Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX, USA
Mitochondrial oxidative stress and dysfunction has been implicated as a possible mechanism for the onset and progression of Parkinson-like neurodegeneration. However, long-term mitochondrial defects in chronic animal neurodegenerative models have not been demonstrated. In this study, we investigated the function of striatal mitochondria 6 weeks after the induction of a chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease (MPD). Although severe depression of mitochondrial respiration was observed immediately after acute administrations of MPTP, we failed to detect a significant mitochondrial inhibition in presence of striatal dopamine (DA) deficit 6 weeks after the chronic MPD induction in young adult mice. In contrast, when aged mice were chronically treated with MPTP and at 6 weeks post-treatment, these animals suffered an inhibition of the basal (state 4) and adenosine 5′-diphosphate-stimulated (state 3) respiration and a fall in adenosine triphosphate level in the striatal mitochondria. The aged chronic MPD also brought about a sustained diminution of striatal anti-oxidant enzyme levels including that of superoxide dismutases and cytochrome c. The mitochondrial deficits in the striatum of aged chronic MPD 6 weeks after treatment were further correlated with significant losses of striatal DA, tyrosine hydroxylase, DA uptake transporter, and with impaired movement when tested on a challenging beam. Our findings suggest that MPTP may trigger the neurodegenerative process by obstructing the mitochondrial function; however, striatal mitochondria in young animals may potentially rejuvenate, whereas mitochondrial dysfunction is sustained in the aged chronic MPD. Therefore, the aged chronic MPD may serve as a suitable investigative model for further elucidating the integral relationship between mitochondrial dysfunction and neurodegenerative disorder, and for assessing the therapeutic efficacy of mitochondrial protective agents as potential neuroprotective drugs.
Mitochondria are dynamic organelles that are present in all mammalian cells. They are functionally well known for generating adenosine triphosphate (ATP) as the source of energy, for sequestering excess cytoplasmic Ca++, for producing and disposing of reactive superoxide free radicals, and for supporting cell survival and preventing cell death (Chan, 2006
). Briefly, the generation of ATP by mitochondria involves oxidative phosphorylation of a number of protein complexes within the electron transport chain system located in the inner mitochondrial membrane. During the process of mitochondrial respiration and ATP production, small amounts of molecular oxygen are naturally reduced to form reactive oxygen species (ROS) as byproducts, like superoxide free radicals. Under healthy conditions, the basal level of ROS produced within cells is quite low and not harmful. Furthermore, a number of mitochondrial anti-oxidant enzymes such as the superoxide dismutase (SOD) and cytochrome c can scavenge any abnormally elevated ROS from internal and external sources and prevent further damages to mitochondrial and cellular DNA, proteins, and lipids.
Conceivably, neurons are extremely active cells that require constant supply of energy in order for them to carry out highly specialized functions such as regulating the activities of neuronal transmission, receptors, ion channels, transporters and synapses. Thus, mitochondria are vital for maintaining the homeostasis and integrity of neuronal functions; and mitochondrial damage and dysfunction may lead to neurological disorders (Schon and Manfredi, 2003
; Chan, 2006
).
With exception of a few well characterized familial cases in connection with specific gene mutations, Parkinson’s disease (PD) is commonly a sporadic, adult-onset, neurological disorder. Pathophysiologically, PD is considered as a movement disorder due to severe loss of dopamine (DA)-producing cells in the substantia nigra pars compacta, although the true origin of PD neurodegeneration is still unclear. Nevertheless, recent advances in PD research have reached a consensus suggesting that PD is not likely caused by a single factor but rather a combination of genetic and environmental factors leading to a complex clinical phenotype with underlying loss of nigrostriatal DA neurons (Bronstein et al., 2009
). Therefore, potential success for preventing and treating PD could not be easily achieved by a monotherapeutic approach.
Many plausible genetic and environmental factors have been investigated and linked to both the initiation and progression of PD neurodegeneration. Amongst them, neuronal oxidative stress and mitochondrial dysfunction have emerged as part reasons for instigating the demise of nigrostriatal DA neurons through a series of deleterious cellular and molecular events. It is generally believed that due to intrinsic genetic and extrinsic environmental factors that may cause ROS accumulation and/or fail to eradicate ROS, neuronal mitochondria will undergo oxidative damage involving depletion of anti-oxidant proteins and activation of a series of caspase-dependent or caspase-independent molecular signaling events leading to apoptotic cell death (Dawson and Dawson, 2003
; Vila and Przedborski, 2003
).
On the genetic basis, a number of published reports have associated point mutations of mitochondrial DNA (mtDNA) with PD neurodegeneration (Wallace, 1997
; Gu et al., 2002
; Bender et al., 2006
; Kraytsberg et al., 2006
). Furthermore, patients who are identified with mtDNA defects show progressive and disabling Parkinsonian features (Filosto et al., 2007
; DiMauro and Schon, 2008
).
