Cytosine methylation of CpG dinucleotides represents a major type of epigenetic modification (
Worm and Guldberg, 2002
). Since CpG dinucleotides cluster in genomic regions called CpG islands that frequently overlap with promoter regions, one of the effects of cytosine methylation is the inhibition of gene transcription by enabling interference via methyl-CpG binding proteins (
Boyes and Bird, 1992
). As a potential mechanism for silencing tumor suppressors or other important genes, aberrant CpG island methylation has gained increasing recognition for its role in cancer (
Jones and Baylin, 2002
). One of the most prominent examples is the recent discovery that promoter methylation of O
6–methylguanine–DNA methyltransferase (MGMT) is associated with improved treatment response of glioblastoma multiforme (GBM) patients to the alkylating agent temozolomide (
Hegi et al., 2005
). Since MGMT is a major repair enzyme for temozolomide-induced DNA damage, the presumed mechanism responsible for the benefit of promoter methylation is loss of repair activity via gene silencing of MGMT. In addition to MGMT, single CpG island/promoter approaches have identified a number of other important genes with aberrantly methylated CpG islands within the GBM genome, such as CD133, RASSF1A, BLU, SOCS1, NDRG2, CDKN2B, MYOD1, LRRC4, and PDGFB (
Uhlmann et al., 2003
;
Bruna et al., 2007
;
Lorente et al., 2008
;
Martini et al., 2008
;
Tepel et al., 2008
;
Yi et al., 2008
;
Zhang et al., 2008
).
Determination of the full extent of aberrant methylation within the GBM methylome using comprehensive genome-wide approaches remains necessary to discover other methylation-based biomarkers. Genome-wide methylation profiling methods have been evolving over the past several years, primarily with the emergence of denser CpG island arrays and next generation sequencing approaches (reviewed in
Zilberman and Henikoff, 2007
;
Gargiulo and Minucci, 2009
). Genome-wide approaches can be broadly grouped into strategies that examine either
unmodified DNA, including restriction enzyme approaches [RLGS, DMH (differential methylation hybridization), DMH variants] and immunoprecipitation approaches (MeDIP, 5-meC, and MIRA), or
bisulfite-modified DNA, including methylation-specific PCR such as Illumina GoldenGate and next generation sequencing. Genome wide methylation profiling has been performed on cancer genomes including osteosarcoma using Me-DIP (
Sadikovic et al., 2008
), pancreatic adenocarcinoma using methylated CpG island amplification (
Omura et al., 2008
), prostate, colon, and breast cancer using methylated CpG island amplification (
Chung et al., 2008
), and lung squamous cell carcinoma using MIRA (
Rauch et al., 2008
).
Thus far, the most high profile genome-wide methylation survey of the glioblastoma genome has been led by The
Cancer Genome Atlas (TCGA) Research Network (2008)
. TCGA surveyed CpG dinucleotide methylation in a large group of GBM patient samples using the Illumina GoldenGate Assay Platform to accompany copy number and gene expression microarray analysis. The major limitation of this platform is the relatively limited coverage, with only 1,500 CpG sites independently evaluated, corresponding to approximately 1,200 genes. More recently, data generated from the Illumina HumanMethylation27 platform providing methylation information on 27,000 CpG sites has been released by TCGA. Additional studies that have surveyed methylation within gliomas have used McrBC-based and DMH-based genome wide approaches (
Waha et al., 2005
;
Ordway et al., 2006
). Indirect examination of the GBM methylome has been performed using microarrays to track gene expression changes in GBM cell lines after pharmacological treatment with demethylating agents (
Foltz et al., 2006
;
Kim et al., 2006
).
Despite the advent of novel approaches using next generation sequencing technology, restriction enzyme-based approaches relying on differential cleavage of recognition sequences containing methylated and unmethylated CpG dinucleotides continue to be widely used for genome-wide methylation detection. The modified MSRE technique used in the present study is a variant of DMH that uses the HpaII/MspI isoschizomer restriction enzyme pair to determine methylation of CpG dinucleotides in the context of CCGG recognition sites (
Yan et al., 2002
;
Adrien et al., 2006
). Similar approaches, such as MIAMI and HELP, have also relied on this isoschizomeric pair combination (
Hatada et al., 2006
;
Khulan et al., 2006
). The major advantage of this strategy is that comparison to a separate reference (control) sample is not necessary, limiting false positive calls from polymorphic variation at restriction sites and incomplete digestion. Aside from DMH, there are other restriction enzyme approaches that do not utilize the isoschizomeric pair of HpaII/MspI, including the fractionation of genomic DNA by McrBC, an enzyme that cleaves methylated sites (
Ordway et al., 2006
;
Pfister et al., 2007
). Recently, this approach was used along with a custom-designed microarray called Comprehensive High-Throughput Arrays for Relative Methylation (CHARM) (
Irizarry et al., 2008
).
In this study, we demonstrate improved genome-wide methylation profiling by coupling the MSRE approach described by
Adrien et al. (2006)
with Agilent high-density CpG island arrays, providing coverage of more than 20,000 CpG islands (
Adrien et al., 2006
).
In silico analysis was used to optimize the assay, and our results pr ovide useful strategies for optimization of restriction enzyme approaches with available or custom microarrays.
Agilent CpG Island Microarray
High-density two-color Human CpG island microarrays (printed using 60-mer SurePrint technology) were purchased from Agilent (G4492A, Agilent Technologies, Santa Clara, CA, USA). Originally designed based on the UCSC genome hg17 but remapped to hg18, each Agilent microarray (Design ID 014791) contains 237,220 probes that tile through each of the ∼27,000 CpG islands (including an extra 95 bp of flanking sequence on each end) with an average probe spacing of 100 bp. Each probe on the array is identified by its location on the genome and its associated gene(s) based on UCSC annotations (Agilent Probe Design ID 014791).
In Silico Simulation of Amplicon Generation and Hybridization
Hybridization to the Agilent CpG island array was simulated
in silico based on the most updated
Homo sapiens Genome hg18 within the UCSC database (
Kuhn et al., 2009
) and the probe design of the Agilent CpG island microarray (eArray, design ID 014791). Most of the
in silico analysis was performed using CRAN R version 2.7.0 in conjunction with BioConductor 2.3 (
Gentleman et al., 2004
). The entire human genome hg18 was imported into R through package BSgenome and was ‘digested’ into fragments based on the location of BfaI restriction sites (C/TAG). Each Agilent CpG island microarray probe was mapped and assigned to a fragment based on its sequence. Probes containing the BfaI recognition site(s) (C/TAG) were determined to be problematic due to possibility of non-specific or dual fragment binding and are thus removed from analysis (designated as Partial binding probes in Table
1
). The compiled list of probes with their hybridizing fragments, fragment size, and number of internal HpaII/MspI restriction sites were exported to a MySQL database and also into a BED file for the UCSC Genome Browser (
Kent et al., 2002
). Fragments without HpaII/MspI restriction sites (C/CGG) were designated as
insensitive fragments and were expected to have identical MspI and HpaII intensities (i.e. log
2 ratio ∼0) (Figure
1
). Therefore, probes corresponding to these insensitive fragments provided a large internal control group because they remain unaffected by the degree of methylation.
