Knowledge in the investigation of A-to-I RNA editing signals
 Giovanni Nigita
Giovanni Nigita Salvatore Alaimo
Salvatore Alaimo Alfredo Ferro
Alfredo Ferro Rosalba Giugno
Rosalba Giugno Alfredo Pulvirenti
Alfredo Pulvirenti- 1Department of Molecular Virology, Immunology and Medical Genetics, Ohio State University, Columbus, OH, USA
- 2Department of Mathematics and Computer Science, University of Catania, Catania, Italy
- 3Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
RNA editing is a post-transcriptional alteration of RNA sequences that is able to affect protein structure as well as RNA and protein expression. Adenosine-to-inosine (A-to-I) RNA editing is the most frequent and common post-transcriptional modification in human, where adenosine (A) deamination produces its conversion into inosine (I), which in turn is interpreted by the translation and splicing machineries as guanosine (G). The disruption of the editing machinery has been associated to various human diseases such as cancer or neurodegenerative diseases. This biological phenomenon is catalyzed by members of the adenosine deaminase acting on RNA (ADAR) family of enzymes and occurs on dsRNA structures. Despite the enormous efforts made in the last decade, the real biological function underlying such a phenomenon, as well as ADAR’s substrate features still remain unknown. In this work, we summarize the major computational aspects of predicting and understanding RNA editing events. We also investigate the detection of short motif sequences potentially characterizing RNA editing signals and the use of a logistic regression technique to model a predictor of RNA editing events. The latter, named AIRlINER, an algorithmic approach to assessment of A-to-I RNA editing sites in non-repetitive regions, is available as a web app at: http://alpha.dmi.unict.it/airliner/. Results and comparisons with the existing methods encourage our findings on both aspects.
Background
In recent times, there has been a change in the range of research on many types of diseases. In the past decades, the principal aim was to add information about the molecular pathways involved in some disease through the study of DNA mutations. Lately, the focus has indeed moved to the analysis of post-transcriptional modification events, such as RNA editing. The knowledge that the activity of RNA editing is higher in mammalian brain than in other tissues (Paul and Bass, 1998), hints that editing may play a crucial role in the central nervous system (Nishikura, 2006). Therefore, malfunctions of RNA editing machineries could lead to serious consequences (Galeano et al., 2012; Tomaselli et al., 2014).
RNA editing is a type of post-transcriptional modification, taking place in eukaryotes, which alters the sequence of primary RNA transcripts by deleting, inserting, or modifying residues. Despite the discovery of several distinct types of RNA editing over the years, adenosine-to-inosine (A-to-I) RNA editing is now considered the most predominant in mammalians (Nishikura, 2010). Through the deamination process, adenosine (A) is converted into inosine (I), which in turn is interpreted as guanosine (G) by both the splicing and the translation machineries (Rueter et al., 1999). Enzymes members of the adenosine deaminase acting on RNA (ADAR) family catalyze this biological phenomenon only on dsRNA structures (Bass, 2002; Jepson and Reenan, 2008; Nishikura, 2010).
Adenosine-to-inosine RNA sites abundantly occur in intronic regions as well as in 3′-UTRs. RNA editing events can modify RNA molecules in several cellular contexts causing: the creation and/or destruction of splicing sites (Rueter et al., 1999); the modulation of gene expression pathways (Bazak et al., 2014b) during translation (Nishikura, 2010); the gain or loss of miRNA recognition elements (MRE) during mRNA targeting (Nishikura, 2006; Borchert et al., 2009) (i.e., MRE can be created or deleted even with a single post-transcriptional modification). As it has been reported in the last few years, RNA editing sites can be found in non-coding RNA molecules, especially within pri-miRNA (Kawahara et al., 2008; Kawahara, 2012), lncRNA (Mitra et al., 2012), and precursor-tRNA (Su and Randau, 2011), the latter deaminated by adenosine deaminases acting on tRNA (ADAT) enzymes.
It is possible to distinguish two forms of A-to-I RNA editing, promiscuous and specific. The promiscuous A-to-I editing occurs within longer duplexes of hundreds of nucleotides, as in the case of stem–loops that are formed by the pairing of repetitive elements (e.g., Alu elements), as seen above. In those cases, up to 60% of adenosines could be edited (Carmi et al., 2011; Bazak et al., 2014b). The specific A-to-I RNA editing occurs in short and/or unstable duplex RNA regions (Wahlstedt and O’Hman, 2011), in which at least 10% of their adenosines selectively could undergo deamination. A-to-I RNA editing events in small non-coding RNAs, such as microRNAs, are perfect examples of specific editing (Nishikura, 2010).
One of the main challenges in the study of the RNA editing phenomenon is certainly RNA editing occurrence. The detection of editing sites in RNA molecules in particular cellular conditions is very difficult considering that RNA editing is a dynamic spatial–temporal process. In the last decade, the application of global approaches to the study of A-to-I editing, including in a first phase bioinformatics methods and, lately, high-throughput sequencing technology (HTS) based pipelines, have led to important advances, allowing the discovery of a large amount of editing sites in the human transcriptome. Despite the enormous efforts made in recent years, the real biological function underlying such a phenomenon, as well as ADAR’s substrate features still remain unknown.
In this work, we give an overview of the current state of knowledge on the editing phenomenon, as well as provide the main features of editing sites as highlighted today. We also investigate, inspired by previous results, methods for the detection of signals characterizing editing events and the prediction of novel A-to-I editing sites in non-repetitive regions. These techniques are based on the analysis of nucleotide profiles within a distance-radius of the probable editing site. Results on the signal detection show that editing sites may not have strong defined signal patterns.