The scientific evidence implicating that exposure to environmental toxic chemicals as a possible contributing factor for causing cellular oxidative damage and mitochondrial dysfunction in association with Parkinson’s disorder has been widely examined (Sherer et al., 2002
). Self-administration of an illicit designer’s drug, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) that results in human Parkinsonism is an early indication of chemically induced toxicity to the nigrostriatal DA neurons (Langston et al., 1983
). Later, MPTP is found to inhibit oxidative phosphorylation involving mitochondrial nicotinamide adenine dinucleotide (NADH): quinone oxidoreductase (complex-1) of the electron transport chain, to increase mitochondrial ROS production, and to deplete ATP (Ramsay and Singer, 1986
; Ali et al., 1994
). Infusion of other complex-1 inhibitors, such as rotenone and trichloroethylene to rodents results in loss of the nigral DA neurons and produces Parkinsonian phenotype suggesting mitochondrial dysfunction in PD pathogenesis (Betarbet et al., 2000
; Gash et al., 2008
). The concept of neuronal oxidative stress and mitochondrial complex-1 inhibition has also been verified in the substantia nigra pars compacta from human PD (Parker Jr. et al., 1989
; Schapira et al., 1990
; Mann et al., 1992
; Floor and Wetzel, 1998
).
Genetically modified animal models are of particular interest as they provide evidence that reducing mtDNA expression and disrupting mitochondrial function can lead to respiratory chain breakdown and neuronal death reminiscent of PD phenotype (Ekstrand et al., 2007
). Over-expression of α-synuclein in cell culture and in transgenic mice impairs mitochondrial function and increases their vulnerability to MPTP neurotoxicity (Hsu et al., 2000
; Song et al., 2004
). On the other hand, α-synuclein knock-out mice are resistant to toxicity produced by respiratory chain inhibitors, such as MPTP (Dauer et al., 2002
; Klivenyi et al., 2006
). These findings strongly divulge a role of mitochondrial deficiency in α-synucleinopathy that is relevant to PD pathogenesis.
As depicted above, most of the findings that associate mitochondrial dysfunction with PD-like DA neuron degeneration have been derived from in vitro experiments with isolated mitochondria and neuronal cells or from short-term investigations using an acute or subacute animal PD model. To further substantiate the functional role of mitochondria for the survival of nigrostriatal DA neurons, the objective of this research was to examine the long-term integrity of striatal mitochondria in a chronic mouse model of Parkinson’s disease (MPD).
Animals
Two different age groups of male, C57BL/6 mice, 6–10 weeks (young adult) and 6–10 months (aged retired-breeder), weighing 25–28 and 35–40 g, respectively were used (Harlan Sprague Dawley, Inc., Indianapolis, IN, USA). Mice were housed in single cages with food pellets and water available ad libitum. The room was maintained at a constant temperature and humidity on a 12-h/12-h light/dark cycle. All animal treatments were carried out strictly in accordance to the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee from the University of Houston. Our experimental procedures did not cause significant animal suffering. A total of 98 mice were used in the present study.
Chronic Mouse Model of Parkinson’s Disease
To prepare the chronic MPD, mice were injected with a total of 10 doses of MPTP hydrochloride (25 and 15 mg/kg/injection in saline, s.c. for young and aged mice, respectively) in combination with an adjuvant, probenecid (250 mg/kg/injection dissolved in dimethyl sulfoxide, i.p.) as previously described (Lau et al., 1990
; Petroske et al., 2001
). The 10-dose regimen was administered on a 5-week schedule with an interval of 3.5 days between injections. MPTP hydrochloride and probenecid were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Experiments were conducted 6 weeks post-chronic treatment. Safety precautions for the use of MPTP during chemical preparation and animal injections were taken according to the procedures previously described (Lau et al., 2005
). Control mice were treated with probenecid only. Probenecid was used to inhibit the rapid clearance and excretion of MPTP and its metabolites from the brain and kidney, and it alone did not produce any significant neurotoxic effect; but in combination it potentiated the neurotoxicity of MPTP (Lau, 2005
; Lau et al., 2005
; Barber-Singh et al., 2009
). For comparison, we also included a group of acute MPTP-treated young mice, which were injected with two doses of MPTP hydrochloride (25 mg/kg/injection in saline, s.c.) 2 h apart. Experiments were conducted 2 h post-acute treatment.
The chronic MPD used in this study has been well characterized. In contrast to the most commonly used acute and subacute MPTP mouse models of PD, in which neurological and behavioral deficits are short-lived and spontaneously reversed soon after the treatment, the chronic MPD has long-term neurological deficits showing many features resembling that of the Parkinson’s syndrome lasting for at least 6 months (Lau, 2005
). The observed features include marked depletion of DA content and terminal DA uptake in association with significant behavioral deficit and loss of DA cells in the substantia nigra (Lau et al., 1990
; Petroske et al., 2001
; Pothakos et al., 2009
). Early neuronal apoptosis and delayed appearance of α-synuclein-positive inclusion bodies along with ultrastructural neuronal damage in the substantia nigra pars compacta have also been demonstrated (Meredith et al., 2002
; Novikova et al., 2006
).