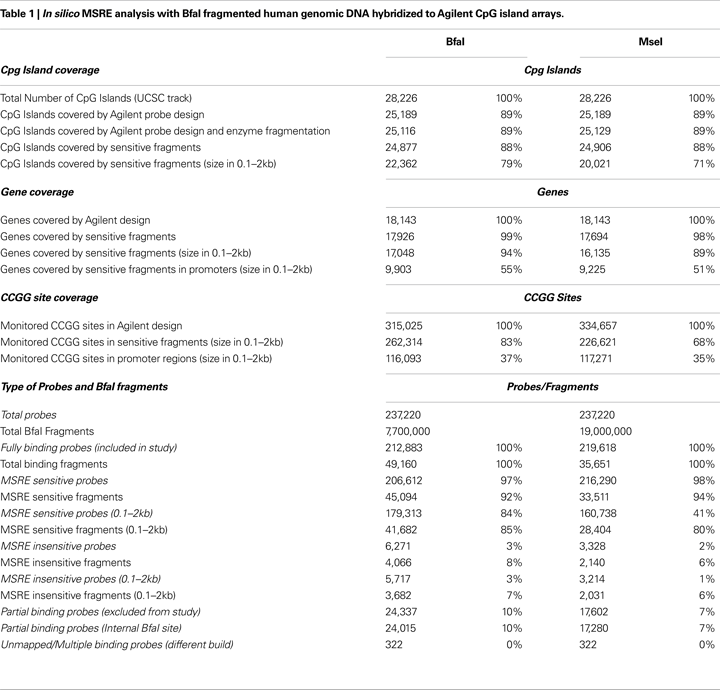
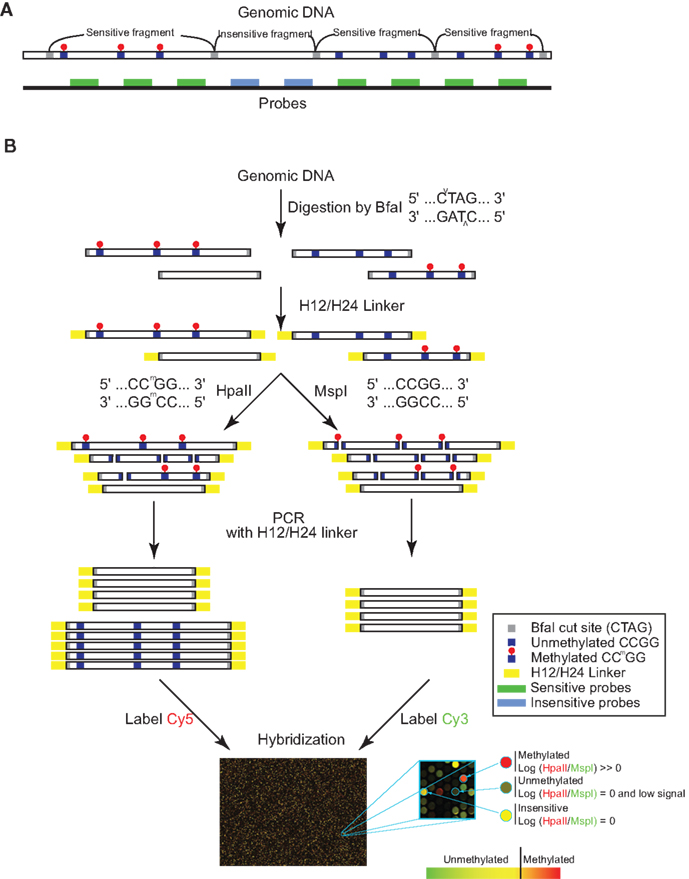
Figure 1. Schematic diagram of in silico annotated genomic DNA and MSRE method. (A) Using in silico simulation, genomic DNA can be separated into MSRE sensitive and insensitive BfaI fragments (flanked by gray boxes) based on the presence or absence of internal HpaII/MspI sites (CCGG) (blue squares). Microarray probes binding to sensitive fragments are represented with green rectangles. Probes binding to insensitive fragments are represented by blue rectangles. Sensitive fragments can have multiple CCGG sites that can be fully methylated, partially methylated, or completely unmethylated in tandem. Methylated CCGG sites (CCmGG) are indicated with a red dot placed above the blue square. (B) Genomic DNA is fragmented with BfaI, ligated to H12/H24 linkers, digested with HpaII and MspI in parallel, amplified, differentially labeled and co-hybridized to Agilent CpG island arrays. Uncleaved fragments will have high intensities compared to cleaved fragments since only uncleaved fragments are amplified. Fully methylated fragments are cut by MspI but not HpaII, resulting in amplification of HpaII digested fragments only with resultant M-value [log2 (HpaII/MspI)] > 0. Completely unmethylated or partially methylated (not all CCGGs methylated in tandem) fragments are cut by both MspI and HpaII. With the absence of amplification, these fragments resulting in low signal intensities in both channels (HpaII and MspI ∼ 0) with M = 0. Insensitive fragments should not be cut by either enzyme, resulting in M = 0.
Acquisition and Derivation of Genomic DNA Samples
Both the GBM tissue sample and normal human brain sample had been collected and archived through an IRB approved protocol and obtained from the UCLA Brain Tumor Translational Resource. The GBM tissue was derived from a treatment-naïve newly diagnosed patient. Normal brain tissue was derived from the right frontal lobe of a trauma patient. Both tissues were stored at −80°C until DNA extraction. Genomic DNA was isolated from the GBM and normal human brain samples and from the U87MG human GBM cell line using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA).
Fully-Methylated and Unmethylated Control Sample Generation
Fully-methylated control
Methylation of all genomic CpG sites in vitro was achieved by treating 4 μg genomic DNA (normal brain) with 16 units of SssI enzyme (NEB, Ipswich, MA, USA) and 160 nmol S-Adenosylmethione for 6 h at 37°C twice. The product was purified using the Zymo Clean and Concentrator Kit (Zymo Research Corp., Orange, CA, USA), in which the DNA was eluted using 20 μL of PCR-grade H2O. The typical yield after purification was 2 μg. Using bisulfite sequencing of uncloned DNA, sample CpG Island regions were verified to have complete methylation of all CpG sites (data not shown).
