Detection of genomic structural variants from next-generation sequencing data
 Lorenzo Tattini
Lorenzo Tattini Romina D’Aurizio
Romina D’Aurizio Alberto Magi
Alberto Magi- 1Department of Neurosciences, Psychology, Pharmacology and Child Health, University of Florence, Florence, Italy
- 2Laboratory of Integrative Systems Medicine (LISM), Institute of Informatics and Telematics and Institute of Clinical Physiology, National Research Council, Pisa, Italy
- 3Department of Clinical and Experimental Medicine, University of Florence, Florence, Italy
Structural variants are genomic rearrangements larger than 50 bp accounting for around 1% of the variation among human genomes. They impact on phenotypic diversity and play a role in various diseases including neurological/neurocognitive disorders and cancer development and progression. Dissecting structural variants from next-generation sequencing data presents several challenges and a number of approaches have been proposed in the literature. In this mini review, we describe and summarize the latest tools – and their underlying algorithms – designed for the analysis of whole-genome sequencing, whole-exome sequencing, custom captures, and amplicon sequencing data, pointing out the major advantages/drawbacks. We also report a summary of the most recent applications of third-generation sequencing platforms. This assessment provides a guided indication – with particular emphasis on human genetics and copy number variants – for researchers involved in the investigation of these genomic events.
Introduction
Structural variants (SVs) are genomic rearrangements affecting more then 50 bp. The average SV size detected by the 1000 Genomes Project is 8 kbp (1000 Genomes Project Consortium et al., 2010), whereas a study based on tiling CGH array (Conrad et al., 2010) reports a four times larger value. SVs comprise balanced as well as unbalanced events, namely, variants altering the total number of base pairs in a genome. Thus, SVs include deletions, insertions, inversions, mobile-element transpositions, translocations, tandem repeats, and copy number variants (CNVs).
Several databases – e.g., the Database of Genomic Variants archive which reports structural variation identified in healthy control samples (DGVa1) – have been created for the collection of SVs data (Lappalainen et al., 2013). Public data resources have been developed with the purpose of supporting the interpretation of clinically relevant variants, e.g., dbVar2, or collecting known disease genes (OMIM3) hit by SVs.
Structural variants account for 1.2% of the variation among human genomes while single nucleotide polymorphisms (SNPs) represent 0.1% (Pang et al., 2010). Notably, unbalanced events provide 99.8% of the entries reported in dbVar (Lin et al., 2014). CNVs may result in benign polymorphic variations or clinical phenotypes due to gene dosage alteration or gene disruption (Zhang et al., 2009). Though the impact of SVs in human genomics was first recognized by their presence in healthy individuals (Zhao et al., 2013), two models account for their association to human disease. Rare large events (<1%, hundreds kbp) have been related to neurological and neurocognitive disorders (Sebat et al., 2007; Girirajan et al., 2013), whereas multicopy gene families, which are commonly copy number variable, contribute to disease susceptibility.
Next-generation sequencing technologies (NGS) have been revolutionizing genome research [for a survey of NGS tools from quality check to variant annotation and visualization, see Pabinger et al. (2014)] as well as the study of CNVs (Duan et al., 2013; Zhao et al., 2013; Samarakoon et al., 2014; Tan et al., 2014; Alkodsi et al., 2015; Kadalayil et al., 2015) and SVs on the whole (Alkan et al., 2011a), replacing microarrays as the leading platform for the investigation of genomic rearrangement (Pinkel et al., 1998; Snijders et al., 2001; Iafrate et al., 2004; Sebat et al., 2007). NGS platforms are based on various implementations of cyclic-array sequencing (Shendure and Ji, 2008; Shendure et al., 2011). They allow for the sequencing of millions of short (few hundreds bp) DNA fragments (reads) simultaneously and may process a whole human genome in three days at 500-fold less cost than previous methods (Voelkerding et al., 2009; Metzker, 2010).
The 1000 Genomes Project applied methods based on all of the four approaches available for the detection of SVs, reporting false discovery rates ranging from 10 to 89%, remarkable differences in terms of genomic regions discovered, size range, and breakpoint precision (Mills et al., 2011; Teo et al., 2012).
Overview of the Approaches
Four strategies for the detection of SV signatures that are diagnostic of different rearrangements have been reported in the literature (Figure 1; Table 1).
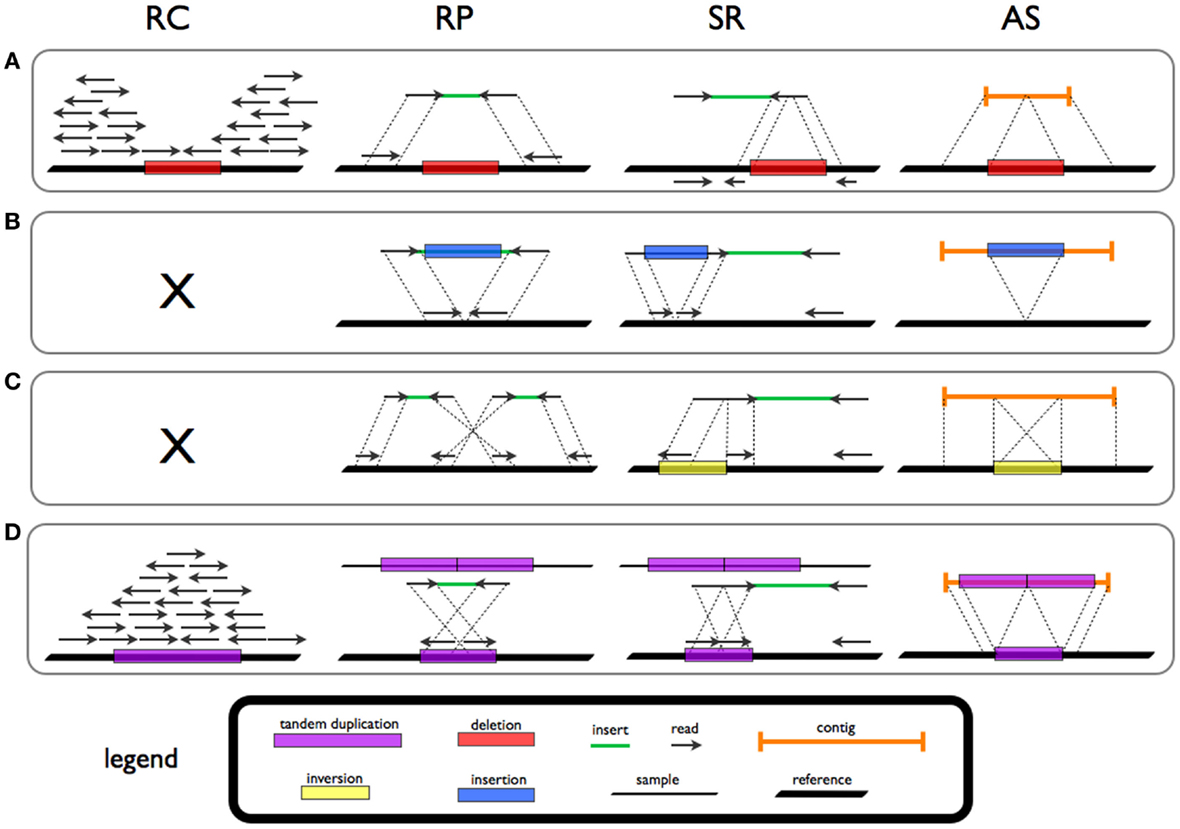
Figure 1. Signatures and patterns of SVs for deletion (A), novel sequence insertion (B), inversion (C), and tandem duplication (D) in read count (RC), read-pair (RP), split-read (SR), and de novo assembly (AS) methods.