Isolation of Striatal Mitochondria
Due to tissue limitation, a crude striatal mitochondrial preparation that was modified according to the protocol of Sullivan et al. (2000
) was used for the mitochondrial respiration assay in this study. Briefly, striata from each animal were isolated and homogenized with a dounce homogenizer in 1 ml of an ice-cold isolation buffer containing mannitol (215 mM), sucrose (75 mM), bovine serum albumin (BSA, 0.1%), EGTA (1 mM), and HEPES (20 mM) at a pH of 7.2. All subsequent procedures were carried out at 4°C. The homogenate was centrifuged at 1,300 rpm for 3 min. The supernatant was transferred to a new tube and the pellet was suspended in 0.5 ml of the isolation buffer and centrifuged again at 1,300 rpm for 3 min. The supernatants from both spins were combined and centrifuged at 13,000 rpm for 10 min. This latter supernatant was discarded and the pellet containing the mitochondrial fraction was suspended in 0.1 ml of a respiration buffer containing mannitol (215 mM), sucrose (75 mM), BSA (0.1%), HEPES (20 mM), MgCl2 (2 mM), and KH2PO4 (2.5 mM) at a pH of 7.2. The mitochondrial protein concentration was determined with the Pierce micro BCA protein assay kit (Rockford, IL, USA) measured at an absorbance of 595 nm with a Beckman DU 640 spectrophotometer (Fullerton, CA, USA).
Mitochondrial Respiration Assay
Since mitochondria are vital organelles responsible for catalyzing the oxidation of various substrates through an electron transport chain system and for generating ATP to support cell function and survival, the respiratory activity of striatal mitochondria was measured polarographically with a Clark-type oxygen electrode in a sealed, thermo-controlled, and continuously stirred chamber (Oxytherm System, Hansatech Instruments, Norfolk, England). Crude mitochondrial protein at a concentration of 0.5 mg/ml in respiration buffer (see above) was initially equilibrated in the electrode chamber at 30°C. The rate of oxygen consumption (nmol/min/mg mitochondrial protein) was calculated based on the slope of polarographic tracings obtained during state 3 and state 4 respiration. All mitochondria preparations had an average respiratory control ratio of at least 4.5 when pyruvate (5 mM) and malate (2.5 mM) were used as NADH-linked substrates. The state 3 respiration was initiated by adding adenosine 5′-diphosphate (ADP, 150 μM).
Measurement of Mitochondrial ATP
Striatal mitochondrial samples were prepared under conditions identical to those of the respiration study. After the completion of respiration experiment, the mitochondrial suspension from the chamber was recovered and mixed with an equal volume of lysing buffer provided in the ATP bioluminescence assay kit (PerkinElmer, Waltham, MA, USA). The mitochondrial content of ATP was measured according to the manufacturer’s instruction. Light emitted from luciferase-mediated reaction was captured in a luminometer (Wallac Victor II, PerkinElmer) and the sample ATP content was extrapolated from a standard curve constructed with a series of known ATP concentrations (Tieu et al., 2003
).
Western Blot Analysis
Besides serving as energy generating organelles, mammalian mitochondria also play a pivotal role in regulating the production and removal of ROS from internal or external sources. Several key anti-oxidant enzymes have been widely studied. Mn SOD (SOD2) is located in the mitochondrial matrix where it protects mitochondrial targets against the internally generated superoxides. Cu–Zn SOD (SOD1) and cytochrome c are capable of removing the superoxides generated by external triggers. These anti-oxidant enzymes are important for scavenging the elevated ROS and preventing them from reaching a toxic level that will cause mitochondrial damage and cell death (McCord, 1993
; Pereverzev et al., 2003
). Therefore, loss of anti-oxidant enzymes and release of cytochrome c in association with mitochondrial dysfunction in the DA neurons may lead to substantial ROS accumulation and cause PD-like neurodegeneration. Thus, we measured the levels of mitochondrial anti-oxidant enzymes in conjunction with the striatal DA synthesizing enzyme and DA uptake protein in the present study.
For western blot analysis of anti-oxidant protein expression, crude striatal mitochondria were prepared according to the procedure as described above, except that mitochondria were finally suspended in Tris–HCl buffer (50 mM, pH 7.4) containing the protease inhibitor cocktail purchased from Sigma Chemical Co. The mitochondrial protein concentration in each sample was determined with the Pierce micro BCA protein assay kit and diluted to an appropriate final concentration in a protein solubilization solution containing SDS (2%), Tris–HCl (62.5 mM, pH 6.8), glycerol (10%), 2-mercaptoethanol (5%), bromophenol blue (0.001%). The sample was boiled for 5 min and 25 μg of each sample was applied to SDS polyacrylamide (12%) gel electrophoresis. The resolved proteins were transferred to a PVDF membrane, which was probed with polyclonal rabbit anti-Mn-SOD (1:1000, Upstate, Lake Placid, NY, USA), polyclonal anti Cu/Zn-SOD (1:1000, Stressgen Bioreagents, Victoria, BC, Canada), monoclonal mouse anti-cytochrome c (1:750, Chemicon, Temecula, CA, USA), mouse anti-tyrosine hydroxylase (anti-TH, 1:1000, Millipore, Temecula, CA, USA), rat anti-DA uptake transporter (anti-DAT, 1:1000, Chemicon) and monoclonal mouse anti-glyceraldehyde 3 phosphate dehydrogenase (anti-GAPDH, 1:3000, Chemicon). Immunoreactivity was visualized by a horseradish peroxidase-conjugated goat anti-rabbit (1:2000, Chemicon) or goat anti-rat (1:2000, Chemicon) or goat anti-mouse IgG (1:1500, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) with an enhanced NuGlo chemiluminescent substrate (Alpha Diagnostic International Inc., San Antonio, TX, USA). The exposure time of the blot on film was confined to the linear scale of detection without exceeding the saturation limit. The intensity of the protein bands was measured by densitometry (Fluorochem 8800, Alpha Innotech Corporation, San Leandro, CA, USA) and expressed as a ratio to that of GAPDH to ensure that changes of protein level are not simply due to differences in the amount of sample loading.