Fully-unmethylated control
‘Removal’ of all methylation markings was achieved by subjecting 10 ng of genomic DNA (normal brain) to whole-genome amplification with the GenomiPhi V2 Amplification Kit (Amersham Biosciences, Piscataway, NJ, USA) according to manufacturer’s instructions with unmodified dCTP. The typical yield was 4–7 μg. Using bisulfite sequencing of uncloned DNA, sample CpG Island regions were verified to have complete absence of methylation of all CpG sites (data not shown).
Amplicon Generation
Two separate experiments were performed for each sample except for the unmethylated control in which only one experiment was performed. Amplicon generation was based on the DMH method with several modifications (
Yan et al., 2002
). Most notably, BfaI was used instead of MseI for genomic fragmentation. Briefly, 750 ng of each genomic sample was digested with 25 units of BfaI (NEB, Ipswich, MA, USA) for 6 h at 37°C. This digested DNA was then ligated to oligonucleotide linkers consisting of annealed H-12 (5′-TAATCCCTCGGA-3′) and H-24 (5′-AGGCAACTGTGCTATCCGAGGGAT-3′) oligonucleotides. Following ligation, the genomic DNA was divided into two equal portions for separate digestion with 20 units of HpaII (methylation-sensitive isoschizomer) or 40 units of MspI (methylation-insensitive isoschizomer) (NEB). Each digest was subjected to PCR amplification using the same H-12/H-24 primers with the following conditions: 72°C for 5 min, followed by 16 cycles of 97°C for 1 min, 62°C for 30 s, 72°C for 3 min, followed by 72°C for 10 min. Amplified HpaII-digested DNA was labeled with Cyanine 5-dUTP (Cy-5) (Perkin-Elmer, Waltham, MA, USA), and amplified MspI-digested DNA was labeled with Cyanine 3-dUTP (Cy-3) (Perkin Elmer) using the Bioprime Array CGH Genomics Labeling Module (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The Cy-5 and Cy-3 labeled amplicons were then co-hybridized at 65°C for 40 h to the Agilent CpG island microarrays according to the manufacturer’s instructions. The hybridized slides were washed using the Agilent Oligo aCGH/ChIP-on-Chip Wash Buffer Kit.
Data Analysis
Hybridized microarrays were scanned with an Agilent DNA Microarray Scanner (UCLA Microarray Core) to create an image file. From this file, Cy-5 (HpaII digested) and Cy-3 (MspI digested) intensities for each probe were determined by the Agilent Feature Extraction 9.1 software using the CGH protocol v4.95 Feb07 (Agilent Technologies). Two channel intensity readings were collected from the Agilent Feature Extraction CGH workflow (Feb 2007, using grid file 20070820) before any normalization was done (up to step 20 in Reference Guide from Feature Extraction 9.1). The tab delimited Feature Extraction output file (.txt file) was loaded into a MySQL database. Data analysis was done in R based on the
limma package (
Smyth, 2005
). As the main tool to analyze the microarray data, MA plots were generated based on the local background correction signals, where
M is the log
2 of the Cy-5/Cy-3 ratio and
A is the average log
2 of the Cy-5 and Cy-3 intensities. A Loess curve was determined from the probes corresponding to the insensitive fragments (insensitive probes) and was used to correct every data point generated from the entire array (
Cleveland et al., 1992
). Application of the Loess correction enabled removal of the intensity dependent error from the two channels and also normalized the readings of insensitive probes, which were expected to have 1:1 ratio (or log
2 ratio equal to 0) between the two digests (
Figure S1 in Supplementary Material). Dye swapping experiments were not performed since it has been shown that such a Loess normalization without background subtraction appears to adequately correct for dye bias particularly in probes with high differential intensities between the dyes (suggesting that methylated calls will have relatively increased reliability) (
Zahurak et al., 2007
). The distribution of signal strength of insensitive fragments was significantly higher than the noise level (measured by technical control spots on Agilent Microarray, which is used as background noise in Feature Extraction software. Refer to
Figure S2 in Supplementary Material for signal distribution).
We then focused our analysis on fragments instead of the individual probes assuming that all probes binding to the same fragment should provide concordant information. To do so, we used the in silico simulation to group all probes binding to the same fragment together and used each group of corrected probe values to calculate the median M value for that fragment. Sensitive fragments were assigned z-scores and p-values based on their M values compared to this insensitive population, assuming a normal distribution for M values. In addition, log2 of Cy-5 channel intensities (HpaII cut – rLog) of each sensitive fragment were also assigned p-values based on the distribution of the insensitive fragments. Overall, a fragment is called positive if it has a high M value and an adequate HpaII digest channel signal (features with signals lower than the bottom 5% of insensitive fragments were removed, p ≤ 0.95). Most positive fragments were in the 0.1–2.0 kb size range as predicted by the typical amplicon size range (Figure S3A in Supplementary Material). This is also apparent from the decreased signal intensity with increased fragment size seen in the insensitive fragments (Figure S3B in Supplementary Material). Various fragments were selected based on their M values and validated with bisulfite sequencing (as described below). An empirical threshold was determined by performing a Receiver Operating Characteristics (ROC) analysis of the validated results. A BED file for the UCSC Genome Browser with detailed results was generated. For replicate experiments, data from M values and rLog values for both experiments were averaged after normalization. p-values were assigned after averaging the two experiments. The data has been uploaded into GEO (Submissions ID GSE19363).
Methylation-Specific (MSP) and Bisulfite Sequencing for Validation
To generate bisulfite modified DNA for manual validation, DNA samples were modified using the EZ DNA Methylation-Gold Kit (D5006, Zymo Research Corp., Orange, CA, USA) following the manufacture protocol. This converts all unmethylated but not methylated cytosines to uracils.
Methylation-specific (MSP)
Primer sequences utilized to analyze the MGMT promoter were 5′-TTTGTGTTTTGATGTT-3′ (upper primer) and 5′ AACTCCACACTCTTCCAA-3′ (lower primer) for detection of unmethylated DNA and 5′-TTTCGACGTTCGTAGGTTTTCGC-3′ (upper primer) and 5′-GCACTCTTCCGAAAACGAAACG-3′ (lower primer) for detection of methylated DNA (
Esteller et al., 1999
). Positive and negative control samples for the MSP reaction were U87MG DNA treated with SssI methyltransferase (NEB, Ipswich, MA, USA) and whole-genome amplification of U87MG DNA using the GenomiPhi V2 Amplification kit (Amersham Biosciences, NJ, USA), respectively.
Bisulfite sequencing validation
Primers were designed with the assistance of the online tool MethPrimer (
Li and Dahiya, 2002
). To validate positive and negative calls, primers were designed to enable coverage of all CCGGs within the BfaI fragment. Sequencing reactions were set up using the designed primers directly on the bisulfite treated sample sent to the UCLA Genotyping and Sequencing Core for sequencing. Any detectable cytosine peak signified a methylated cytosine in genomic DNA.