Finally, by using a logistic regression technique we developed AIRlINER, an algorithmic approach for the prediction of A-to-I RNA editing sites in non-repetitive regions. This method has been compared with InosinePredict (Eggington et al., 2011), a similar technique, which analyzes the nucleotides flanking the editing site. InosinePredict assumes a multiplicative relationship between the coefficients necessary to compute the percentage of editing. Our results clearly show that AIRlINER improves the quality of predictions with respect to InosinePredict and suggest further research directions. AIRlINER is available at the following address: http://alpha.dmi.unict.it/airliner/.
Knowledge and Features of Editing Sites Signals
At the end of 80s, ADARs, initially identified as associated with an unknown dsRNA-unwinding activity (Bass and Weintraub, 1987; Rebagliati and Melton, 1987), were discovered as RNA editing machineries able to alter adenosine into inosine through deamination, especially in dsRNA structures (Bass and Weintraub, 1988; Wagner et al., 1989). In the next 10 years, three members of the ADAR gene family were identified in humans: two isoforms of ADAR1 (N-terminally truncated ADAR1p110 and a full-length ADAR1p150) (Kim et al., 1994; Patterson and Samuel, 1995), ADAR2 (Lai et al., 1997) (both these members expressed in many tissues), and ADAR3 (Chen et al., 2000) present only in the central nervous system. While for ADAR1 and ADAR2 the enzymatic activity was established, for ADAR3 it remains unknown. Unlike ADAR1 and ADAR2, an interesting feature about ADAR3 is the presence of the R domain, which enables the enzyme to bind to single strand structures. ADAR1 and ADAR2 have two common functional regions, an N-terminal dsRNA-binding domain (dsRBD) and a C-terminal deaminase domain, but only ADAR1 contains two Z-DNA-binding domains, Zα and Zβ. Some editing events are edited only by ADAR1 or ADAR2, showing a significant difference in their RNA-substrate interactions (Wong et al., 2001; Riedmann et al., 2008). For instance, the serotonin B site is deaminated not only by ADAR1, while the serotonin D and the GluR-B Q/R sites are deaminated exclusively by ADAR2 (Burns et al., 1997; Yang et al., 1997), but also ADAR1 and ADAR2 can edited the same target, as in the cases of serotonin A and C editing sites (Burns et al., 1997). Subsequently, the characterization of the neighborhood profiles of both ADAR1 and ADAR2 were established. In particular, ADAR1 has 5′ neighboring base preference consisting of uracil, adenosine, cytosine, and guanosine in order (U ≈ A > G > C), but not 3′neighbor preference has been identified (Polson and Bass, 1994). Similarly, ADAR2 has a 5′ neighbor preference, but, differently from ADAR1, ADAR2 has a 3′ neighboring base preference (U = G > C = A) forming particular trinucleotide sequences with an adenosine at the second base (UAU, AAG, UAG, AAU) (Lehmann and Bass, 2000).
In 2003, Hoopengardner et al. (2003) discovered that highly conserved regions, which in turn form a dsRNA structure, surround many editing sites. Later, by considering these findings, bioinformatics methods mapping ESTs against a reference genome were able to discover tens of thousands of A-to-I RNA editing sites, with more than 90% of them occurring within Alu repeats (Athanasiadis et al., 2004; Kim et al., 2004; Levanon et al., 2004). A significant problem in all the bioinformatics approaches for RNA editing detection, as described above, still remains the limitations posed by sequencing technologies, specifically, the inability to distinguish a guanosine originating from an I-to-G replacement from a guanosine as a product of noise, sequencing errors or SNP. A solution to this issue was proposed by Sakurai et al. (2010) who designed a biochemical method, called inosine chemical erasing (ICE), able to identify inosine sites on RNA molecules by employing inosine-specific cyanoethylation with reverse transcription. This is a reliable and accurate biochemical method to detect inosines in RNA strands.
The recent years have been characterized by the development of several approaches for editing discovery based on deep sequencing. It was recently hypothesized that more than 100 million editing sites could be found in human Alu repeats, located mainly in genic regions (Bazak et al., 2014a). Although these recent methods prove to be more accurate than previous ones, some of them nonetheless present limitations in terms of false positives produced (Kleinman and Majewski, 2012; Lin et al., 2012; Pickrell et al., 2012). In recent years, a considerable number of RNAseq based methods have emerged (Li et al., 2009; Ju et al., 2011; Bahn et al., 2012; Peng et al., 2012; Picardi et al., 2012; Ramaswami et al., 2012, 2013; Bazak et al., 2014a), gradually improved the accuracy in discovering new editing sites, leading, in addition, to the identification of a set of human editing sites orders of magnitude larger than before. Recently, Sakurai et al. (2014) combined the ICE method with HTS (ICE seq) for an unbiased genome-wide screening of novel A-to-I editing sites. ICE seq is able to detect editing sites in both repeat elements and short hairpins, rendering this a currently unique method for genome-wide identification of A-to-I editing events in both tissues and clinical specimens without genomic DNAs.
The application of HTS technology to RNA editing discovery has not only brought improvements in the editing discovery but also helped to increase the knowledge about the features inherent to the phenomenon. In fact, thanks to the analysis of a large RNA-seq data, Bazak et al. (2014b) studied the global characteristics that affect the editability at the Alu level, uncovering some important features. An important parameter that influences the editing of the Alu is the distance to the nearest complementary inverse sequence. Indeed, the editing, on average, exponentially decays with this distance, with a typical length of about 800 nt. Another aspect is that the editing levels are positively correlated with the number of reversely complementary repeats in the flanking regions of the Alu. Instead, they are negatively correlated with the number of same-strand repeats. Furthermore, the editing level depends on both the lengths of the Alu repeats and their closest reversely oriented sequence, additionally to whether the latter resides in the same intron/exon. Finally, the consensus strand of the Alus is more edited than the reverse strand.
