1
Laboratory of Neural Microcircuitry, Brain and Mind Institute, EPFL, Lausanne, Switzerland
2
Department of Integrative Neurophysiology, Faculty of Earth and Life Sciences, Vrije Universiteit, Amsterdam, The Netherlands
3
Cellular and Molecular Biology Department, Harvard University, Cambridge, MA, USA
A single intra-peritoneal injection of valproic acid (VPA) on embryonic day (ED) 11.5 to pregnant rats has been shown to produce severe autistic-like symptoms in the offspring. Previous studies showed that the microcircuitry is hyperreactive due to hyperconnectivity of glutamatergic synapses and hyperplastic due to over-expression of NMDA receptors. These changes were restricted to the dimensions of a minicolumn (<50 μm). In the present study, we explored whether Long Term Microcircuit Plasticity (LTMP) was altered in this animal model. We performed multi-neuron patch-clamp recordings on clusters of layer 5 pyramidal cells in somatosensory cortex brain slices (PN 12–15), mapped the connectivity and characterized the synaptic properties for connected neurons. Pipettes were then withdrawn and the slice was perfused with 100 μM sodium glutamate in artificial cerebrospinal fluid in the recording chamber for 12 h. When we re-patched the same cluster of neurons, we found enhanced LTMP only at inter-somatic distances beyond minicolumnar dimensions. These data suggest that hyperconnectivity is already near its peak within the dimensions of the minicolumn in the treated animals and that LTMP, which is normally restricted to within a minicolumn, spills over to drive hyperconnectivity across the dimensions of a minicolumn. This study provides further evidence to support the notion that the neocortex is highly plastic in response to new experiences in this animal model of autism.
Autism is a neurodevelopmental disorder that results in a severe handicap in social interactions, verbal communication, and flexible behavior (DSM-IV, 1994
) in addition to severe cognitive alterations in the domains of perception (Bertone et al., 2003
; Boddaert et al., 2004
; Dakin and Frith, 2005
), attention (Landry and Bryson, 2004
; Noterdaeme et al., 2001
; Rinehart et al., 2001
) and memory (Ben Shalom, 2003
; Bennetto et al., 1996
). These disabilities have traditionally been interpreted as resulting from neurological deficits manifested in alterations of brain areas and pathways that lead to hypo-function and mental retardation (Fombonne, 2005
; Lord and Spence, 2006
; Rapin and Dunn, 2003
; Rapin and Katzman, 1998
). Behavioral as well as pharmaceutical therapies usually aim to enhance cognitive capabilities and neural plasticity (Bethea and Sikich, 2007
; Chugani, 2005
; Rubenstein and Merzenich, 2003
; Sikich, 2001
). Most animal models of autism therefore assume a priori a cognitive impairment and neural under-functioning as the basis for autism (Hayashi et al., 2007
; Moy and Nadler, 2008
; Sadamatsu et al., 2006
).
These interpretations however have not taken into account the numerous studies that indicate early hypertrophy of the brain during brain development, hypertrophy of pathways, and excessive perception, attention and memory in autism (Grandin, 1996
; Pring, 2005
; Treffert, 2006
). These observations are usually regarded as interesting exceptions to the norm (Morgan et al., 2007
; Thioux et al., 2006
; Young and Nettelbeck, 1995
).
We recently put forward a diametrically opposite hypothesis for autism, the Intense World Syndrome (Markram et al., 2007a
), based on results obtained from the valproic acid (VPA) rat model of autism (Markram et al., 2008
; Rinaldi et al., 2007
, 2008a
,b
) which is also supported by a large number of studies in the human autism literature (reviewed in Markram et al., 2007a
). The Intense World hypothesis potentially provides a new paradigm for understanding autism by suggesting that excessive neuronal functioning is the core pathology of autism. Studies with the VPA rat model of autism revealed on the cellular level hyperconnected, hyperreactive and hyperplastic microcircuits in different brain areas, which was reflected on the molecular level in massive and selective over-expression of the NMDA receptor subunits NR2A and NR2B in the neocortex (Rinaldi et al., 2007
).
These hyperfunctioning microcircuits were observed in the somatosensory cortex (Rinaldi et al., 2008a
), prefrontal cortex (Rinaldi et al., 2008b
) and in the amygdala (Markram et al., 2008
). The neocortex mediates higher function cognitive processes, including perception, attention and memory (Allman et al., 2001
), while the amygdala is an integral part of the emotional (LeDoux, 2003
; Murray, 2007
) and social brain (Baron-Cohen et al., 2000
; Brothers, 1990
) and has been linked to anxiety (Davis, 1992
; Davis and Whalen, 2001
; Gross and Hen, 2004
) and the storage of fear memories (Phelps, 2006
). Indeed, we found that neocortical hyperplasticity was reflected on the behavioral level by enhanced whisker discrimination learning and memory (Markram et al., 2007b
), a barrel cortex mediated form of learning (Krupa et al., 2001
), while amygdaloid hyperplasticity was reflected in greatly amplified conditioned fear memory (Markram et al., 2008
), an amygdala-mediated form of learning (Dolan, 2002
; Rosen and Donley, 2006
). These evidences corroborated the notion that hyperreactive microcircuits could amplify stimuli from the outside world which could lead to abnormally intense perceptions. Indeed, hypersensitivity to sensory stimulation is an often described autistic symptom (Charman, 2005
; Grandin, 1996
; Volkmar and Pauls, 2003
) and lead to withdrawal and anxiety attacks observed in autism (Amaral and Corbett, 2003
; Gillott and Standen, 2007
). Neocortical hyperplasticity can translate intense perceptions into potentially debilitating associations by forming extraordinary strong memories, causing a rapid lock down of behavioral routines to a minute fraction of possibilities, which are then repeated excessively. Amygdala hyperfunctionality may amplify the sensory intensity and memory formation and render some stimuli aversive as observed in VPA-exposed offspring (Markram et al., 2008
), and reported in humans (Bernier et al., 2005
; Gaigg and Bowler, 2007
; Kanner, 1943
).