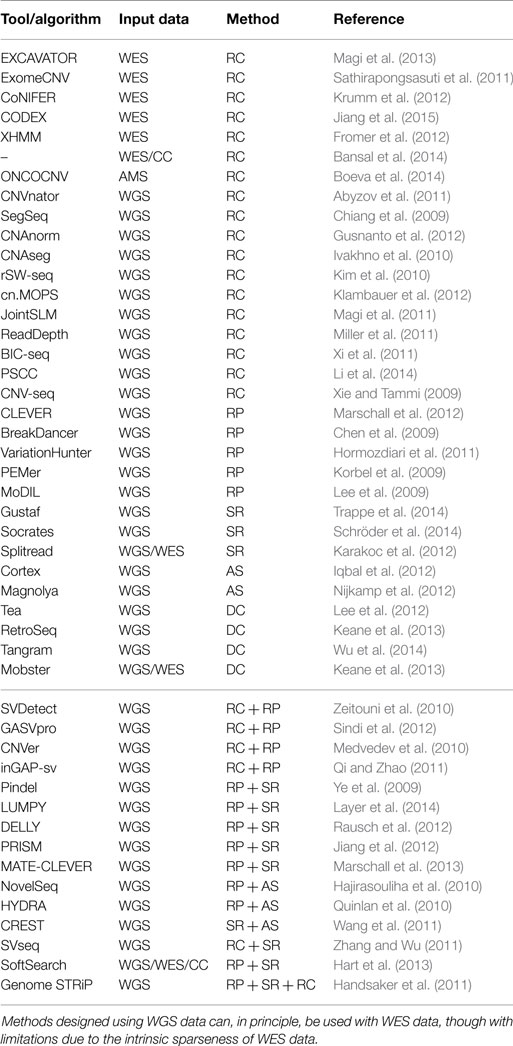
Table 1. A non-exhaustive summary of the tools/algorithms for the investigation of SVs, their input data (WGS, whole-genome sequencing; WES, whole-exome sequencing; CC, custom capture; AMS, amplicon sequencing), and their underling approach.
Read-pair (RP) methods are based on the evaluation of the span and orientation of paired-end reads. Discordant pairs in which the mapping span and/or orientation of the read pairs are inconsistent with the expected insert size are collected. Several classes of SVs can be investigated by means of this approach. Read pairs mapping too far apart are associated to deletions while those found closer than expected are indicative of insertions. Furthermore, orientation inconsistencies can represent inversions and a specific class of tandem duplications.
Read-depth (or read count, RC) approaches assume a random (Poisson or modified Poisson) distribution in mapping depth and investigate the divergence from this distribution to highlight duplications and deletions (Magi et al., 2012). Sequencing of duplicated/amplified regions results in higher read depth while deleted regions show reduced read depth when compared to normal (e.g., diploid) regions.
Split-read (SR) methods allow for the detection of SVs with single base-pair resolution. The presence of a SV breakpoint is investigated on the basis of a split sequence-read signature breaking the alignment to the reference. A gap in the read is a marker of a deletion while stretches in the reference reflect insertions.
Theoretically, all forms of structural variation could be investigated by means of de novo assembly (AS) methods. De novo assembly refers to merging and ordering short fragments to reassemble the original sequence from which the short fragments were sampled (Earl et al., 2011). NGS data intrinsic characteristics, such as (short) read length, limit the use of AS approaches for variant investigation.
Moreover, a specific class of SV, mobile elements (ME) insertions, can be investigated exploiting discordant and clipped (DC) read information.
Read Count Methods
Read count is suitable for the investigation of CNVs. RC methods comprise four steps: RC data preparation, data normalization, SV regions identification, and copy number estimation. Reads mapping to windows/bins of fixed size are counted (Yoon et al., 2009; Magi et al., 2011) and the results are normalized for the mitigation of local GC content and mappability effects.
The correlation between local GC content and read coverage has been detected through the analysis of data from several platforms (Harismendy et al., 2009). Mappability bias is due to repetitive regions within the human genome (Miller et al., 2011).
A segmentation step is necessary to split RC signal into segments characterized by a constant DNA copy number. Algorithms conceived for aCGH data such as the circular binary segmentation (CBS) algorithm (Campbell et al., 2008; Miller et al., 2011) and those based on hidden Markov models (HMM) (Magi et al., 2010) are used with this scope.
Copy number estimation can be tackled by means of two strategies. Both assume that the sequencing process is uniform. Thus, the number of reads mapping to a genomic region is expected to be proportional to the number of times the regions appears in the DNA sample. Three methods (Campbell et al., 2008; Yoon et al., 2009; Magi et al., 2011) estimate DNA copy number of all the detected regions rounding the median RCs (normalized to copy number 2) to the nearest integer, while CNVnator (Abyzov et al., 2011) uses RC signal normalized to the genomic average for the regions of the same length.
A considerable number of methods for the detection of CNV in whole-genome sequencing (WGS) data have been reported in the literature, including CNVnator, CNAnorm, CNAseg, rSW-seq, cn.MOPS, JointSLM, ReadDepth, and BIC-seq (Ivakhno et al., 2010; Kim et al., 2010; Abyzov et al., 2011; Magi et al., 2011; Miller et al., 2011; Xi et al., 2011; Gusnanto et al., 2012; Klambauer et al., 2012). Recently, PSCC (Li et al., 2014) has been compared with SegSeq (Chiang et al., 2009) and ReadDepth (Miller et al., 2011).