Assay for Striatal Dopamine and 3,4-Dihydroxyphenylacetic Acid Content
The striatal levels of DA and its metabolite, 3,4-dihydroxyphenylacetic acid (DOPAC) were determined as previously described (Lau et al., 2005
). Briefly, striata from each animal were isolated, weighed and suspended in 0.5 ml of 0.2 N perchloric acid. Each sample was sonicated and centrifuged at 11,000×g for 15 min at 4°C. The supernatant was filtered through a 4-mm nylon syringe filter with a pore size of 0.45 μm (National Scientific, Rockwood, TN, USA). An aliquot of the filtrate was injected into a high performance liquid chromatography (HPLC Model 1525, Waters Corporation, Milford, MA, USA) equipped with a C18 reverse phase, 3 μ LUNA column (100 mm × 2.0 mm, Phenomenex, Torrance, CA, USA). The sample was eluted by a mobile phase made of 25 mM NaH2PO4, 50 mM Na-citrate, 0.03 mM EDTA, 10 mM diethylamine HCl, and 2.2 mM sodium octyl sulfate (pH 3.2), 30 ml/l methanol and 22 ml/l dimethylacetamide at a flow rate of 0.4 ml/min. DA and DOPAC peaks were determined by the Coulometric electrochemical detector (Model Coulochem III, ESA, Inc., Chelmsford, MA, USA) and were calculated by extrapolating the peak area from a standard curve (ranging 0.05–1 ng of each chemical standard) constructed under the same conditions during each run.
Animal Balance Performance on a Challenging Beam
We monitored the ability of the aged chronic MPD in maintaining balance on a challenging beam 6 weeks post-chronic treatment as previously described with the young adult mice (Pothakos et al., 2009
). The challenging beam was a 1-m long wooden beam suspended 23 cm above a bench top, which was covered with soft pads to protect the mouse in case of a fall. The beam was divided in four gradually narrowing sections (25 cm/section) leading to the mouse’s home cage. The beam widths of the four sections were 3.5, 2.5, 1.5, and 0.5 cm in decreasing order. The beam was covered with surgical tape that provided sufficient surface traction for the animals to walk on. There were 1 cm wide ledges hanging 1 cm below each side of the beam to encourage the mice to use their normal gait strategies even when their limbs slipped. All mice were pre-trained for two consecutive days (five trials per day) with an inter-trial interval (ITI) of 10–12 s on traversing the beam. On the third day, each mouse was given five trials (ITI = 10–12 s). The average number of videotaped limb slips per trial and the time latency for returning to the home cage were recorded and compared with control mice for statistical analysis. Slips were counted only while the mouse was in forward motion.
Statistical Analysis
Statistical comparisons of values between control and treated groups were carried out by Student’s t-test. In all cases, a P value of <0.05 was considered to be significant. All data are represented as mean ± SEM.
For studying the hypothesis of striatal mitochondrial dysfunction in the chronic MPD, we initially used young adult mice at the age of 6–10 weeks. We first replicated the animal model by demonstrating striatal dopaminergic deficit resulting from a 5-week, 10-dose treatment with MPTP (25 mg/kg) and probenecid (250 mg/kg) as shown in previous studies (Lau et al., 1990
; Petroske et al., 2001
). Thus, 6 weeks after chronic MPD treatment, we detected a sustained 74.9 and 70.9% loss of striatal DA and DOPAC, respectively (Table 1
). Acute treatment in young mice with two doses of MPTP (25 mg/kg) caused a rapid depletion of 54.1% DA and 48.3% DOPAC in the striatum 2 h later (Table 1
). These results indicate that MPTP has an acute effect in depleting the vesicular DA stores; and when administered chronically with probenecid, MPTP produces a persistent loss of DA in the striatum.
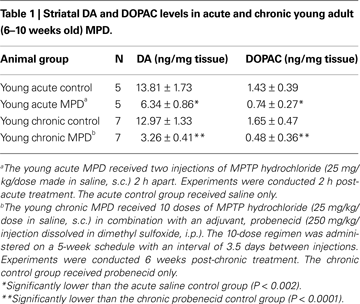
Mitochondrial respiration in the striatum was measured and compared between acute and chronic MPD. The basal rates of mitochondrial respiration (state 4 respiration) in young adult mice were not significantly different between acute control, acute MPD, chronic control, and chronic MPD groups (Table 2
). The ADP-stimulated mitochondrial respiration (state 3 respiration) in young acute MPD was markedly reduced when compared with the acute control animals (Table 2
). To our surprise, the young chronic MPD showed no signs of respiratory depression in the striatal mitochondria when compared with that in the chronic control mice (Table 2
). These data demonstrate that although MPTP has an immediate toxic effect on mitochondrial respiration, this defect is not sustained 6 weeks after the chronic MPTP/probenecid treatment.