The goal of this study was to investigate the phenomenon of hyperplastic microcircuits in VPA-exposed rat offspring in more detail. Previous studies revealed that hyperconnectivity was confined to the minicolumnar range (Rinaldi et al., 2008a
). The minicolumn has been proposed to be the basic and the smallest processing unit of the mature neocortex (Davies et al., 1955
; Mountcastle, 1957
, 1978
, 1997
, 2003
) and has been implicated in autism (Casanova, 2006
). Minicolumns are the basic building blocks of a column, the generic processing unit of the neocortex (Bruno et al., 2003
; Favorov and Diamond, 1990
; Georgopoulos et al., 2006
; Kohn et al., 1997
; Tommerdahl et al., 1993
). They are formed by a chain of about around 120 neurons and extend through layers II–VI. Minicolumns are thought to have a diameter of about 20–60 μm (Hubel and Wiesel, 1974
, 1977
; Kaas et al., 1981
) and are connected by dense, short-ranged, horizontal connections forming a column with a cross-sectional diameter of 100–500 μm.
We examined cortical Long Term Microcircuit Plasticity (LTMP) alterations in VPA-exposed rats offspring using in vitro paired recordings and induced LTMP as reported by Le Be and Markram (2006)
. The technique involves patching clusters of neurons before and after prolonged network stimulation with sodium glutamate. We examined the consequences of VPA treatment on the change in connectivity, strength of connections and probability of release as a function of somata distances. We confirm that prenatally VPA-exposed offspring results in hyperconnectivity within minicolumnar dimensions and further show that the induced LTMP occurs beyond the minicolumnar range.
Animals, Preparation and Anesthesia
Young (12–15 days old) male Wistar rats, pre-natally exposed to VPA through intra-peritoneal injection in pregnant rats at precisely ED12.5, were anesthetized in an induction chamber with air and isofluorane, the cardiac rhythm being monitored. Depth of anesthesia was accessed by monitoring pinch withdrawal and vibrissae movements. Once anesthetized, animals were decapitated, the brain immediately removed and sliced in artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 25 mM D-glucose, 25 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, and 1 mM MgCl2, at ice cold temperature. 300 μm sagittal somatosensory slices were cut from the brain and then incubated for 30 min at 34°C and for 30 min at room temperature. All experimental procedures were carried out according to the Swiss Federation rules for animal experimentation.
Electrophysiology Recordings
Clusters of four to seven layer 5 pyramidal cells (L5PCs) were stimulated and recorded using multiple whole-cell patch clamp. Direct synaptic connections were examined by eliciting short trains of precisely timed action potentials (APs) at 30 Hz followed by a recovery test response 500 ms later (Figure 1
A). After establishing the connectivity between cells, the pipettes were withdrawn and the slice was left in the recording chamber in a quiescent state for 12 h, while ACSF with 100 μM sodium glutamate (Sigma-Aldrich) was perfused in order to evoke appearances of new connections (Figure 1
B). After 12 h the recording chamber was washed with regular ACSF perfusion, the same cells were re-patched and the same stimulation protocol was executed to monitor the connectivity in the new state (‘after’; Figure 1
C) (Le Be and Markram, 2006
).
