bamm ‘05 Tutorials. J.M. Bower and D. Beeman (eds.). Brains Minds Media, Vol. 1, bmm228 (urn:nbn:de:0009-3-2282). http://www.
brains-minds-media.org/archive/228
Ekeberg, ö., and Djurfeldt, M. (2008). MUSIC – multisimulation coordinator: request for comments. Nature Proceedings. Available at: http://dx. doi.org/10.1038/npre.2008.1830.1.
Le Novère, N., Finney, A., Hucka, M., Bhalla, U. S., Campagne, F., Collado-Vides, J., Crampin, E. J., Halstead, M., Klipp, E., Mendes, P., Nielsen, P., Sauro, H., Shapiro, B., Snoep, J. L., Spence, H. D., and Wanner, B. L. (2005). Minimum information requested in the annotation of biochemical models (MIRIAM). Nat. Biotechnol. 23, 1509–1515.