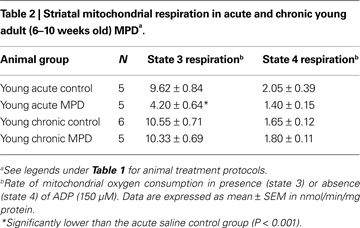
We further examined the expression of striatal mitochondrial anti-oxidant proteins by western blot analysis. In young acute MPD, the levels of Mn SOD, Cu–Zn SOD, and cytochrome c were not significantly altered in the striatal mitochondria (Figures 1
A–C). Similarly, the contents of these three proteins 6 weeks after chronic MPTP/probenecid treatment were not different from either control or acute MPD group of young mice (Figures 1
A–C). Since the basal levels of these proteins in acute saline-treated and chronic probenecid-treated young mice were not statistically different, only the control data from chronic probenecid-treated group were presented.
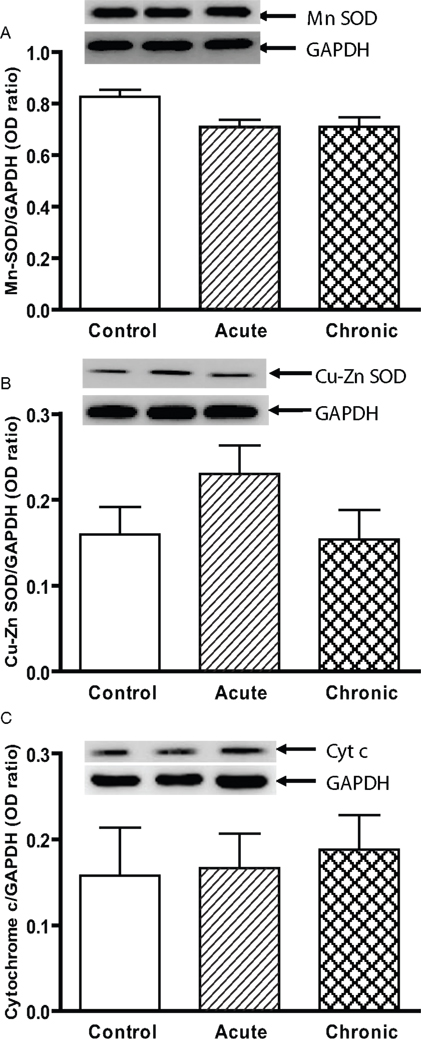
Figure 1. Protein expression of (A) Mn SOD, (B) Cu–Zn SOD, and (C) cytochrome c in the striatum of young control, acute MPTP, and 6-week post-chronic MPTP/probenecid-treated parkinsonian (MPD) mice. Western blot technique and densitometry analysis were used to detect and quantify the respective anti-oxidant protein bands and GAPDH. The protein contents of Mn SOD, Cu–Zn SOD, and cytochrome c were expressed as a ratio to that of GAPDH to ensure that the changes in protein levels were not due to sample loading variances. A representative image of western blot protein bands was correspondingly shown above each set of bar graph. Each data point represents mean ± SEM, N = 6 per group of animals. The levels of Mn SOD, Cu–Zn SOD, and cytochrome c in the striatum of young control, acute MPTP, and 6-week post-chronic MPD mice were not statistically different from each other.
It is possible that the functionality of mitochondria may gradually recover in young adult mice after the toxic species of MPTP are dissipated from the brain. Before ruling out the possible association between Parkinson-like neurodegeneration and mitochondrial disorder, we were curious to investigate whether mitochondrial dysfunction may exist and can be demonstrated in aged chronic MPD. Since aged mice are known to be more sensitive to MPTP neurotoxicity (Ricaurte et al., 1987
) and also based on our own experience that aged mice will not survive the same chronic MPTP/probenecid regimen as in young adult mice, therefore we treated the aged chronic MPD with a lesser dose of MPTP (15 mg/kg) in the present study. Indeed, the 6- to 10-month-old mice when treated with 10 doses of MPTP (15 mg/kg, s.c.) and probenecid (250 mg/kg, i.p.) over 5 weeks had all survived and they displayed 64.6 and 53.3% loss of DA and DOPAC, respectively 6 weeks after the cessation of treatment (Table 3
). Analyses of TH and DAT expression in the striatal tissue further revealed that there were significant losses of TH and DAT contents in the aged chronic MPD 6 weeks after treatment when compared with that of the control animals (Figures 2
A,B). These results implicate that this modified chronic MPD treatment in aged mice generates a moderate and sustained loss of dopaminergic transmission and function in the striatum.
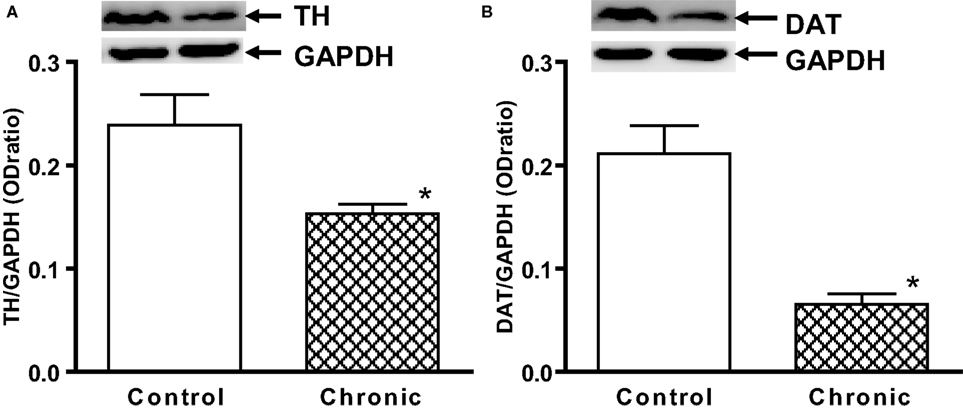
Figure 2. Western blot analysis of (A) TH and (B) DAT expression in the striatum of aged chronic probenecid control and aged chronic MPD mice. A representative image of western blot protein bands was correspondingly shown above each set of bar graph. The protein contents of TH and DAT were expressed as a ratio to that of GAPDH and each data point represents mean ± SEM (N = 5 for chronic control and N = 8 for chronic MPD). Western blots revealed that there were significant reductions of TH and DAT content in the striatum of aged MPD 6 weeks after chronic treatment when compared with that of the control mice (*P < 0.001).