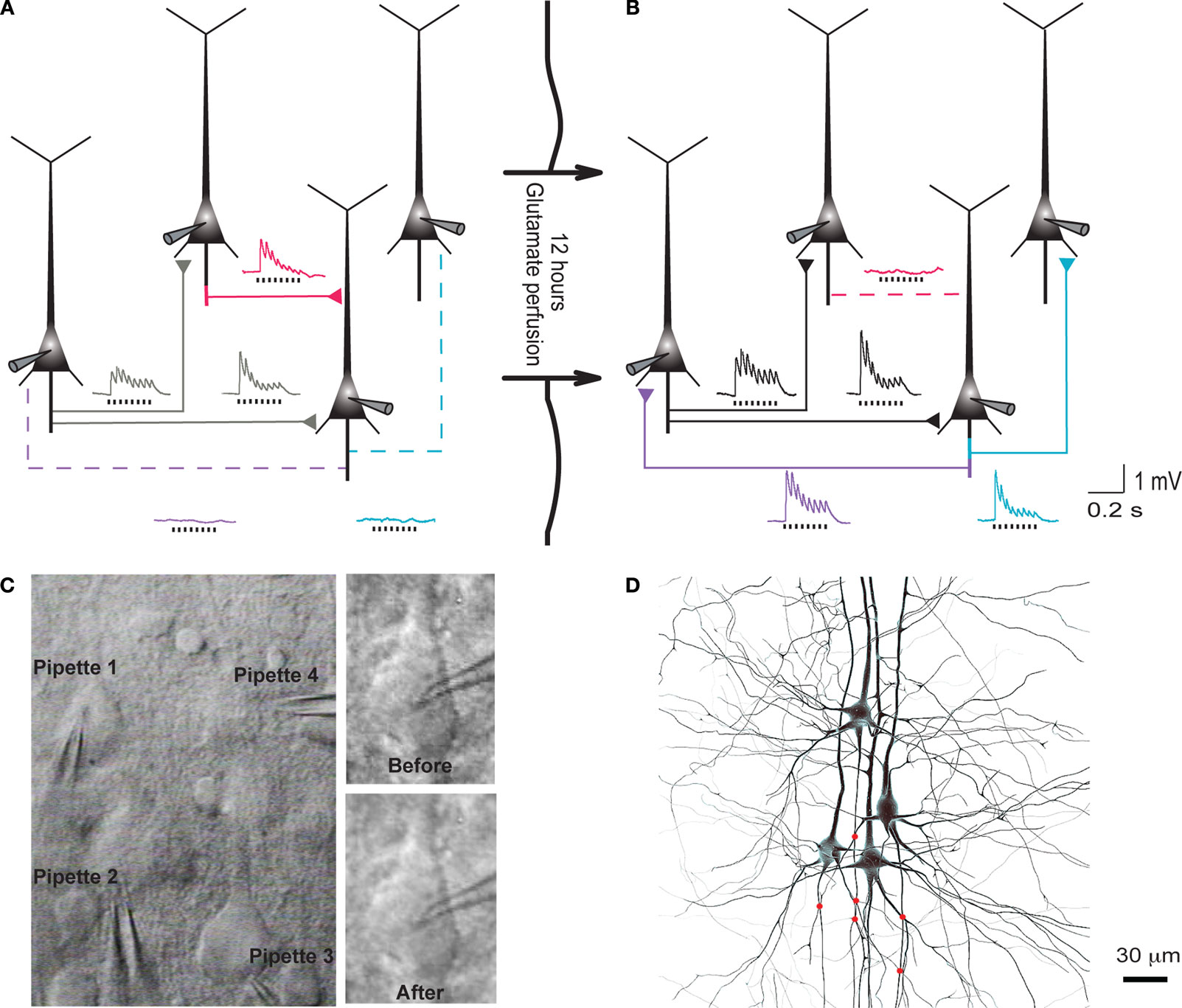
Figure 1. Evoked emergence of connections following 12 h network stimulation. (A) Recording of layer 5 pyramidal cells (PC) using multiple whole-cell patch-clamp. Direct synaptic connections were examined by eliciting short trains of precisely timed action potentials at 30 Hz. (B) After 12 h of network stimulation by sodium glutamate. The red trace indicates a disappearance, blue and purple traces indicate emergences, grey and black traces indicate stable connections (Le Be and Markram, 2006
). (C) Cluster of four cells viewed by infra-red differential interference contrast microscope. (Upper right) Neurons during first patch. (Lower right) Neurons after 12 h of glutamate stimulation. (D) Mesh rendering on camera lucida of anatomical reconstructions of two neurons where a connection appeared after evoking with sodium glutamate. (Red Dots): Putative synaptic contact sites.
Recordings were made with long-taper patch pipettes with DC resistances of 5–7 MΩ pulled from borosilicate glass tubing on a Sutter puller in a three-stage pull. Pipettes were filled with a solution of potassium gluconate 100 mM, Hepes 10 mM, phosphocreatine 10 mM, MgATP 4 mM, GTP 0.3 mM, KCl 10 mM and 0.5 biocytin, which pH was adjusted at 7.3.
To establish the whole-cell recording configuration we applied a conventional current-clamp technique. Pipettes were advanced into the slice in small steps while pulsing ±5 mV steps with an interval of 8 ms. Positive pressure (25–40 mbar) was applied to the pipette while it was being lowered. After touching the target cell membrane, positive pressure was removed when an increase in pipette resistance was detected as a sudden decrease in the current pulse amplitude. This usually resulted in a seal resistance of 1 GΩ or higher. Occasionally, slight negative pressure was applied to establish a giga-ohm seal. The whole-cell configuration was established by applying a slow ramp of negative pressure. Voltage recordings were amplified using an Axoclamp-200B amplifier (Axon instruments, Foster City, CA, USA), filtered at 3 kHz and digitized at 5–20 kHz (ITC-16; Instrutech, New York, NY, USA).
Histological Procedures
Following physiological recordings, the 300 μm slices were fixed for at least 24 h at 4°C in 0.1 M phosphate buffer (100 mM, pH 7.4) containing 2% paraformaldehyde. Slices were then rinsed and processed with the avidin-biotin-peroxidase method (Horikawa and Armstrong, 1988
) to reveal cell morphology, and mounted on slides using an aqueous mounting medium. The biocytin labeling could then be used to double-check that the same neurons were patched for the two, 12-h separated recordings (Figure 1
D).
