Mitochondrial dysfunction in inflammatory bowel disease
 Elizabeth A. Novak
Elizabeth A. Novak Kevin P. Mollen
Kevin P. Mollen- Department of Surgery, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Inflammatory Bowel Disease (IBD) represents a group of idiopathic disorders characterized by chronic or recurring inflammation of the gastrointestinal tract. While the exact etiology of disease is unknown, IBD is recognized to be a complex, multifactorial disease that results from an intricate interplay of genetic predisposition, an altered immune response, changes in the intestinal microbiota, and environmental factors. Together, these contribute to a destruction of the intestinal epithelial barrier, increased gut permeability, and an influx of immune cells. Given that most cellular functions as well as maintenance of the epithelial barrier is energy-dependent, it is logical to assume that mitochondrial dysfunction may play a key role in both the onset and recurrence of disease. Indeed several studies have demonstrated evidence of mitochondrial stress and alterations in mitochondrial function within the intestinal epithelium of patients with IBD and mice undergoing experimental colitis. Although the hallmarks of mitochondrial dysfunction, including oxidative stress and impaired ATP production are known to be evident in the intestines of patients with IBD, it is as yet unclear whether these processes occur as a cause of consequence of disease. We provide a current review of mitochondrial function in the setting of intestinal inflammation during IBD.
Introduction: Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a complex, chronic, relapsing, and remitting inflammatory condition of the gastrointestinal tract characterized by symptoms such as diarrhea, bloody stools, abdominal pain, and weight loss (Greco et al., 2011; Indriolo et al., 2011; Rigoli and Caruso, 2014). There are two diseases which fall under the heading of IBD: Crohn's disease and ulcerative colitis. Crohn's disease is characterized by transmural inflammation that may affect any part of the gastrointestinal tract, and presentation of disease is dependent upon both location and severity of inflammation (Podolsky, 2002; Indriolo et al., 2011). Inflammation in ulcerative colitis is limited to the mucosa of the colon and rectum. The pattern of clinical disease in IBD is often cyclical with periods of active inflammation and subsequent remissions (Indriolo et al., 2011). Additionally, there is a strong association between IBD and development of colorectal cancer (Persson et al., 1994; Canavan et al., 2006; Grivennikov, 2013). Although there is no cure for IBD, a range of therapeutics (e.g., corticosteroids, immunomodulators, antibiotics, aminosalicylates, and biologic therapies) is employed to help manage the symptoms of disease. The results of medical treatment are highly variable, and the potential exists for significant morbidity over a long lifetime with disease.
In the United States, it is currently estimated that approximately 1.6 million people suffer from IBD, with as many as 70,000 new cases reported each year (Crohn's and Colitis Foundation of America, 2014). The peak age of onset is between 15 and 35 years with approximately 5–10% of patients diagnosed during childhood (< 20 years). IBD is a chronic, lifelong disease that creates a vast financial burden. Previous studies have estimated that the annual direct health care cost for a patient with Crohn's disease is $8265–18,963 and $5066–15,020 per patient diagnosed with ulcerative colitis (Gibson et al., 2008). Extrapolating these figures onto the current prevalence estimates of IBD reveals an estimated annual total direct cost of between 11 and 28 billion dollars (Crohn's and Colitis Foundation of America, 2014).
While the exact pathophysiology of IBD is not yet understood, it is known that the disease is triggered by a complex interaction between genetic, environmental, and immunoregulatory factors. Studies have delineated a clear genetic link to disease. Children of parents affected by IBD have an increased risk of developing the disease (Noble and Arnott, 2008). The risk is significantly higher when both parents have IBD, with disease developing in up to 36% of people with two parents previously diagnosed with IBD (Bennett et al., 1991). Other studies have demonstrated a much higher disease frequency (5–20% increase) amongst first-degree relatives of affected individuals compared to the general population (Russell and Satsangi, 2008). The familial link appears to be stronger in Crohn's disease compared to ulcerative colitis (Tysk et al., 1988; Thompson et al., 1996; Orholm et al., 2000; Halfvarson et al., 2003). Although genetics clearly play a role in disease, the exact nature of genetic predisposition is quite complex, and it is possible that susceptibility to IBD may involve the interaction of several genes. To date, genome-wide association studies (GWAS) have identified more than 160 genetic loci that confer susceptibility to disease (Liu et al., 2015). The fact that genetic polymorphism alone does not predict disease, but merely confers risk of developing IBD highlights the importance that other elements, such as environmental constituents, must also be a contributing factor (Cho and Brant, 2011). Interestingly, the majority of the genetic loci confer susceptibility to both ulcerative colitis and Crohn's disease, calling into question the rigid categorization of IBD subsets.
Although the precise environmental factors that trigger IBD are not known, several risk factors, including antibiotic exposure, stress, dysbiosis, and nonsteroidal anti-inflammatory drug exposure (NSAIDs), are thought to play a role in disease onset and progression (Loftus, 2004; Bernstein et al., 2006; Bernstein, 2008; Molodecky and Kaplan, 2010). Research has demonstrated that IBD is more common in developed countries compared to developing countries, which suggests that many factors associated with the “westernized” lifestyle, such as diet, decreased exposure to sunlight, exposure to pollution and industrial chemicals, may be associated with disease development (Hanauer, 2006). Interestingly, the incidence of IBD in some developing countries (e.g., India and China) is beginning to rise as they become more industrialized (Desai and Gupte, 2005; Zheng et al., 2005). Likewise, migrant studies have revealed that when populations relocate from regions of low IBD prevalence to regions of higher prevalence, they acquire an increased risk of developing disease (Bernstein and Shanahan, 2008; Mikhailov and Furner, 2009). This highlights the importance of environmental factors in the onset and progression of disease in susceptible hosts (Hanauer, 2006).
An evolving body of literature would suggest that predisposing factors converge, resulting in a breakdown of the intestinal barrier and the translocation of luminal antigens. In genetically susceptible individuals, this bacterial translocation triggers a dysfunctional mucosal immune response and promotes inflammation. Although the theory of increased intestinal epithelial permeability as a primary cause of IBD has yet to be proven, it is supported by murine models of experimental colitis (Madsen et al., 1999; Resta-Lenert et al., 2005; Turner, 2009) and some human studies (Söderholm et al., 1999; Zeissig et al., 2007). Since the maintenance of epithelial junction integrity is energy-dependent, it would suggest that mitochondrial function might be central for the appropriate preservation of epithelial barrier function. Interestingly, constituents that have the potential to contribute to IBD susceptibility, such as gastrointestinal infection, and nonsteroidal anti-inflammatory drugs, have also been shown to affect mitochondrial function (Roediger, 1980a; Singh et al., 2009b; Schoultz et al., 2011). Additionally, structurally abnormal mitochondria have been observed in both animal models of intestinal disease (Rodenburg et al., 2008) and in tissues from patients with intestinal inflammation (Nazli et al., 2004). Moreover, processes which influence mitochondrial function, such as autophagy (Travassos et al., 2010), endoplasmic reticulum (ER) stress (Kaser et al., 2008), and the dysregulated production of reactive oxygen species (ROS) (Pavlick et al., 2002; Restivo et al., 2004; Beltrán et al., 2010) have all been implicated in IBD. Despite the present interest in mitochondrial function in the pathophysiology of diabetes (Chowdhury et al., 2013), obesity (Rath and Haller, 2011), and neuromuscular disease (Tarnopolsky and Raha, 2005), little is known about the biological behavior of mitochondria in intestinal inflammation. Here, we summarize the current literature that implicates mitochondrial dysfunction in the pathogenesis of IBD.
Mitochondrial Homeostasis
Mitochondria are membrane-bound organelles that maintain cellular energy production through oxidative phosphorylation (Mitchell and Moyle, 1967). Mitochondria contain a circular genome that encodes 13 proteins and the 22 tRNAs and 2 rRNAs needed to translate those proteins within the mitochondrial matrix. All 13 proteins encoded by mitochondrial DNA (mtDNA) form essential subunits of the respiratory complexes I, III, IV, and V (Anderson et al., 1981; Taanman, 1999). The small mitochondrial genome necessitates that nuclear-encoded genes provide the majority of proteins required for the respiratory apparatus as well as all of the enzymes involved in other cellular biosynthetic and oxidative functions (Anderson et al., 1981; Taanman, 1999). Despite the limited coding-capacity of the mtDNA, mitochondria regulate vital cellular functions aside from energy production, such as the generation of ROS and reactive nitrogen species (RNS), the induction of programmed cell death, and the transduction of stress and metabolic signals (Galluzzi et al., 2012; Tait and Green, 2012). Thus, it is necessary that mitochondrial dynamics be tightly controlled in order to maintain overall cellular homeostasis.
Mitochondrial biogenesis, the process by which new mitochondria are generated and repaired, plays a significant role in maintaining cellular metabolic homeostasis. Through the growth and division of established mitochondria, the transcription and assembly of new mitochondrial proteins, or the de novo synthesis of new mitochondria, mitochondrial biogenesis provides the cell with an adequate pool of healthy mitochondria. This process is influenced by numerous cellular environmental stresses, such as caloric restriction, hypothermia, exercise, cell division, and oxidative stress (Wenz, 2013). Variations in mitochondrial number, size, and mass exist between all cells and are reflective of the current cellular metabolic state (Leary et al., 1998; Leverve and Fontaine, 2001; Pfeiffer et al., 2001; Kunz, 2003). Mitochondrial biogenesis is a complex process, utilizing mitochondrial proteins encoded by both the mitochondrial and nuclear genomes; thus, precise communication between the mitochondria and nucleus is extremely important. Peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α) is a co-transcriptional regulation factor that is a central modulator of mitochondrial biogenesis (Puigserver et al., 1998). It drives biogenesis by activating various transcription factors, such as nuclear respiratory factor-1 (NRF-1) and nuclear respiratory factor-2 (NRF-2), which not only control the expression of nuclear genes that encode mitochondrial proteins, but also interact with mitochondrial transcription factor A (Tfam) (Jornayvaz and Shulman, 2010), which promotes the transcription and replication of the mitochondrial genome (Virbasius and Scarpulla, 1994).
The competing processes of mitochondrial fusion and fission operate to preserve mitochondrial function or eliminate irreparably damaged mitochondria, respectively. Through their role in regulating mitochondrial dynamics, fusion and fission events fine-tune biological processes central to cell survival, such as ATP generation, calcium homeostasis, and ROS generation. Consequently, they also play a role in apoptosis, mitophagy, cell-cycle progression, and oxygen sensing (Archer, 2013). Highly conserved guanosine triphosphates (GTPases) regulate both processes of fusion and fission (Youle and van der Bliek, 2012; Ishihara et al., 2013). Fusion is regulated by isoforms of two proteins in the outer mitochondrial membrane (OMM), mitofusion-1 and mitofusion-2, and by a dynamin family member, optic atrophy 1 (Opa1) protein, in the inner mitochondrial membrane (IMM) (Youle and van der Bliek, 2012). Mitofusions initiate fusion between neighboring mitochondria through the formation of homodimeric or heterodimeric linkages (Santel and Fuller, 2001; Chen et al., 2003; Hoppins et al., 2007). Opa1 then facilitates the merging of the IMMs (Alexander et al., 2000; Hoppins et al., 2007). Mitofusion-2 also localizes to the ER, where it alters mitochondrial and ER morphology and encourages ER-mitochondria tethering, which enhances calcium signaling (Rojo et al., 2002; de Brito and Scorrano, 2008). Fusion allows for mitochondrial complementation by permitting two mitochondria to fuse and compensate for the defects of each other, thereby generating all of the compulsory machineries for a functional mitochondrial organelle (Archer, 2013). Mitochondria with mtDNA mutations are allowed to fuse with other mitochondria as long as the total mutation burden remains below 80–90% for the cell (Yoneda et al., 1994; Nakada et al., 2001). Mitochondrial fusion is an attempt to buffer brief stresses and fractional defects through the exchange of components in the matrix and intermembrane space (Nunnari et al., 1997; Ono et al., 2001; Chan, 2006; Youle and van der Bliek, 2012).
When mitochondrial damage extends beyond a critical threshold, the quality control mechanisms of fission are initiated. Both ER-mitochondria interactions (Friedman et al., 2011) and the cytosolic protein dynamin-related protein 1 (Drp1) (Chen et al., 2003; Cribbs and Strack, 2009) are conserved features of mitochondrial fission. ER-mitochondria contact points mark the location of mitochondrial division where ER tubules physically wrap around and constrict the mitochondria, presumably to a diameter comparable to the Drp1 helices (Ingerman et al., 2005; Friedman et al., 2011). After ER constriction and upon activation, Drp1 translocates to and multimerizes around the OMM, where it pinches and severs both the IMM and OMM (Legesse-Miller et al., 2003; Lee et al., 2004; Zhu et al., 2004). Fission functions to isolate damaged components of mitochondria by segregating the damaged components of the organelle. After fission, the healthy mitochondrion is able to reincorporate into the network while the damaged mitochondrion is inhibited from reincorporation by a reduction in expression of fusion mediators, such as Opa1. This allows the damaged mitochondria to then be packaged into autophagic vacuoles that are delivered to the lysosome for disposal by the autophagic mechanism of mitophagy (Archer, 2013).
Severely damaged or superfluous mitochondria are degraded by the mitophagy—a specialized form of autophagy that targets individual mitochondria. During mitophagy, whole mitochondria are sequestered into autophagosomes and sent to lysosomes for degradation. Mitophagy is regulated by both the mitochondrial phosphatase and tensin homolog (PTEN)-induced kinase 1 (Pink1) and the cytosolic E3 ubiquitin ligase Parkin (Pellegrino and Haynes, 2015). In healthy mitochondria, expression of Pink1 is repressed by its transport into the IMM and subsequent degradation (Yamano and Youle, 2013; Thomas et al., 2014). However, in damaged mitochondria, Pink1 fails to be imported into the IMM, and instead integrates into the OMM (Geisler et al., 2010; Narendra et al., 2010) with its kinase domain exposed to the cytosol (Zhou et al., 2008), which subsequently recruits Parkin from the cytosol (Geisler et al., 2010; Narendra et al., 2010). Once recruited, Parkin ubiquitinates proteins on the OMM, targeting the mitochondrion for autophagic elimination (Narendra et al., 2008). This mitophagy pathway is also intimately connected with mitochondrial mobility. A major component of mitochondrial transport is mitochondria Rho-GTPase (Miro), a mitochondrial adaptor protein that attaches kinesin motors to the surface of mitochondria. Pink1 and Parkin associate with Miro upon depolarization of the mitochondrial membrane potential, triggering Pink1 to phosphorylate the mitochondrial adaptor protein, subsequently resulting in Parkin-dependent proteosomal degradation of Miro. Degradation of Miro causes the mitochondrion and the kinesin motor complex to separate, arresting mitochondrial motility (Wang et al., 2011). Arrest of mitochondrial motility, like degradation of mitochondrial fusion proteins, potentially functions to quarantine damaged mitochondria from reincorporating into the mitochondrial network, since static mitochondria are less prone to undergo fusion with other mitochondria (Twig et al., 2010). Homeostasis of the mitochondrial network as well as the proper functionality of the mitochondria is dependent on the cooperation of these cellular functions.