CNV Detection from Whole-Exome Data
Due to the costs associated to WGS, the investigation of CNVs using whole-exome sequencing (WES) data is definitely an attractive perspective. Nevertheless, the sparse nature of the target and the non-uniform read-depth among captured regions make CNV detection from WES data awkward with respect to WGS [in particular, regarding the segmentation step as reported in Magi et al. (2013)].
Several tools have been reported in the literature for this purpose including ExomeCNV (Sathirapongsasuti et al., 2011), CoNIFER (Krumm et al., 2012), CNV-seq (Xie and Tammi, 2009), XHMM (Fromer et al., 2012), and recently EXCAVATOR (Magi et al., 2013) and CODEX (Jiang et al., 2015). Notably, the method developed by Bansal and co-workers (Bansal et al., 2014) allows for the analysis of NGS data generated from small subsets of the exome, namely custom capture (CC) data.
Amplicon Sequencing Data
Amplicon sequencing (AMS) techniques have been reported in the literature in particular for clinical applications (Desai and Jere, 2012; Beadling et al., 2013).
Amplicon sequencing data show different biases in respect of WES data (Boeva et al., 2014). Data normalization can be less effective due to the limited number of target regions. Furthermore, protocols involved in the preparation of amplicon libraries result in high depth of coverage at the expense of coverage homogeneity.
The first method designed for the investigation of CNV from AMS data is ONCOCNV. Duplicate sequences are not removed, while RC is performed assigning “each read to only one amplicon region, the one with which the read alignment has the maximum overlap” (Boeva et al., 2014).
Data are then normalized with respect to library size assuming a similar efficiency of PCR amplification for all the targeted regions. GC content and amplicon length biases are corrected by means of a local polynomial regression fitting. Principal component analysis (PCA) is employed to construct a baseline reflecting the technological bias in control samples. The baseline is calculated by means of the first three principal components (calculated from control samples data). In order to define a significant threshold to call a copy number change, the standard deviation of the normalized RCs for each amplicon region is calculated.
This procedure is applied to data from test samples keeping the residuals of the linear regression of normalized RCs over the baseline calculated for the control samples.
Segmentation of the resulting signal profile is performed with CBS method (Venkatraman and Olshen, 2007). A segmentation and clustering approach (SCA) is used to define the copy number state (neutral, gain, or loss) of the segmented regions.
Read-Pair Algorithms
As already mentioned, RP methods, as well as SR approaches, are suitable for the detection of several classes of SV including insertions of novel sequences and inversions. Notably, RP algorithms cannot detect the signatures of novel sequence insertions larger than the average insert size. Several tools based on the detection of SV signatures from clusters of read-pairs have been reported in the literature including BreakDancer, VariationHunter, PEMer, and GASV (Chen et al., 2009; Hormozdiari et al., 2009, 2011; Korbel et al., 2009; Sindi et al., 2009). Remarkably, PEMer can be exploited for the identification of linked insertions (Medvedev et al., 2010).
Clusters can be defined according to two strategies. The standard clustering strategy relies on two parameters: the minimum number of pairs with similar signature and the maximum value of the mean insert size standard deviation for a pair to be considered concordant. The maximum standard deviation value is fixed and events spanning the same locus, resulting in a small value of the insert size standard deviation, may be missed.
Distribution-based approaches, e.g., MoDIL (Lee et al., 2009), exploit the local distribution of all the mappings spanning a particular location on the genome. A read cluster is generated when the local distribution is shifted in respect to the typical insert size distribution. This approach allows for the detection of smaller events (e.g., compared with VariationHunter). The presence of two superimposed insert size distributions can be also detected, thus allowing for the discrimination of homozygous and heterozygous variants.
In the first implementations of the approach, e.g., BreakDancer (Chen et al., 2009), reads with multiple mappings were discarded. Thus, repetitive regions of the genome (including segmental duplications and copy-number amplifications) could not be investigated. Notably, BreakDancer allows for the identification of inter- and intra-chromosomal translocations. Tools such as MoDIL and VariationHunter or, more recently, CLEVER (Marschall et al., 2012) deal with multiple mapping reads [aligned, for instance, with mrFast (Alkan et al., 2009), Mosaik (Lee et al., 2014), BWA (Li and Durbin, 2010), or Bowtie (Langmead et al., 2009)]. CLEVER uses an insert size-based approach to build a graph with all reads and evaluates SV from maximal cliques. It is particularly well-tuned for the investigation of insertions and deletions of 50–100 bp.
Split-Read Approaches
Though SR methods were conceived for Sanger sequencing reads (Mills et al., 2006), algorithms such as Pindel, Splitread, and Gustaf (Ye et al., 2009; Karakoc et al., 2012; Trappe et al., 2014) use paired-end NGS reads to identify SVs (or indel) events. SR approaches take advantage of one-end anchored reads, namely those pairs in which “one end is anchored to the reference genome and the other end maps imprecisely owing to the presence of an underlying structural variant or indel breakpoint” (Karakoc et al., 2012). SR-based tools can be applied solely to unique reference regions.
Pindel uses pattern growth for optimal matching in target regions, exploiting reads mapped with SSAHA2 [Sequence Search and Alignment by Hashing Algorithm, Ning et al. (2001)], BWA, or Mosaik. It must be stressed that the latest version of Pindel integrates RP to the SR information (Lin et al., 2014). Splitread searches for clusters of split reads using balanced splits as seeds. Splitread can detect, at least in theory, deletions without size limitation, while for insertions the size spectrum depends on the sequencing library. Insertions shorter than the read length can be accurately identified but larger insertions can only be approximately characterized within the insert size (Karakoc et al., 2012). Splitread is suitable for WGS/WES reads aligned using mrsFAST (Hach et al., 2010) to discover indels, SVs, de novo events, and pseudogenes.
Recently, Socrates (a SR method designed for cancer genomics) was compared to several tools (Schröder et al., 2014), including BreakDancer, CLEVER, CREST (Wang et al., 2011), DELLY (Rausch et al., 2012), Pindel, and PRISM (Jiang et al., 2012).
Assembly Based Tools
De novo assembly allows – at least in principle – for the detection of all the forms of structural variation but the application of this approach is still challenging due to the limited length of NGS reads (Alkan et al., 2011a; O’Rawe et al., 2015).