In Silico Simulation of MSRE Method Applied to Agilent CPG Island Microarray Indicates that BfaI is Superior to MseI for Genomic Fragmentation
As diagrammed in Figure
1
, we used Agilent CpG island arrays in conjunction with the MSRE method described by
Adrien et al. (2006)
. In terms of amplicon generation, the main difference was the use of BfaI (suggested by P. Yan, personal communication) instead of MseI for initial genomic fragmentation. To compare the theoretical CpG island coverage characteristics achievable with BfaI or MseI fragmentation, we performed
in silico determination of all ‘evaluable’ CpG islands using the following criteria: (1) the CpG island has a hybridizing genomic fragment(s) tiled by Agilent probe(s) and (2) the fragment contains one or more HpaII/MspI (C/CGG) recognition site(s). The results of the
in silico simulation are shown in Table
1
.
First, we determined all CpG islands (retrieved from UCSC database, hg18) that could hybridize to probes and found 25,189 of 28,226 total CpG islands were tiled by the Agilent CpG island array at 100 bp intervals.
In silico BfaI digestion of the UCSC hg18 human genome resulted in generation of 49,160 fragments (comprising ∼41 million base pairs, 832.4 bp average size) hybridizing to one or more of the ‘useable’ Agilent probe sequences, whereas MseI digestion resulted in generation of 35,651 fragments (comprising ∼43 million base pairs, 1,200 bp average size). Probes (designated as partial binding) containing internal fragmentation (BfaI or MseI) were omitted due to potential poor or multiple binding. These fragments were then subdivided into sensitive fragments, which have one or more internal HpaII/MspI (CCGG) sites, and insensitive fragments, which have no internal CCGG sites (Figure
1
A). For BfaI, this resulted in 45,094 ‘sensitive’ fragments and 4,066 ‘insensitive’ fragments; for MseI, this resulted in 33,511 ‘sensitive’ fragments and 2,140 ‘insensitive’ fragments. In order to provide a conservative estimate of the effective coverage, only fragments with size in the range of 0.1–2 kb were deemed usable (Materials and Methods). We found by matching ‘sensitive’ fragments restricted to a range of 0.1–2 kb in length with the entire list of CpG islands tracked by UCSC that 22,362 CpG islands for BfaI versus 20,021 CpG islands for MseI met the criteria for ‘evaluability’ by our method. For BfaI, this number represents 89% the original 25,189 CpG islands tiled by the Agilent probe library design. Further demonstrating superiority of BfaI fragmentation over MseI fragmentation, we determined that BfaI enabled increased coverage per CpG island: 1.86 fragments per CpG island for BfaI compared to 1.42 for MseI (restricting to fragments 0.1–2.0 kb in size). We also surveyed the %GC content of fragments generated in both fragmentation schemes and found no significant difference in distribution (
Figure S4 in Supplementary Material). Based on this theoretical superiority of BfaI versus MseI, we conducted MSRE experiments with BfaI fragmentation.
MSRE on Fully-Methylated SSSI Treated Brain Sample Confirms Distinct M-Value Distributions between Predicted Insensitive and Sensitive Fragments
As shown in Table
1
, BfaI fragmentation results in generation of 4,066 fragments designated as ‘insensitive’ based on the lack of internal HpaII/MspI sites. To confirm that these fragments would remain ‘insensitive’ in a fully methylated sample, we analyzed fully-methylated (SssI-treated) and fully-unmethylated (whole genome amplified) genomic normal brain DNA using MSRE as described in Materials and Methods. For the fully-methylated sample, we observed the expected shift in
M-values between the sensitive fragments compared to the insensitive fragments. For the fully-unmethylated DNA sample, sensitive and insensitive fragments showed no significant difference in their
M-values (Figure
2
). Closer inspection of the
M distribution (Figure
2
C) indicates that there is a slight skew towards
M > 0 in the methylated sample versus the unmethylated sample. This indicates that a small subset of the
in silico identified insensitive fragments may not actually be ‘insensitive’, possibly due to the presence of polymorphisms in generating HpaII sites or altering BfaI sites. Given the negligibly small subset of these discrepant fragments, we utilized this ‘insensitive’ group of fragments by identifying corresponding probes (∼6,000) and using their
M-values as an internal control for normalization of our experimental data and quality control (described in Materials and Methods).
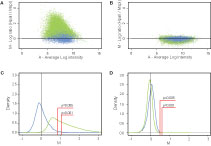
Figure 2. MSRE analysis of SssI treated and whole-genome amplified normal human brain DNA samples. (A) MA plot (log2 ratio vs. average log intensity of both channels) of SssI-treated (completely methylated) normal brain DNA. Each dot represents average data from two experiments for a single fragment calculated based on the data from all available probes binding to that fragment (Materials and Methods). Insensitive fragments are shown in blue and sensitive fragments in green. (B) MA plot of whole-genome amplified (completely unmethylated) DNA. Average data from two experiments is plotted as in (A) showing significantly lower M values compared to SssI treated DNA. (C) M-value density plot of SssI-treated DNA. The blue curve represents density of M values for insensitive fragments, and the green curve represents density of M values for sensitive fragments. Area under both curves has been normalized to 100%. The ‘sensitive’ fragment curve is shifted to the right of the ‘insensitive’ fragment curve. p ≤ 0.005 and p ≤ 0.001 thresholds are indicated by red lines calculated relative to median of insensitive fragments (black vertical line). The area under the green curve to the right of the red line represents methylated fragments by MSRE analysis. (D) M-value density plot of whole-genome amplified DNA. Sensitive and insensitive curves are closely superimposed.
We analyzed the positive control data to investigate the ability to detect methylated fragments based on their size and their number of internal HpaII sites (Figure
3
). This result confirmed our proposed criteria for fragment size limitation between 0.1–2.0 kb (Figure
3
A), with the optimal number of internal HpaII sites between 1–10 (Figure
3
B). We suggest that every fragment result can be annotated with the fragment length and number of internal CCGG to guide the interpretation.
Figure 3. Distribution of fragments by number of fragment length and HpaII sites correlated with positive control results. The histograms were generated from the in silico determined (A) number of fragment length sites and (B) internal HpaII sites. Positive fragments for SssI treated human brain DNA were identified using a p ≤ 0.005 threshold. Experimental results (gray bars) were compared with in silico prediction (100% methylated – white bars). The percentage of observed positives is depicted as red dots for each increment. In (A) the length of fragments was separated by 100 bp increments up to 5000 bp. In both plots, the red dot values drop off with increasing number of HpaII sites or fragment length.