The mitochondrial population must be sustained in order to maintain cellular bioenergetic homeostasis and ensure cellular energy demands are being fulfilled. The plasticity of mitochondrial function and structure is an essential feature to maintaining cellular homeostasis, and indeed, changes in mitochondrial mass have been documented in both health and disease. For example, mitochondrial biogenesis increases in muscle cells upon exercise (Holloszy, 1967). Conversely, research has shown that as mammals age, there is a general decline in both mitochondrial mass and function (Yan and Sohal, 1998; Liu et al., 2002; Chistiakov et al., 2014). There is a wide range of clinical conditions that result from mitochondrial dysfunction, including muscular disorders, cardiomyopathy, diabetes, cancer, deafness, lactic acidosis, and skeletal myopathy (Vafai and Mootha, 2012). In addition, studies show that 1 in every 5000 individuals is affected by a mitochondrial disease (Pfeffer et al., 2012). Mitochondrial dysfunction can affect cell signaling through ROS and metabolites, and can interrupt the intimate physical connections between the mitochondria and other organelles (e.g., ER, etc.). Additionally, mitochondrial dysfunction has severe consequences on the bioenergetics of the cell. Understanding the complex responsibility mitochondria carry in the biology of cell processes and how mitochondrial dysfunction leads to disease can help target specific cellular mechanisms for the treatment and/or prevention of disease.
A variety of conditions and stimuli can alter mitochondrial function. Any disruption of mitochondrial performance can affect overall cellular function and eventually tissue/organ function. Here we review data supporting a role for mitochondrial dysfunction in the development and/or progression of IBD (Figure 1). Although a causative role of mitochondrial stress in IBD has not yet been established, the current literature would support a key correlation between mitochondrial function and intestinal inflammation. While we discuss several potential mechanisms by which mitochondrial function may impact disease, it is important to note that all of these processes are interconnected themselves. For example, an alteration in mitochondrial morphology can lead to defective mitochondrial function and communication, build-up of ROS, and activation of the inflammasome, potentially culminating in a disruption of the intestinal barrier, increased permeability, and ultimately intestinal inflammation. Additionally, some of the mechanistic links of mitochondrial dysfunction discussed are more strongly supported by scientific and clinical research (e.g., ROS generation, NLRP3 inflammasome, and autophagy). Nonetheless, it is important to understand how any alteration in the multifaceted functionality of the mitochondrion may contribute to the initiation and propagation of an inflammatory insult.
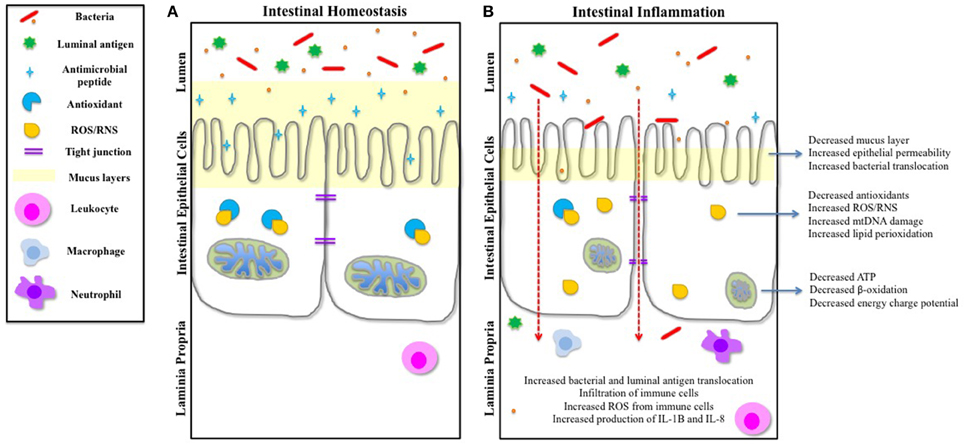
Figure 1. IEC function and intestinal homeostasis can be influenced by mitochondrial dysfunction. (A) During intestinal homeostasis, goblet cells produce a healthy mucus layer that protects the IECs from the contents of the lumen, and Paneth cells produce and release antimicrobial peptides to protect IECs. Mitochondria are dense and contain well-formed cristae. The tight junctions inhibit translocation of luminal antigens across the epithelial barrier. Any basal ROS produced is negated by cellular antioxidants. Leukocytes survey the laminia propria for threats. (B) Studies have shown that during the inflammatory conditions of IBD, the mucus layer is reduced and production of antimicrobial peptides is decreased, exposing the intestinal epithelium to the intestinal microbiota and luminal antigens. Mitochondria are swollen and abnormal, and cristae are irregular, resulting in a reduction in ATP production and an increase in ROS. Cellular antioxidants are also decreased, causing a buildup of cellular ROS. There is an increase in epithelial permeability (both transcellular and paracellular) and translocation of bacteria and luminal antigens. This results in an infiltration of immune cells, which also causes an increase of ROS. Both IL-8 and IL-1B are released by immune cells, and immune cell-bacterial interactions further instigates the release of pro-inflammatory mediators, which can feedback onto the IECs and influence other cellular components of the intestinal epithelium.
Mitochondrial Dysfunction in IBD
Form and Function
Mitochondrial form and function are intimately connected in normal cells. The mitochondria are compartmentalized organelles surrounded by two protein-containing phospholipid bilayers. The OMM encloses the entire organelle, and in conjunction with the IMM, separates the IMS and matrix compartments (Perkins and Frey, 2000; Strauss et al., 2008). Both the OMM and IMM contain translocases that function as mitochondrial protein entry ports, directing proteins to the correct subcompartment. The OMM also serves as a central signaling hub for several signal transduction pathways in the cell (Nunnari and Suomalainen, 2012). For example, innate antiviral immunity modulated by mitochondria is dependent upon mitochondrial antiviral signaling (MAVS), an OMM adaptor protein (Seth et al., 2005; Koshiba, 2013); and mitochondria-associated membranes (MAMs) are areas on the OMM where the ER and mitochondrion physically and functionally interact (Pizzo and Pozzan, 2007). The OMM is freely permeable to small molecules, and as such, the intermembrane space contains the same concentration of small molecules (e.g., ions, sugars, etc.) that are also present in the cytosol (Stowe and Camara, 2009). Cytochrome c, a protein that is integral to respiration as well as the induction of apoptosis, localizes to the intermembrane space (Koehler et al., 2006; Webb et al., 2006). The IMM, an impermeable membrane that allows for the gated exchanged of metabolites and proteins and undergoes intense folding into cristae to increase the membrane surface area, encloses the matrix compartment. The IMM facilitates lipid trafficking and respiratory complex formation (five complexes in mammals) (Perkins and Frey, 2000; Strauss et al., 2008), which are involved in oxidative phosphorylation and ATP production (Arco and Satrústegui, 2005). The matrix houses numerous copies of the circular mitochondrial genome as well as the machinery needed for its replication, transcription, and subsequent translation of the encoded proteins. Additionally within the matrix is a diverse set of enzymes required for cellular metabolic processes, including fatty-acid synthesis, the Tricarboxylic Acid Cycle (TCA), heme-synthesis, and iron-sulfur cluster formation (Ryan and Hoogenraad, 2007).
There are five primary functions of the mitochondria that are pivotal to mitochondrial form-function dynamics. First, mitochondrial biogenesis regulates the mitochondrial population in order to meet the energy requirements of the cell (Archer, 2013). Second, the cell maintains the health of mitochondria through the process of mitophagy, which eliminates damaged, depolarized mitochondria via lysosomal vacuoles. Mitophagy is facilitated by both the process of fission, which isolates depolarized mitochondria and suppresses fusion mediators, and by inhibiting the reorganization of the damaged mitochondria back into the network (Archer, 2013). Third, mitochondria are mobile organelles that transverse a dynamic network of dynein and kinesin within the cytosol (Wang et al., 2011). While the relationship between mitochondrial mobility and form and function is not clear, the dynein/dynactin complex is known to regulate the process of fission by recruiting Drp1 to the OMM (Varadi et al., 2004). Fourth, mitochondria are important oxygen-sensing beacons in the cell, and the initial steps in the mechanism of redox signaling depend upon mitochondrial dynamics (Marsboom et al., 2012; Hong et al., 2013). Lastly, mitochondria are linked to the ER through MAMs, enabling these two organelles to communicate through calcium signaling, which has affects on oxidative metabolism and apoptosis (Szabadkai et al., 2004; Denton, 2009; Patergnani et al., 2011). Hence, a minor perturbation in mitochondrial structure or function can lead to mitochondrial dysfunction, which can have deleterious effects on the cell.
Supporting the importance of mitochondrial form and function, enterocytes isolated from patients with IBD have been reported to exhibit swollen mitochondria with irregular cristae (Delpre et al., 1989; Söderholm et al., 2002b; Nazli et al., 2004). Abnormal mitochondrial structure is also seen in intestinal epithelial cells (IECs) from mice subjected to experimental models of colitis (Rodenburg et al., 2008). These morphological changes are suggestive of cellular stress and bioenergetic failure. Indeed, patients with IBD have reduced ATP levels within the intestine (Roediger, 1980a; Kameyama et al., 1984; Schürmann et al., 1999). As would be expected, morphological changes in mitochondria have been shown to result in deficiencies in the β-oxidation of short-chain fatty acids (SCFA) (Halestrap and Dunlop, 1986). It remains unclear, however whether observed changes in mitochondrial structure come as a result of disease or whether they may play a role in the pathogenesis of inflammation.
Intestinal Epithelial Barrier Function
Numerous cellular processes are dependent upon healthy mitochondria for an adequate energy supply. The intestinal mucosa of IBD patients has been demonstrated to be in a state of energy deficiency characterized by low ATP levels and low energy charge potential, (Roediger, 1980a; Kameyama et al., 1984; Söderholm et al., 2002a), calling into question the functionality of this organelle during disease. Indeed, the colonic epithelial cells of patients with ulcerative colitis exhibit mitochondrial alterations before other ultrastructural abnormalities in the epithelium are apparent and before the onset of mucosal inflammation (Delpre et al., 1989; Hsieh et al., 2006). The integrity of the intestinal epithelium, tight junction maintenance, and β-oxidation are key cellular processes within the intestinal epithelium that are not only dependent upon properly functioning mitochondria, but are also known to be altered in animal models of intestinal inflammation and in humans with IBD.
Intestinal Epithelial Cells (IECs)
It is known that IBD is a multifactorial disease, involving the interplay of immune dysregulation, genetic susceptibility, environmental factors, and microbial dysbiosis. The intestinal epithelium comprises the interface between these factors, and thus, may play a vital role in governing this interplay. A key feature of IBD is recurrent damage of the intestinal epithelium concomitant with disruption of the intestinal barrier function (Roda et al., 2010; Henderson et al., 2011; Salim and Söderholm, 2011). The intestinal epithelium is the host's defensive barrier against the luminal microenvironment with discriminatory absorption of nutrients and antigen permeability. The intestinal epithelium is in a constant state of turnover, renewing every 4–5 days and necessitating a considerable supply of energy. The epithelium is comprised of a single layer of different subtypes of IECs, including absorptive enterocytes, mucus-producing goblet cells, enteroendocrine cells, and defensin-producing Paneth cells—all of which differentiate from intestinal epithelial Lgr5+ stem cells (Gibson et al., 1996; Crosnier et al., 2006; van der Flier and Clevers, 2009). The intestinal stem cells are believed to undergo asymmetric division to give rise to transit amplifying (TA) progenitor cells, which are rapidly cycling cells that amplify the progeny of the stem cells, undergoing a limited number of divisions before terminally differentiating into a mature cell lineage and being sloughed off at the villus tip. The cellular structure of the epithelium is organized in space, such that the proliferating stem cells are buried in the crypts and the differentiated mature cells migrate up the surface of the villi (Gibson et al., 1996; Crosnier et al., 2006; van der Flier and Clevers, 2009). Each subset of IECs serves a unique purpose within the epithelium, yet all are critical for intestinal homeostasis and modulating the crosstalk between the microbial community and the circulating immune cells (van der Flier and Clevers, 2009; Noah et al., 2011; Dupaul-Chicoine et al., 2013). Consequently, dysregulation of IEC differentiation has serious effects on the pathogenesis of IBD, and several genes for IEC differentiation have been shown to be perversely expressed in the setting of inflammation (Ahn et al., 2008; Zheng et al., 2011; Coskun et al., 2012). Indeed, depletion of mucus and goblet cells is a characteristic of patients with ulcerative colitis (Jass and Walsh, 2001; Danese and Fiocchi, 2011). Muc2-deficient mice, which lack the gene encoding the major component of mucin, spontaneously develop colitis (Van der Sluis et al., 2006). Likewise, several IBD susceptibility genes are associated with Paneth cell dysfunction. For example, the Nod2 risk allele for Crohn's disease is associated with a decrease in α-defensin production by Paneth cells in humans (Wehkamp et al., 2004, 2005), and NOD2-deficient mice also exhibit a decrease in α-defensin production (Kobayashi et al., 2005). Paneth cell dysfunction in both humans and mice is also associated with autophagy related 16-like 1 (ATG16L1) (Cadwell et al., 2008, 2009) and X-box binding protein 1 (XBP1) (Kaser et al., 2008), both of which are associated with increased risk of Crohn's disease (Rioux et al., 2007; Kaser et al., 2008). Adolf et al. has shown that by deleting both ATG16L1 and XBP1, mice develop spontaneous CD-like ileitis, which may be a consequence of Paneth cell dysfunction (Adolph et al., 2013). Furthermore, mice lacking caspase-8, a cysteine protease involved in mediating cellular apoptosis, had reduced numbers of goblet cells, no Paneth cells, and also spontaneously developed ileitis (Günther et al., 2011). Thus, defects in intestinal epithelial homeostasis results in an inadequate intestinal barrier defense, which may allow luminal antigens and/or microbes to interact with or violate the intestinal epithelium and consequently cause inflammation (Gersemann et al., 2011). However, the role of mitochondrial dysfunction during IEC differentiation needs to be further evaluated in order to understand the role it may play in the development of intestinal inflammation. Interestingly, Bär et al. demonstrated that altered mitochondrial oxidative phosphorylation activity influences intestinal inflammation in models of experimental colitis using strains of conplastic mice, which have identical nuclear genomes but diverse mitochondrial genomes (Bär et al., 2013). Two strains of conplastic mice, which had increased concentrations of intestinal ATP and augmented oxidative phosphorylation complex activity, were protected from dextran sodium sulfate (DSS)- and trinitrobenzene sulfonate (TNBS)-induced colitis. These mice also had increased proliferation of enterocytes, suggesting that increased intestinal ATP levels (which were due to mtDNA polymorphisms) caused a surge in the turnover rate of the intestinal epithelium—a process that is central to the renewal of the epithelium after exposure to harsh conditions and noxious provocations, such as DSS and TNBS (Bär et al., 2013). This study suggests that increased regeneration of the intestinal epithelium (by means of increased mitochondrial function) is a key factor in combating intestinal inflammation. Indeed, recent clinical evidence has demonstrated that complete mucosal healing is associated with long-term remissions and decreased risk of operative intervention in IBD patients. Mucosal healing also results in the improved mitochondrial structure in the IECs of patients with ulcerative colitis (Fratila and Craciun, 2010). Aside from the providing energy supplies for cell differentiation, there is accumulating evidence that mitochondria play additional roles in cellular differentiation (Maeda and Chida, 2013; Xu et al., 2013; Weinberg et al., 2015). Thus, it is possible that mitochondrial dysfunction could impact IEC differentiation either through energy production or signaling networks, adversely affecting the integrity of the epithelial cell barrier and potentially influencing the development of disease
Tight Junctions
The capacity of the intestinal epithelium to function as a physical, protective barrier is dependent upon tight junctions (TJs), which seal the paracellular space between epithelial cells and polarize the cell membrane. TJs contribute to the integrity of the gut barrier by controlling paracellular permeability and barrier competence of the intestinal epithelium as well as contributing to mucus layer production and infection control (Peterson and Artis, 2014). There are studies that provide a strong link between the development of IBD and altered expression and structural modifications of TJs. Indeed, evidence shows an association between aberrant intestinal permeability and intestinal mucosal inflammation in IBD (Schmitz et al., 1999; Heller et al., 2005; Zeissig et al., 2007). Reports have correlated increased intestinal permeability in first-degree relatives of patients with IBD, and interestingly, studies have also demonstrated that the spouses of patients with IBD can experience increased gut permeability (May et al., 1993; Söderholm et al., 1999; Breslin et al., 2001; Thjodleifsson et al., 2003). Maintenance of TJ integrity is energy-dependent, and it is not surprising that disruption of the barrier by toxins, pathogens, or noxious stimuli can be initiated by damaged mitochondria (Dickman et al., 2000; He et al., 2000). Certain insults, such as NSAID exposure, are known to disrupt the structure and function of mitochondria, and at least transiently, increase gut permeability (Somasundaram et al., 1997, 2000; Söderholm et al., 1999; Zamora et al., 1999; Basivireddy et al., 2002). Additionally, it has been reported that some patients with Crohn's disease develop immune reactivity against components of their gut microbiome (Pirzer et al., 1991; Duchmann et al., 1996). Consistent with these reports, Nazli et al. demonstrated that treating a cell monolayer with dinitrophenol (an oxidative phosphorylation uncoupler) resulted in cellular internalization of a non-invasive strain of Escherichia coli. From this, the authors hypothesized that under metabolic stress resulting from mitochondrial dysfunction, the enteric epithelium loses its ability to distinguish between commensals and pathogens, and as a result, begins internalizing commensal organisms, which can lead to an exacerbated intestinal inflammatory response (Nazli et al., 2004). The mechanism behind developing reactivity to one's own microbiota is not understood, and more research is needed to delineate the role of metabolic stress (e.g., energy deprivation as a result of decreased mitochondrial function) in this process. Studies do suggest that both mitochondrial dysfunction (Lewis and McKay, 2009) and increased gut permeability (De-Souza and Greene, 2005; Deitch et al., 2006) affect the overall competence of the intestinal epithelial barrier, but the stimuli that initiates either process is not known. Nonetheless, these studies lend credence to the implication of epithelial mitochondrial dysfunction as a predisposing factor for an increase in gut epithelial permeability and a loss of gut barrier function, resulting in intestinal inflammation.