AS methods were first exploited for Sanger sequencing data (characterized by read length between 300 and 1000 bp). The original string graph approach has been extended to de Bruijn graphs. The Assemblathon competition (Earl et al., 2011) produced a detailed comparison among de novo assemblers, including Phusion2 (Mullikin and Ning, 2003), SGA (Simpson and Durbin, 2010, 2012), Quake (Kelley et al., 2010), the first implementation of SOAPdenovo (Li et al., 2010; Luo et al., 2012), and ALLPATHS-LG (Gnerre et al., 2011), based on simulated data.
Two AS based callers have been reported in the literature for the investigation of SVs. Magnolya (Nijkamp et al., 2012) uses a Poisson mixture model (PMM) for CNV detection from contigs co-assembled from NGS sequencing data. The authors use an overlap-layout-consensus assembler to generate a contig string graph. Contig string graphs are characterized by nodes representing reads and edges representing an overlap. The final form of the graph is produced by transitive reduction – which removes redundant edges – and by unitigging (i.e., collapsing simple paths without branches) (Myers, 2005). In the resulting contig string graph, each node represents a collapsed set of reads called contig. Finally, the PMM approach for modeling read count is introduced to estimate the copy number of a contig. Once the model has been corrected for the presence of repetitive regions in the genome and prior knowledge on ploidy has been included, the model with the optimal number of Poisson distributions is selected by means of the lowest Bayesian information criterion. Integer copy numbers can be thus inferred by maximum a posteriori estimation. Remarkably, the method can be applied when no reference is available but – as already stressed – it is limited by the short read length typical of NGS platforms.
Cortex uses colored de Bruijn graphs with colors of both edges and nodes representing different samples and, possibly, reference sequences or known variants to assemble NGS reads. “The graph consists of a set of nodes representing words of length k (k-mers). Directed edges join k-mers seen consecutively in the input” (Iqbal et al., 2012). The package includes four algorithms for variant discovery. For example, the bubble calling algorithm may be exploited for the detection of variant bubbles in a colored de Bruijn graph from a single diploid individual. It must be stressed that using a reference genome aids the identification of variants while it is indispensable for the investigation of homozygous variant sites. Nevertheless, the sensitivity of the method decreases with the size of the variant. The tool has been extensively tested on human data.
Combined Methods
None of the aforementioned approaches is capable of capturing the full spectrum of SV events with high sensitivity and specificity. RC methods can accurately predict absolute copy numbers but the breakpoint resolution is often inadequate and events such as inversions and novel sequence insertions cannot be detected. On the other hand, RP and SR approaches show low sensitivity in repetitive regions. Several packages combining different approaches for the investigation of SVs have been reported.
Combining RC for the detection of large events and RP for accurate identification of breakpoints can reduce the number of false positive calls [SVDetect (Zeitouni et al., 2010), CNVer (Medvedev et al., 2010), GASVPro (Sindi et al., 2012), and inGAP-sv (Qi and Zhao, 2011)]. Genome STRiP (Handsaker et al., 2011) exploits RP, RC, SR, and population-scale patterns to detect genome structural polymorphisms.
Packages implementing RP and (local) AS have been also reported [NovelSeq (Hajirasouliha et al., 2010), HYDRA (Quinlan et al., 2010)] as well as tools exploiting SR and RC/RP such as SVseq, MATE-CLEVER, and PRISM (Zhang and Wu, 2011; Jiang et al., 2012; Marschall et al., 2013). PRISM was tested on simulated data and compared with Pindel, SVseq, Splitread, and CREST. Notably, DELLY is suitable for detecting copy-number variable deletion and tandem duplication events as well as balanced rearrangements such as inversions or reciprocal translocations (Rausch et al., 2012), while SoftSearch (Hart et al., 2013) is designed for WGS, WES, and CC data. Recently, LUMPY has been shown to be “especially pronounced when evidence is scarce, either due to low coverage data or low variant allele frequency” (Layer et al., 2014). LUMPY is designed to integrate signals rather then refining primary signal with a secondary one. Furthermore, the tool combines different types of evidence from multiple samples.
Detection of Mobile Elements
Mobile elements are repetitive DNA sequences that can change position within the genome (Lander et al., 2001). Due to this intrinsic characteristic, their detection is challenging. Latest estimates suggest that more than half of the human genome is repetitive or repeat-derived (de Koning et al., 2011). Though the DC approach can be ascribed to RP and SR methods, “the mates of the anchoring reads are then mapped to a custom but configurable library of known active ME consensus sequences” (Thung et al., 2014).
Among WGS tools, Tangram (Wu et al., 2014), a tool developed using Mosaik (Lee et al., 2014) alignments (though it may use alignments produced by other mappers), Next-Generation VariationHunter (Hormozdiari et al., 2010), Tea (Lee et al., 2012), RetroSeq eKeane:2013kq, and Mobster (Thung et al., 2014) have been reported in the literature.
Conclusion
Overall, all the approaches discussed are fairly limited with respect to repeated regions of the reference genome (Alkan et al., 2009, 2011b). The complete range of structural DNA variation cannot be investigated with a single tool (Mills et al., 2011), though combined methods may aid the discovery of SV. Three pipelines integrating different tools exploiting WGS data have been reported in the literature (Wong et al., 2010; Lam et al., 2012; Mimori et al., 2013). WES data can be exploited for the investigation of SVs by means of RC, SR, and RP methods – though with limitations due to the intrinsic sparseness of exomic data.
Each method for the detection of SVs shows advantages/drawback. RC methods are particularly well-suited for the investigation of a particular class of SV, namely CNV. Notably, RC can be used to predict absolute copy number. A major drawback of RC tools is the poor breakpoint resolution. Furthermore, they cannot distinguish tandem from interspersed duplications. SR algorithms can accurately predict SV breakpoint (down to single-base resolution) as well as AS methods. Finally, the RP and SR approaches can be applied for the investigation of the widest range of SV classes (i.e., deletions, inversions, novel sequence insertions, tandem duplications), though both cannot be exploited for the calculation of absolute copy number.