Lately, Pinto et al. (2014) conducted a study with the scope to find mammalian conserved editing sites. Surprisingly, only a very small fraction (0.004%) of human editing sites is conserved in mammals. Noteworthy, by considering the nucleotide frequency, the 10-nt upstream and downstream regions of conserved editing sites are stronger than the ones of all non-Alu human editing sites.
The large number of editing sites discovered by these methodologies has given rise to the need for public databases to record such information in order to further elucidate the biological functions underlying the RNA editing phenomenon. The first centralized repository was DARNED1 (Kiran and Baranov, 2010), whose last release contains more than 300,000 editing sites (Kiran and Baranov, 2010; Kiran et al., 2013). Later, Ramaswami and Li (2014) built RADAR2, a rigorously manually curated database of annotated A-to-I editing sites, amounting to about 1.4 million editing events. Unfortunately, both DARNED and RADAR do not offer a grade of confidence for each editing site due to the heterogeneity of the discovering methods applied, making the creation of a standard measure of confidence necessary in the future.
Investigation of Motifs Characterizing the RNA Editing Events
It is well known that the vast majority of editing events occur in repetitive regions. Recently, Ramaswami et al. (2012) developed a computational framework to identify editing events both Alu and non-Alu regions (repetitive non-Alu and non-repetitive regions) by analyzing the genomic DNA and RNA sequences. Through this method they found that more than 97% of the discovered editing events occur in Alu regions, also speculating that the remaining non-Alu editing sites are related to nearby edited Alu ones. This makes the identification of sequence motifs able to characterize RNA editing a very challenging problem. Therefore, any approach aimed at the search of sequence or structural motifs associated to RNA editing events should take into account the bias introduced by repetitive regions. Consequently, the searching should be done outside of repetitive regions in order to detect signals independent of the background.
Our strategy has been the following. First, we selected a set of non-Alu editing events and then generated edited regions (ERs) based on the distances between non-Alu editing site, as described below. Next, we applied MEME (Bailey et al., 2009) in order to discover motifs within such a set of sequences. MEME analyzes the input data and searches for significant ungapped sequence patterns shared among the sequences.
In order to obtain the ERs, considering the human editing sites listed in the RADAR database (Ramaswami and Li, 2014), we firstly filtered the A-to-I editing sites, which resulted to be SNPs, as compared to dbSNP141 (Solomon et al., 2014). We then computed δ as the weighted average distance between the editing sites. We obtained that on average there are 6,057 nt between two editing events. This value has been considered as a breakpoint during the construction of ERs. In particular, starting from a generic editing site x, we searched for the next one y. When y falls within a distance less than or equal to δ, the editing site y is included in the ER and the process continues. Otherwise, if the next site is found at a distance greater than δ, the ER is no longer extended. As a result, a total of 55,952 ERs have been defined. Additionally, we separated ERs containing repetitive elements from those, which do not contain any, obtaining a total of 48,164 repetitive ERs and 7,788 non-repetitive ERs. The fact that ERs possess different lengths could allow us to take into account the possibility that they may contain motifs close to the editing sites in secondary structures.