MSRE Analysis of Normal Brain, U87MG, and GBM Patient Samples can Identify Differentially Methylated Fragments
To apply MSRE to actual human genomic DNA samples, we performed two independent MSRE experiments on each of three samples – genomic DNA from normal brain (right frontal), the U87MG cell line and a GBM patient tissue – and averaged the results (
M and
A values). A list of differentially methylated fragments possibly relevant to brain cancer was selected for bisulfite sequencing (Table
2
).
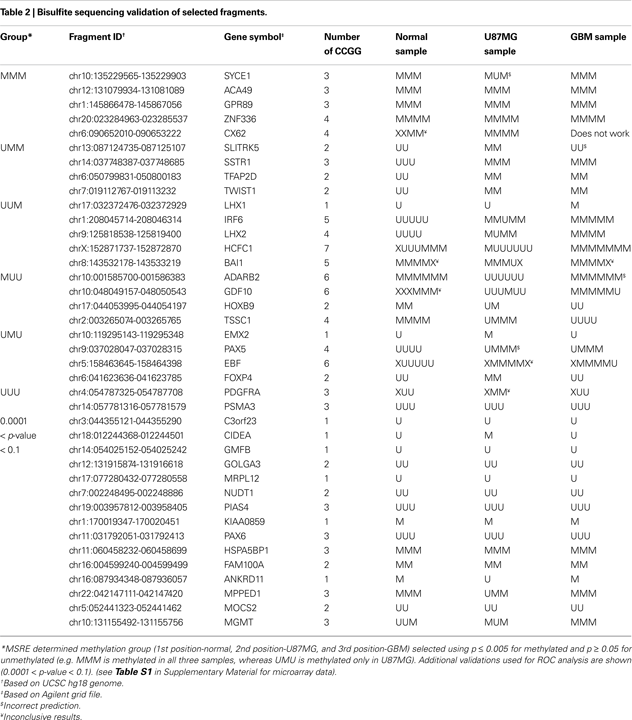
p-values for each fragment were assigned based on the
M-value relative to the distribution of ‘insensitive’ fragment
M-values as described in Materials and Methods. We initially chose fragments most likely to be differentially methylated in the three samples by applying arbitrary thresholds of
p ≤ 0.005 for methylated and
p ≥ 0.05 for unmethylated (
p values were assigned using the insensitive population as described in Materials and Methods). Depending on whether we predicted the fragment to be methylated or unmethylated in normal, U87MG and GBM genomic DNA samples, fragments were grouped into MMM (i.e. methylated in normal, methylated in U87MG and methylated in GBM), UMM, UUM, MUU, UMU, and UUU categories. For each selected fragment, bisulfite sequencing primers were designed, and uncloned DNA samples were subjected to bisulfite sequencing as described in Materials and Methods. The DNA bisulfite sequencing results were tabulated in Table
2
. All CCGGs within a fragment were sequenced with a few exceptions (labeled X). For confirmation of fragment methylation, we required methylation to be found at all fragment CCGG sites; in contrast, for confirmation of lack of fragment methylation, we required that at least one CCGG was unmethylated even if other sites were methylated or unable to be sequenced. Out of the targeted 72 validations, we were able to generate 65 such confirmations. Based on the initial arbitrary
p-value thresholds, only 4 of 65 fragments were miscalled (3 false positive and 1 false negative). As a measure of the agreement between the replicate experiments, from the six total experiments on the three samples, we found that 7 of 72 validations has discordant methylation calls between the technical replicates using the threshold of
p ≤ 0.005 (
Table S1 in Supplementary Material). In addition, unsupervised clustering using dChip (
Li and Wong, 2001
) based on
z-scores of log
2 ratio of all fragments monitored by MSRE showed similarity of technical replicates (Figure S5 in Supplementary Material).
Determination of p-value Threshold by ROC Analysis
To determine the correct threshold for methylation discrimination, we used the validation results to perform a receiver operating characteristic (ROC) analysis. However, due to the selection criteria applied during the first round of validation, we lacked data points corresponding to 0.005 <
p < 0.05. Therefore, we selected another 15 fragments with
p-values encompassing this region and performed uncloned bisulfite sequencing on each sample. Results from these 15 fragments provided 45 more points for our analysis (bottom portion of Table
2
). ROC analysis was performed by calculating True Positive Rate (TPR) vs. False Positive Rate (FPR) and Positive Predictive Value (PPV) and Negative Predictive Value (NPV) at different
p-values(Figure
4
).
Figure 4. ROC analysis of bisulfite sequencing validation results. (A) ROC curve of True Positive Rate vs. False Positive Rate. Red square indicates p = 0.005; red triangle indicates p = 0.001. (B) Plot of Negative (green) and Positive (red) Predictive Value versus Log2 (p-value). Intersection at NPV = PPV = 87% and p = 0.001.
Inspection of these graphs suggests that the optimal threshold lies between 0.001 ≤
p ≤ 0.005. At
p ≤ 0.001, NPV = PPV = 87%, and at
p ≤ 0.005 (Figure
4
A), the FPR plateaus (Figure
4
B). All validation results were indicated in the MA plots (Figure
5
).
Figure 5. Bisulfite sequencing validation results superimposed on MA plots. Bisulfite sequencing results (Table
2
) were mapped on the corresponding MA plots for normal brain, U87MG and GBM DNA samples. Red dots represent validated methylated fragments (all CCGG sites are methylated in tandem), black dots represent fully unmethylated fragments, and blue dots represent mixed methylation of CCGG sites which are expected to be called unmethylated by MSRE. Light blue dots represent insensitive fragments, and light green dotes represent sensitive fragments. Light red highlighted sensitive fragments have
M-values with
p ≤ 0.001 and HpaII channel intensity with
p ≤ 0.95. Also, the position of MGMT Fragment 2 (265 bp) has been indicated with a blue triangle.
Using the threshold of
p ≤ 0.001, we found that 5,521 fragments were methylated in the normal brain sample, representing 4,256 CpG islands, whereas we found 7,047 and 6,782 methylated fragments (5,174 and 5,045 CpG islands) for the U87MG and GBM patient DNA, respectively (Table
3 and Figure
6
A). At this threshold, 3,125 fragments covering 2,417 CpG islands were commonly methylated in all three samples. To compare differentially methylated CpG islands between samples, we used a more stringent positive threshold (
p ≤ 0.001, PPV = 93%) and negative threshold (
p ≥ 0.01, NPV = 98%) to lower false positive/negative rate. With this comparison, we were able identify 70, 480, and 145 uniquely methylated CpG islands in normal, U87MG, and GBM respectively (Figure
6
B).