β-oxidation
IBD has been suggested to involve a state of energy-deficiency, whereby oxidative metabolism is altered within IECs (Fukushima and Fiocchi, 2004; Saitoh et al., 2008). The SCFA butyrate is the preferred energy source of colonic epithelial cells (Roediger, 1980a; Hamer et al., 2008) and also plays a role in maintaining colonic mucosal health (Hamer et al., 2008). It is a natural nutrient both found in food and produced as an intestinal fermentation by-product of dietary fiber by gut bacteria (Santhanam et al., 2007). Butyrate undergoes catabolic degradation through β-oxidation in the mitochondrial matrix of colonocytes, providing over 70% of the energy demand of the colonic epithelium (Roediger, 1980b). Butyrate metabolism was demonstrated to be impaired in an animal model of colitis (Ahmad et al., 2000), and numerous studies have reported impaired metabolism in the intestinal mucosa of patients with IBD (Roediger, 1980a; Kameyama et al., 1984; Harig et al., 1989; Ramakrishna et al., 1991; Chapman et al., 1994). Similarly, intestinal mucosal inflammation results when butyrate oxidation is inhibited in experimental animals (Roediger and Nance, 1986). Santhanam et al. showed that the mitochondrial acetoacetyl CoA thiolase, which catalyzes the critical last step in butyrate oxidation, was significantly impaired in the colonic mucosa of patients with ulcerative colitis. Furthermore, they conclude that an increase in mitochondrial ROS may trigger this enzymatic defect (Santhanam et al., 2007). Polymorphisms in SLC22A5, the gene that encodes for the carnitine transporter OCTN2, is a known risk factor for IBD (Barrett et al., 2008; Singh et al., 2009a). IECs utilize carnitine as a transporter of long-chain fatty acids into the mitochondria for β-oxidation (Rinaldo et al., 2002). Furthermore, genetic ablation of OCTN2 as well as pharmacologic inhibition of intestinal fatty acid β-oxidation results in murine experimental colitis (Roediger and Nance, 1986; Shekhawat et al., 2007). Studies involving the treatment of epithelia cells with dinitrophenol to induce mitochondrial stress resulted in decreased transepithelial resistance and increased bacterial translocation (Lewis et al., 2010)—both of which are features of gut barrier dysfunction. Thus, defective β-oxidation in the mitochondria has deleterious effects beyond energy requirements. Likewise, a dysfunctional gut microbiome or a poor diet may also result in a decrease of butyrate metabolism in the colonic epithelium. Enhanced production of butyrate may potentially benefit the colonic epithelial cells by stimulating an enhancement in cellular homeostasis, including antioxidant and anti-inflammatory roles as well as protective gut-barrier functions.
Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS)
Oxidative stress within the intestinal epithelium is thought to play a key role in the pathogenesis of intestinal inflammation (Grisham, 1994; Elson et al., 1995; Conner et al., 1996). Although ROS and RNS are important signaling intermediates involved in a variety of homeostatic molecular pathways (Brown and Griendling, 2009; Gillespie et al., 2009), excessive oxidative stress can provoke cellular damage through the oxidation of proteins, lipids, and DNA, altering their biological functions and potentiating cell death (Andersen, 2004). At baseline, the deleterious effects of ROS generation are negated by a plethora of endogenous antioxidants (Haddad, 2002; Gillespie et al., 2009). The intestinal lumen and epithelium are continuously exposed to noxious stimuli, such as ingested nutrients, local microbes or infections, gastric acid production, and periods of ischemia/reperfusion that have the potential to stimulate the generation oxygen and nitrogen radicals (Parks et al., 1988; Parks, 1989; Young and Woodside, 2001; Sánchez et al., 2002; Biswas et al., 2003; Mazalli and Bragagnolo, 2009). Additionally, the infiltration of leukocytes, monocytes, and neutrophils during inflammation can further enhance intestinal ROS production through both respiratory burst enzymes and prostaglandin and leukotriene metabolism (Babbs, 1992). Several studies have demonstrated increased ROS/RNS levels within the intestinal epithelium of patients with IBD (Kruidenier and Verspaget, 2002; Pravda, 2005; Rezaie et al., 2007) and in murine models of experimental colitis (Girgin et al., 1999; Tham et al., 2002; Narushima et al., 2003; Sundaram et al., 2003; Oz et al., 2005; Siddiqui et al., 2006; dos Reis et al., 2009; Kajiya et al., 2009; Abdolghaffari et al., 2010; Yao et al., 2010; Lenoir et al., 2011; Ock et al., 2011; Sengül et al., 2011; Borrelli et al., 2013; Arab et al., 2014). High concentrations of oxidized molecules have also been measured in the plasma, serum, exhaled air, and saliva of patients with IBD (Tüzün et al., 2002; Rezaie et al., 2006). Others have shown a positive correlation between oxidative stress and disease severity, suggesting a role in the development and potentiation of inflammation (Rachmilewitz et al., 1995, 1998; Herulf et al., 1998).
In addition to changes in the generation of reactive species, several studies have shown an overall reduction in endogenous antioxidants, such as ascorbate, β-carotene, α-tocopherol, and reduced glutathione, in patients with IBD (Buffinton and Doe, 1995; McKenzie et al., 1996; Schorah, 1998; Sido et al., 1998; Geerling et al., 2000). Interestingly, mice lacking an important antioxidant enzyme, glutathione peroxidase, spontaneously develop symptoms and histologic features similar to those in IBD patients (Esworthy et al., 2001). Moreover, in a murine DSS-induced colitis model, mice subjected to DSS exhibited diminished blood levels of reduced glutathione, which were restored to normal after treatment with various antioxidants (Oz et al., 2005). Levels of catalase, glutathione peroxidase, and superoxide dismutase at baseline have been shown to be lower in the human colonic mucosa, submucosa, and serosa as compared to the human liver (Grisham et al., 1990; Mulder et al., 1991; Buffinton and Doe, 1995) and small intestine (Blau et al., 1999), suggesting a limited capacity to combat oxidative stress in the setting of inflammation. Furthermore, treatment with a mitochondria-targeted antioxidant, MitQ, reduced mitochondrial ROS and protected against experimental colitis in mice subjected to DSS (Dashdorj et al., 2013). Likewise, Wang et al. reported that mitochondrial superoxide was the principal initiator of internalization and transcytosis of a commensal microbes across metabolically stressed epithelium in cell lines and human colonic tissue analyzed ex vivo, and that treatment with mitochondrially-targeted antioxidants countered this epithelial barrier defect (Wang et al., 2014). However, it is not yet fully understood if the correlation between ROS/RNS and IBD predicts an actual etiologic relationship for oxidative stress in intestinal inflammation, or if reactive molecular species are merely a consequence of the inflammatory process.
Oxidative stress is thought to exert deleterious effects largely though direct DNA damage and lipid oxidation. Multiple studies have reported increased oxidative DNA damage in the blood and mucosa of patients with IBD (Lih-Brody et al., 1996; D'Incà et al., 2004; Dincer et al., 2007). It has been suggested that these changes may contribute to the increased susceptibility to colorectal cancer that is seen in IBD later in life (Persson et al., 1994; Canavan et al., 2006; Grivennikov, 2013). Colonic mucosal biopsies and plasma from IBD patients also demonstrate an increase in lipid peroxidation products, implying increased ROS production (Pereira et al., 2015). Mice deficient in the antioxidant not only show evidence of increased lipid peroxidation products in both the colon and ileum, but also spontaneously develop colitis (Esworthy et al., 2001), indicating that the ability to combat the oxidative degradation of lipids may be critical in maintaining intestinal homeostasis.
Mitochondria are the most abundant source of ROS in the cell (Beltrán et al., 2010). Under healthy cellular conditions, low levels of ROS are generated and neutralized by the endogenous antioxidant machinery (Haddad, 2002; Gillespie et al., 2009). However, when mitochondria are destabilized by damage or mutations, excessive oxidative stress may result, leading to a reduction of ATP, inhibition of the respiratory chain, and mtDNA damage (Du et al., 1998). Prolonged oxidative stress reduces mitochondrial bioenergetics and homeostasis, promoting cellular damage and ultimately cell death (Scherz-Shouval and Elazar, 2007; Chen and Gibson, 2008). Recent studies have proposed mitochondria as significant cellular drivers and mediators of the inflammatory process (Lee and Hüttemann, 2014). The mitochondria are also a major target of the deleterious effects of oxidative stress, but little is known about how this may lead to inflammation. Understanding the role of mitochondrially-derived ROS in the pathogenesis of IBD may offer key insights into the initiation and propagation of disease.
NLRP3 Inflammasome
Mitochondria are capable of regulating the pro-inflammatory response of the cell through activation of a molecular complex known as the inflammasome. The inflammasome is a multi-protein, caspase-1 activating complex. NLRP3 (NLR family, pyrin domain containing 3) has emerged as critical regulator of intestinal homeostasis (Davis et al., 2014). Formation of the NLRP3 inflammasome is activated by pathogen-associated molecular patterns (PAMPs) as well as damage-associated molecular patterns (DAMPs) that signify cellular stress, such as extracellular ATP, mtDNA, and ROS (Martinon et al., 2002; López-Armada et al., 2013). Once activated, NLRP3 associates with the adaptor molecule ASC (apoptosis-associated speck-like protein), which contains a caspase recruitment domain (CARD). The associated NLRP3-ASC complex oligomerizes and recruits procapase-1, resulting in formation of the active inflammasome, which in turn causes autocleavage of caspase-1 and release of activated inflammatory cytokines IL-8 and IL-1β (Tschopp, 2011; Zitvogel et al., 2012). Data from murine experimental colitis models and human intestinal specimens reveal that elevated expression of IL-8 and IL-1β is central to the pathogenesis of IBD (Sartor, 1994; Ishiguro, 1999; Monteleone et al., 1999; Kwon et al., 2005; Maeda et al., 2005; Ishihara et al., 2013). IL-8 induces expression of IL-1β and other pro-inflammatory cytokines (Kim et al., 2010), which results in intense intestinal inflammation. IL-1β has been shown to increase gut permeability, which then allows for increased bacterial translocation (Al-Sadi et al., 2012). Furthermore, the secretion of biologically active IL-8 and IL-1β is mediated by caspase-1, which has been reported to play a role in DSS-induced colitis (Siegmund et al., 2001). Bauer et al. demonstrate that DSS induces activation of caspase-1 through NLRP3 inflammasome activation. They further show that NLRP3-deficient mice are protected from DSS-induced colitis, exhibiting a significantly reduced production of pro-inflammatory cytokines as well as improved clinical assessments and histological scores (Bauer et al., 2010). Additionally, polymorphisms in the Nlrp3 gene are associated with an increased susceptibility to Crohn's disease (Villani et al., 2009). Schoultz et al. has reported that polymorphisms in the genes encoding both Nlrp3 and Card8, a potent component of the NLRP3 inflammasome, confer increased susceptibility to developing Crohn's disease in Swedish men (Schoultz et al., 2009).