Figure 1 shows that repetitive ERs are longer than non-repetitive ones, with the largest number of editing sites found in regions containing some repetitive elements, as confirmed in the literature (Wahlstedt and O’Hman, 2011). We built a training set of non-repetitive ERs by selecting those regions with a length of 2,000–6,000 nt, containing at least 10 editing sites. Hence, we obtained a final dataset of 47 ERs, in particular, 29 regions are in positive strand with 479 editing sites and 18 ones are in negative strand with 319 editing sites.
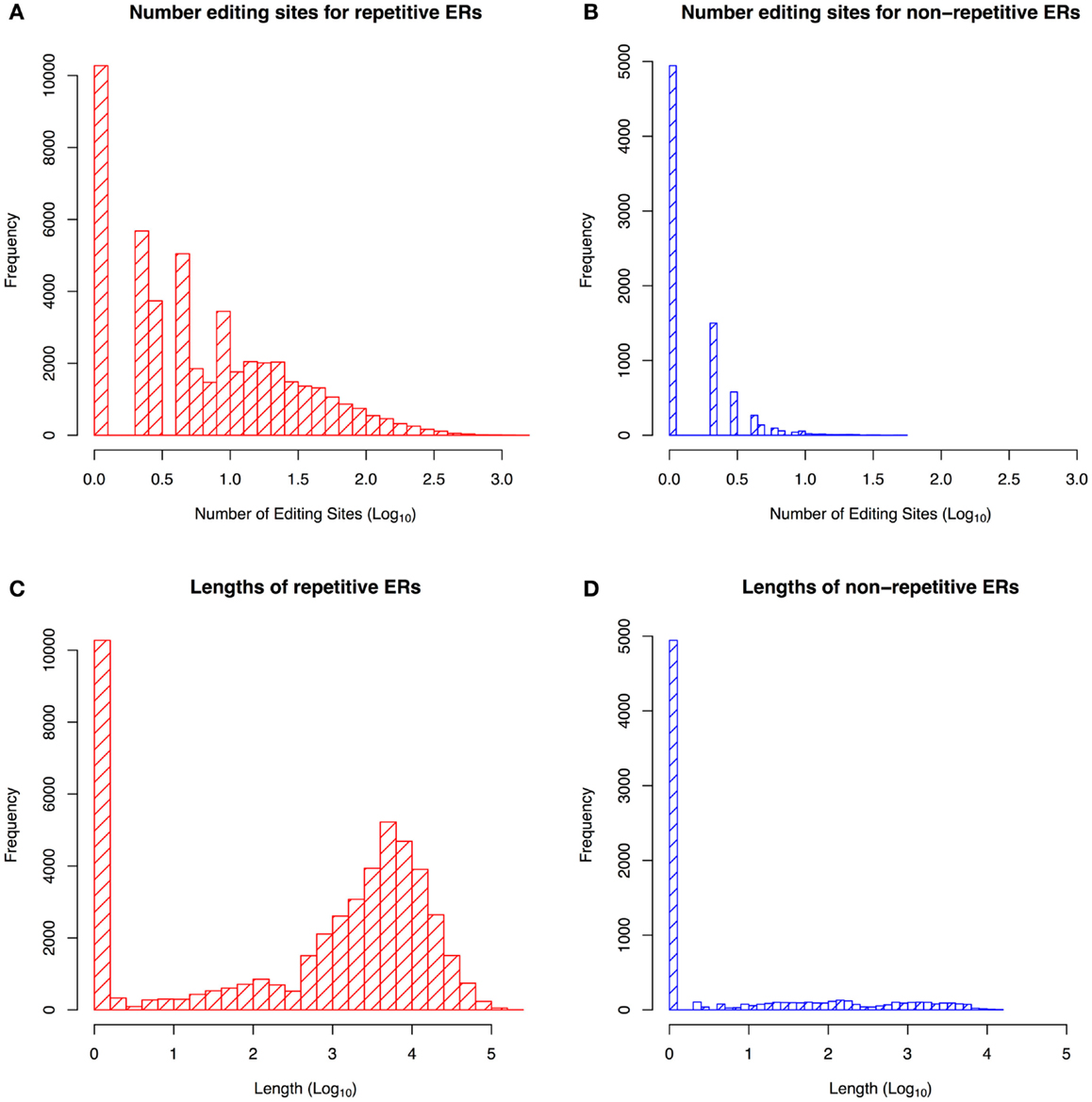
Figure 1. Statistics about the repetitive and non-repetitive edited regions (ER). Distribution of editing sites frequency in repetitive ERs (A) and non-repetitive ERs (B). Distribution of repetitive ERs sequence length (C) and non-repetitive ERs sequence length (D). The figure shows that the non-repetitive ERs are shorter than repetitive ones and contain fewer editing sites.
We ran MEME on such dataset by searching both palindromic and non-palindromic motifs with a length ranging from 6 to 50 nt. We bound the number of motifs to 50 palindromic and 50 non-palindromic.
From these 100 motifs we took only those with an E-value <0.05. Next, we filter out motifs that were contained in a set of human ultra-conserved sequences having no known editing site (Bejerano et al., 2004), with respect to DARNED and RADAR databases. Finally, a total of 16 motifs (4 palindromic and 12 non-palindromic) have been discovered.
In order to validate the filtered motifs, we performed a permutation test using 100 samples of 1,000 randomly taken 3′ UTR sequences (hg19) with masked repetitive regions. As shown in Table 1, only 13 motifs were significant (p-value <0.01).
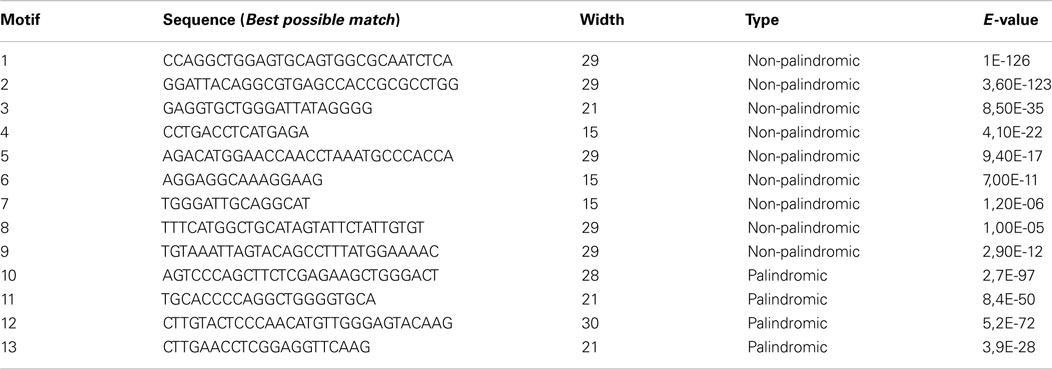
Table 1. Filtered motifs in ERs (47 edited regions).
From Nucleotide Frequency to an Approach to Assessment of A-to-I RNA Editing Sites
Starting from the idea proposed by Pinto et al. (2014), we used a logistic regression technique to determine a model from which we can compute the probability that an adenosine in a non-repetitive region of the genome is affected by the A-to-I editing phenomenon. Our method, called AIRlINER, determines the editing probability of an adenosine by analyzing its flanking region of 10 nt. Such pattern is then combined with a similar model calculated from un-edited sequences, resulting in the estimation of an unbiased editing probability.
In order to train our method, we built a dataset composed of 30,280 sequences of 21 nt centered on an adenosine, from the human genome (hg19). According to their provenance, our dataset can be divided equally into two sets: known editing sites and random sites. For the purpose of retrieving known editing sites in non-repetitive regions, only human sites which do not have any repetitive elements in their flanking regions of 2,000 nt were selected from the RADAR database (Ramaswami and Li, 2014). Random sites were chosen by randomly selecting a number of sequences equal to that of the known editing sites. From such a selection, we excluded known editing sites in both repetitive and non-repetitive regions.