Analysis of Synaptic Dynamics
Peak amplitudes of excitatory postsynaptic potentials (EPSPs) evoked by a train of 7 presynaptic action potentials were measured. Waveforms were averaged over several repetitions and labeled as ‘before’ or ‘after’ when referred to recordings preceding or following chemical network stimulation by sodium glutamate perfusion. For the population analysis, only recordings with a high signal-to-noise ratio were considered (i.e. peak amplitude of the first EPSP above 1.5 mV) and each waveform was normalized to its maximal amplitude within the response train. The plot of the peak EPSP amplitudes after the chemical stimulation, as a function of the corresponding peak in the train before stimulation, indicates which one of a temporal redistribution or a uniform rescaling of synaptic efficacy occurred.
The connection dynamics was decomposed with the TM phenomenological model of dynamic synapses (Tsodyks and Markram, 1997
). The model assumes that a synapse is characterized by a finite amount of resources. Each presynaptic spike (arriving at time tsp) activates a fraction (USE, utilization of synaptic efficacy) of resources, which then quickly inactivate with a time constant of a few milliseconds (τin) and recovers with a time constant of about 1 s (τrec). The corresponding kinetic equations are:
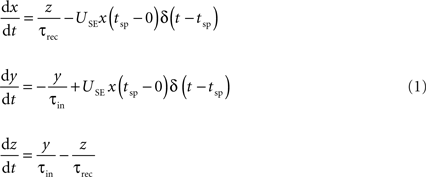
where x, y and z are the fraction of resources in the recovered, active and inactive states, respectively. The postsynaptic current is proportional to the fraction of resources in the active state, Is(t) = A y(t). The two main parameters defining the model are A, the absolute synaptic efficacy and USE, the utilization of synaptic efficacy (Tsodyks and Markram, 1997
; Tsodyks et al., 1998
, 2000
).
The absolute synaptic efficacy of a connection is the maximal response that can be produced by a connection if the probability of transmitter release would be maximal (i.e. 1) at all possible release sites. The utilization of synaptic efficacy describes the average fraction of the absolute synaptic efficacy that each action potential (AP) uses and is analogous to the probability of transmitter release (Pr) if the mechanism of frequency dependence is located purely presynaptically (Markram et al., 1998b
).
Connectivity Indices and Statistical Analysis
The control group consisted of 13 animals (12–15 postnatal day old, PN), while the VPA group consisted of 16 animals. Only one cluster was obtained from each animal for only one slice. The significance of differences among connection rates was determined using two-sided chi-square tests. Because there was a single neuronal cluster per animal experiment, each with a variable number of neurons per cluster, the Mantel–Haenzel test was used to control for between-animal variance introduced by this stratification.
Two indices were used to quantify appearances of connections and to compare values along inter-somatic distances:
1. The connection probability (CP), a measure of connection likelihood over all possible connections (assuming that they are independent events), defined as:

Where NC stands for the total number of connections found and NPC the number of possible connections, which depends on the number of cells patched. By performing paired-recordings on a cluster of C cells, the number of possible connections between these cells is NPC = C * (C − 1), i.e. the number of all possible combinations of monosynaptic connections between C cells.
2. The emergence index (Ei), that gives the likelihood of appearances of new connections is defined as:

Where NA is the total number of connections that appeared after evoking by sodium glutamate perfusion and NC number of connections found in the first recording (‘before’).
The position of each cell patched was recorded in (x,y,z) coordinates and the above indices are then calculated as function of the Euclidian distance.
All indices data is presented as mean ± standard error of the mean (SEM), which was calculated assuming binomial data distribution by,

For the comparison of CPs before and after sodium glutamate perfusion and Ei along the defined distance ranges, χ2 test was used. To compare the absolute synaptic efficacy and the utilization of synaptic efficacy before and after sodium glutamate perfusion, the Kolmogorov–Smirnov (K–S) test was performed between the respective samples. Attributed levels of significance were (*p ≤ 0.05) and (**p ≤ 0.01). Throughout the text, when referring to CP ‘n’ stands for the number of connections probed, while for Ei ‘n’ stands for remaining connections, i.e. number of possible pairs after establishing connectivity with the first recording.
Connection Probability
We first examined the connection probabilities, as a function of inter-somatic distance, before and after 12 h of sodium glutamate perfusion (Figures 2
A1,A2). As previously reported (Rinaldi et al., 2008a
), we found an increased probability of direct connections between L5PCs (hyperconnectivity) for inter-somatic distances smaller than 50 μm before the glutamate perfusion in VPA-exposed offspring slices (CP = 10 ± 2%, n = 146 for control group, CP = 19 ± 3%, n = 134 for VPA group, p = 0.03). After 12 h of glutamate perfusion, no significant difference between the two groups was observed at distances smaller than 50 μm (CP = 20 ± 3% for control group, CP = 28 ± 4% for VPA group, p = 0.17). This suggests that, for the prenatally VPA-exposed group the number of possible functional connections may already be ‘saturated’,, in the range of less than 50 μm preventing the appearances of new connections. In other words, the baseline connectivity levels seem to be saturated in VPA-exposed offspring in the minicolumnar range and cannot be further enhanced by network stimulation through sodium glutamate perfusion.