Interestingly, both the basal (state 4) and ADP-stimulated (state 3) mitochondrial respiration in the striatum of aged chronic MPD mice were significantly impaired when compared to the chronic control animals (Table 4
). There was a 50.8 and 41.0% reduction of state 3 and state 4 respiration, respectively 6 weeks after the chronic MPD treatment (Table 4
). The mitochondrial ATP level was also decreased by 46.9% (Table 4
). These data demonstrate that 6 weeks after chronic MPTP/probenecid treatment in the aged mice, there is a sustained attenuation of mitochondrial respiration and ATP production in the striatum. Western blot analyses of anti-oxidant proteins additionally showed that the levels of Mn SOD, Cu–Zn SOD, and cytochrome c were significantly reduced in the striatal mitochondria of the aged chronic MPD 6 weeks after treatment (Figures 3
A–C).
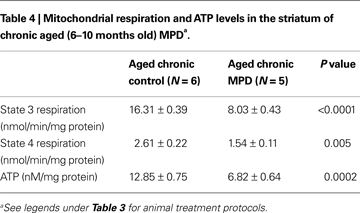
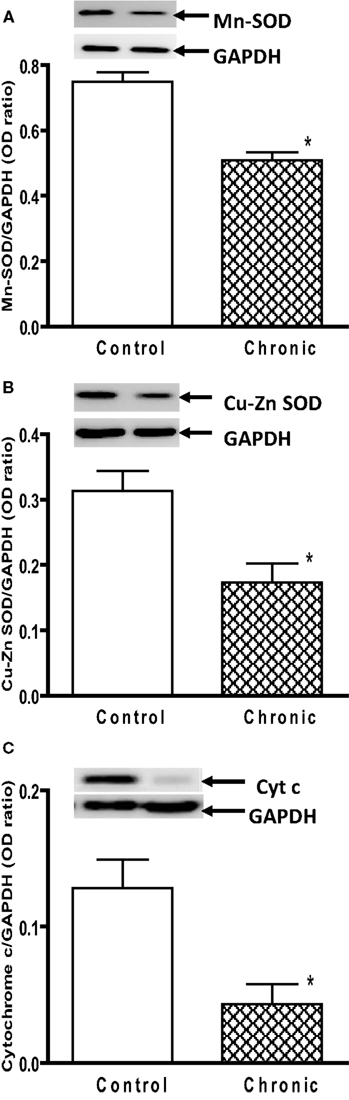
Figure 3. Protein expression of (A) Mn SOD, (B) Cu–Zn SOD, and (C) cytochrome c in the striatum of aged chronic probenecid control and aged chronic MPD mice. A representative image of western blot protein bands was correspondingly shown above each set of bar graph. The protein contents of Mn SOD, Cu–Zn SOD, and cytochrome c were expressed as a ratio to that of GAPDH and each data point represents mean ± SEM, N = 6 per group of animals. Statistical analyses revealed that the levels of Mn SOD, Cu–Zn SOD, and cytochrome c in the striatum of aged chronic MPD 6 weeks after chronic treatment were significantly lower than that of chronic control animals (*P < 0.05).
Although the absolute value of the state 3 respiration in aged mice (Table 4
) appeared to be higher than that in the young mice (Table 2
), statistical comparison between them was not performed because these two sets of experiments were not performed at the same time and under the same conditions. Furthermore, the chronic MPD in young and aged animals were treated with a different dose of MPTP due to age-related sensitivity. Thus, the aim and design of the present study was to directly compare mitochondrial respiration between age-matched control and chronic MPD groups under the same experimental conditions.
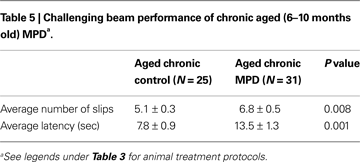
We used the challenging beam in this study to examine the animal’s balance and motor coordination skills. Six weeks after MPTP/probenecid treatment, the chronic MPD group exhibited a significantly greater number of foot slips on the challenging beam than that of the control group (Table 5
). In addition, the aged chronic MPD mice were more hesitant and took longer time than the control animals on the balance beam before returning to the home cage (Table 5
).