From such a dataset, two probabilities P(j, i) and P’(j, i) can be computed: the first one corresponds to the probability of finding nucleotide j in position i of a region affected by editing, while the second one represents the probability of finding nucleotide j in position i of an un-ER. Starting from these probabilities, we computed the graphs in Figure 2, which represent the distributions of the nucleotides for the two types of regions.
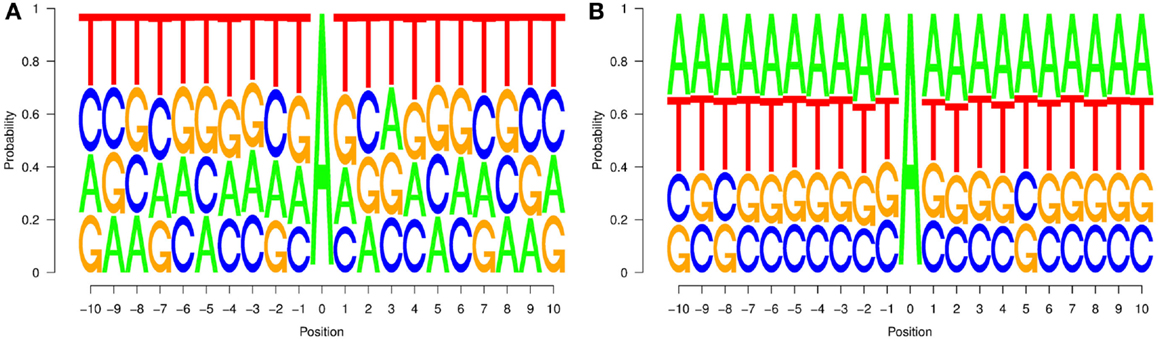
Figure 2. Neighborhood preferences that we computed for experimentally verified editing sites in non-repetitive regions (A) and random sites (B) chosen among those for which no editing event is reported. Neighborhood preferences are coherent with the upstream nucleotide distribution of editing site sequence contexts reported in Eggington et al. (2011).
Therefore, let s be a nucleotide sequence and P(s) its editing probability, using the previously defined probabilities we are able to train a logistic regression model such as:
where s[i] is the i-th nucleotide in a sequence. Now we can use this model to estimate the editing probability of any sequence of 21 nt centered on an adenosine, and if such probability is >0.5, we can say that such a sequence may be affected by editing.
To tune and validate our method, we applied a 10-fold cross validation procedure and computed a mean error. To compare our method with InosinePredict, we used a threshold to establish the presence or absence of editing in a specific sequence. Such a threshold was set to 9.6% for InosinePredict, as shown in Eggington et al. (2011). For our algorithm, we choose all sites for which an editing probability >0.5 is computed. We also took into account the fact that InosinePredict can produce predictions for both hADAR1 and hADAR2. We do not have this information in our dataset, so we chose to select the maximum score produced by InosinePredict for editing sites, and the minimum score for random sequences. Consequently, we are able to ensure a fair comparison with our method despite the absence of information on which ADAR affects each editing site.
In Tables 2 and 3, we show the confusion matrices computed using the previously described procedure. The two algorithms were applied to the dataset and the values computed for the central adenosines in each sequence were used to determine the presence or absence of editing. Our method significantly reduces the number of false negatives compared to InosinePredict, thus resulting in a better editing sites prediction quality. AIRlINER is also able to achieve a substantial reduction of false positives, even if nothing can be stated with certainty about them, as the absence of editing in these sites can also be determined by lack of experimental tests. The best quality in predicting editing sites, however, may reflect the fact that the random sequences classified as non-edited could be with high probability considered as such.

Table 2. Confusion matrix computed by applying InosinePredict (Eggington et al., 2011) to our dataset.

Table 3. Confusion matrix computed by applying AIRlINER to our dataset.
Further confirmation of the quality of our methodology is represented by the receiver operating characteristic curves (ROCs), Figure 3, computed from the results produced by the two algorithms. The curves demonstrate a significant improvement in performance. Such curves also show that the threshold chosen to distinguish editing sites from non-editing ones does not affect the performance difference between the two algorithms. As a confirmation of this, InosinePredict obtains an average area under the ROC curve (AUC) of 0.5072, while AIRlINER reaches 0.7466. In Figure 3, we also compare a variant of our method, AIRlINER 4 nt, with InosinePredict. Such a variant computes the editing probability of an adenosine by considering its flanking region of 4 nt. This comparison shows that our strategy is superior to InosinePredict even when the prediction is calculated from this same region around an adenosine.
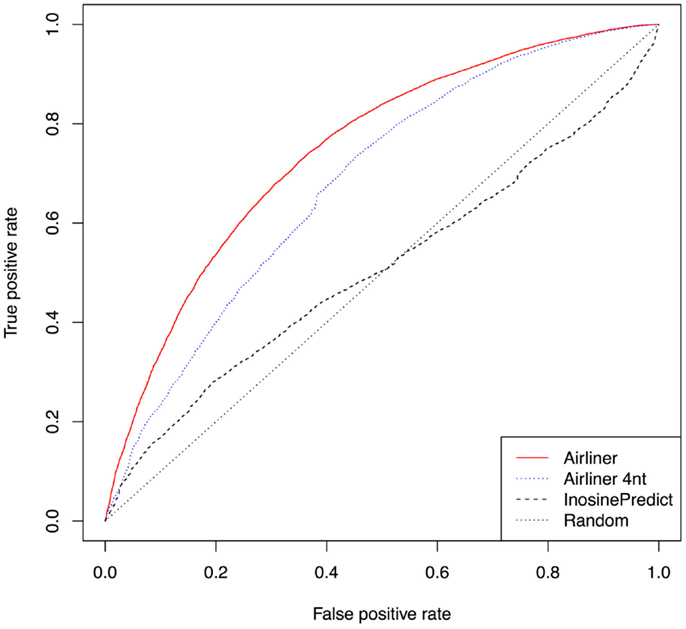
Figure 3. Receiver operating characteristic curve (ROC) computed for the two prediction algorithms. We also provide a ROC curve for a variant of our algorithm (AIRlINER 4 nt), which takes into account only the flanking region of 4 nt around an adenosine. Such a curve is useful to compare the performance with our algorithm using the same flanking region. AIRlINER shows an average area under the ROC curve (AUC) equal to 0.7466, while InosinePredict gets an AUC of 0.5072. AIRlINER 4 nt has an AUC of 0.7464.