Figure 6. Comparison of methylation between U87MG, and GBM DNA samples. (A) Estimation of methylated fragments in all samples determined by using p ≤ 0.001 for each sample identifies 3,125 fragments (covering 2,417 CpG Islands). 4,414 fragments (3,321 CpG Islands) were common to both GBM and U87MG, 4,471 fragments (3,452 CpG Islands) common to both GBM and normal, and 3,518 fragments (2,704 CpG Islands) common to both normal and U87MG. (B) Estimation of uniquely methylated fragments using methylation determination based on p ≤ 0.001 and unmethylated determination based on p > 0.01. Fragments within the intermediate region with 0.001 ≤ p ≤ 0.01 are not categorized.
MRSE can Detect MGMT Promoter Methylation
To examine whether
MGMT promoter methylation could be detected by our approach, we compared the MSRE profiling results with methylation-specific PCR (MSP) on
MGMT (
Hegi et al., 2004
) and bisulfite sequencing determination of the three samples. The
MGMT CpG Island is covered by two BfaI fragments (Figure
7
A). One fragment (Fragment 1) is 502 bp and contains 12 CCGG sites. The other fragment (Fragment 2), coinciding with the MSP primers, is 265 bp in size and contains 3 CCGG sites. Using a threshold of
p ≤ 0.001, the 502-bp fragment was not found to be methylated in any of the samples by MSRE whereas methylation of the 265-bp fragment (Fragment 2) was detected for the GBM sample, but not for U87MG or normal brain (Figure
5
). However, by MSP, both the U87MG and GBM sample but not normal brain are methylated (Figure
7
B). When this fragment was subjected to bisulfite sequencing, we found that the GBM fragment was methylated at each of the three CCGG sites (Figure
7
C). In contrast, the U87MG fragment was unmethylated at one of the three CCGG sites within the fragment, whereas the normal brain sample was unmethylated at two of the three CCGG sites.
Figure 7. BfaI fragment coverage of the MGMT CpG island and bisulfite validation. (A) UCSC browser representation of MGMT CpG island showing two MSRE sensitive fragments in gray, predicted CpG island in blue, Agilent probes in green (note partial binding probes *), and methylation-specific PCR primers in red. Fragment 1 is 502 bp and contains 12 CCGG sites. Fragment 2 is 265 bp and contains 3 CCGG sites and binds MSP primers. Fragment 2 contains a portion of exon 1. (B) MSP results of human DNA samples confirm bisulfite sequencing with normal brain showing an unmethylated band, and GBM and U87MG showing a methylated band (as well as a unmethylated band). For controls, methylated SssI-treated DNA (pos.), and unmethylated whole genome amplified DNA (neg.) were analyzed in parallel. (C) Schematic diagram of bisulfite sequencing results on all CpG sites in Fragment 2 of uncloned GBM, U87MG, and normal DNA. Open circles indicate unmethylated CGs, and closed circles indicated methylated CGs. CGs in context of CCGG are enclosed in rectangles. Half-filled circles indicated sites where C peak (methylated) and T peak (unmethylated) from bisulfite sequencing were detected evenly
.
In this study, we have described a genome-wide methylation profiling approach utilizing MSRE combined with Agilent 244K CpG island microarrays that has been optimized via in silico restriction enzyme analysis of the human genome sequence. In silico comparison between MseI and BfaI fragmentation demonstrated that BfaI provides better coverage and fragment characteristics for MSRE when reasonably amplified fragments sizes (<2.0 kb) are considered. To explore the feasibility of the method, we applied MSRE to genomic DNA from normal brain tissue, U87MG GBM cell line, and a GBM patient tissue sample. Uncloned bisulfite sequencing was used to cross validate ∼120 fragments to determine the sensitivity and specificity of the calls from our analysis. The validation results also provided the basis to estimate a threshold separating methylated and unmethylated fragments.
The MSRE approach is derived from DMH, one of the most commonly used techniques for large-scale methylation profiling of cancer genomes (
Huang et al., 1999
;
Yan et al., 2009
). The main advantage MSRE has over DMH is that methylation determination relative to an independent reference sample is not necessary since parallel HpaII and MspI digestions of the same sample are compared simultaneously (
Adrien et al., 2006
;
Hatada et al., 2006
). However, since such methylation sensitive and insensitive isoschizomers are essentially limited to this single pair, a possible advantage of DMH compared to MSRE is the potential to use an enzyme cocktail selected from the entire set of methylation sensitive restriction enzymes. However, in terms of coverage, our
in silico simulation indicates that solely monitoring CCGG restriction sites enables coverage of nearly all CpG islands covered with only a small number of predicted insensitive fragments (8.3%). Furthermore, increasing the number of monitored restriction sites per fragment by using additional methylation sensitive restriction enzymes appears to be disadvantageous based on recognition that the major limitation of both DMH and MSRE is the necessity for
every CCGG restriction site within a fragment to be methylated in tandem in order to detect methylation. Another important benefit of the
in silico analysis was identification of MSRE insensitive fragments based on the lack of the internal CCGG sites. This small group of fragments can be easily tracked in every assay, and provided an internal control group that was used for normalization of our two-color data in order to determine the
M-value threshold indicative of fragment methylation. In addition, this insensitive population provides the signal intensity range of binding fragments. Utilizing such information can reduce dye bias and batch effects between microarray experiments.
From our bisulfite sequencing validation of individual CpGs, we most commonly observed that fragments contained either complete methylation or absence of methylation at all CpGs (including those outside of CCGG sites). This observation provided additional support for the utility of restriction based approaches to detect ‘native’ genomic methylation. However, in a minority of instances, we observed patchy methylation (not full tandem methylation) of the CCGG sites within the fragment such that a predominantly methylated fragment would remain undetected by MSRE. One strategy to lessen the impact of this limitation is to use a restriction enzyme for fragmentation that generates more sensitive fragments with lower numbers of internal HpaII sites. We performed in silico simulations with various enzymes to determine numbers of sensitive fragments containing one to three HpaII sites. We found that AluI (34,474) and HpyCH4IV (29,320) could generate a significantly increased number of sensitive fragments with low numbers of HpaII sites (1–3) compared to BfaI (15,868) or even BfaI/MseI (21,110) (Table S2 in Supplementary Material). However when enzymes such as AluI and HpyCH4IV are utilized, compared to BfaI, two disadvantages are apparent: (1) decreased probe redundancy and (2) more ‘partial binding probes’. Both of these problems can be addressed by custom array design tailored to the particular fragmentation scheme. Ultimately, this issue of patchy methylation demonstrates where ‘massively parallel next-generation bisulfite sequencing’ will be advantageous in assessing not only CGs outside of a CCGG context, but also each CG independently of others within the same fragment.