The advent of third-generation sequencing (TGS) technology may contribute to overcome these issues (Schadt et al., 2010; Niedringhaus et al., 2011; Pareek et al., 2011; Venkatesan and Bashir, 2011). TGS single-end reads, characterized by read length up to thousands base pairs, may boost AS methods and the application of mapping algorithms allowing for split alignment such as BWA (Li and Durbin, 2010), LAST (Kiełbasa et al., 2011) and BLASR (Chaisson and Tesler, 2012). Though TGS platforms rely on different chemistry, reads produced by platforms, such as PacBio RS (Kim et al., 2014) and Oxford Nanopore MinION (Bayley, 2015), show similar read length and base-calling accuracy (~85%) (Quail et al., 2012; Quick et al., 2014; Ashton et al., 2015; Chaisson et al., 2015). Recent works have demonstrated that these technologies allow for the investigation of complex repetitive regions of the human genome (Chaisson et al., 2015) as well as the structure of complex antibiotic resistance islands in Salmonella typhi (Ashton et al., 2015) and tandem repeats in human bacterial artificial chromosome (Jain et al., 2015).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
1000 Genomes Project Consortium, Abecasis, G. R., Altshuler, D., Auton, A., Brooks, L. D., Durbin, R. M., et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. doi: 10.1038/nature09534
Abyzov, A., Urban, A. E., Snyder, M., and Gerstein, M. (2011). Cnvnator: an approach to discover, genotype, and characterize typical and atypical cnvs from family and population genome sequencing. Genome Res. 21, 974–984. doi:10.1101/gr.114876.110
Alkan, C., Coe, B. P., and Eichler, E. E. (2011a). Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376. doi:10.1038/nrg2958
Alkan, C., Sajjadian, S., and Eichler, E. E. (2011b). Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65. doi:10.1038/nmeth.1527
Alkan, C., Kidd, J. M., Marques-Bonet, T., Aksay, G., Antonacci, F., Hormozdiari, F., et al. (2009). Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41, 1061–1067. doi:10.1038/ng.437
Alkodsi, A., Louhimo, R., and Hautaniemi, S. (2015). Comparative analysis of methods for identifying somatic copy number alterations from deep sequencing data. Brief. Bioinform. 16, 242–254. doi:10.1093/bib/bbu004
Ashton, P. M., Nair, S., Dallman, T., Rubino, S., Rabsch, W., Mwaigwisya, S., et al. (2015). MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 33, 296–300. doi:10.1038/nbt.3103
Bansal, V., Dorn, C., Grunert, M., Klaassen, S., Hetzer, R., Berger, F., et al. (2014). Outlier-based identification of copy number variations using targeted resequencing in a small cohort of patients with tetralogy of fallot. PLoS ONE 9:e85375. doi:10.1371/journal.pone.0085375
Bayley, H. (2015). Nanopore sequencing: from imagination to reality. Clin. Chem. 61, 25–31. doi:10.1373/clinchem.2014.223016
Beadling, C., Neff, T. L., Heinrich, M. C., Rhodes, K., Thornton, M., Leamon, J., et al. (2013). Combining highly multiplexed pcr with semiconductor-based sequencing for rapid cancer genotyping. J. Mol. Diagn. 15, 171–176. doi:10.1016/j.jmoldx.2012.09.003
Boeva, V., Popova, T., Lienard, M., Toffoli, S., Kamal, M., Le Tourneau, C., et al. (2014). Multi-factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinformatics 30, 3443–3450. doi:10.1093/bioinformatics/btu436
Campbell, P. J., Stephens, P. J., Pleasance, E. D., O’Meara, S., Li, H., Santarius, T., et al. (2008). Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 40, 722–729. doi:10.1038/ng.128
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (blasr): application and theory. BMC Bioinformatics 13:238. doi:10.1186/1471-2105-13-238
Chaisson, M. J. P., Huddleston, J., Dennis, M. Y., Sudmant, P. H., Malig, M., Hormozdiari, F., et al. (2015). Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611. doi:10.1038/nature13907
Chen, K., Wallis, J. W., McLellan, M. D., Larson, D. E., Kalicki, J. M., Pohl, C. S., et al. (2009). Breakdancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681. doi:10.1038/nmeth.1363
Chiang, D. Y., Getz, G., Jaffe, D. B., O’Kelly, M. J. T., Zhao, X., Carter, S. L., et al. (2009). High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods 6, 99–103. doi:10.1038/nmeth.1276
Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y., et al. (2010). Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712. doi:10.1038/nature08516
de Koning, A. P. J., Gu, W., Castoe, T. A., Batzer, M. A., and Pollock, D. D. (2011). Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7:e1002384. doi:10.1371/journal.pgen.1002384
Desai, A. N., and Jere, A. (2012). Next-generation sequencing: ready for the clinics? Clin. Genet. 81, 503–510. doi:10.1111/j.1399-0004.2012.01865.x
Duan, J., Zhang, J.-G., Deng, H.-W., and Wang, Y.-P. (2013). Comparative studies of copy number variation detection methods for next-generation sequencing technologies. PLoS ONE 8:e59128. doi:10.1371/journal.pone.0059128
Earl, D., Bradnam, K., St John, J., Darling, A., Lin, D., Fass, J., et al. (2011). Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 21, 2224–2241. doi:10.1101/gr.126599.111
Fromer, M., Moran, J. L., Chambert, K., Banks, E., Bergen, S. E., Ruderfer, D. M., et al. (2012). Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 91, 597–607. doi:10.1016/j.ajhg.2012.08.005
Girirajan, S., Dennis, M. Y., Baker, C., Malig, M., Coe, B. P., Campbell, C. D., et al. (2013). Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 92, 221–237. doi:10.1016/j.ajhg.2012.12.016
Gnerre, S., Maccallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518. doi:10.1073/pnas.1017351108
Gusnanto, A., Wood, H. M., Pawitan, Y., Rabbitts, P., and Berri, S. (2012). Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics 28, 40–47. doi:10.1093/bioinformatics/btr593
Hach, F., Hormozdiari, F., Alkan, C., Hormozdiari, F., Birol, I., Eichler, E. E., et al. (2010). mrsfast: a cache-oblivious algorithm for short-read mapping. Nat. Methods 7, 576–577. doi:10.1038/nmeth0810-576