Furthermore, we investigated that ADAR acts on each editing site in our training set by building an additional data set from editing sites experimentally identified in (Bahn et al., 2012). Using human cell lines U87MG in which the gene expression of ADAR1 was repressed, the authors were able to identify about 4,000 ADAR1-specific editing sites. Four hundreds of such sites were identified in non-repetitive regions. From the latter, we have built a training set using the same procedure described above and trained our model. In Figure 4, we show the results of this experiment by means of ROC curves. Even in this case, the AIRlINER methodology is significantly better than InosinePredict. As further confirmation, we also computed the AUC, which amounts to 0.6763 for AIRlINER, and 0.4498 for InosinePredict.
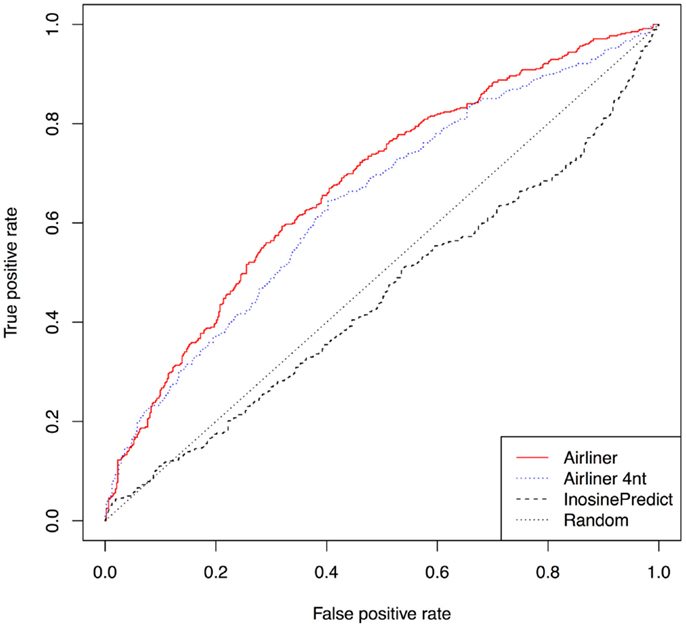
Figure 4. Comparison between AIRlINER and InosinePredict by means of receiver operating characteristic curve (ROC) computed using the data set built from Bahn et al. (2012). Here we also show a ROC curve for a variant of the proposed algorithm (AIRlINER 4 nt), which takes into account only the flanking region of 4 nt around an adenosine. AIRlINER shows an average area under the ROC curve (AUC) equal to 0.6763, while InosinePredict gets an AUC of 0.4498. AIRlINER 4 nt has an AUC of 0.6435.
Finally, to verify the quality of the editing sites predicted by our algorithm, we selected from the literature 52 experimentally validated sites by Sanger method and 7 sites validated as non-edited (as shown in Table S1 in Supplementary Material). We then applied the two methodologies and checked how many of them are correctly identified. AIRlINER is able to predict 42 of 52 editing sites and 5 of 7 non-editing sites while InosinePredict identifies 26 editing sites and 4 non-editing ones. More details can be found in the Table S1 in Supplementary Material.
AIRlINER is available as a web app at the following URL: http://alpha.dmi.unict.it/airliner/.
Conclusion and Future Directions
RNA editing is a post-transcriptional phenomenon that occurs in eukaryotes and contributes to the diversity of transcriptome. A-to-I is the most common form of RNA editing in mammals, altering the sequence of primary RNA transcripts by adenosine deamination. In this last decade, computational methods and RNAseq based approaches to RNA editing discovery have emerged, contributing to the identification of more than a million editing events in human, many of which located close to or within Alu repeats. Despite the enormous efforts made so far, the biological significance of the editing phenomenon remains largely unknown.
In the first part of this work, we summarized some of the most important characteristics discovered for RNA editing. Inspired by literature, we investigated the presence of motifs in non-repetitive regions characterizing the editing events, finding a small set of candidates. Moreover, we considered the frequency of the 20 nt centered on each RNA editing site to compute the probability that an adenosine in a non-repetitive region of the genome may be affected by the A-to-I editing phenomenon. Our method, available on line, significantly reduces the number of false negatives with respect to existing methods, thus indicating a better editing-site prediction quality.