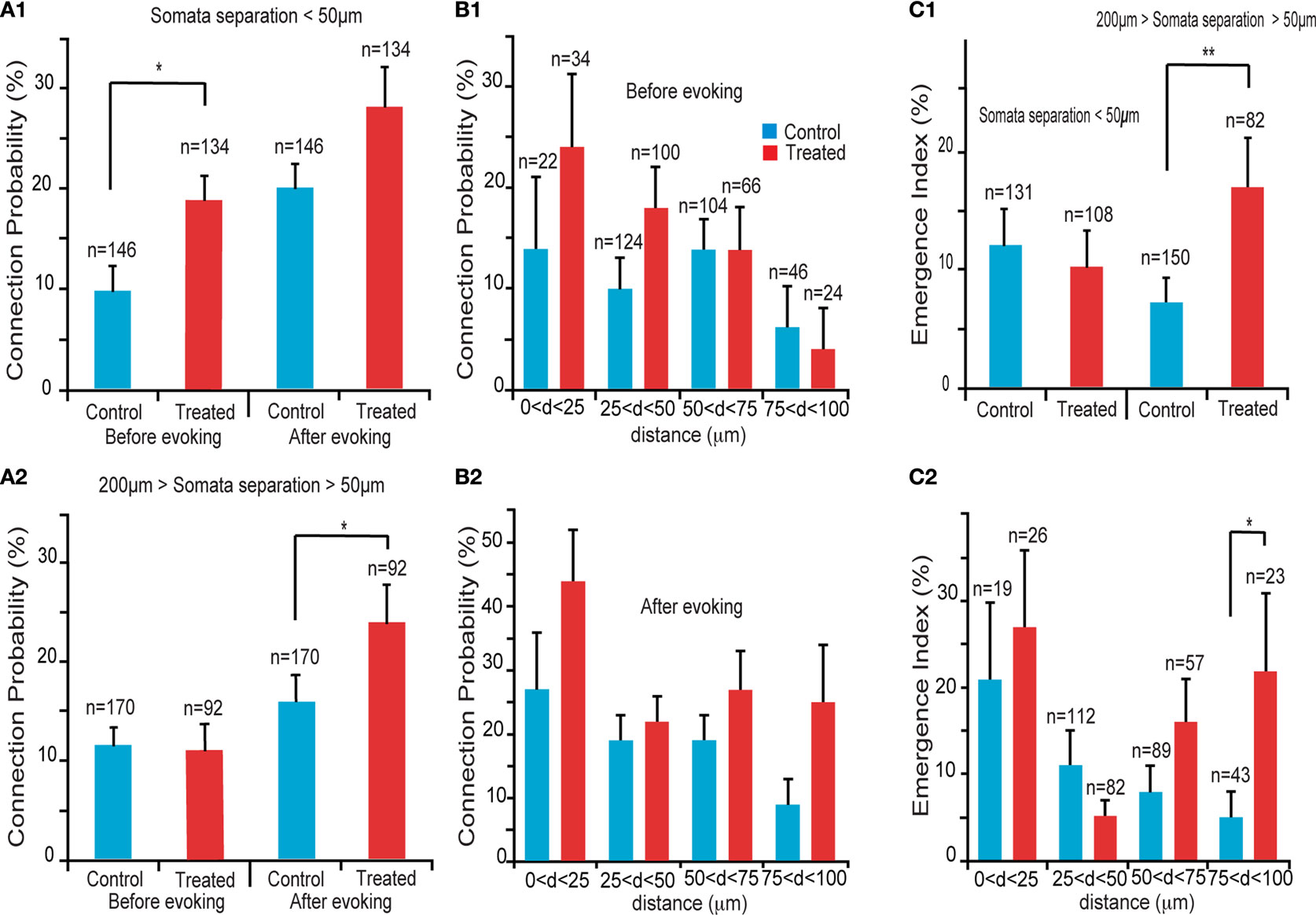
Figure 2. Connectivity indices. (A1) Hyperconnectivity for inter-somatic distances smaller than 50 μm. (A2) Hyperplasticity for inter-somatic distances between 50 and 200 μm. (B1) Connection probability in (25 μm) discrete zones, before sodium glutamate perfusion. (B2) Connection probability in (25 μm) discrete zones, after sodium glutamate perfusion. (C1) Emergence index, showing higher tendency for appearances of new connections at somata separation between 50 and 200 μm. (C2) Emergence index calculated along (25 μm) discrete inter-soma distances, showing significant difference at somatic separation between 75 and 100 μm.
For inter-somatic distances between 50 and 200 μm (Figure 2
A2) we found no CP differences between VPA and control groups before the perfusion of sodium glutamate, (CP = 12 ± 2%, n = 170 for control group, CP = 11 ± 3%, n = 92 for VPA group, p = 0.83) in agreement with a previous report by Rinaldi et al. (2008a)
. However, after 12 h of sodium glutamate perfusion, slices of pre-natally VPA exposed rats exhibited significantly higher CP levels than controls (CP = 16 ± 3% for control group, CP = 26 ± 4% for VPA group, p = 0.05). This indicated that VPA-exposed offspring form many more new connections between minicolumns than within minicolumns after being exposed to continuous network stimulation as compared to control microcircuits. Therefore, in the region of ‘no hyperconnectivity’, i.e. outside the minicolumnar but confined to the columnar cross-sectional radius, we observed enhanced LTMP in the neocortical microcircuit of pre-natally VPA-exposed rats.