Hajirasouliha, I., Hormozdiari, F., Alkan, C., Kidd, J. M., Birol, I., Eichler, E. E., et al. (2010). Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics 26, 1277–1283. doi:10.1093/bioinformatics/btq152
Handsaker, R. E., Korn, J. M., Nemesh, J., and McCarroll, S. A. (2011). Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 43, 269–276. doi:10.1038/ng.768
Harismendy, O., Ng, P. C., Strausberg, R. L., Wang, X., Stockwell, T. B., Beeson, K. Y., et al. (2009). Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 10, R32. doi:10.1186/gb-2009-10-3-r32
Hart, S. N., Sarangi, V., Moore, R., Baheti, S., Bhavsar, J. D., Couch, F. J., et al. (2013). Softsearch: integration of multiple sequence features to identify breakpoints of structural variations. PLoS ONE 8:e83356. doi:10.1371/journal.pone.0083356
Hormozdiari, F., Alkan, C., Eichler, E. E., and Sahinalp, S. C. (2009). Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 19, 1270–1278. doi:10.1101/gr.088633.108
Hormozdiari, F., Hajirasouliha, I., Dao, P., Hach, F., Yorukoglu, D., Alkan, C., et al. (2010). Next-generation variationhunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics 26, i350–i357. doi:10.1093/bioinformatics/btq216
Hormozdiari, F., Hajirasouliha, I., McPherson, A., Eichler, E. E., and Sahinalp, S. C. (2011). Simultaneous structural variation discovery among multiple paired-end sequenced genomes. Genome Res. 21, 2203–2212. doi:10.1101/gr.120501.111
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y., et al. (2004). Detection of large-scale variation in the human genome. Nat. Genet. 36, 949–951. doi:10.1038/ng1416
Iqbal, Z., Caccamo, M., Turner, I., Flicek, P., and McVean, G. (2012). De novo assembly and genotyping of variants using colored de bruijn graphs. Nat. Genet. 44, 226–232. doi:10.1038/ng.1028
Ivakhno, S., Royce, T., Cox, A. J., Evers, D. J., Cheetham, R. K., and Tavaré, S. (2010). Cnaseg – a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics 26, 3051–3058. doi:10.1093/bioinformatics/btq587
Jain, M., Fiddes, I. T., Miga, K. H., Olsen, H. E., Paten, B., and Akeson, M. (2015). Improved data analysis for the minion nanopore sequencer. Nat. Methods 12, 351–356. doi:10.1038/nmeth.3290
Jiang, Y., Oldridge, D. A., Diskin, S. J., and Zhang, N. R. (2015). Codex: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 43, e39. doi:10.1093/nar/gku1363
Jiang, Y., Wang, Y., and Brudno, M. (2012). Prism: pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants. Bioinformatics 28, 2576–2583. doi:10.1093/bioinformatics/bts484
Kadalayil, L., Rafiq, S., Rose-Zerilli, M. J. J., Pengelly, R. J., Parker, H., Oscier, D., et al. (2015). Exome sequence read depth methods for identifying copy number changes. Brief. Bioinform. 16, 380–392. doi:10.1093/bib/bbu027
Karakoc, E., Alkan, C., O’Roak, B. J., Dennis, M. Y., Vives, L., Mark, K., et al. (2012). Detection of structural variants and indels within exome data. Nat. Methods 9, 176–178. doi:10.1038/nmeth.1810
Keane, T. M., Wong, K., and Adams, D. J. (2013). Retroseq: transposable element discovery from next-generation sequencing data. Bioinformatics 29, 389–390. doi:10.1093/bioinformatics/bts697
Kelley, D. R., Schatz, M. C., and Salzberg, S. L. (2010). Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 11, R116. doi:10.1186/gb-2010-11-11-r116
Kiełbasa, S. M., Wan, R., Sato, K., Horton, P., and Frith, M. C. (2011). Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493. doi:10.1101/gr.113985.110
Kim, K. E., Peluso, P., Babayan, P., Yeadon, P. J., Yu, C., Fisher, W. W., et al. (2014). Long-read, whole-genome shotgun sequence data for five model organisms. Sci. Data 1, 140045. doi:10.1038/sdata.2014.45
Kim, T.-M., Luquette, L. J., Xi, R., and Park, P. J. (2010). rsw-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinformatics 11:432. doi:10.1186/1471-2105-11-432
Klambauer, G., Schwarzbauer, K., Mayr, A., Clevert, D.-A., Mitterecker, A., Bodenhofer, U., et al. (2012). cn.mops: mixture of poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40, e69. doi:10.1093/nar/gks003
Korbel, J. O., Abyzov, A., Mu, X. J., Carriero, N., Cayting, P., Zhang, Z., et al. (2009). Pemer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 10, R23. doi:10.1186/gb-2009-10-2-r23
Krumm, N., Sudmant, P. H., Ko, A., O’Roak, B. J., Malig, M., Coe, B. P., et al. (2012). Copy number variation detection and genotyping from exome sequence data. Genome Res. 22, 1525–1532. doi:10.1101/gr.138115.112
Lam, H. Y. K., Pan, C., Clark, M. J., Lacroute, P., Chen, R., Haraksingh, R., et al. (2012). Detecting and annotating genetic variations using the hugeseq pipeline. Nat. Biotechnol. 30, 226–229. doi:10.1038/nbt.2134
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi:10.1038/35057062
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi:10.1186/gb-2009-10-3-r25
Lappalainen, I., Lopez, J., Skipper, L., Hefferon, T., Spalding, J. D., Garner, J., et al. (2013). Dbvar and dgva: public archives for genomic structural variation. Nucleic Acids Res. 41, D936–D941. doi:10.1093/nar/gks1213
Layer, R. M., Chiang, C., Quinlan, A. R., and Hall, I. M. (2014). Lumpy: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84. doi:10.1186/gb-2014-15-6-r84
Lee, E., Iskow, R., Yang, L., Gokcumen, O., Haseley, P., Luquette, L. J. III, et al. (2012). Landscape of somatic retrotransposition in human cancers. Science 337, 967–971. doi:10.1126/science.1222077
Lee, S., Hormozdiari, F., Alkan, C., and Brudno, M. (2009). Modil: detecting small indels from clone-end sequencing with mixtures of distributions. Nat. Methods 6, 473–474. doi:10.1038/nmeth.f.256
Lee, W.-P., Stromberg, M. P., Ward, A., Stewart, C., Garrison, E. P., and Marth, G. T. (2014). Mosaik: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS ONE 9:e90581. doi:10.1371/journal.pone.0090581
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi:10.1093/bioinformatics/btp698
Li, R., Fan, W., Tian, G., Zhu, H., He, L., Cai, J., et al. (2010). The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317. doi:10.1038/nature08696