Future work will concern the use of different motif-detecting algorithms to confirm the consistency of our current findings. Motif detection methods may make use of information from the secondary structure of the editing regions with respect also to the different classes of ADAR. Finally, further investigation is needed to highlight any significant combination of motif patterns.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
GN has been supported by Italian Foundation for Cancer Research (NG 15046). We also wish to thank Dario Veneziano for reviewing the English of the final version of the article. AP, RG and AF have been partially supported by a PON 2007–2013 grant, SIGMA – PON01_00683 – CUP B61H11000380005.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Journal/10.3389/fbioe.2015.00018/abstract
Footnotes
References
Athanasiadis, A., Rich, A., and Maas, S. (2004). Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2:e391. doi: 10.1371/journal.pbio.0020391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bahn, J. H., Lee, J.-H., Li, G., Greer, C., Peng, G., and Xiao, X. (2012). Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 22, 142–150. doi:10.1101/gr.124107.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi:10.1093/nar/gkp335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bass, B. L. (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846. doi:10.1146/annurev.biochem.71.110601.135501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bass, B. L., and Weintraub, H. (1987). A developmentally regulated activity that unwinds RNA duplexes. Cell 48, 607–613. doi:10.1016/0092-8674(87)90239-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bass, B. L., and Weintraub, H. (1988). An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098. doi:10.1016/0092-8674(88)90253-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bazak, L., Haviv, A., Barak, M., Jacob-Hirsch, J., Deng, P., Zhang, R., et al. (2014a). A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 24, 365–376. doi:10.1101/gr.164749.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bazak, L., Levanon, E. Y., and Eisenberg, E. (2014b). Genome-wide analysis of Alu editability. Nucleic Acids Res. 42, 6876–6884. doi:10.1093/nar/gku414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S., et al. (2004). Ultraconserved elements in the human genome. Science 304, 1321–1325. doi:10.1126/science.1098119
Borchert, G. M., Gilmore, B. L., Spengler, R. M., Xing, Y., Lanier, W., Bhattacharya, D., et al. (2009). Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum. Mol. Genet. 18, 4801–4807. doi:10.1093/hmg/ddp443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burns, C. M., Chu, H., Rueter, S. M., Hutchinson, L. K., Canton, H., Sanders-Bush, E., et al. (1997). Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387, 303–308. doi:10.1038/387303a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carmi, S., Borukhov, I., and Levanon, E. Y. (2011). Identification of widespread ultra-edited human RNAs. PLoS Genet. 7:e1002317. doi:10.1371/journal.pgen.1002317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, C. X., Cho, D. S., Wang, Q., Lai, F., Carter, K. C., and Nishikura, K. (2000). A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6, 755–767. doi:10.1017/S1355838200000170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eggington, J. M., Greene, T., and Bass, B. L. (2011). Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2, 319. doi:10.1038/ncomms1324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galeano, F., Tomaselli, S., Locatelli, F., and Gallo, A. (2012). A-to-I RNA editing: the “ADAR” side of human cancer. Sem. Cell Dev. Biol. 23, 244–250. doi:10.1016/j.semcdb.2011.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoopengardner, B., Bhalla, T., Staber, C., and Reenan, R. (2003). Nervous system targets of RNA editing identified by comparative genomics. Science 301, 832–836. doi:10.1126/science.1086763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jepson, J. E. C., and Reenan, R. A. (2008). RNA editing in regulating gene expression in the brain. Biochim. Biophys. Acta 1779, 459–470. doi:10.1016/j.bbagrm.2007.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ju, Y. S., Kim, J.-I., Kim, S., Hong, D., Park, H., Shin, J.-Y., et al. (2011). Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 43, 745–752. doi:10.1038/ng.872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawahara, Y. (2012). Quantification of adenosine-to-inosine editing of microRNAs using a conventional method. Nat. Protoc. 7, 1426–1437. doi:10.1038/nprot.2012.073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kawahara, Y., Megraw, M., Kreider, E., Iizasa, H., Valente, L., Hatzigeorgiou, A. G., et al. (2008). Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 36, 5270–5280. doi:10.1093/nar/gkn479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, D. D. Y., Kim, T. T. Y., Walsh, T., Kobayashi, Y., Matise, T. C., Buyske, S., et al. (2004). Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res. 14, 1719–1725. doi:10.1101/gr.2855504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, U., Wang, Y., Sanford, T., Zeng, Y., and Nishikura, K. (1994). Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. U.S.A. 91, 11457–11461. doi:10.1073/pnas.91.24.11457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiran, A., and Baranov, P. V. (2010). DARNED: a database of RNa editing in humans. Bioinformatics 26, 1772–1776. doi:10.1093/bioinformatics/btq285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiran, A. M., O’Mahony, J. J., Sanjeev, K., and Baranov, P. V. (2013). Darned in 2013: inclusion of model organisms and linking with Wikipedia. Nucleic Acids Res. 41, D258–D261. doi:10.1093/nar/gks961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kleinman, C. L., and Majewski, J. (2012). Comment on “widespread RNA and DNA sequence differences in the human transcriptome”. Science 335, 1302–1302. doi:10.1126/science.1209658
Lai, F., Chen, C. X., Carter, K. C., and Nishikura, K. (1997). Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol. Cell. Biol. 17, 2413–2424.
Lehmann, K. A., and Bass, B. L. (2000). Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities †. Biochemistry 39, 12875–12884. doi:10.1021/bi001383g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levanon, E. Y., Eisenberg, E., Yelin, R., Nemzer, S., Hallegger, M., Shemesh, R., et al. (2004). Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005. doi:10.1038/nbt996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, J. B., Levanon, E. Y., Yoon, J.-K., Aach, J., Xie, B., Leproust, E., et al. (2009). Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324, 1210–1213. doi:10.1126/science.1170995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, W., Piskol, R., Tan, M. H., and Li, J. B. (2012). Comment on “widespread RNA and DNA sequence differences in the human transcriptome”. Science 335, 1302. doi:10.1126/science.1210624
Mitra, S. A., Mitra, A. P., and Triche, T. J. (2012). A central role for long non-coding RNA in cancer. Front. Genet. 3:17. doi:10.3389/fgene.2012.00017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishikura, K. (2006). Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat. Rev. Mol. Cell Biol. 7, 919–931. doi:10.1038/nrm2061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishikura, K. (2010). Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 79, 321–349. doi:10.1146/annurev-biochem-060208-105251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patterson, J. B., and Samuel, C. E. (1995). Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 15, 5376–5388.