One issue that poses a limitation for methylation profiling techniques such as MSRE is the inability to determine the relative abundance of methylation at a particular loci especially in the context of tissues containing a mixture of cell types. Our MSRE analysis enables only the categorical detection of methylation as being either present or absent. In theory, if individual fragments are not subject to any amplification bias, then the HpaII intensity or the magnitude of the ratio may be useful to determine relative abundance of methylation. In future studies, quantitative bisulfite validation techniques can be used to determine whether MSRE data can be used to give information on abundance of methylation. At this point, the major recommendation is to screen tissue for example to make sure the sample is predominantly tumor versus normal prior to DNA isolation minimize the mixture of cell types. A strategy of cell selection using laser capture dissection is limited by the DNA requirements of the assay.
In light of the new TCGA methylation data set using Illumina Infinium HumanMethylation27 (Illumina, San Diego, CA, USA) for GBM, we attempted to compare our MSRE result of single GBM sample against TCGA methylation results of GBM samples (118 samples). We mapped all 27,578 loci monitored by HumanMethylation27 to our MSRE fragmentation scheme and found 14,402 loci overlapping with 11,337 ‘evaluable’ BfaI fragments (Figure S6 in Supplementary Material). To determine overlapping methylated GBM loci/fragments in both platforms, we filtered the TCGA data for loci methylated in more than 50% of 118 samples (using beta-value ≥ 0.6 for methylated call). Of the 4,854 methylated loci satisfying these criteria, 718 loci are located within our BfaI fragments and 175 loci are located in methylated fragments in our GBM sample (p ≤ 0.001). This indicates that MSRE and HumanMethylation27 can detect methylation in not completely overlapping set of CpG islands.
In summary, this method provides a cost-effective tool for broad genome-wide CpG island methylation analysis of cancer genomes such as GBM that utilizes as little as 1 μg of sample DNA. In contrast to approaches using sonication for genomic fragmentation or MeDIP/anti-MeC antibodies for methylation discrimination, we could exploit the assay’s use of restriction enzymes for both fragmentation and methylation determination to perform
in silico simulations in the context of the Agilent CpG island probe design. In this way, we were able to predict overall coverage, identify an internal ‘control’ population of BfaI fragments not containing internal HpaII/MspI sites, understand fragment features influencing relative reliability of methylation determination such as fragment size, number of internal HpaII/MspI sites or probe reliability, and provide a rationale for selection of fragmentation enzymes and array design that enable increased ‘effective’ coverage. These data provide critical guidance to individuals seeking to determine the methylation status of individual CpG islands using restriction enzyme-based approaches. The methods can be adapted for other restriction enzyme approaches and can be applied to other array designs to indicate beneficial compositional design alterations. For example, the importance of gene regulation via methylation of ‘CpG island shores’ has recently been identified (
Irizarry et al., 2009
). MSRE coverage can be expanded to include these areas by altering the probe design and fragmentation enzyme.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was made possible by funding from Art of the Brain, Cancer Center of Santa Barbara, Stein-Oppenheimer Foundation, and KO8 CA124479-01 award (A.L.). We also thank Dr. Pearlly Yan, Ohio State University for input on the restriction enzyme selection.
Adrien, L. R., Schlecht, N. F., Kawachi, N., Smith, R. V., Brandwein-Gensler, M., Massimi, A., Chen, S., Prystowsky, M. B., Childs, G., and Belbin, T. J. (2006). Classification of DNA methylation patterns in tumor cell genomes using a CpG island microarray.
Cytogenet. Genome Res. 114, 16–23.
Boyes, J., and Bird, A. (1992). Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein.
EMBO J. 11, 327–333.
Bruna, A., Darken, R. S., Rojo, F., Ocana, A., Penuelas, S., Arias, A., Paris, R., Tortosa, A., Mora, J., Baselga, J., and Seoane, J. (2007). High TGFbeta Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene.
Cancer Cell 11, 147–160.
Cancer Genome Atlas Research Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways.
Nature 455, 1061–1068.
Chung, W., Kwabi-Addo, B., Ittmann, M., Jelinek, J., Shen, L., Yu, Y., and Issa, J. P. (2008). Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling.
PLoS ONE 3, e2079. doi: 10.1371/journal.pone.0002079.
Cleveland, W. S., Grosse, E., and Shyu, M. J. (1992). Local regression models. In Statistical Models in S, J. M. Chambers and T. Hastie, eds (New York, Chapman and Hall), pp. 309–376.
Esteller, M., Hamilton, S. R., Burger, P. C., Baylin, S. B., and Herman, J. G. (1999). Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia.
Cancer Res. 59, 793–797.
Foltz, G., Ryu, G. Y., Yoon, J. G., Nelson, T., Fahey, J., Frakes, A., Lee, H., Field, L., Zander, K., Sibenaller, Z., Ryken, T. C., Vibhakar, R., Hood, L., and Madan, A. (2006). Genome-wide analysis of epigenetic silencing identifies BEX1 and BEX2 as candidate tumor suppressor genes in malignant glioma.
Cancer Res. 66, 6665–6674.
Gargiulo, G., and Minucci, S. (2009). Epigenomic profiling of cancer cells.
Int. J. Biochem. Cell Biol. 41, 127–135.
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A. J., Sawitzki, G., Smith, C., Smyth, G., Tierney, L., Yang, J. Y., and Zhang, J. (2004). Bioconductor: open software development for computational biology and bioinformatics.
Genome Biol. 5, R80.
Hatada, I., Fukasawa, M., Kimura, M., Morita, S., Yamada, K., Yoshikawa, T., Yamanaka, S., Endo, C., Sakurada, A., Sato, M., Kondo, T., Horii, A., Ushijima, T., and Sasaki, H. (2006). Genome-wide profiling of promoter methylation in human.
Oncogene 25, 3059–3064.
Hegi, M. E., Diserens, A. C., Godard, S., Dietrich, P. Y., Regli, L., Ostermann, S., Otten, P., Van Melle, G., de Tribolet, N., and Stupp, R. (2004). Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide.
Clin. Cancer Res. 10, 1871–1874.
Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., de Tribolet, N., Weller, M., Kros, J. M., Hainfellner, J. A., Mason, W., Mariani, L., Bromberg, J. E., Hau, P., Mirimanoff, R. O., Cairncross, J. G., Janzer, R. C., and Stupp, R. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma.
N. Engl. J. Med. 352, 997–1003.
Huang, T. H., Perry, M. R., and Laux, D. E. (1999). Methylation profiling of CpG islands in human breast cancer cells.
Hum. Mol. Genet. 8, 459–470.
Irizarry, R. A., Ladd-Acosta, C., Carvalho, B., Wu, H., Brandenburg, S. A., Jeddeloh, J. A., Wen, B., and Feinberg, A. P. (2008). Comprehensive high throughput arrays for relative methylation (CHARM).
Genome Res. 18, 780–790.
Irizarry, R. A., Ladd-Acosta, C., Wen, B., Wu, Z., Montano, C., Onyango, P., Cui, H., Gabo, K., Rongione, M., Webster, M., Ji, H., Potash, J. B., Sabunciyan, S., and Feinberg, A. P. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores.