By further refining the inter-somatic distance categorization in steps of 25 μm, it appears that the strongest tendency for hyperconnectivity is in the 25–50 μm inter-somatic distance (10 ± 3%, n = 124 vs. 18 ± 4%, n = 100 for control and VPA respectively, p = 0.07; Figure 2
B1). In contrast, after 12 h of glutamate stimulation the tendency for hyperconnectivity was only observed for inter-somatic distances of 70–100 μm (9 ± 4%, n = 46 vs. 25 ± 9%, n = 24, p = 0.06; Figure 2
B2). This shows hyperLTMP in the cross-minicolumnar and intra-columnar ranges for pre-natally VPA exposed rats.
Changes in the Emergence Index
Following the CP analysis, we calculated the emergence index (Ei), which measures the probability of appearance of new connections after 12 h of network stimulation by sodium glutamate perfusion. No significant difference between emergence indices for inter-somatic distances smaller than 50 μm were found. (Ei = 12 ± 3%, n = 131 for control group, Ei = 10 ± 3%, n = 108 for VPA group – p = 0.62; Figure 2
C1). However, significant differences between emergence indices for inter-somatic distances between 50 and 200 μm were observed (Ei = 7 ± 2%, n = 150 for control group Ei = 17 ± 4%, n = 82 for VPA group – p = 0.01; Figure 2
C1).
Comparing the emergence indices across bins of 25 μm, (Figure 2
C2), we verified the trend that the higher emergence index for pre-natally VPA exposed animals tends to be in the non-hyperconnected, cross minicolumnar region (0 < d < 25 μm, Ei = 21 ± 9%, n = 19 for control group and Ei = 27 ± 9%, n = 26 for VPA group – p = 0.65; 25 < d < 50 μm, Ei = 11 ± 3%, n = 112 for control group and Ei = 5 ± 2%, n = 82 for VPA group – p = 0.14; 50 < d < 75 μm, Ei = 8 ± 3%, n = 89 for control group and Ei = 16 ± 5%, n = 57 for VPA group – p = 0.12; 75 < d < 100 μm, Ei = 5 ± 3%, n = 43 for control group, significantly smaller than Ei = 22 ± 9%, n = 23 for VPA group – p = 0.03). Thus, there is a significantly higher tendency for rewiring of synaptic connections across minicolumns and within the dimensions of a neocortical column.
Biophysical Property Changes
The 12 h chemical stimulation results in a uniform rescaling of each EPSP evoked by a train of presynaptic action potentials (Figure 3
A). This indicates that no change occurs in recovery time constants associated with short-term depression nor with transmitter release probability. This is further supported by the linear relationship between normalized EPSPs before and after glutamate stimulation for both control (n = 9) and VPA treated (n = 14) rats (Figure 3
B; R = 99) All normalized data points scattered across a unitary line suggest a variation in EPSP amplitude, but not in other EPSP parameters.
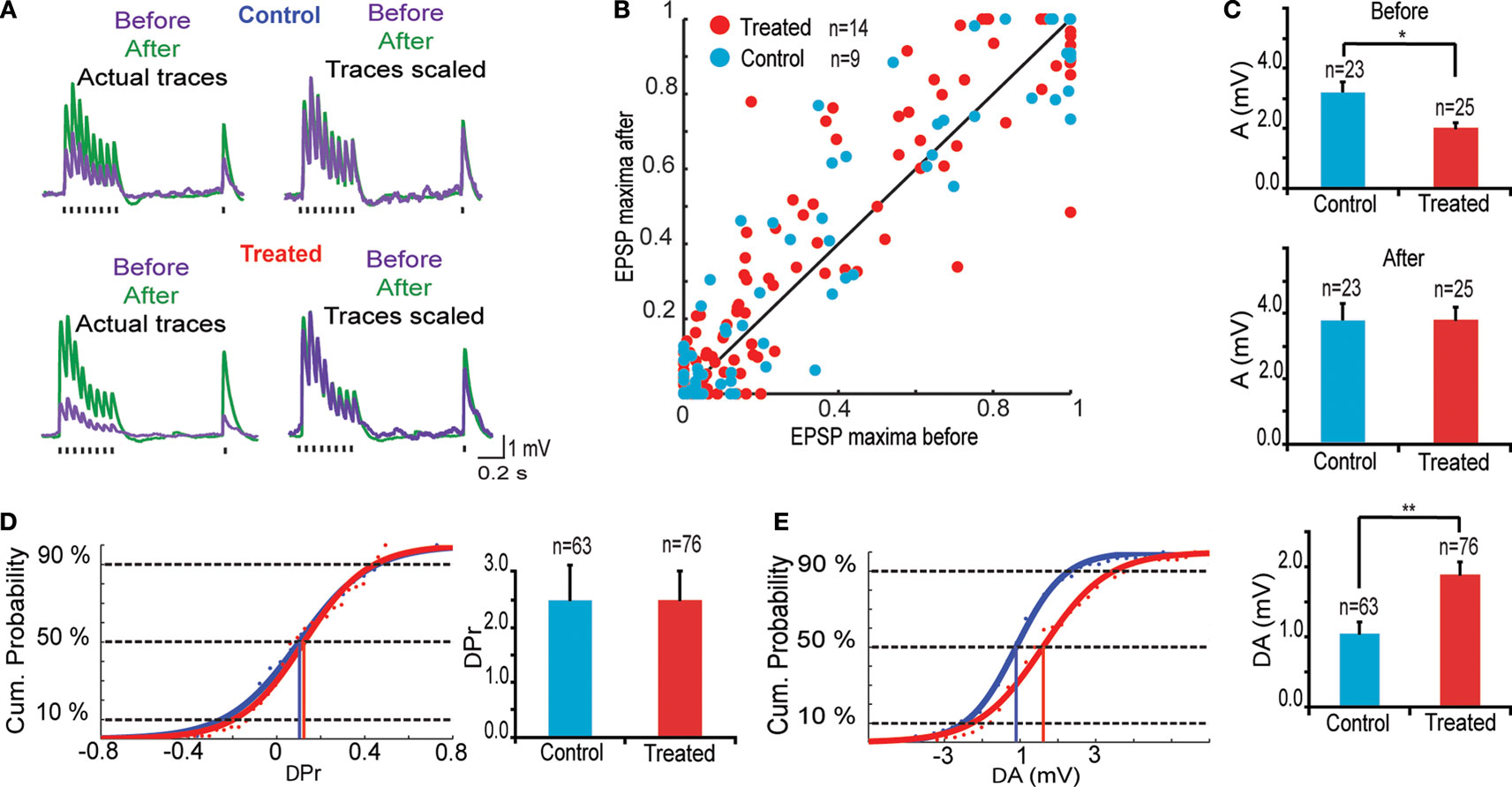
Figure 3. Changes in synaptic efficacy. (A) (Upper) Trains of EPSPs evoked by somatic current injection before and after 12 h of sodium glutamate perfusion of control population. (Lower) Trains of EPSPs evoked by somatic current injection before and after 12 h of glutamate perfusion of treated population. (B) EPSP maxima scattered around unitary line showing trend for voltage rescaling after glutamate perfusion. (C) Absolute values of synaptic efficacy (A), before and after network stimulation by sodium glutamate. (D) Cumulative probability histogram of ΔPr = Prafter − Prbefore. Highlighted points are those at 50% and correspond to the mean values for ΔPr. No significant difference in Pr between the two groups. (E) Cumulative probability histogram of ΔA = Aafter − Abefore. Highlighted points are those at 50% and correspond to the mean values for ΔA. Global increase in synaptic efficacy significantly higher for treated population.