The exact mechanism of NLRP3 inflammasome activation in IBD is not yet known. Several studies have revealed that the NLRP3 inflammasome is involved in murine experimental colitis, and that stimulation of the inflammasome was modulated by mitochondrial ROS (Shimada et al., 2012). However, there remains controversy about the source of ROS that activates intestinal inflammation. Mitochondria are a major, but not the only source of ROS production in the cell. Dashdorj et al. recently published a study implicating mitochondrial ROS as the instigator of inflammation activation (Dashdorj et al., 2013). Consistent with this, other studies have demonstrated that inflammasome activation occurs in mice deficient in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits—a membrane-bound enzymatic complex that functions to generate superoxide, a type of ROS. Additionally, stimulation of the inflammasome also occurs in the mononuclear phagocytes from patients with chronic granulomatous disease, a condition that stems from mutations in the NADPH oxidase subunits (Meissner et al., 2010). Thus, it seems likely that mitochondrial ROS production plays a key role in the intestinal inflammation associated with IBD. However, more research is needed to delineate the extent to which the mitochondria and its concomitant ROS and oxidized mtDNA are involved in the development and progression of intestinal inflammation. Recent studies have begun to appreciate the communication that occurs between mitochondria and pathogen recognition receptors (PRRs) (West et al., 2011). It is interesting to consider the role this communication may play in maintaining immune-microbial homeostasis in the intestinal tract, and how mitochondrial dysfunction may affect the development of intestinal inflammation. Studying the molecular behavior of the inflammasome and its downstream effectors in IBD should add important insights into the mechanistic pathways relevant to the pathogenesis and treatment of disease.
Mitochondrial Communication
Sustaining the mitochondrial population in the cell is central to maintaining cellular bioenergetic homeostasis. Mitochondria have a dedicated repertoire of quality control machinery dedicated to maintaining protein-folding homeostasis. They are composed of four compartments, each of which is a separate protein-folding environment that must be maintained for proper function. Chaperone proteins are localized in the matrix and are required for protein import and promote proper protein folding. In addition to matrix-localized chaperones, there are proteases located in the matrix and IMM, which function to recognize and degrade proteins that fail to fold or assemble properly (Tatsuta and Langer, 2008). Only approximately 10% of the proteins that comprise the electron transport chain are encoded by the mitochondria. The remainder are encoded by the nucleus, translated in the cytosol, and then transported into the mitochondria, where they subsequently assemble into stoichiometric complexes with mitochondrial-encoded proteins (Haynes and Ron, 2010). Thus, it is apparent how cellular stress, such as excessive ROS, mutated proteins, or environmental stress, can negatively affect the protein-folding capacity of the mitochondria, resulting in an accumulation of misfolded proteins or misassembled protein complexes (Ron and Walter, 2007; Ryan and Hoogenraad, 2007). The cell has evolved several quality control pathways to monitor mitochondrial homeostasis and prevent mitochondrial dysfunction. One of these pathways, the mitochondrial unfolded protein response (UPRmt), is a protective response that fosters survival during times of mitochondrial dysfunction or stress by functioning to lessen proteotoxic stress and re-establish protein homeostasis by increasing the population of mitochondrial quality control proteases and chaperones. The UPRmt is a mitochondrial-nuclear cross-talk pathway that, upon communication of unfolded protein stress, activates the transcription factor C/EBP homologous protein (CHOP) (Papa and Germain, 2014), which in turn induces expression of UPRmt-responsive genes (Haynes et al., 2007; Horibe and Hoogenraad, 2007; Baker et al., 2011). It this thought that the UPRmt functions to stabilize and promote the recovery of those mitochondria that are not beyond repair, whereas those organelles that are not salvageable are targeted for mitophagy (Haynes et al., 2013).
Numerous diseases, particularly metabolic and neurogenerative diseases are associated with mitochondrial dysfunction. Some diseases, such as spastic paraplegia, stem directly from mutations that impair mitochondrial function and homeostasis (Casari et al., 1998; Hansen et al., 2002). Most diseases that are associated with mitochondrial dysfunction, though, display characteristics, such as an accrual of mtDNA mutations, augmented ROS generation, and a reduction in ATP output (Haynes and Ron, 2010). All of these features secondarily affect the protein-folding environment of the mitochondria and are common to IBD. However, the exact role UPRmt plays in IBD is just beginning to be uncovered. Rath et al. has recently demonstrated that UPRmt signaling interfaces with the ER unfolded protein response (UPRER) pathway via double-stranded-RNA-activated protein kinase (PKR). Additionally, IECs were unable to activate cpn60, an UPRmt target gene, in PKR-deficient mice subjected to DSS, resulting in resistance to DSS-induced colitis (Rath et al., 2012). This study suggests that the UPRmt has a role in the pathogenesis of IBD, and since it seems that PKR integrates UPRmt signaling into UPRER, then both mitochondrial and ER protein homeostatic responses might contribute to intestinal inflammation.
The UPRmt is similar to the well-known UPRER. While both processes contain their own set of chaperones and proteases, and seem to be two distinct pathways, both signaling pathways share the transcription factor CHOP (Horibe and Hoogenraad, 2007) and converge together at PKR (Rath et al., 2012). Moreover, the mitochondria and ER are not only functionally connected, but also physically connected via MAMs, which play a role in calcium homeostasis and lipid biosynthesis. Calcium release at MAMs may advise mitochondria to future apoptotic events (Szabadkai et al., 2004; Denton, 2009; Patergnani et al., 2011). Accumulation of misfolded proteins in the ER has been suggested to contribute to the development of IBD (Kaser and Blumberg, 2009), and various UPRER modulators have been correlated to the pathogenesis of IBD (Maloy and Powrie, 2011). Likewise, patients with active IBD normally express augmented ER stress markers in the epithelium of ileum and/or colon (Hu et al., 2007; Shkoda et al., 2007; Heazlewood et al., 2008; Kaser et al., 2008). Furthermore, mice with IEC-specific expression of a dysfunctional UPRER signaling protein displayed fragmented ER and deteriorated mitochondria (Cao et al., 2014), implying both ER and mitochondrial dysfunction. Both mitochondrial stress and ER stress have been implicated in a set of diseases associated with mitochondrial dysfunction (Fukushima and Fiocchi, 2004; Ozcan et al., 2004; Zhang and Kaufman, 2008; Lim et al., 2009; Haga et al., 2010; Rath and Haller, 2011). It is not known if mitochondrial dysfunction is a result of ER stress and dysfunction; or if a separate, external signal (e.g., diet, microbiome, ROS, etc.) damages the mitochondria, which then consequently, influences the functionality of the ER. It is also remarkable to note that butyrate has been shown to impact mitochondrial pathways and UPRER signaling in IECs (Fung et al., 2011; Kolar et al., 2011). Since protein homeostasis is sensitive to environmental conditions, it is attractive to speculate that a collaborative UPR (both ER and mitochondria) functions as an innate response to detect harsh changes in the fluctuating intestinal environment. Additionally, given that butyrate is a by-product of the microbial fermentation of SCFAs, it is possible that the composition of the gut microbiota (as well as other luminal antigens) may influence mitochondrial-ER signaling pathways. Nonetheless, it is imperative to consider the contributing role of mitochondria-ER communication in intestinal inflammation.
Mitophagy and Autophagy
Defective autophagy pathways have also been associated with several diseases, including IBD. Cells defective in autophagy accumulate ROS as well as deformed mitochondria (Mizushima and Klionsky, 2007; Saitoh et al., 2008). GWAS have implicated several autophagy genes, including Atg16l1, Lrrk2, and Irgm in the genetic susceptibility to Crohn's disease (Rioux et al., 2007; Barrett et al., 2008; Lees et al., 2011; Umeno et al., 2011). Additionally, previous studies demonstrate that a deficiency in Atg16l1 results in an increased susceptibility to experimental colitis, abnormal appearance and distribution of Paneth cell granules, and altered mitochondria (Cadwell et al., 2008; Saitoh et al., 2008). Furthermore, Liu et al. showed Irgm1-deficient mice exhibited a higher frequency of tubular and swollen mitochondria and increased LC3-positive autophagic vacuoles (Liu et al., 2013). This is consistent with studies that report in humans IRGM localizes to the mitochondria, where it plays a role in mitophagy (Singh et al., 2010). Furthermore, a defect in either ATG16L1 or IRGM has been associated with reduced Paneth cell function, increased susceptibility to bacterial infection, and development of colitis (Cadwell et al., 2008; Saitoh et al., 2008; Liu et al., 2013).
Additionally, prohibitin 1 (PHB), a protein that is important in maintaining normal mitochondrial respiratory function, has been implicated in modulating autophagy. Kathiria et al. demonstrated that PHB regulates autophagy in IECs via intracellular ROS signaling. Moreover, diminished expression of PHB and inhibition of autophagy aggravated mitochondrial depolarization and reduced cell survival, suggesting PHB is an indicator that signals inflammatory stress to the cell, which stimulates autophagy in order to maintain cellular homeostasis and viability (Kathiria et al., 2012). PHB is primarily located on the mitochondria in IECs, and several lines of evidence imply it functions in regulating mitochondrial morphology and function (Artal-Sanz and Tavernarakis, 2009). Interestingly, PHB is decreased in patients with active IBD as well as in animals subjected to experimental colitis (Hsieh et al., 2006; Theiss et al., 2007). Restoration of PHB expression in colonic epithelial cells protected mice from experimental colitis and also exhibited antioxidant properties (Theiss et al., 2009, 2011). Recently, PHB has been shown to interact with the transcription factor STAT3 in colonic epithelial cells and mediate its downstream apoptotic effects. Interestingly, STAT3 has been shown to reside in the mitochondria where it promotes optimal electron transport chain activity, and its activity as a signal transducer has been implicated in IBD (Han et al., 2014).
Mitophagy has also been implicated in the pathogenesis of IBD by a study that revealed an association between single nucleotide polymorphisms in the gene SMAD specific E3 ubiquitin protein ligase 1 (SMURF1) and IBD (Franke et al., 2010). As a regulator of mitophagy, SMURF1 is recruited to damaged mitochondria, where it promotes degradation of the mitochondria by modulating the transport of the autophagic substrate to the autophagosome (Ni et al., 2015). SMURF1 was identified as a crucial mediator of viral autophagy and mitophagy (Orvedahl et al., 2011). However, further studies are needed in order to unravel the part mitophagy plays, beyond normal functions, in IBD pathogenesis. Taken together, these findings suggest autophagy as an important mediator of intestinal homeostasis. Further research is needed in order to delineate the mechanisms of autophagy and their role in intestinal inflammation. Likewise, the interrelation of mitochondrial dysfunction, autophagy, and IBD is still elusive.
Conclusion
Mitochondrial function is undoubtedly crucial to the maintenance of the intestinal epithelium (Figure 1). IECs undergo a constant process of cellular turnover and, as such, necessitate a high-energy production at baseline. Aside from supplying the cell with energy, mitochondria also contribute to a plethora of cellular processes, rendering mitochondria central to cell and ultimately organ survival. There are several intestinal inflammatory diseases that involve mitochondrial dysfunction. For example, several clinically significant enteric pathogens that cause intestinal inflammation, including enteropathogenic E. coli (EPEC) (Nagai et al., 2005; Kozjak-Pavlovic et al., 2008), Helicobacter pylori (Ashktorab et al., 2004; Kozjak-Pavlovic et al., 2008), and Salmonella typhimurium (Hernandez et al., 2003; Layton et al., 2005), target effector proteins to the mitochondria of the host cell. Infection with Citrobacter rodentium has been shown to be result in a disruption in mitochondrial function and structure in mice (Ma et al., 2006). Mutations in mtDNA and reductions in cytochrome c oxidase activity have also been reported in human colorectal cancer (Heerdt et al., 1990; Alonso et al., 1997; Polyak et al., 1998; Payne et al., 2005; Namslauer and Brzezinski, 2009). IBD, hypothesized to be an energy-deficient disease of the intestinal epithelium, has been demonstrated to be associated with mitochondrial abnormalities of the intestinal epithelium, which occur before the onset of inflammation. Although there is no evidence for a causative association between mitochondrial dysfunction and IBD, here we provide several studies that demonstrate potential links connecting the two. A variety of stimuli and environmental conditions can perturb mitochondrial function, yet the primary stimuli of intestinal mitochondrial stress in IBD have yet to be determined. Since IBD is theorized to require two or more “hits” for the development of disease, it is not illogical to suggest that mitochondrial dysfunction in the intestinal epithelium is an integral component of the intestinal inflammatory process, potentially through effects on epithelial permeability, host-microbiota interactions, or effects on the signaling processes mitochondria are involved in. Nonetheless, the findings reviewed here suggest that the intestinal mitochondria may serve as a novel pharmacological target in the treatment and prevention of IBD, which is consistent with studies published recently using mitochondrial-targeted antioxidants to treat experimental colitis in mice (Dashdorj et al., 2013; Wang et al., 2014). Our understanding of mitochondrial dysfunction in intestinal inflammation is still in its infancy, and there are many more questions than answers. Elucidating the link between mitochondria and IBD will enable the development of new therapeutic strategies aimed at treating the cause of mitochondrial dysfunction, which may potentially prevent and/or treat disease by maintaining both mitochondrial health and homeostasis of the intestinal epithelium.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Garret Vincent and Dr. Kellie Cunningham for reviewing the manuscript. We apologize to anyone whose work we left out due to space requirements. This work is supported by a NIH K-award (DK101753) to KPM.