There is a large body of in vitro and acute in vivo research findings suggesting a possible link between mitochondrial dysfunction and neurodegenerative disorders. However, very few investigations have been conducted and results are inconclusive involving the long term defects of mitochondria in chronic neurodegenerative models. In the present study, we investigated mitochondrial respiration and measured mitochondrial anti-oxidant enzyme levels 6 weeks after the induction of Parkinsonism in a chronic MPD. In contrast to our initial hypothesis stating that long-term mitochondrial deficiency is associated with chronic Parkinsonism, we failed to detect a significant inhibition of either the basal (state 4) or ADP-stimulated (state 3) mitochondrial respiration in the striatum of young adult chronic MPD mice at 6–10 weeks of age, although considerable loss of striatal DA neurotransmitter persisted in these animals. Furthermore, we did not ascertain any noticeable change in the levels of mitochondrial anti-oxidant enzymes including Mn SOD, Cu–Zn SOD, and cytochrome c. Our results may be interpreted as either mitochondrial impairment is not an obligatory pathophysiological factor for chronic neurodegenerative process or MPTP may instigate oxidative stress and inflammation to the mitochondria in other types of striatal cells in addition to the DA neurons; consequently, striatal mitochondria in the non-DA cells plus or minus DA neurons in young animals may have the capability to rejuvenate their functionality over time when the neurotoxin clears away from the brain. The latter possibility on cell specificity was not tested in the present study due to tissue limitation.
For comparison, we confirmed that acute injections of MPTP caused an immediate depletion of striatal DA and simultaneously suppressed the state 3 mitochondrial respiration. These observations are in line with the premise that neurodegeneration may be provoked by MPTP causing accumulation of excitotoxic mediators, production of oxidative free radicals, disruption of Ca++ homeostasis, and ATP depletion in association with an inhibition of the neuronal mitochondrial complex I component of the electron transport chain (Przedborski and Jackson-Lewis, 1998
; Blum et al., 2001
). However, whether neurodegeneration follows a progressive course or not would depend on how rapid and how long the brain is exposed to neurotoxic chemicals. Indeed, the DA depleting effect of MPTP, when it is administered acutely or subacutely in mice, tends to be short-lived and reversible (Lau, 2005
). In addition, as expected and shown in our results, we did not detect an immediate action of MPTP on turning over the anti-oxidant enzyme levels of SODs and cytochrome c in acutely treated young mice. We did not further carry out the long-term investigation of acute MPTP on mitochondrial respiration, SODs and cytochrome c contents. We do not anticipate that acute MPTP will cause any sustained mitochondrial changes, because even chronically repeated administration of MPTP/probenecid in young mice did not generate any significant alterations in mitochondrial respiration and anti-oxidant enzyme levels at 6 weeks after treatment.
To address the possibility that striatal mitochondria in young animals may have the potential for functional recovery over time when the neurotoxin is eliminated from the brain, we decided to examine the long-term neuronal and mitochondrial toxic effects of chronic MPTP/probenecid in an aged group of animals (6–10 months old). Resembling the observations from young adult animals, even with a reduced dose of MPTP (15 mg/kg/injection), the aged chronic MPD acquired significant losses of striatal DA, DOPAC, TH and DAT to a moderate level at 6 weeks post-treatment. In contrast to the young animals, the aged chronic MPD consistently suffered an inhibition of the basal and ADP-stimulated respiration and a fall in ATP production in the striatal mitochondria. Chronic MPTP/probenecid treatment in aged mice also brought about a sustained diminution of striatal anti-oxidant enzyme levels including that of Mn SOD, Cu–Zn SOD, and cytochrome c. The striatal neurochemical and mitochondrial deficits in aged chronic MPD 6 weeks after treatment were further supported by behavioral observations showing that these animals made more foot-slip errors and it took twice as long for them to complete the challenging beam test.
It should be emphasized that a close correlation between neuronal, mitochondrial, and behavioral deficits 6 weeks after chronic Parkinsonism in the aged animals would suggest an important role of mitochondrial function in maintaining neuronal plasticity. However, such a positive correlation alone is not sufficient for establishing a cause-and-effect relationship between mitochondrial disorder and Parkinson-like neurodegeneration. Furthermore, our failure for demonstrating long-term mitochondrial deficiency in the young chronic MPD would not dispute the possibility that neurotoxins like MPTP can cause mitochondrial oxidative dysfunction and trigger the neurodegenerative process, but the mitochondria may potentially revive from the initial insult in young animals. This is well demonstrated in the present study that mitochondrial functions are fully intact whereas neuronal functions are severely impaired 6 weeks after chronic MPD treatment in young mice. The main significance of this investigation is the finding of coexisted phenotype between neurodegenerative and mitochondrial defects in the aged chronic MPD. This will provide an appropriate model for continuously establishing whether these two disorders are cause-and-effect related and for using these altered neurobiological indicators as biomarkers for potential neuroprotective targets in preclinical tests. One of the approaches that we currently undertake using this model in our laboratory is to elucidate whether long-term physical exercise could simultaneously offer mitochondrial as well as neuronal protection in the neostriatum.