Li, X., Chen, S., Xie, W., Vogel, I., Choy, K. W., Chen, F., et al. (2014). Pscc: sensitive and reliable population-scale copy number variation detection method based on low coverage sequencing. PLoS ONE 9:e85096. doi:10.1371/journal.pone.0085096
Lin, K., Smit, S., Bonnema, G., Sanchez-Perez, G., and de Ridder, D. (2014). Making the difference: integrating structural variation detection tools. Brief. Bioinform. doi:10.1093/bib/bbu047
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). Soapdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi:10.1186/2047-217X-1-18
Magi, A., Benelli, M., Marseglia, G., Nannetti, G., Scordo, M. R., and Torricelli, F. (2010). A shifting level model algorithm that identifies aberrations in array-cgh data. Biostatistics 11, 265–280. doi:10.1093/biostatistics/kxp051
Magi, A., Benelli, M., Yoon, S., Roviello, F., and Torricelli, F. (2011). Detecting common copy number variants in high-throughput sequencing data by using jointslm algorithm. Nucleic Acids Res. 39, e65. doi:10.1093/nar/gkr068
Magi, A., Tattini, L., Cifola, I., D’Aurizio, R., Benelli, M., Mangano, E., et al. (2013). Excavator: detecting copy number variants from whole-exome sequencing data. Genome Biol. 14, R120. doi:10.1186/gb-2013-14-10-r120
Magi, A., Tattini, L., Pippucci, T., Torricelli, F., and Benelli, M. (2012). Read count approach for dna copy number variants detection. Bioinformatics 28, 470–478. doi:10.1093/bioinformatics/btr707
Marschall, T., Costa, I. G., Canzar, S., Bauer, M., Klau, G. W., Schliep, A., et al. (2012). Clever: clique-enumerating variant finder. Bioinformatics 28, 2875–2882. doi:10.1093/bioinformatics/bts566
Marschall, T., Hajirasouliha, I., and Schönhuth, A. (2013). Mate-clever: Mendelian-inheritance-aware discovery and genotyping of midsize and long indels. Bioinformatics 29, 3143–3150. doi:10.1093/bioinformatics/btt556
Medvedev, P., Fiume, M., Dzamba, M., Smith, T., and Brudno, M. (2010). Detecting copy number variation with mated short reads. Genome Res. 20, 1613–1622. doi:10.1101/gr.106344.110
Metzker, M. L. (2010). Sequencing technologies – the next generation. Nat. Rev. Genet. 11, 31–46. doi:10.1038/nrg2626
Miller, C. A., Hampton, O., Coarfa, C., and Milosavljevic, A. (2011). Readdepth: a parallel r package for detecting copy number alterations from short sequencing reads. PLoS ONE 6:e16327. doi:10.1371/journal.pone.0016327
Mills, R. E., Luttig, C. T., Larkins, C. E., Beauchamp, A., Tsui, C., Pittard, W. S., et al. (2006). An initial map of insertion and deletion (indel) variation in the human genome. Genome Res. 16, 1182–1190. doi:10.1101/gr.4565806
Mills, R. E., Walter, K., Stewart, C., Handsaker, R. E., Chen, K., Alkan, C., et al. (2011). Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65. doi:10.1038/nature09708
Mimori, T., Nariai, N., Kojima, K., Takahashi, M., Ono, A., Sato, Y., et al. (2013). isvp: an integrated structural variant calling pipeline from high-throughput sequencing data. BMC Syst. Biol. 7(Suppl. 6):S8. doi:10.1186/1752-0509-7-S6-S8
Mullikin, J. C., and Ning, Z. (2003). The phusion assembler. Genome Res. 13, 81–90. doi:10.1101/gr.731003
Myers, E. W. (2005). The fragment assembly string graph. Bioinformatics 21(Suppl. 2), ii79–ii85. doi:10.1093/bioinformatics/bti1114
Niedringhaus, T. P., Milanova, D., Kerby, M. B., Snyder, M. P., and Barron, A. E. (2011). Landscape of next-generation sequencing technologies. Anal. Chem. 83, 4327–4341. doi:10.1021/ac2010857
Nijkamp, J. F., van den Broek, M. A., Geertman, J.-M. A., Reinders, M. J. T., Daran, J.-M. G., and de Ridder, D. (2012). De novo detection of copy number variation by co-assembly. Bioinformatics 28, 3195–3202. doi:10.1093/bioinformatics/bts601
Ning, Z., Cox, A. J., and Mullikin, J. C. (2001). Ssaha: a fast search method for large dna databases. Genome Res. 11, 1725–1729. doi:10.1101/gr.194201
O’Rawe, J. A., Ferson, S., and Lyon, G. J. (2015). Accounting for uncertainty in dna sequencing data. Trends Genet. 31, 61–66. doi:10.1016/j.tig.2014.12.002
Pabinger, S., Dander, A., Fischer, M., Snajder, R., Sperk, M., Efremova, M., et al. (2014). A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinformatics 15, 256–278. doi:10.1093/bib/bbs086
Pang, A. W., MacDonald, J. R., Pinto, D., Wei, J., Rafiq, M. A., Conrad, D. F., et al. (2010). Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 11, R52. doi:10.1186/gb-2010-11-5-r52
Pareek, C. S., Smoczynski, R., and Tretyn, A. (2011). Sequencing technologies and genome sequencing. J. Appl. Genet. 52, 413–435. doi:10.1007/s13353-011-0057-x
Pinkel, D., Segraves, R., Sudar, D., Clark, S., Poole, I., Kowbel, D., et al. (1998). High resolution analysis of dna copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20, 207–211. doi:10.1038/2524
Qi, J., and Zhao, F. (2011). ingap-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res. 39, W567–W575. doi:10.1093/nar/gkr506
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of ion torrent, pacific biosciences and illumina miseq sequencers. BMC Genomics 13:341. doi:10.1186/1471-2164-13-341
Quick, J., Quinlan, A. R., and Loman, N. J. (2014). A reference bacterial genome dataset generated on the minion™ portable single-molecule nanopore sequencer. Gigascience 3, 22. doi:10.1186/2047-217X-3-22
Quinlan, A. R., Clark, R. A., Sokolova, S., Leibowitz, M. L., Zhang, Y., Hurles, M. E., et al. (2010). Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 20, 623–635. doi:10.1101/gr.102970.109
Rausch, T., Zichner, T., Schlattl, A., Stütz, A. M., Benes, V., and Korbel, J. O. (2012). Delly: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. doi:10.1093/bioinformatics/bts378
Samarakoon, P. S., Sorte, H. S., Kristiansen, B. E., Skodje, T., Sheng, Y., Tjønnfjord, G. E., et al. (2014). Identification of copy number variants from exome sequence data. BMC Genomics 15:661. doi:10.1186/1471-2164-15-661