References
Abdolghaffari, A. H., Baghaei, A., Moayer, F., Esmaily, H., Baeeri, M., Monsef-Esfahani, H. R., et al. (2010). On the benefit of Teucrium in murine colitis through improvement of toxic inflammatory mediators. Hum. Exp. Toxicol. 29, 287–295. doi: 10.1177/0960327110361754
Adolph, T. E., Tomczak, M. F., Niederreiter, L., Ko, H. J., Bock, J., Martinez-Naves, E., et al. (2013). Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276. doi: 10.1038/nature12599
Ahmad, M. S., Krishnan, S., Ramakrishna, B. S., Mathan, M., Pulimood, A. B., and Murthy, S. N. (2000). Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut 46, 493–499. doi: 10.1136/gut.46.4.493
Ahn, S. H., Shah, Y. M., Inoue, J., Morimura, K., Kim, I., Yim, S., et al. (2008). Hepatocyte nuclear factor 4alpha in the intestinal epithelial cells protects against inflammatory bowel disease. Inflamm. Bowel Dis. 14, 908–920. doi: 10.1002/ibd.20413
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Alonso, A., Martin, P., Albarran, C., Aquilera, B., Garcia, O., Guzman, A., et al. (1997). Detection of somatic mutations in the mitochondrial DNA control region of colorectal and gastric tumors by heteroduplex and single-strand conformation analysis. Electrophoresis 18, 682–685. doi: 10.1002/elps.1150180504
Al-Sadi, R., Guo, S., Dokladny, K., Smith, M. A., Ye, D., Kaza, A., et al. (2012). Mechanism of interleukin-1beta induced-increase in mouse intestinal permeability in vivo. J. Interferon Cytokine Res. 32, 474–484. doi: 10.1089/jir.2012.0031
Andersen, J. K. (2004). Oxidative stress in neurodegeneration: cause or consequence? Nat. Med. 10(Suppl.), S18–S25. doi: 10.1038/nrn1434
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi: 10.1038/290457a0
Arab, H. H., Al-Shorbagy, M. Y., Abdallah, D. M., and Nassar, N. N. (2014). Telmisartan attenuates colon inflammation, oxidative perturbations and apoptosis in a rat model of experimental inflammatory bowel disease. PLoS ONE 9:e97193. doi: 10.1371/journal.pone.0097193
Archer, S. L. (2013). Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251. doi: 10.1056/NEJMra1215233
Arco, A. D., and Satrústegui, J. (2005). New mitochondrial carriers: an overview. Cell. Mol. Life Sci. 62, 2204–2227. doi: 10.1007/s00018-005-5197-x
Artal-Sanz, M., and Tavernarakis, N. (2009). Prohibitin and mitochondrial biology. Trends Endocrinol. Metab. 20, 394–401. doi: 10.1016/j.tem.2009.04.004
Ashktorab, H., Frank, S., Khaled, A. R., Durum, S. K., Kifle, B., and Smoot, D. T. (2004). Bax translocation and mitochondrial fragmentation induced by Helicobacter pylori. Gut 53, 805–813. doi: 10.1136/gut.2003.024372
Babbs, C. F. (1992). Oxygen radicals in ulcerative colitis. Free Radic. Biol. Med. 13, 169–181. doi: 10.1016/0891-5849(92)90079-V
Baker, M. J., Tatsuta, T., and Langer, T. (2011). Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 3:a007559. doi: 10.1101/cshperspect.a007559
Bär, F., Bochmann, W., Widok, A., von Medem, K., Pagel, R., Hirose, M., et al. (2013). Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology 145, 1055–1063.e1053. doi: 10.1053/j.gastro.2013.07.015
Barrett, J. C., Hansoul, S., Nicolae, D. L., Cho, J. H., Duerr, R. H., Rioux, J. D., et al. (2008). Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 40, 955–962. doi: 10.1038/ng.175
Basivireddy, J., Vasudevan, A., Jacob, M., and Balasubramanian, K. A. (2002). Indomethacin-induced mitochondrial dysfunction and oxidative stress in villus enterocytes. Biochem. Pharmacol. 64, 339–349. doi: 10.1016/S0006-2952(02)01067-5
Bauer, C., Duewell, P., Mayer, C., Lehr, H. A., Fitzgerald, K. A., Dauer, M., et al. (2010). Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199. doi: 10.1136/gut.2009.197822
Beltrán, B., Nos, P., Dasi, F., Iborra, M., Bastida, G., Martínez, M., et al. (2010). Mitochondrial dysfunction, persistent oxidative damage, and catalase inhibition in immune cells of naive and treated Crohn's disease. Inflamm. Bowel Dis. 16, 76–86. doi: 10.1002/ibd.21027
Bennett, R. A., Rubin, P. H., and Present, D. H. (1991). Frequency of inflammatory bowel disease in offspring of couples both presenting with inflammatory bowel disease. Gastroenterology 100, 1638–1643.
Bernstein, C. N. (2008). Assessing environmental risk factors affecting the inflammatory bowel diseases: a joint workshop of the Crohn's & Colitis Foundations of Canada and the USA. Inflamm. Bowel Dis. 14, 1139–1146. doi: 10.1002/ibd.20494
Bernstein, C. N., and Shanahan, F. (2008). Disorders of a modern lifestyle: reconciling the epidemiology of inflammatory bowel diseases. Gut 57, 1185–1191. doi: 10.1136/gut.2007.122143
Bernstein, C. N., Walker, J. R., and Graff, L. A. (2006). On studying the connection between stress and IBD. Am. J. Gastroenterol. 101, 782–785. doi: 10.1111/j.1572-0241.2006.00474.x
Biswas, K., Bandyopadhyay, U., Chattopadhyay, I., Varadaraj, A., Ali, E., and Banerjee, R. K. (2003). A novel antioxidant and antiapoptotic role of omeprazole to block gastric ulcer through scavenging of hydroxyl radical. J. Biol. Chem. 278, 10993–11001. doi: 10.1074/jbc.M210328200
Blau, S., Rubinstein, A., Bass, P., Singaram, C., and Kohen, R. (1999). Differences in the reducing power along the rat GI tract: lower antioxidant capacity of the colon. Mol. Cell. Biochem. 194, 185–191. doi: 10.1023/A:1006994800272
Borrelli, F., Fasolino, I., Romano, B., Capasso, R., Maiello, F., Coppola, D., et al. (2013). Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem. Pharmacol. 85, 1306–1316. doi: 10.1016/j.bcp.2013.01.017
Breslin, N. P., Nash, C., Hilsden, R. J., Hershfield, N. B., Price, L. M., Meddings, J. B., et al. (2001). Intestinal permeability is increased in a proportion of spouses of patients with Crohn's disease. Am. J. Gastroenterol. 96, 2934–2938. doi: 10.1111/j.1572-0241.2001.04684.x
Brown, D. I., and Griendling, K. K. (2009). Nox proteins in signal transduction. Free Radic. Biol. Med. 47, 1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023
Buffinton, G. D., and Doe, W. F. (1995). Depleted mucosal antioxidant defences in inflammatory bowel disease. Free Radic. Biol. Med. 19, 911–918. doi: 10.1016/0891-5849(95)94362-H
Cadwell, K., Liu, J. Y., Brown, S. L., Miyoshi, H., Loh, J., Lennerz, J. K., et al. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263. doi: 10.1038/nature07416
Cadwell, K., Patel, K. K., Komatsu, M., Virgin, H. W.T., and Stappenbeck, T. S. (2009). A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 5, 250–252. doi: 10.4161/auto.5.2.7560
Canavan, C., Abrams, K. R., and Mayberry, J. (2006). Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment. Pharmacol. Ther. 23, 1097–1104. doi: 10.1111/j.1365-2036.2006.02854.x
Cao, S. S., Wang, M., Harrington, J. C., Chuang, B. M., Eckmann, L., and Kaufman, R. J. (2014). Phosphorylation of eIF2alpha is dispensable for differentiation but required at a posttranscriptional level for paneth cell function and intestinal homeostasis in mice. Inflamm. Bowel Dis. 20, 712–722. doi: 10.1097/MIB.0000000000000010
Casari, G., De Fusco, M., Ciarmatori, S., Zeviani, M., Mora, M., Fernandez, P., et al. (1998). Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93, 973–983. doi: 10.1016/S0092-8674(00)81203-9
Chan, D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252. doi: 10.1016/j.cell.2006.06.010
Chapman, M. A., Grahn, M. F., Boyle, M. A., Hutton, M., Rogers, J., and Williams, N. S. (1994). Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis. Gut 35, 73–76. doi: 10.1136/gut.35.1.73
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, Y., and Gibson, S. B. (2008). Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy 4, 246–248. doi: 10.4161/auto.5432
Chistiakov, D. A., Sobenin, I. A., Revin, V. V., Orekhov, A. N., and Bobryshev, Y. V. (2014). Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res. Int. 2014:238463. doi: 10.1155/2014/238463
Cho, J. H., and Brant, S. R. (2011). Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 140, 1704–1712. doi: 10.1053/j.gastro.2011.02.046
Chowdhury, S. K., Smith, D. R., and Fernyhough, P. (2013). The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 51, 56–65. doi: 10.1016/j.nbd.2012.03.016
Conner, E. M., Brand, S. J., Davis, J. M., Kang, D. Y., and Grisham, M. B. (1996). Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease: toxins, mediators, and modulators of gene expression. Inflamm. Bowel Dis. 2, 133–147.
Coskun, M., Olsen, A. K., Holm, T. L., Kvist, P. H., Nielsen, O. H., Riis, L. B., et al. (2012). TNF-alpha-induced down-regulation of CDX2 suppresses MEP1A expression in colitis. Biochim. Biophys. Acta 1822, 843–851. doi: 10.1016/j.bbadis.2012.01.012
Cribbs, J. T., and Strack, S. (2009). Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol. 457, 231–253. doi: 10.1016/S0076-6879(09)05013-7
Crohn's Colitis Foundation of America. (2014). The Facts about Inflammatory Bowel Disease. Available online at: http://www.ccfa.org/assets/pdfs/updatedibdfactbook.pdf (Accessed July 21, 2015).
Crosnier, C., Stamataki, D., and Lewis, J. (2006). Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat. Rev. Genet. 7, 349–359. doi: 10.1038/nrg1840
Danese, S., and Fiocchi, C. (2011). Ulcerative colitis. N. Engl. J. Med. 365, 1713–1725. doi: 10.1056/NEJMra1102942
Dashdorj, A., Jyothi, K. R., Lim, S., Jo, A., Nguyen, M. N., Ha, J., et al. (2013). Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 11:178. doi: 10.1186/1741-7015-11-178
Davis, B. K., Philipson, C., Hontecillas, R., Eden, K., Bassaganya-Riera, J., and Allen, I. C. (2014). Emerging significance of NLRs in inflammatory bowel disease. Inflamm. Bowel Dis. 20, 2412–2432. doi: 10.1097/MIB.0000000000000151
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
Deitch, E. A., Xu, D., and Kaise, V. L. (2006). Role of the gut in the development of injury- and shock induced SIRS and MODS: the gut-lymph hypothesis, a review. Front. Biosci. 11:1816. doi: 10.2741/1816
Delpre, G., Avidor, I., Steinherz, R., Kadish, U., and Ben-Bassat, M. (1989). Ultrastructural abnormalities in endoscopically and histologically normal and involved colon in ulcerative colitis. Am. J. Gastroenterol. 84, 1038–1046.
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. doi: 10.1016/j.bbabio.2009.01.005
Desai, H. G., and Gupte, P. A. (2005). Increasing incidence of Crohn's disease in India: is it related to improved sanitation? Indian J. Gastroenterol. 24, 23–24.
De-Souza, D. A., and Greene, L. J. (2005). Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit. Care Med. 33, 1125–1135. doi: 10.1097/01.CCM.0000162680.52397.97
Dickman, K. G., Hempson, S. J., Anderson, J., Lippe, S., Zhao, L., Burakoff, R., et al. (2000). Rotavirus alters paracellular permeability and energy metabolism in Caco-2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 279, G757–G766.
D'Incà, R., Cardin, R., Benazzato, L., Angriman, I., Martines, D., and Sturniolo, G. C. (2004). Oxidative DNA damage in the mucosa of ulcerative colitis increases with disease duration and dysplasia. Inflamm. Bowel Dis. 10, 23–27. doi: 10.1097/00054725-200401000-00003
Dincer, Y., Erzin, Y., Himmetoglu, S., Gunes, K. N., Bal, K., and Akcay, T. (2007). Oxidative DNA damage and antioxidant activity in patients with inflammatory bowel disease. Dig. Dis. Sci. 52, 1636–1641. doi: 10.1007/s10620-006-9386-8
dos Reis, S. B., de Oliveira, C. C., Acedo, S. C., Miranda, D. D., Ribeiro, M. L., Pedrazzoli, J., et al. (2009). Attenuation of colitis injury in rats using Garcinia cambogia extract. Phytother. Res. 23, 324–329. doi: 10.1002/ptr.2626
Du, G., Mouithys-Mickalad, A., and Sluse, F. E. (1998). Generation of superoxide anion by mitochondria and impairment of their functions during anoxia and reoxygenation in vitro. Free Radic. Biol. Med. 25, 1066–1074. doi: 10.1016/S0891-5849(98)00148-8
Duchmann, R., Schmitt, E., Knolle, P., Meyer zum Büschenfelde, K. H., and Neurath, M. (1996). Tolerance towards resident intestinal flora in mice is abrogated in experimental colitis and restored by treatment with interleukin-10 or antibodies to interleukin-12. Eur. J. Immunol. 26, 934–938. doi: 10.1002/eji.1830260432
Dupaul-Chicoine, J., Dagenais, M., and Saleh, M. (2013). Crosstalk between the intestinal microbiota and the innate immune system in intestinal homeostasis and inflammatory bowel disease. Inflamm. Bowel Dis. 19, 2227–2237. doi: 10.1097/MIB.0b013e31828dcac7
Elson, C. O., Sartor, R. B., Tennyson, G. S., and Riddell, R. H. (1995). Experimental models of inflammatory bowel disease. Gastroenterology 109, 1344–1367. doi: 10.1016/0016-5085(95)90599-5
Esworthy, R. S., Aranda, R., Martín, M. G., Doroshow, J. H., Binder, S. W., and Chu, F. F. (2001). Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G848–G855.
Franke, A., Balschun, T., Sina, C., Ellinghaus, D., Häsler, R., Mayr, G., et al. (2010). Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat. Genet. 42, 292–294. doi: 10.1038/ng.553
Fratila, O. C., and Craciun, C. (2010). Ultrastructural evidence of mucosal healing after infliximab in patients with ulcerative colitis. J. Gastrointestin. Liver Dis. 19, 147–153.
Friedman, J. R., Lackner, L. L., West, M., Dibenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fukushima, K., and Fiocchi, C. (2004). Paradoxical decrease of mitochondrial DNA deletions in epithelial cells of active ulcerative colitis patients. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G804–G813. doi: 10.1152/ajpgi.00398.2003
Fung, K. Y., Brierley, G. V., Henderson, S., Hoffmann, P., McColl, S. R., Lockett, T., et al. (2011). Butyrate-induced apoptosis in HCT116 colorectal cancer cells includes induction of a cell stress response. J. Proteome Res. 10, 1860–1869. doi: 10.1021/pr1011125
Galluzzi, L., Kepp, O., and Kroemer, G. (2012). Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788. doi: 10.1038/nrm3479
Geerling, B. J., Badart-Smook, A., van Deursen, C., van Houwelingen, A. C., Russel, M. G., Stockbrugger, R. W., et al. (2000). Nutritional supplementation with N-3 fatty acids and antioxidants in patients with Crohn's disease in remission: effects on antioxidant status and fatty acid profile. Inflamm. Bowel Dis. 6, 77–84. doi: 10.1097/00054725-200005000-00002
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Gersemann, M., Stange, E. F., and Wehkamp, J. (2011). From intestinal stem cells to inflammatory bowel diseases. World J. Gastroenterol. 17, 3198–3203. doi: 10.3748/wjg.v17.i27.3198
Gibson, P. R., Anderson, R. P., Mariadason, J. M., and Wilson, A. J. (1996). Protective role of the epithelium of the small intestine and colon. Inflamm. Bowel Dis. 2, 279–302.