The parameters A(mV) and Pr were averaged over stable and evoked connections using the TM model (Markram et al., 1998a
,b
; Tsodyks and Markram, 1997
). As previously reported (Rinaldi et al., 2007
), absolute synaptic efficacy (A) was significantly smaller in pre-natally VPA-exposed rats than controls at baseline levels (Abefore = 3.2 ± 0.4 mV, n = 23 for control group, Abefore = 2.4 ± 0.3 mV, n = 25 for VPA group, p = 0.05; Figure 3
C). After network stimulation by sodium glutamate a global increase in A was observed for both groups but there was no difference anymore between control and VPA treated rats (Aafter = 3.8 ± 0.5 mV, n = 23 for control group, Aafter = 3.8 ± 0.4 mV, n = 25 for VPA group, p = 0.90; Figure 3
C).
The difference in absolute synaptic efficacy before and after glutamate stimulation (ΔA = Aafter − Abefore) shows that synapses of pre-natally VPA exposed rats experienced a significantly higher change in strength than those of saline exposed rats (ΔA = 1.0 ± 0.2 mV, n = 63 for control group, ΔA = 1.8 ± 0.2 mV, n = 76 for VPA group, p = 0.002; Figure 3
E). Showing that alterations are extended into the domain of kinetic synaptic properties.
As suggested by the linear relationship of scaled EPSPs before and after stimulation, there is no difference in the probability of release (Pr) after glutamate stimulation both for control and VPA treated rats (data not shown). These results show that the processes governing neurotransmitter release probability (Pr) are the same for both groups while the synaptic strength or absolute synaptic efficacy (A) is modulated to a greater extent in the VPA group than in controls (Figure 3
D).
This study reveals that VPA-treated offspring display enhanced LTMP across neocortical minicolumnar and within columnar dimensions. The study also reveals hyperplasticty of synaptic strength (LTP) due to prolonged excitation of the microcircuitry, which is consistent with a previous report of hyperplasticity caused by acute electrical stimulation. This study also provides confirmation of another previous study demonstrating hyperconnectivity in the VPA-treated rat neocortex.
Hyperconnectivity
The VPA animal model of autism displays an intriguing neuropathology of glutamateric synapses. Pyramidal neurons are hyperconnected to their neighbors, but this hyperconnectivity does not seem to be the consequence of generalized hypertrophy since the number of putative synaptic contacts deployed to form each connection is reduce while more neurons are contacted (Rinaldi et al., 2008a
). The hyperconnectivity was also found previously to be restricted to the dimensions of a minicolumn (<50 μm). The current study verifies hyperconnectivity that has been proposed to cause the hyperreactivity in the neocortex of this animal model, which in turn could underlie many of the hypersensory abnormalities in autism.