The topic on deterioration of mitochondrial functions associated with normal aging or correlated with age-related diseases has been widely reviewed (Calabrese et al., 2001
; Bertoni-Freddari et al., 2004
; Crompton, 2004
; Martin, 2006
; Arduino et al., 2009
; Swerdlow, 2009
). In the laboratory, neurons isolated from the brains of aged rats show deficiency in cytochrome c and inner mitochondrial membrane-associated cardiolipin levels that would preempt normal mitochondrial respiration and oxidative phosphorylation (Jones and Brewer, 2009
). A single dose of MPTP (40 mg/kg) does not acutely generate ROS in young mice, but significantly releases ROS in old mice (Ali et al., 1994
). Moreover, although attempts have been made to link mutations of mtDNA directly to normal aging or age-related disease states, it has not been confirmed that mtDNA mutations are either the primary cause or the result of aging or disease process (Arnheim and Cortopassi, 1992
; Wallace, 1997
; Kang and Hamasaki, 2005
; Reeve et al., 2008
). For translational consideration, one can envision that the capacity of mitochondria for carrying out oxidative respiration and generating ATP and for coping with acute oxidative insult in the neuron of a 20-year-old would be much greater than that of a 70-year-old individual. In other words, the mitochondria and neurons of an aged person would be more subject to irreparable injuries following chronic exposure to endogenous or exogenous toxic chemical species. As implicated in the present study, our results are in agreement with the notion that the anti-oxidant capacity and the recovery potential for mitochondria and neurons post-injury are expected to decline with increase in age (Beal, 2005
).
It is reasonable to presume that normal physiological release of ROS from mitochondria is insignificantly low and is unlikely to cause neuronal death. Therefore, intentional reduction of endogenously produced ROS below its basal physiological level would not be considered health beneficial. However, if chronic exposure to neurotoxic chemicals like MPTP results in lasting depletion of anti-oxidant enzymes such as SODs and cytochrome c in the aged subject, as demonstrated in the current study, the loss of mitochondrial anti-oxidant protection can certainly exacerbate neurotoxicity and cell death initiated by the elevated ROS. Thus, developing new approaches for protecting the capacity of mitochondrial defense system, for eradicating the elevated ROS level, and for restoring the mitochondrial respiratory functions would be important strategies for preventing and treating progressive cell death under oxidative stress conditions.
In summary, the present study examined the role of striatal mitochondrial function in the chronic MPD. Although we confirmed that MPTP acutely suppressed mitochondrial respiration, it did not cause a long-lasting inhibitory effect or deplete anti-oxidant enzymes in the young chronic MPD. These data suggest that MPTP may initiate neurodegenerative process by blocking mitochondrial respiration, yet the mitochondria in the striatum of young animals have the potential for recovery. However, we demonstrated that aged mice were more vulnerable to chronic MPTP toxicity. Long-term inhibition of mitochondrial respiration, depletion of anti-oxidant enzymes and ATP were displayed in aged mice coincided with marked loss of striatal DA neuronal functions and impaired motor response. These results indicate that aged chronic MPD may serve as a suitable investigative model for further elucidating the integral relationship between mitochondrial dysfunction and neurodegenerative disorder, and for assessing the therapeutic efficacy of mitochondrial protective agents as potential neuroprotective drugs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Drs. Mostafa Badr, Patrick Sullivan, and Donald Fox for their advice on the preparation of striatal mitochondria and on the measurement of mitochondrial respiration. We appreciate the advice and help of Dr. Konstantinos Pothakos on the assessment of animal’s balance behavior. Preliminary results of this study were published in abstract form at the Experimental Biology meeting in San Diego, CA, USA, April 5–9, 2008. This study was supported by a grant from the National Institute of Neurological Disorders and Stroke (NS 47920).
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., Jaros, E., Hersheson, J. S., Betts, J., Klopstock, T., Taylor, R. W., and Turnbull, D. M. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517.
Bronstein, J., Carvey, P., Chen, H., Cory-Slechta, D., DiMonte, D., Duda, J., English, P., Goldman, S., Grate, S., Hansen, J., Hoppin, J., Jewell, S., Kamel, F., Koroshetz, W., Langston, J. W., Logroscino, G., Nelson, L., Ravina, B., Rocca, W., Ross, G. W., Schettler, T., Schwarzschild, M., Scott, B., Seegal, R., Singleton, A., Steenland, K., Tanner, C. M., Van Den Eeden, S., and Weisskopf, M. (2009). Meeting report: consensus statement-Parkinson’s disease and the environment: collaborative on health and the environment and Parkinson’s Action Network (CHE PAN) conference 26–28 June 2007. Environ. Health Perspect. 117, 117–121.
Dauer, W., Kholodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., Staal, R., Tieu, K., Schmitz, Y., Yuan, C. A., Rocha, M., Jackson-Lewis, V., Hersch, S., Sulzer, D., Przedborski, S., Burke, R., and Hen, R. (2002). Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 99, 14524–14529.
Ekstrand, M. I., Terzioglu, M., Galter, D., Zhu, S., Hofstetter, C., Lindqvist, E., Thams, S., Bergstrand, A., Hansson, F. S., Trifunovic, A., Hoffer, B., Cullheim, S., Mohammed, A. H., Olson, L., and Larsson, N. G. (2007). Progressive Parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. U.S.A. 104, 1325–1330.
Gash, D. M., Rutland, K., Hudson, N. L., Sullivan, P. G., Bing, G., Cass, W. A., Pandya, J. D., Liu, M., Choi, D. Y., Hunter, R. L., Gerhardt, G. A., Smith, C. D., Slevin, J. T., and Prince, T. S. (2008). Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann. Neurol. 63, 184–192.