Gibson, T. B., Ng, E., Ozminkowski, R. J., Wang, S., Burton, W. N., Goetzel, R. Z., et al. (2008). The direct and indirect cost burden of Crohn's disease and ulcerative colitis. J. Occup. Environ. Med. 50, 1261–1272. doi: 10.1097/JOM.0b013e318181b8ca
Gillespie, M. N., Pastukh, V., and Ruchko, M. V. (2009). Oxidative DNA modifications in hypoxic signaling. Ann. N.Y. Acad. Sci. 1177, 140–150. doi: 10.1111/j.1749-6632.2009.05036.x
Girgin, Karaoglu, Tüzün, Erkus, Ozütemiz, Dinçer. (1999). Effects of trimetazidine in ethanol- and acetic acid-induced colitis: oxidant/anti-oxidant status. Colorectal Dis. 1, 338–346. doi: 10.1046/j.1463-1318.1999.00078.x
Greco, C. A., Maurer-Spurej, E., Scott, M. D., Kalab, M., Nakane, N., and Ramírez-Arcos, S. M. (2011). PEGylation prevents bacteria-induced platelet activation and biofilm formation in platelet concentrates. Vox Sang. 100, 336–339. doi: 10.1111/j.1423-0410.2010.01419.x
Grisham, M. B. (1994). Oxidants and free radicals in inflammatory bowel disease. Lancet 344, 859–861. doi: 10.1016/S0140-6736(94)92831-2
Grisham, M. B., Macdermott, R. P., and Deitch, E. A. (1990). Oxidant defense mechanisms in the human colon. Inflammation 14, 669–680. doi: 10.1007/BF00916370
Grivennikov, S. I. (2013). Inflammation and colorectal cancer: colitis-associated neoplasia. Semin. Immunopathol. 35, 229–244. doi: 10.1007/s00281-012-0352-6
Günther, C., Martini, E., Wittkopf, N., Amann, K., Weigmann, B., Neumann, H., et al. (2011). Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339. doi: 10.1038/nature10400
Haddad, J. J. (2002). Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell. Signal. 14, 879–897. doi: 10.1016/S0898-6568(02)00053-0
Haga, N., Saito, S., Tsukumo, Y., Sakurai, J., Furuno, A., Tsuruo, T., et al. (2010). Mitochondria regulate the unfolded protein response leading to cancer cell survival under glucose deprivation conditions. Cancer Sci. 101, 1125–1132. doi: 10.1111/j.1349-7006.2010.01525.x
Halestrap, A. P., and Dunlop, J. L. (1986). Intramitochondrial regulation of fatty acid beta-oxidation occurs between flavoprotein and ubiquinone. A role for changes in the matrix volume. Biochem. J. 239, 559–565. doi: 10.1042/bj2390559
Halfvarson, J., Bodin, L., Tysk, C., Lindberg, E., and Järnerot, G. (2003). Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology 124, 1767–1773. doi: 10.1016/S0016-5085(03)00385-8
Hamer, H. M., Jonkers, D., Venema, K., Vanhoutvin, S., Troost, F. J., and Brummer, R. J. (2008). Review article: the role of butyrate on colonic function. Aliment. Pharmacol. Ther. 27, 104–119. doi: 10.1111/j.1365-2036.2007.03562.x
Han, J., Yu, C., Souza, R. F., and Theiss, A. L. (2014). Prohibitin 1 modulates mitochondrial function of Stat3. Cell. Signal. 26, 2086–2095. doi: 10.1016/j.cellsig.2014.06.006
Hanauer, S. B. (2006). Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm. Bowel Dis. 12(Suppl. 1), S3–S9. doi: 10.1097/01.MIB.0000195385.19268.68
Hansen, J. J., Dürr, A., Cournu-Rebeix, I., Georgopoulos, C., Ang, D., Nielsen, M. N., et al. (2002). Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 70, 1328–1332. doi: 10.1086/339935
Harig, J. M., Soergel, K. H., Komorowski, R. A., and Wood, C. M. (1989). Treatment of diversion colitis with short-chain-fatty acid irrigation. N. Engl. J. Med. 320, 23–28. doi: 10.1056/NEJM198901053200105
Haynes, C. M., and Ron, D. (2010). The mitochondrial UPR—protecting organelle protein homeostasis. J. Cell Sci. 123, 3849–3855. doi: 10.1242/jcs.075119
Haynes, C. M., Fiorese, C. J., and Lin, Y. F. (2013). Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 23, 311–318. doi: 10.1016/j.tcb.2013.02.002
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480. doi: 10.1016/j.devcel.2007.07.016
He, D., Hagen, S. J., Pothoulakis, C., Chen, M., Medina, N. D., Warny, M., et al. (2000). Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology 119, 139–150. doi: 10.1053/gast.2000.8526
Heazlewood, C. K., Cook, M. C., Eri, R., Price, G. R., Tauro, S. B., Taupin, D., et al. (2008). Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 5:e54. doi: 10.1371/journal.pmed.0050054
Heerdt, B. G., Halsey, H. K., Lipkin, M., and Augenlicht, L. H. (1990). Expression of mitochondrial cytochrome c oxidase in human colonic cell differentiation, transformation, and risk for colonic cancer. Cancer Res. 50, 1596–1600.
Heller, F., Florian, P., Bojarski, C., Richter, J., Christ, M., Hillenbrand, B., et al. (2005). Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129, 550–564. doi: 10.1016/j.gastro.2005.05.002
Henderson, P., van Limbergen, J. E., Schwarze, J., and Wilson, D. C. (2011). Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm. Bowel Dis. 17, 382–395. doi: 10.1002/ibd.21379
Hernandez, L. D., Pypaert, M., Flavell, R. A., and Galán, J. E. (2003). A Salmonella protein causes macrophage cell death by inducing autophagy. J. Cell Biol. 163, 1123–1131. doi: 10.1083/jcb.200309161
Herulf, M., Ljung, T., Hellström, P. M., Weitzberg, E., and Lundberg, J. O. (1998). Increased luminal nitric oxide in inflammatory bowel disease as shown with a novel minimally invasive method. Scand. J. Gastroenterol. 33, 164–169. doi: 10.1080/00365529850166897
Holloszy, J. O. (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 242, 2278–2282.
Hong, Z., Kutty, S., Toth, P. T., Marsboom, G., Hammel, J. M., Chamberlain, C., et al. (2013). Role of dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ. Res. 112, 802–815. doi: 10.1161/CIRCRESAHA.111.300285
Hoppins, S., Lackner, L., and Nunnari, J. (2007). The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 76, 751–780. doi: 10.1146/annurev.biochem.76.071905.090048
Horibe, T., and Hoogenraad, N. J. (2007). The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2:e835. doi: 10.1371/journal.pone.0000835
Hsieh, S. Y., Shih, T. C., Yeh, C. Y., Lin, C. J., Chou, Y. Y., and Lee, Y. S. (2006). Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 6, 5322–5331. doi: 10.1002/pmic.200500541
Hu, S., Ciancio, M. J., Lahav, M., Fujiya, M., Lichtenstein, L., Anant, S., et al. (2007). Translational inhibition of colonic epithelial heat shock proteins by IFN-gamma and TNF-alpha in intestinal inflammation. Gastroenterology 133, 1893–1904. doi: 10.1053/j.gastro.2007.09.026
Indriolo, A., Greco, S., Ravelli, P., and Fagiuoli, S. (2011). What can we learn about biofilm/host interactions from the study of inflammatory bowel disease. J. Clin. Periodontol. 38(Suppl. 11), 36–43. doi: 10.1111/j.1600-051X.2010.01680.x
Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., McCaffery, J. M., Hinshaw, J. E., et al. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027. doi: 10.1083/jcb.200506078
Ishiguro, Y. (1999). Mucosal proinflammatory cytokine production correlates with endoscopic activity of ulcerative colitis. J. Gastroenterol. 34, 66–74. doi: 10.1007/s005350050218
Ishihara, N., Otera, H., Oka, T., and Mihara, K. (2013). Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid. Redox Signal. 19, 389–399. doi: 10.1089/ars.2012.4830
Jass, J. R., and Walsh, M. D. (2001). Altered mucin expression in the gastrointestinal tract: a review. J. Cell. Mol. Med. 5, 327–351. doi: 10.1111/j.1582-4934.2001.tb00169.x
Jornayvaz, F. R., and Shulman, G. I. (2010). Regulation of mitochondrial biogenesis. Essays Biochem. 47, 69–84. doi: 10.1042/bse0470069
Kajiya, M., Silva, M. J., Sato, K., Ouhara, K., and Kawai, T. (2009). Hydrogen mediates suppression of colon inflammation induced by dextran sodium sulfate. Biochem. Biophys. Res. Commun. 386, 11–15. doi: 10.1016/j.bbrc.2009.05.117
Kameyama, J., Narui, H., Inui, M., and Sato, T. (1984). Energy level in large intestinal mucosa in patients with ulcerative colitis. Tohoku J. Exp. Med. 143, 253–254. doi: 10.1620/tjem.143.253
Kaser, A., and Blumberg, R. S. (2009). Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin. Immunol. 21, 156–163. doi: 10.1016/j.smim.2009.01.001
Kaser, A., Lee, A. H., Franke, A., Glickman, J. N., Zeissig, S., Tilg, H., et al. (2008). XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756. doi: 10.1016/j.cell.2008.07.021
Kathiria, A. S., Butcher, L. D., Feagins, L. A., Souza, R. F., Boland, C. R., and Theiss, A. L. (2012). Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS ONE 7:e31231. doi: 10.1371/journal.pone.0031231
Kim, G. Y., Lee, J. W., Ryu, H. C., Wei, J. D., Seong, C. M., and Kim, J. H. (2010). Proinflammatory cytokine IL-1beta stimulates IL-8 synthesis in mast cells via a leukotriene B4 receptor 2-linked pathway, contributing to angiogenesis. J. Immunol. 184, 3946–3954. doi: 10.4049/jimmunol.0901735
Kobayashi, K. S., Chamaillard, M., Ogura, Y., Henegariu, O., Inohara, N., Nunez, G., et al. (2005). Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734. doi: 10.1126/science.1104911
Koehler, C. M., Beverly, K. N., and Leverich, E. P. (2006). Redox pathways of the mitochondrion. Antioxid. Redox Signal. 8, 813–822. doi: 10.1089/ars.2006.8.813
Kolar, S., Barhoumi, R., Jones, C. K., Wesley, J., Lupton, J. R., Fan, Y. Y., et al. (2011). Interactive effects of fatty acid and butyrate-induced mitochondrial Ca(2)(+) loading and apoptosis in colonocytes. Cancer 117, 5294–5303. doi: 10.1002/cncr.26205
Koshiba, T. (2013). Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta 1833, 225–232. doi: 10.1016/j.bbamcr.2012.03.005
Kozjak-Pavlovic, V., Ross, K., and Rudel, T. (2008). Import of bacterial pathogenicity factors into mitochondria. Curr. Opin. Microbiol. 11, 9–14. doi: 10.1016/j.mib.2007.12.004
Kruidenier, L., and Verspaget, H. W. (2002). Review article: oxidative stress as a pathogenic factor in inflammatory bowel disease–radicals or ridiculous? Aliment. Pharmacol. Ther. 16, 1997–2015. doi: 10.1046/j.1365-2036.2002.01378.x
Kunz, W. S. (2003). Different metabolic properties of mitochondrial oxidative phosphorylation in different cell types—important implications for mitochondrial cytopathies. Exp. Physiol. 88, 149–154. doi: 10.1113/eph8802512
Kwon, K. H., Murakami, A., Hayashi, R., and Ohigashi, H. (2005). Interleukin-1beta targets interleukin-6 in progressing dextran sulfate sodium-induced experimental colitis. Biochem. Biophys. Res. Commun. 337, 647–654. doi: 10.1016/j.bbrc.2005.09.107
Layton, A. N., Brown, P. J., and Galyov, E. E. (2005). The Salmonella translocated effector SopA is targeted to the mitochondria of infected cells. J. Bacteriol. 187, 3565–3571. doi: 10.1128/JB.187.10.3565-3571.2005
Leary, S. C., Battersby, B. J., and Moyes, C. D. (1998). Inter-tissue differences in mitochondrial enzyme activity, RNA and DNA in rainbow trout (Oncorhynchus mykiss). J. Exp. Biol. 201(Pt 24), 3377–3384.
Lee, I., and Hüttemann, M. (2014). Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta 1842, 1579–1586. doi: 10.1016/j.bbadis.2014.05.031
Lee, Y. J., Jeong, S. Y., Karbowski, M., Smith, C. L., and Youle, R. J. (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011. doi: 10.1091/mbc.E04-04-0294
Lees, C. W., Barrett, J. C., Parkes, M., and Satsangi, J. (2011). New IBD genetics: common pathways with other diseases. Gut 60, 1739–1753. doi: 10.1136/gut.2009.199679
Legesse-Miller, A., Massol, R. H., and Kirchhausen, T. (2003). Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol. Biol. Cell 14, 1953–1963. doi: 10.1091/mbc.E02-10-0657
Lenoir, L., Rossary, A., Joubert-Zakeyh, J., Vergnaud-Gauduchon, J., Farges, M. C., Fraisse, D., et al. (2011). Lemon verbena infusion consumption attenuates oxidative stress in dextran sulfate sodium-induced colitis in the rat. Dig. Dis. Sci. 56, 3534–3545. doi: 10.1007/s10620-011-1784-x
Leverve, X. M., and Fontaine, E. (2001). Role of substrates in the regulation of mitochondrial function in situ. IUBMB Life 52, 221–229. doi: 10.1080/15216540152846037
Lewis, K., and McKay, D. M. (2009). Metabolic stress evokes decreases in epithelial barrier function. Ann. N.Y. Acad. Sci. 1165, 327–337. doi: 10.1111/j.1749-6632.2009.04036.x
Lewis, K., Lutgendorff, F., Phan, V., Söderholm, J. D., Sherman, P. M., and McKay, D. M. (2010). Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate. Inflamm. Bowel Dis. 16, 1138–1148. doi: 10.1002/ibd.21177
Lih-Brody, L., Powell, S. R., Collier, K. P., Reddy, G. M., Cerchia, R., Kahn, E., et al. (1996). Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig. Dis. Sci. 41, 2078–2086. doi: 10.1007/BF02093613
Lim, J. H., Lee, H. J., Ho Jung, M., and Song, J. (2009). Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 21, 169–177. doi: 10.1016/j.cellsig.2008.10.004
Liu, B., Gulati, A. S., Cantillana, V., Henry, S. C., Schmidt, E. A., Daniell, X., et al. (2013). Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G573–G584. doi: 10.1152/ajpgi.00071.2013
Liu, J. Z., van Sommeren, S., Huang, H., Ng, S. C., Alberts, R., Takahashi, A., et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. doi: 10.1038/ng.3359
Liu, J., Killilea, D. W., and Ames, B. N. (2002). Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L- carnitine and/or R-alpha -lipoic acid. Proc. Natl. Acad. Sci. U.S.A. 99, 1876–1881. doi: 10.1073/pnas.261709098
Loftus, E. V. Jr. (2004). Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 126, 1504–1517. doi: 10.1053/j.gastro.2004.01.063
López-Armada, M. J., Riveiro-Naveira, R. R., Vaamonde-Garcia, C., and Valcárcel-Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13, 106–118. doi: 10.1016/j.mito.2013.01.003
Ma, C., Wickham, M. E., Guttman, J. A., Deng, W., Walker, J., Madsen, K. L., et al. (2006). Citrobacter rodentium infection causes both mitochondrial dysfunction and intestinal epithelial barrier disruption in vivo: role of mitochondrial associated protein (Map). Cell. Microbiol. 8, 1669–1686. doi: 10.1111/j.1462-5822.2006.00741.x
Madsen, K. L., Malfair, D., Gray, D., Doyle, J. S., Jewell, L. D., and Fedorak, R. N. (1999). Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm. Bowel Dis. 5, 262–270. doi: 10.1097/00054725-199911000-00004
Maeda, S., Hsu, L. C., Liu, H., Bankston, L. A., Iimura, M., Kagnoff, M. F., et al. (2005). Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science 307, 734–738. doi: 10.1126/science.1103685
Maeda, Y., and Chida, J. (2013). Control of cell differentiation by mitochondria, typically evidenced in dictyostelium development. Biomolecules 3, 943–966. doi: 10.3390/biom3040943
Maloy, K. J., and Powrie, F. (2011). Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306. doi: 10.1038/nature10208
Marsboom, G., Toth, P. T., Ryan, J. J., Hong, Z., Wu, X., Fang, Y. H., et al. (2012). Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 110, 1484–1497. doi: 10.1161/CIRCRESAHA.111.263848
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
May, G. R., Sutherland, L. R., and Meddings, J. B. (1993). Is small intestinal permeability really increased in relatives of patients with Crohn's disease? Gastroenterology 104, 1627–1632.