Paul, M. S., and Bass, B. L. (1998). Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 17, 1120–1127. doi:10.1093/emboj/17.4.1120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peng, Z., Cheng, Y., Tan, B. C.-M., Kang, L., Tian, Z., Zhu, Y., et al. (2012). Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 30, 253–260. doi:10.1038/nbt.2122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Picardi, E., Gallo, A., Galeano, F., Tomaselli, S., and Pesole, G. (2012). A novel computational strategy to identify A-to-I RNA editing sites by RNA-Seq data: de novo detection in human spinal cord tissue. PLoS One 7:e44184. doi:10.1371/journal.pone.0044184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pickrell, J. K., Gilad, Y., and Pritchard, J. K. (2012). Comment on “widespread RNA and DNA sequence differences in the human transcriptome”. Science 335, 1302. doi:10.1126/science.1210484
Pinto, Y., Cohen, H. Y., and Levanon, E. Y. (2014). Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 15, R5. doi:10.1186/gb-2014-15-1-r5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Polson, A. G., and Bass, B. L. (1994). Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 13, 5701–5711.
Ramaswami, G., and Li, J. B. (2014). RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–D113. doi:10.1093/nar/gkt996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramaswami, G., Lin, W., Piskol, R., Tan, M. H., Davis, C., and Li, J. B. (2012). Accurate identification of human Alu and non-Alu RNA editing sites. Nat Meth 9, 579–581. doi:10.1038/nmeth.1982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramaswami, G., Zhang, R., Piskol, R., Keegan, L. P., Deng, P., O’Connell, M. A. A., et al. (2013). Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 10, 128–132. doi:10.1038/nmeth.2330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rebagliati, M. R., and Melton, D. A. (1987). Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell 48, 599–605. doi:10.1016/0092-8674(87)90238-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Riedmann, E. M., Schopoff, S., Hartner, J. C., and Jantsch, M. F. (2008). Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14, 1110–1118. doi:10.1261/rna.923308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rueter, S. M., Dawson, T. R., and Emeson, R. B. (1999). Regulation of alternative splicing by RNA editing. Nature 399, 75–80. doi:10.1038/19992
Sakurai, M., Ueda, H., Yano, T., Okada, S., Terajima, H., Mitsuyama, T., et al. (2014). A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 24, 522–534. doi:10.1101/gr.162537.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakurai, M., Yano, T., Kawabata, H., Ueda, H., and Suzuki, T. (2010). Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat. Chem. Biol. 6, 733–740. doi:10.1038/nchembio.434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Solomon, O., Bazak, L., Levanon, E. Y., Amariglio, N., Unger, R., Rechavi, G., et al. (2014). Characterizing of functional human coding RNA editing from evolutionary, structural, and dynamic perspectives. Proteins 82, 3117–3131. doi:10.1002/prot.24672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Su, A. A. H., and Randau, L. (2011). A-to-I and C-to-U editing within transfer RNAs. Biochemistry(Mosc.) 76, 932–937. doi:10.1134/S0006297911080098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tomaselli, S., Locatelli, F., and Gallo, A. (2014). The RNA editing enzymes ADARs: mechanism of action and human disease. Cell Tissue Res. 356, 527–532. doi:10.1007/s00441-014-1863-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagner, R. W., Smith, J. E., Cooperman, B. S., and Nishikura, K. (1989). A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl. Acad. Sci. U.S.A. 86, 2647–2651. doi:10.1073/pnas.86.8.2647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wahlstedt, H., and O’Hman, M. (2011). Site-selective versus promiscuous A-to-I editing. Wiley Interdiscip Rev RNA 2, 761–771. doi:10.1002/wrna.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, S. K., Sato, S., and Lazinski, D. W. (2001). Substrate recognition by ADAR1 and ADAR2. RNA 7, 846–858. doi:10.1017/S135583820101007X
Yang, J. H., Sklar, P., Axel, R., and Maniatis, T. (1997). Purification and characterization of a human RNA adenosine deaminase for glutamate receptor B pre-mRNA editing. Proc. Natl. Acad. Sci. U.S.A. 94, 4354–4359. doi:10.1073/pnas.94.9.4354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: A-to-I RNA editing, motif analysis, prediction, ADARs, logistic regression
Citation: Nigita G, Alaimo S, Ferro A, Giugno R and Pulvirenti A (2015) Knowledge in the investigation of A-to-I RNA editing signals. Front. Bioeng. Biotechnol. 3:18. doi: 10.3389/fbioe.2015.00018
Received: 14 November 2014; Accepted: 07 February 2015;
Published online: 24 February 2015.
Edited by:
Marco Pellegrini, Consiglio Nazionale delle Ricerche, ItalyCopyright: © 2015 Nigita, Alaimo, Ferro, Giugno and Pulvirenti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosalba Giugno and Alfredo Pulvirenti, Department of Clinical and Experimental Medicine, University of Catania, Via Santa Sofia, Catania 78 | I-95122, Italy e-mail: giugno@dmi.unict.it; apulvirenti@dmi.unict.it
†Giovanni Nigita and Salvatore Alaimo have contributed equally to this work.
‡Rosalba Giugno and Alfredo Pulvirenti have contributed equally to this work.