Sathirapongsasuti, J. F., Lee, H., Horst, B. A. J., Brunner, G., Cochran, A. J., Binder, S., et al. (2011). Exome sequencing-based copy-number variation and loss of heterozygosity detection: Exomecnv. Bioinformatics 27, 2648–2654. doi:10.1093/bioinformatics/btr462
Schadt, E. E., Turner, S., and Kasarskis, A. (2010). A window into third-generation sequencing. Hum. Mol. Genet. 19, R227–R240. doi:10.1093/hmg/ddq416
Schröder, J., Hsu, A., Boyle, S. E., Macintyre, G., Cmero, M., Tothill, R. W., et al. (2014). Socrates: identification of genomic rearrangements in tumour genomes by re-aligning soft clipped reads. Bioinformatics. 30, 1064–1072. doi:10.1093/bioinformatics/btt767
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. doi:10.1126/science.1138659
Shendure, J., and Ji, H. (2008). Next-generation dna sequencing. Nat. Biotechnol. 26, 1135–1145. doi:10.1038/nbt1486
Shendure, J. A., Porreca, G. J., Church, G. M., Gardner, A. F., Hendrickson, C. L., Kieleczawa, J., et al. (2011). Overview of dna sequencing strategies. Curr. Protoc. Mol. Biol. Chapter 7, Unit7.1. doi:10.1002/0471142727.mb0701s96
Simpson, J. T., and Durbin, R. (2010). Efficient construction of an assembly string graph using the fm-index. Bioinformatics 26, i367–i373. doi:10.1093/bioinformatics/btq217
Simpson, J. T., and Durbin, R. (2012). Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556. doi:10.1101/gr.126953.111
Sindi, S., Helman, E., Bashir, A., and Raphael, B. J. (2009). A geometric approach for classification and comparison of structural variants. Bioinformatics 25, i222–i230. doi:10.1093/bioinformatics/btp208
Sindi, S. S., Onal, S., Peng, L. C., Wu, H.-T., and Raphael, B. J. (2012). An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol. 13, R22. doi:10.1186/gb-2012-13-3-r22
Snijders, A. M., Nowak, N., Segraves, R., Blackwood, S., Brown, N., Conroy, J., et al. (2001). Assembly of microarrays for genome-wide measurement of dna copy number. Nat. Genet. 29, 263–264. doi:10.1038/ng754
Tan, R., Wang, Y., Kleinstein, S. E., Liu, Y., Zhu, X., Guo, H., et al. (2014). An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum. Mutat. 35, 899–907. doi:10.1002/humu.22537
Teo, S. M., Pawitan, Y., Ku, C. S., Chia, K. S., and Salim, A. (2012). Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics 28, 2711–2718. doi:10.1093/bioinformatics/bts535
Thung, D., de Ligt, J., Vissers, L., Steehouwer, M., Kroon, M., de Vries, P., et al. (2014). Mobster: accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 15, 488. doi:10.1186/s13059-014-0488-x
Trappe, K., Emde, A.-K., Ehrlich, H.-C., and Reinert, K. (2014). Gustaf: detecting and correctly classifying SVs in the NGS twilight zone. Bioinformatics 30, 3484–3490. doi:10.1093/bioinformatics/btu431
Venkatesan, B. M., and Bashir, R. (2011). Nanopore sensors for nucleic acid analysis. Nat. Nanotechnol. 6, 615–624. doi:10.1038/nnano.2011.129
Venkatraman, E. S., and Olshen, A. B. (2007). A faster circular binary segmentation algorithm for the analysis of array cgh data. Bioinformatics 23, 657–663. doi:10.1093/bioinformatics/btl646
Voelkerding, K. V., Dames, S. A., and Durtschi, J. D. (2009). Next-generation sequencing: from basic research to diagnostics. Clin. Chem. 55, 641–658. doi:10.1373/clinchem.2008.112789
Wang, J., Mullighan, C. G., Easton, J., Roberts, S., Heatley, S. L., Ma, J., et al. (2011). Crest maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 8, 652–654. doi:10.1038/nmeth.1628
Wong, K., Keane, T. M., Stalker, J., and Adams, D. J. (2010). Enhanced structural variant and breakpoint detection using svmerge by integration of multiple detection methods and local assembly. Genome Biol. 11, R128. doi:10.1186/gb-2010-11-12-r128
Wu, J., Lee, W.-P., Ward, A., Walker, J. A., Konkel, M. K., Batzer, M. A., et al. (2014). Tangram: a comprehensive toolbox for mobile element insertion detection. BMC Genomics 15:795. doi:10.1186/1471-2164-15-795
Xi, R., Hadjipanayis, A. G., Luquette, L. J., Kim, T.-M., Lee, E., Zhang, J., et al. (2011). Copy number variation detection in whole-genome sequencing data using the bayesian information criterion. Proc. Natl. Acad. Sci. U.S.A. 108, E1128–E1136. doi:10.1073/pnas.1110574108
Xie, C., and Tammi, M. T. (2009). Cnv-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10:80. doi:10.1186/1471-2105-10-80
Ye, K., Schulz, M. H., Long, Q., Apweiler, R., and Ning, Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. doi:10.1093/bioinformatics/btp394
Yoon, S., Xuan, Z., Makarov, V., Ye, K., and Sebat, J. (2009). Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 19, 1586–1592. doi:10.1101/gr.092981.109
Zeitouni, B., Boeva, V., Janoueix-Lerosey, I., Loeillet, S., Legoix-né, P., Nicolas, A., et al. (2010). Svdetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 26, 1895–1896. doi:10.1093/bioinformatics/btq293
Zhang, F., Gu, W., Hurles, M. E., and Lupski, J. R. (2009). Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 10, 451–481. doi:10.1146/annurev.genom.9.081307.164217
Zhang, J., and Wu, Y. (2011). Svseq: an approach for detecting exact breakpoints of deletions with low-coverage sequence data. Bioinformatics 27, 3228–3234. doi:10.1093/bioinformatics/btr563
Keywords: next generation sequencing, structural variants, copy number variants, statistical methods, whole-exome sequencing, whole-genome sequencing, amplicon sequencing
Citation: Tattini L, D’Aurizio R and Magi A (2015) Detection of genomic structural variants from next-generation sequencing data. Front. Bioeng. Biotechnol. 3:92. doi: 10.3389/fbioe.2015.00092
Received: 10 December 2014; Accepted: 10 June 2015;
Published: 25 June 2015
Edited by:
Marco Pellegrini, Consiglio Nazionale delle Ricerche, ItalyReviewed by:
Christian Cole, University of Dundee, UKAlexander Schönhuth, Centrum Wiskunde & Informatica, Netherlands
Copyright: © 2015 Tattini, D’Aurizio and Magi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo Tattini, Department of Neurosciences, Psychology, Pharmacology and Child Health, University of Florence, Viale Pieraccini, 6, Florence 50139, Italy, lorenzo.tattini@unifi.it