Mazalli, M. R., and Bragagnolo, N. (2009). Increase of cholesterol oxidation and decrease of PUFA as a result of thermal processing and storage in eggs enriched with n-3 fatty acids. J. Agric. Food Chem. 57, 5028–5034. doi: 10.1021/jf901187j
McKenzie, S. J., Baker, M. S., Buffinton, G. D., and Doe, W. F. (1996). Evidence of oxidant-induced injury to epithelial cells during inflammatory bowel disease. J. Clin. Invest. 98, 136–141. doi: 10.1172/JCI118757
Meissner, F., Seger, R. A., Moshous, D., Fischer, A., Reichenbach, J., and Zychlinsky, A. (2010). Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 116, 1570–1573. doi: 10.1182/blood-2010-01-264218
Mikhailov, T. A., and Furner, S. E. (2009). Breastfeeding and genetic factors in the etiology of inflammatory bowel disease in children. World J. Gastroenterol. 15, 270–279. doi: 10.3748/wjg.15.270
Mitchell, P., and Moyle, J. (1967). Chemiosmotic hypothesis of oxidative phosphorylation. Nature 213, 137–139. doi: 10.1038/213137a0
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40. doi: 10.1146/annurev.nutr.27.061406.093749
Molodecky, N. A., and Kaplan, G. G. (2010). Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. (N.Y.) 6, 339–346.
Monteleone, G., Trapasso, F., Parrello, T., Biancone, L., Stella, A., Iuliano, R., et al. (1999). Bioactive IL-18 expression is up-regulated in Crohn's disease. J. Immunol. 163, 143–147.
Mulder, T. P., Verspaget, H. W., Janssens, A. R., de Bruin, P. A., Peña, A. S., and Lamers, C. B. (1991). Decrease in two intestinal copper/zinc containing proteins with antioxidant function in inflammatory bowel disease. Gut 32, 1146–1150.
Nagai, T., Abe, A., and Sasakawa, C. (2005). Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J. Biol. Chem. 280, 2998–3011. doi: 10.1074/jbc.M411550200
Nakada, K., Inoue, K., Ono, T., Isobe, K., Ogura, A., Goto, Y. I., et al. (2001). Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 7, 934–940. doi: 10.1038/90976
Namslauer, I., and Brzezinski, P. (2009). A mitochondrial DNA mutation linked to colon cancer results in proton leaks in cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 106, 3402–3407. doi: 10.1073/pnas.0811450106
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Narushima, S., Spitz, D. R., Oberley, L. W., Toyokuni, S., Miyata, T., Gunnett, C. A., et al. (2003). Evidence for oxidative stress in NSAID-induced colitis in IL10-/- mice. Free Radic. Biol. Med. 34, 1153–1166. doi: 10.1016/S0891-5849(03)00065-0
Nazli, A., Yang, P. C., Jury, J., Howe, K., Watson, J. L., Söderholm, J. D., et al. (2004). Epithelia under metabolic stress perceive commensal bacteria as a threat. Am. J. Pathol. 164, 947–957. doi: 10.1016/S0002-9440(10)63182-3
Ni, H. M., Williams, J. A., and Ding, W. X. (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. doi: 10.1016/j.redox.2014.11.006
Noah, T. K., Donahue, B., and Shroyer, N. F. (2011). Intestinal development and differentiation. Exp. Cell Res. 317, 2702–2710. doi: 10.1016/j.yexcr.2011.09.006
Noble, C. L., and Arnott, I. D. (2008). What is the risk that a child will develop inflammatory bowel disease if 1 or both parents have IBD? Inflamm. Bowel Dis. 14(Suppl. 2), S22–S23. doi: 10.1002/ibd.20575
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Nunnari, J., Marshall, W. F., Straight, A., Murray, A., Sedat, J. W., and Walter, P. (1997). Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell 8, 1233–1242. doi: 10.1091/mbc.8.7.1233
Ock, C. Y., Kim, E. H., Hong, H., Hong, K. S., Han, Y. M., Choi, K. S., et al. (2011). Prevention of colitis-associated colorectal cancer with 8-hydroxydeoxyguanosine. Cancer Prev. Res. (Phila.) 4, 1507–1521. doi: 10.1158/1940-6207.CAPR-11-0161
Ono, T., Isobe, K., Nakada, K., and Hayashi, J. I. (2001). Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28, 272–275. doi: 10.1038/90116
Orholm, M., Binder, V., Sørensen, T. I., Rasmussen, L. P., and Kyvik, K. O. (2000). Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand. J. Gastroenterol. 35, 1075–1081. doi: 10.1080/003655200451207
Orvedahl, A., Sumpter, R. Jr., Xiao, G., Ng, A., Zou, Z., Tang, Y., et al. (2011). Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117. doi: 10.1038/nature10546
Oz, H. S., Chen, T. S., McClain, C. J., and de Villiers, W. J. (2005). Antioxidants as novel therapy in a murine model of colitis. J. Nutr. Biochem. 16, 297–304. doi: 10.1016/j.jnutbio.2004.09.007
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi, N. N., Ozdelen, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. doi: 10.1126/science.1103160
Papa, L., and Germain, D. (2014). SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34, 699–710. doi: 10.1128/MCB.01337-13
Parks, D. A. (1989). Oxygen radicals: mediators of gastrointestinal pathophysiology. Gut 30, 293–298. doi: 10.1136/gut.30.3.293
Parks, D. A., Williams, T. K., and Beckman, J. S. (1988). Conversion of xanthine dehydrogenase to oxidase in ischemic rat intestine: a reevaluation. Am. J. Physiol. 254, G768–G774.
Patergnani, S., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., et al. (2011). Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 9:19. doi: 10.1186/1478-811X-9-19
Pavlick, K. P., Laroux, F. S., Fuseler, J., Wolf, R. E., Gray, L., Hoffman, J., et al. (2002). Role of reactive metabolites of oxygen and nitrogen in inflammatory bowel disease. Free Radic. Biol. Med. 33, 311–322. doi: 10.1016/S0891-5849(02)00853-5
Payne, C. M., Holubec, H., Bernstein, C., Bernstein, H., Dvorak, K., Green, S. B., et al. (2005). Crypt-restricted loss and decreased protein expression of cytochrome C oxidase subunit I as potential hypothesis-driven biomarkers of colon cancer risk. Cancer Epidemiol. Biomarkers Prev. 14, 2066–2075. doi: 10.1158/1055-9965.EPI-05-0180
Pellegrino, M. W., and Haynes, C. M. (2015). Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 13:22. doi: 10.1186/s12915-015-0129-1
Pereira, C., Grácio, D., Teixeira, J. P., and Magro, F. (2015). Oxidative stress and DNA damage: implications in inflammatory bowel disease. Inflamm. Bowel Dis. 21, 2403–2417. doi: 10.1097/MIB.0000000000000506
Perkins, G. A., and Frey, T. G. (2000). Recent structural insight into mitochondria gained by microscopy. Micron 31, 97–111. doi: 10.1016/S0968-4328(99)00065-7
Persson, P. G., Karlén, P., Bernell, O., Leijonmarck, C. E., Broström, O., Ahlbom, A., et al. (1994). Crohn's disease and cancer: a population-based cohort study. Gastroenterology 107, 1675–1679.
Peterson, L. W., and Artis, D. (2014). Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14, 141–153. doi: 10.1038/nri3608
Pfeffer, G., Majamaa, K., Turnbull, D. M., Thorburn, D., and Chinnery, P. F. (2012). Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 4:CD004426. doi: 10.1002/14651858.CD004426.pub3
Pfeiffer, T., Schuster, S., and Bonhoeffer, S. (2001). Cooperation and competition in the evolution of ATP-producing pathways. Science 292, 504–507. doi: 10.1126/science.1058079
Pirzer, U., Schönhaar, A., Fleischer, B., Hermann, E., and Meyer zum Büschenfelde, K. H. (1991). Reactivity of infiltrating T lymphocytes with microbial antigens in Crohn's disease. Lancet 338, 1238–1239. doi: 10.1016/0140-6736(91)92104-A
Pizzo, P., and Pozzan, T. (2007). Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol. 17, 511–517. doi: 10.1016/j.tcb.2007.07.011
Podolsky, D. K. (2002). Inflammatory bowel disease. N. Engl. J. Med. 347, 417–429. doi: 10.1056/NEJMra020831
Polyak, K., Li, Y., Zhu, H., Lengauer, C., Willson, J. K., Markowitz, S. D., et al. (1998). Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 20, 291–293. doi: 10.1038/3108
Pravda, J. (2005). Radical induction theory of ulcerative colitis. World J. Gastroenterol. 11, 2371–2384. doi: 10.3748/wjg.v11.i16.2371
Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M., and Spiegelman, B. M. (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. doi: 10.1016/S0092-8674(00)81410-5
Rachmilewitz, D., Eliakim, R., Ackerman, Z., and Karmeli, F. (1998). Direct determination of colonic nitric oxide level–a sensitive marker of disease activity in ulcerative colitis. Am. J. Gastroenterol. 93, 409–412.
Rachmilewitz, D., Stamler, J. S., Bachwich, D., Karmeli, F., Ackerman, Z., and Podolsky, D. K. (1995). Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn's disease. Gut 36, 718–723. doi: 10.1136/gut.36.5.718
Ramakrishna, B. S., Roberts-Thomson, I. C., Pannall, P. R., and Roediger, W. E. (1991). Impaired sulphation of phenol by the colonic mucosa in quiescent and active ulcerative colitis. Gut 32, 46–49. doi: 10.1136/gut.32.1.46
Rath, E., and Haller, D. (2011). Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur. J. Nutr. 50, 219–233. doi: 10.1007/s00394-011-0197-0
Rath, E., Berger, E., Messlik, A., Nunes, T., Liu, B., Kim, S. C., et al. (2012). Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61, 1269–1278. doi: 10.1136/gutjnl-2011-300767
Resta-Lenert, S., Smitham, J., and Barrett, K. E. (2005). Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G153–G162. doi: 10.1152/ajpgi.00395.2004
Restivo, N. L., Srivastava, M. D., Schafer, I. A., and Hoppel, C. L. (2004). Mitochondrial dysfunction in a patient with crohn disease: possible role in pathogenesis. J. Pediatr. Gastroenterol. Nutr. 38, 534–538. doi: 10.1097/00005176-200405000-00014
Rezaie, A., Ghorbani, F., Eshghtork, A., Zamani, M. J., Dehghan, G., Taghavi, B., et al. (2006). Alterations in salivary antioxidants, nitric oxide, and transforming growth factor-beta 1 in relation to disease activity in Crohn's disease patients. Ann. N.Y. Acad. Sci. 1091, 110–122. doi: 10.1196/annals.1378.060
Rezaie, A., Parker, R. D., and Abdollahi, M. (2007). Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig. Dis. Sci. 52, 2015–2021. doi: 10.1007/s10620-006-9622-2
Rigoli, L., and Caruso, R. A. (2014). Inflammatory bowel disease in pediatric and adolescent patients: a biomolecular and histopathological review. World J. Gastroenterol. 20, 10262–10278. doi: 10.3748/wjg.v20.i30.10262
Rinaldo, P., Matern, D., and Bennett, M. J. (2002). Fatty acid oxidation disorders. Annu. Rev. Physiol. 64, 477–502. doi: 10.1146/annurev.physiol.64.082201.154705
Rioux, J. D., Xavier, R. J., Taylor, K. D., Silverberg, M. S., Goyette, P., Huett, A., et al. (2007). Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604. doi: 10.1038/ng2032
Roda, G., Sartini, A., Zambon, E., Calafiore, A., Marocchi, M., Caponi, A., et al. (2010). Intestinal epithelial cells in inflammatory bowel diseases. World J. Gastroenterol. 16, 4264–4271. doi: 10.3748/wjg.v16.i34.4264
Rodenburg, W., Keijer, J., Kramer, E., Vink, C., van der Meer, R., and Bovee-Oudenhoven, I. M. (2008). Impaired barrier function by dietary fructo-oligosaccharides (FOS) in rats is accompanied by increased colonic mitochondrial gene expression. BMC Genomics 9:144. doi: 10.1186/1471-2164-9-144
Roediger, W. E. (1980a). The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet 2, 712–715. doi: 10.1016/S0140-6736(80)91934-0
Roediger, W. E. (1980b). Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 21, 793–798. doi: 10.1136/gut.21.9.793
Roediger, W. E., and Nance, S. (1986). Metabolic induction of experimental ulcerative colitis by inhibition of fatty acid oxidation. Br. J. Exp. Pathol. 67, 773–782.
Rojo, M., Legros, F., Chateau, D., and Lombès, A. (2002). Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115, 1663–1674.
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Russell, R. K., and Satsangi, J. (2008). Does IBD run in families? Inflamm. Bowel Dis. 14(Suppl. 2), S20–S21. doi: 10.1002/ibd.20573
Ryan, M. T., and Hoogenraad, N. J. (2007). Mitochondrial-nuclear communications. Annu. Rev. Biochem. 76, 701–722. doi: 10.1146/annurev.biochem.76.052305.091720
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268. doi: 10.1038/nature07383
Salim, S. Y., and Söderholm, J. D. (2011). Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 362–381. doi: 10.1002/ibd.21403
Sánchez, S., Martín, M. J., Ortiz, P., Motilva, V., and Alarcón de la Lastra, C. (2002). Effects of dipyrone on inflammatory infiltration and oxidative metabolism in gastric mucosa: comparison with acetaminophen and diclofenac. Dig. Dis. Sci. 47, 1389–1398. doi: 10.1023/A:1015395103160
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114, 867–874.
Santhanam, S., Venkatraman, A., and Ramakrishna, B. S. (2007). Impairment of mitochondrial acetoacetyl CoA thiolase activity in the colonic mucosa of patients with ulcerative colitis. Gut 56, 1543–1549. doi: 10.1136/gut.2006.108449
Sartor, R. B. (1994). Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology 106, 533–539.