HyperLTP
The previous study of Rinaldi et al. (2007)
showed that long term potentiation was also enhanced in the VPA offspring. The expression of LTP was in the form of a significant increase of the absolute strength of synaptic connections and could be due to potentiation or addition of AMPA receptors or to the formation of additional synaptic contacts at existing synapses. These glutamatergic synapses also express more NMDA receptors that mediate more NMDA current during synaptic activation (Rinaldi et al., 2007
), which is the most likely cause of the observed hyperplasticity. The current study confirms the finding of hyperLTP and extends this finding to include a more chronic induction – long-term perfusion of glutamate. Both acute and chronic stimulation can therefore lead to exaggerated synaptic plasticity in the neocortex of this animal model of autism.
HyperLTMP
Le Be and Markram (2006)
demonstrated a novel form of microcircuit rewiring where new connections can be formed and existing connections can be eliminated within hours of continuous stimulation with glutamate. The current study confirms the finding of LTMP in control animals and further found that LTMP is enhanced in the VPA offspring for connections separated by more than the minicolumnar dimensions. The lack of an enhancement of LTMP within minicolumnar dimensions seems to be because the pyramidal neurons are already maximally hyperconnected within these dimensions. HyperLTMP could cause tighter coupling between minicolumns further amplifying already hyperexcitable columns. Le Be and Markram (2006)
also showed that the key receptors involved in LTMP where NMDA and mGluR5 receptors. In a recent large-scale glutamatergic screening study we found a significant enhancement also of the expression of mGluR5 receptors in the somatosensory cortex of VPA-treated offspring (in preparation). Thus, the enhanced LTMP in the animal model of autism could be due to the hyperNMDA and hypermGluR5 receptor expression.
Loss of Connections
Le Be and Markram (2006)
showed that small but matured (in terms of probability of release) connections are vulnerable to elimination during LTMP. In the current study, we could not obtain sufficient data to determine whether there is also a greater elimination of synaptic connections. Such a study would be important for future studies because the rate of connection formation and elimination could be at a different equilibrium in autism to account for the higher basal connection probability.
Minicolumnar Pathology
A previous study on human brain tissue suggested a minicolumnar pathology where the minicolumnar width is shown to be 27.2 mm in controls and 25.7 mm in autistic patients (Casanova, 2006
; Casanova, et al., 2002
). Neuron densities in autism were also found to exceed the comparison group by 23%. In a previous study we did not find changes in neuronal densities (Rinaldi et al., 2008a
), but changes in the packing of neurons within the dimensions of a minicolumn cannot be ruled out. In rat brain tissue, minicolumnar (and columnar) dimensions are more difficult to assess than in human tissue, and a difference of 5.5% (as described in the above mentioned results from Casanova, 2006
) would therefore be difficult to detect. With the functional analysis conducted in the current study and previous studies we found both connectivity and plasticity alterations within very narrow dimensions comparable to mincolumns (much less that 3–500 μm), which does however further support the hypothesis of a minicolumnar pathology in autism.
Implications for the Inhibitory System
A detailed study of the inhibitory system of the VPA model of autism has not been carried out yet. However, in a previous study (Rinaldi et al., 2008a
,b
), we found that hyperreactivity was also observed in terms of the inhibitory responses and we found a significant increase in the probability of disynaptic connections. Although this measure does not reflect the involvement of the full spectrum of interneuron subtypes, it does show that the hyperconnectivity extends beyond the direct connections between pyramidal neurons. This finding could therefore be sufficiently explained by the hyperconnectivity of glutamatergic extending also to the inhibitory neurons and thereby allowing the inhibitory system to simply follow and apply matching inhibition to the more excitable excitatory system. Indeed, in this previous study, we also did not find an imbalance in the excitation and inhibition further suggesting that the inhibitory system of the neocortex at this age is hardly affected. However, the hyperreactive excitatory system may still bring the microcircuit very close to seizure threshold, which may then occur if additional (perhaps even minor) alterations in inhibition system take place to upset the balance between excitation and inhibition. Seizures are common in autism, but do not occur in all cases (around 30% of cases) and such cases may therefore reflect a complication of the neuropathological changes in autism. These alterations restricted to the excitatory system seem to differ from alterations in the amygdala where a loss of inhibition seems to account for the hyperreactivity (Markram et al., 2008
).
The previous findings that excitatory connectivity is enhanced, giving rise to hyperreactivity to stimulation, and that NMDA receptors are over-expressed, giving rise to hyperplasticity in the microcircuit has demanded a revision of the possible neuropathology of autism. The Intense World Syndrome provides a new perspective on the potential circuit pathology in autism. In this hypothesis, modular neocortical arrangement (such as columns and minicolumns) are overly sensitive to stimulation, but once triggered, they become autonomous with run-away processing that is difficult to command from higher brain regions and thereby lead to a deficit in executive and other higher brain functions. The core cognitive pathology that has been hypothesized to arise is hyperperception, hyperattention, hypermemory and hyperemotionality. The current study suggests that hypermemory is not only due to hyperplasticity of existing synaptic connections, but also due to hyperplasticity of the wiring of neurons in the microcircuit, at least for early experiences during development.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Deborah La Mendola, Michele Giugliano, Michele Pignatelli, Rajnish Ranjan, Rodrigo Perin, Sébastien Lasserre, Barry Sefton and Srikanth Ramaswamy. This work was supported by the EUSynapse Consortium grant.