Nat. Genet. 41, 178–186.
Jones, P. A., and Baylin, S. B. (2002). The fundamental role of epigenetic events in cancer.
Nat. Rev. Genet. 3, 415–428.
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., and Haussler, D. (2002). The human genome browser at UCSC.
Genome Res. 12, 996–1006.
Khulan, B., Thompson, R. F., Ye, K., Fazzari, M. J., Suzuki, M., Stasiek, E., Figueroa, M. E., Glass, J. L., Chen, Q., Montagna, C., Hatchwell, E., Selzer, R. R., Richmond, T. A., Green, R. D., Melnick, A., and Greally, J. M. (2006). Comparative isoschizomer profiling of cytosine methylation: the HELP assay.
Genome Res. 16, 1046–1055.
Kim, B., Kim, H., Song, B. J., Cha, S. H., Lee, M. O., and Park, S. H. (2006). Oligonucleotide DNA chips are useful adjuncts in epigenetic studies of glioblastomas.
Neuropathology 26, 409–416.
Kuhn, R. M., Karolchik, D., Zweig, A. S., Wang, T., Smith, K. E., Rosenbloom, K. R., Rhead, B., Raney, B. J., Pohl, A., Pheasant, M., Meyer, L., Hsu, F., Hinrichs, A. S., Harte, R. A., Giardine, B., Fujita, P., Diekhans, M., Dreszer, T., Clawson, H., Barber, G. P., Haussler, D., and Kent, W. J. (2009). The UCSC Genome Browser Database: update 2009.
Nucleic Acids Res. 37, D755–D761.
Li, C., and Wong, W. H. (2001). Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection.
Proc. Natl. Acad. Sci. U.S.A. 98, 31–36.
Li, L. C., and Dahiya, R. (2002). MethPrimer: designing primers for methylation PCRs.
Bioinformatics 18, 1427–1431.
Lorente, A., Mueller, W., Urdangarin, E., Lazcoz, P., Lass, U., von Deimling, A., and Castresana, J. S. (2008). RASSF1A, BLU, NORE1A, PTEN and MGMT expression and promoter methylation in gliomas and glioma cell lines and evidence of deregulated expression of de novo DNMTs.
Brain Pathol. 19, 279–292.
Martini, M., Pallini, R., Luongo, G., Cenci, T., Lucantoni, C., and Larocca, L. M. (2008). Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme.
Int. J. Cancer 123, 2955–2960.
Omura, N., Li, C. P., Li, A., Hong, S. M., Walter, K., Jimeno, A., Hidalgo, M., and Goggins, M. (2008). Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma.
Cancer Biol. Ther. 7, 1146–1156.
Ordway, J. M., Bedell, J. A., Citek, R. W., Nunberg, A., Garrido, A., Kendall, R., Stevens, J. R., Cao, D., Doerge, R. W., Korshunova, Y., Holemon, H., McPherson, J. D., Lakey, N., Leon, J., Martienssen, R. A., and Jeddeloh, J. A. (2006). Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets.
Carcinogenesis 27, 2409–2423.
Pfister, S., Schlaeger, C., Mendrzyk, F., Wittmann, A., Benner, A., Kulozik, A., Scheurlen, W., Radlwimmer, B., and Lichter, P. (2007). Array-based profiling of reference-independent methylation status (aPRIMES) identifies frequent promoter methylation and consecutive downregulation of ZIC2 in pediatric medulloblastoma.
Nucleic Acids Res. 35, e51.
Rauch, T. A., Zhong, X., Wu, X., Wang, M., Kernstine, K. H., Wang, Z., Riggs, A. D., and Pfeifer, G. P. (2008). High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer.
Proc. Natl. Acad. Sci. U.S.A. 105, 252–257.
Sadikovic, B., Yoshimoto, M., Al-Romaih, K., Maire, G., Zielenska, M., and Squire, J. A. (2008). In vitro analysis of integrated global high-resolution DNA methylation profiling with genomic imbalance and gene expression in osteosarcoma.
PLoS ONE 3, e2834. doi: 10.1371/journal.pone.0002834.
Smyth, G. K. (2005). Limma: linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor, R. Gentleman, V. Carey, S. Dudoit, R. Irizarry, and W. Huber, eds (New York, Springer), pp. 397–420.
Tepel, M., Roerig, P., Wolter, M., Gutmann, D. H., Perry, A., Reifenberger, G., and Riemenschneider, M. J. (2008). Frequent promoter hypermethylation and transcriptional downregulation of the NDRG2 gene at 14q11.2 in primary glioblastoma.
Int. J. Cancer 123, 2080–2086.
Uhlmann, K., Rohde, K., Zeller, C., Szymas, J., Vogel, S., Marczinek, K., Thiel, G., Nurnberg, P., and Laird, P. W. (2003). Distinct methylation profiles of glioma subtypes.
Int. J. Cancer 106, 52–59.
Waha, A., Guntner, S., Huang, T. H., Yan, P. S., Arslan, B., Pietsch, T., and Wiestler, O. D. (2005). Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas.
Neoplasia 7, 193–199.
Worm, J., and Guldberg, P. (2002). DNA methylation: an epigenetic pathway to cancer and a promising target for anticancer therapy.
J. Oral Pathol. Med. 31, 443–449.
Yan, P. S., Potter, D., Deatherage, D. E., Huang, T. H., and Lin, S. (2009). Differential methylation hybridization: profiling DNA methylation with a high-density CpG island microarray.
Methods Mol. Biol. 507, 89–106.
Yan, P. S., Wei, S. H., and Huang, T. H. (2002). Differential methylation hybridization using CpG island arrays.
Methods Mol. Biol. 200, 87–100.
Yi, J. M., Tsai, H. C., Glockner, S. C., Lin, S., Ohm, J. E., Easwaran, H., James, C. D., Costello, J. F., Riggins, G., Eberhart, C. G., Laterra, J., Vescovi, A. L., Ahuja, N., Herman, J. G., Schuebel, K. E., and Baylin, S. B. (2008). Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors.
Cancer Res. 68, 8094–8103.
Zahurak, M., Parmigiani, G., Yu, W., Scharpf, R. B., Berman, D., Schaeffer, E., Shabbeer, S., and Cope, L. (2007). Pre-processing Agilent microarray data.
BMC Bioinformatics 8, 142.
Zhang, Z., Li, D., Wu, M., Xiang, B., Wang, L., Zhou, M., Chen, P., Li, X., Shen, S., and Li, G. (2008). Promoter hypermethylation-mediated inactivation of LRRC4 in gliomas.
BMC Mol. Biol. 9, 99.
Zilberman, D., and Henikoff, S. (2007). Genome-wide analysis of DNA methylation patterns.
Development 134, 3959–3965.