Scherz-Shouval, R., and Elazar, Z. (2007). ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427. doi: 10.1016/j.tcb.2007.07.009
Schmitz, H., Barmeyer, C., Fromm, M., Runkel, N., Foss, H. D., Bentzel, C. J., et al. (1999). Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology 116, 301–309. doi: 10.1016/S0016-5085(99)70126-5
Schorah, C. J. (1998). Antioxidants in children with inflammatory bowel disease. Am. J. Clin. Nutr. 67, 151–152.
Schoultz, I., Söderholm, J. D., and McKay, D. M. (2011). Is metabolic stress a common denominator in inflammatory bowel disease? Inflamm. Bowel Dis. 17, 2008–2018. doi: 10.1002/ibd.21556
Schoultz, I., Verma, D., Halfvarsson, J., Törkvist, L., Fredrikson, M., Sjöqvist, U., et al. (2009). Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn's disease in Swedish men. Am. J. Gastroenterol. 104, 1180–1188. doi: 10.1038/ajg.2009.29
Schürmann, G., Brüwer, M., Klotz, A., Schmid, K. W., Senninger, N., and Zimmer, K. P. (1999). Transepithelial transport processes at the intestinal mucosa in inflammatory bowel disease. Int. J. Colorectal Dis. 14, 41–46. doi: 10.1007/s003840050181
Sengül, N., Işik, S., Aslim, B., Uçar, G., and Demirbağ, A. E. (2011). The effect of exopolysaccharide-producing probiotic strains on gut oxidative damage in experimental colitis. Dig. Dis. Sci. 56, 707–714. doi: 10.1007/s10620-010-1362-7
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Shekhawat, P. S., Srinivas, S. R., Matern, D., Bennett, M. J., Boriack, R., George, V., et al. (2007). Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (OCTN2(-/-)) mice. Mol. Genet. Metab. 92, 315–324. doi: 10.1016/j.ymgme.2007.08.002
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Shkoda, A., Ruiz, P. A., Daniel, H., Kim, S. C., Rogler, G., Sartor, R. B., et al. (2007). Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology 132, 190–207. doi: 10.1053/j.gastro.2006.10.030
Siddiqui, A., Ancha, H., Tedesco, D., Lightfoot, S., Stewart, C. A., and Harty, R. F. (2006). Antioxidant therapy with N-acetylcysteine plus mesalamine accelerates mucosal healing in a rodent model of colitis. Dig. Dis. Sci. 51, 698–705. doi: 10.1007/s10620-006-3194-z
Sido, B., Hack, V., Hochlehnert, A., Lipps, H., Herfarth, C., and Dröge, W. (1998). Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut 42, 485–492. doi: 10.1136/gut.42.4.485
Siegmund, B., Lehr, H. A., Fantuzzi, G., and Dinarello, C. A. (2001). IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 98, 13249–13254. doi: 10.1073/pnas.231473998
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009a). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Singh, S., Graff, L. A., and Bernstein, C. N. (2009b). Do NSAIDs, antibiotics, infections, or stress trigger flares in IBD? Am. J. Gastroenterol. 104, 1298–1313; quiz 1314. doi: 10.1038/ajg.2009.15
Singh, S. B., Ornatowski, W., Vergne, I., Naylor, J., Delgado, M., Roberts, E., et al. (2010). Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 12, 1154–1165. doi: 10.1038/ncb2119
Söderholm, J. D., Olaison, G., Lindberg, E., Hannestad, U., Vindels, A., Tysk, C., et al. (1999). Different intestinal permeability patterns in relatives and spouses of patients with Crohn's disease: an inherited defect in mucosal defence? Gut 44, 96–100. doi: 10.1136/gut.44.1.96
Söderholm, J. D., Olaison, G., Peterson, K. H., Franzén, L. E., Lindmark, T., Wirén, M., et al. (2002a). Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn's disease. Gut 50, 307–313. doi: 10.1136/gut.50.3.307
Söderholm, J. D., Yang, P. C., Ceponis, P., Vohra, A., Riddell, R., Sherman, P. M., et al. (2002b). Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology 123, 1099–1108. doi: 10.1053/gast.2002.36019
Somasundaram, S., Rafi, S., Hayllar, J., Sigthorsson, G., Jacob, M., Price, A. B., et al. (1997). Mitochondrial damage: a possible mechanism of the “topical” phase of NSAID induced injury to the rat intestine. Gut 41, 344–353. doi: 10.1136/gut.41.3.344
Somasundaram, S., Sigthorsson, G., Simpson, R. J., Watts, J., Jacob, M., Tavares, I. A., et al. (2000). Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment. Pharmacol. Ther. 14, 639–650. doi: 10.1046/j.1365-2036.2000.00723.x
Stowe, D. F., and Camara, A. K. (2009). Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid. Redox Signal. 11, 1373–1414. doi: 10.1089/ars.2008.2331
Strauss, M., Hofhaus, G., Schröder, R. R., and Kühlbrandt, W. (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160. doi: 10.1038/emboj.2008.35
Sundaram, U., Hassanain, H., Suntres, Z., Yu, J. G., Cooke, H. J., Guzman, J., et al. (2003). Rabbit chronic ileitis leads to up-regulation of adenosine A1/A3 gene products, oxidative stress, and immune modulation. Biochem. Pharmacol. 65, 1529–1538. doi: 10.1016/S0006-2952(03)00067-4
Szabadkai, G., Simoni, A. M., Chami, M., Wieckowski, M. R., Youle, R. J., and Rizzuto, R. (2004). Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 16, 59–68. doi: 10.1016/j.molcel.2004.09.026
Taanman, J. W. (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123. doi: 10.1016/S0005-2728(98)00161-3
Tait, S. W., and Green, D. R. (2012). Mitochondria and cell signalling. J. Cell Sci. 125, 807–815. doi: 10.1242/jcs.099234
Tarnopolsky, M. A., and Raha, S. (2005). Mitochondrial myopathies: diagnosis, exercise intolerance, and treatment options. Med. Sci. Sports Exerc. 37, 2086–2093. doi: 10.1249/01.mss.0000177341.89478.06
Tatsuta, T., and Langer, T. (2008). Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306–314. doi: 10.1038/sj.emboj.7601972
Tham, D. M., Whitin, J. C., and Cohen, H. J. (2002). Increased expression of extracellular glutathione peroxidase in mice with dextran sodium sulfate-induced experimental colitis. Pediatr. Res. 51, 641–646. doi: 10.1203/00006450-200205000-00016
Theiss, A. L., Idell, R. D., Srinivasan, S., Klapproth, J. M., Jones, D. P., Merlin, D., et al. (2007). Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 21, 197–206. doi: 10.1096/fj.06-6801com
Theiss, A. L., Jenkins, A. K., Okoro, N. I., Klapproth, J. M., Merlin, D., and Sitaraman, S. V. (2009). Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol. Biol. Cell 20, 4412–4423. doi: 10.1091/mbc.E09-05-0361
Theiss, A. L., Laroui, H., Obertone, T. S., Chowdhury, I., Thompson, W. E., Merlin, D., et al. (2011). Nanoparticle-based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm. Bowel Dis. 17, 1163–1176. doi: 10.1002/ibd.21469
Thjodleifsson, B., Sigthorsson, G., Cariglia, N., Reynisdottir, I., Gudbjartsson, D. F., Kristjansson, K., et al. (2003). Subclinical intestinal inflammation: an inherited abnormality in Crohn's disease relatives? Gastroenterology 124, 1728–1737. doi: 10.1016/S0016-5085(03)00383-4
Thomas, R. E., Andrews, L. A., Burman, J. L., Lin, W. Y., and Pallanck, L. J. (2014). PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 10:e1004279. doi: 10.1371/journal.pgen.1004279
Thompson, N. P., Driscoll, R., Pounder, R. E., and Wakefield, A. J. (1996). Genetics versus environment in inflammatory bowel disease: results of a British twin study. BMJ 312, 95–96. doi: 10.1136/bmj.312.7023.95
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhães, J. G., et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62. doi: 10.1038/ni.1823
Tschopp, J. (2011). Mitochondria: Sovereign of inflammation? Eur. J. Immunol. 41, 1196–1202. doi: 10.1002/eji.201141436
Turner, J. R. (2009). Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809. doi: 10.1038/nri2653
Tüzün, A., Erdil, A., Inal, V., Aydin, A., Bağci, S., Yeşilova, Z., et al. (2002). Oxidative stress and antioxidant capacity in patients with inflammatory bowel disease. Clin. Biochem. 35, 569–572. doi: 10.1016/S0009-9120(02)00361-2
Twig, G., Liu, X., Liesa, M., Wikstrom, J. D., Molina, A. J., Las, G., et al. (2010). Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am. J. Physiol. Cell Physiol. 299, C477–C487. doi: 10.1152/ajpcell.00427.2009
Tysk, C., Lindberg, E., Järnerot, G., and Flodérus-Myrhed, B. (1988). Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 29, 990–996. doi: 10.1136/gut.29.7.990
Umeno, J., Asano, K., Matsushita, T., Matsumoto, T., Kiyohara, Y., Iida, M., et al. (2011). Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn's disease and ulcerative colitis. Inflamm. Bowel Dis. 17, 2407–2415. doi: 10.1002/ibd.21651
Vafai, S. B., and Mootha, V. K. (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. doi: 10.1038/nature11707
van der Flier, L. G., and Clevers, H. (2009). Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 71, 241–260. doi: 10.1146/annurev.physiol.010908.163145
Van der Sluis, M., De Koning, B. A., De Bruijn, A. C., Velcich, A., Meijerink, J. P., Van Goudoever, J. B., et al. (2006). Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129. doi: 10.1053/j.gastro.2006.04.020
Varadi, A., Johnson-Cadwell, L. I., Cirulli, V., Yoon, Y., Allan, V. J., and Rutter, G. A. (2004). Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J. Cell Sci. 117, 4389–4400. doi: 10.1242/jcs.01299
Villani, A. C., Lemire, M., Fortin, G., Louis, E., Silverberg, M. S., Collette, C., et al. (2009). Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat. Genet. 41, 71–76. doi: 10.1038/ng.285
Virbasius, J. V., and Scarpulla, R. C. (1994). Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. U.S.A. 91, 1309–1313. doi: 10.1073/pnas.91.4.1309
Wang, A., Keita, Å. V., Phan, V., McKay, C. M., Schoultz, I., Lee, J., et al. (2014). Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am. J. Pathol. 184, 2516–2527. doi: 10.1016/j.ajpath.2014.05.019
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Webb, C. T., Gorman, M. A., Lazarou, M., Ryan, M. T., and Gulbis, J. M. (2006). Crystal structure of the mitochondrial chaperone TIM9.10 reveals a six-bladed alpha-propeller. Mol. Cell 21, 123–133. doi: 10.1016/j.molcel.2005.11.010
Wehkamp, J., Harder, J., Weichenthal, M., Schwab, M., Schaffeler, E., Schlee, M., et al. (2004). NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut 53, 1658–1664. doi: 10.1136/gut.2003.032805
Wehkamp, J., Salzman, N. H., Porter, E., Nuding, S., Weichenthal, M., Petras, R. E., et al. (2005). Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc. Natl. Acad. Sci. U.S.A. 102, 18129–18134. doi: 10.1073/pnas.0505256102
Weinberg, S. E., Sena, L. A., and Chandel, N. S. (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. doi: 10.1016/j.immuni.2015.02.002
Wenz, T. (2013). Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 13, 134–142. doi: 10.1016/j.mito.2013.01.006
West, A. P., Shadel, G. S., and Ghosh, S. (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402. doi: 10.1038/nri2975
Xu, X., Duan, S., Yi, F., Ocampo, A., Liu, G. H., and Izpisua Belmonte, J. C. (2013). Mitochondrial regulation in pluripotent stem cells. Cell Metab. 18, 325–332. doi: 10.1016/j.cmet.2013.06.005
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Yan, L. J., and Sohal, R. S. (1998). Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. U.S.A. 95, 12896–12901. doi: 10.1073/pnas.95.22.12896
Yao, J., Wang, J. Y., Liu, L., Li, Y. X., Xun, A. Y., Zeng, W. S., et al. (2010). Anti-oxidant effects of resveratrol on mice with DSS-induced ulcerative colitis. Arch. Med. Res. 41, 288–294. doi: 10.1016/j.arcmed.2010.05.002
Yoneda, M., Miyatake, T., and Attardi, G. (1994). Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell. Biol. 14, 2699–2712. doi: 10.1128/MCB.14.4.2699
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Young, I. S., and Woodside, J. V. (2001). Antioxidants in health and disease. J. Clin. Pathol. 54, 176–186. doi: 10.1136/jcp.54.3.176
Zamora, S. A., Hilsden, R. J., Meddings, J. B., Butzner, J. D., Scott, R. B., and Sutherland, L. R. (1999). Intestinal permeability before and after ibuprofen in families of children with Crohn's disease. Can. J. Gastroenterol. 13, 31–36.
Zeissig, S., Bürgel, N., Günzel, D., Richter, J., Mankertz, J., Wahnschaffe, U., et al. (2007). Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut 56, 61–72. doi: 10.1136/gut.2006.094375
Zhang, K., and Kaufman, R. J. (2008). From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462. doi: 10.1038/nature07203
Zheng, J. J., Zhu, X. S., Huangfu, Z., Gao, Z. X., Guo, Z. R., and Wang, Z. (2005). Crohn's disease in mainland China: a systematic analysis of 50 years of research. Chin. J. Dig. Dis. 6, 175–181. doi: 10.1111/j.1443-9573.2005.00227.x
Zheng, X., Tsuchiya, K., Okamoto, R., Iwasaki, M., Kano, Y., Sakamoto, N., et al. (2011). Suppression of hath1 gene expression directly regulated by hes1 via notch signaling is associated with goblet cell depletion in ulcerative colitis. Inflamm. Bowel Dis. 17, 2251–2260. doi: 10.1002/ibd.21611
Zhou, C., Huang, Y., Shao, Y., May, J., Prou, D., Perier, C., et al. (2008). The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 105, 12022–12027. doi: 10.1073/pnas.0802814105
Zhu, P. P., Patterson, A., Stadler, J., Seeburg, D. P., Sheng, M., and Blackstone, C. (2004). Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J. Biol. Chem. 279, 35967–35974. doi: 10.1074/jbc.M404105200
Keywords: mitochondrial dysfunction, inflammatory bowel disease, intestinal inflammation, metabolic stress, reactive oxygen species, inflammasome, gut-barrier function, autophagy
Citation: Novak EA and Mollen KP (2015) Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 3:62. doi: 10.3389/fcell.2015.00062
Received: 01 August 2015; Accepted: 14 September 2015;
Published: 01 October 2015.
Edited by:
Seppo J. Vainio, University of Oulu, FinlandCopyright © 2015 Novak and Mollen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin P. Mollen, Division of Pediatric General and Thoracic Surgery, Children's Hospital of Pittsburgh of UPMC, University of Pittsburgh School of Medicine, 4401 Penn Avenue, Faculty Pavilion Suite 7000, Pittsburgh, PA 15224-1334, USA, kevin.mollen@chp.edu