Major host factors involved in epithelial cell invasion of Campylobacter jejuni: role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1
- 1 School for Biomedical and Biomolecular Science, Belfield Campus, University College Dublin, Dublin, Ireland
- 2 Department of Microbiology, Otto von Guericke University, Magdeburg, Germany
- 3 Department of Medical Microbiology, Helmholtz Center for Infection Research, Braunschweig, Germany
- 4 Department of Molecular Medicine, Max-Planck-Institute for Biochemistry, Martinsried, Germany
- 5 Department of Biological Sciences, Alabama State University, Montgomery, AL, USA
Host cell entry by the food-borne pathogen Campylobacter jejuni has been reported as one of the primary reasons of tissue damage in infected humans, however, molecular invasion mechanisms and cellular factors involved in this process are widely unclear. Here we used knockout cell lines derived from fibronectin−/−, integrin beta1−/−, and focal adhesion kinase (FAK)−/− deficient mice and corresponding wild-type (WT) controls, to study C. jejuni-induced signaling cascades involved in the bacterial invasion process. Using high resolution scanning electron microscopy, GTPase pull-downs, G-LISA, and gentamicin protection assays we found that each of these host cell factors is indeed required for activation of the small Rho GTPase member Rac1 and maximal host cell invasion of this pathogen. Interestingly, membrane ruffling, tight engulfment of bacteria and invasion were only seen during infection of WT control cells, but not in fibronectin−/−, integrin beta1−/−, and FAK−/− knockout cell lines. We also demonstrate that C. jejuni activates FAK autophosphorylation activity at Y-397 and phosphorylation of Y-925, which is required for stimulating two downstream guanine exchange factors, DOCK180 and Tiam-1, which are upstream of Rac1. Small interfering (si) RNA studies further show that DOCK180 and Tiam-1 act cooperatively to trigger Rac1 activation and C. jejuni invasion. Moreover, mutagenesis data indicate that the bacterial fibronectin-binding protein CadF and the intact flagellum are involved in Rho GTPase activation and host cell invasion. Collectively, our results suggest that C. jejuni infection of host epithelial target cells hijacks a major fibronectin → integrin beta1 → FAK → DOCK180/Tiam-1 signaling cascade, which has a crucial role for Rac1 GTPase activity and bacterial entry into host target cells.
Introduction
Despite the sophisticated human immune system, some microbial food-borne pathogens have co-evolved with their hosts during evolution to overcome protective cellular barriers and to establish short- or long-term infections in the gut. Salmonella, Campylobacter, Escherichia, Shigella, Listeria, and other bacteria as well as some parasites and enteric viruses are the most common food-borne pathogens (Fang et al., 1991; Salyers and Whitt, 1994; Sougioultzis and Pothoulakis, 2003; Eckmann and Kagnoff, 2005; Lamps, 2007). Importantly, infections with these organisms represent one of the leading causes of morbidity and death in the human population worldwide. Reports by the World Health Organization (WHO) revealed that humans suffer from 4.5 billion incidences of diarrhea causing about 1.8 million deaths annually (World Health Organization, 2004). These infections are especially problematic in infants, young children, or immunocompromised patients, while the majority of enteric infections in healthy adults seem to be self-limiting. Upon ingestion, such pathogens commonly pass through the stomach in sufficient numbers in order to infect the small intestine or colon. To establish and maintain a successful infection in the gastrointestinal tract, microbial pathogens have evolved various strategies to invade tissues, avoid or resist the innate immunity, disturb the normal gut flora, damage the cells, and multiply in specific niches (Thanassi and Hultgren, 2000; Burns et al., 2003; Alouf and Popoff, 2005). Interestingly, most but not all food-borne bacterial pathogens can be classified with invasive phenotypes, which are able to induce their own uptake into epithelial cells that are normally non-phagocytic. According to specific features of the invasion process, we distinguish between the “zipper”- and “trigger”-mechanism, respectively (Cossart and Sansonetti, 2004). Whereas the “zipper”-mechanism is initiated by a bacterial surface protein (commonly by an adhesin) which binds to a specific host cell receptor followed by internalization of the bacterium, the “trigger”-mechanism involves injected bacterial factors by type III/IV secretion systems which often mimic or highjack specific host cell factors to trigger the bacterial uptake process.
Campylobacter infections of the human gastrointestinal tract have been recognized as the leading causes of enteric bacterial infection (Nachamkin et al., 2008). They may be responsible for as many as 400–500 million bacterial gastroenteritis cases per year worldwide (Friedman et al., 2000). Statistical data indicate that Campylobacter infections of humans cause considerable use of medication and health service burden. In the USA, it has been estimated that Campylobacter-associated illnesses cost up to 6.2 billion dollar per year (Forsythe, 2000). Remarkably, in many studies in the USA and other industrialized countries, Campylobacters were found to cause diarrheal disease more than 2–7 times as frequently as Salmonella and Shigella species or pathogenic E. coli (Allos, 2001; Tam, 2001). In particular, two Campylobacter species, Campylobacter jejuni and C. coli, are most frequently isolated from infected humans. C. jejuni is a typical zoonotic pathogen as it can be found as part of the normal intestinal flora in numerous mammals and birds. Thus, C. jejuni can contaminate poultry, beef, veal, pork, water, and milk during food processing, and is mainly transmitted by the fecal–oral route (Potturi-Venkata et al., 2007). Campylobacters remain highly motile in the intestinal mucus, and their microaerophilic nature ensures its survival in the mucus layer. As a consequence of infection, the bacteria colonize the ileum and colon, where they can interfere with the normal functions of the gastrointestinal tract. This may cause some intestinal diseases typically associated with fever, malaise, abdominal pain, and watery diarrhea, often containing blood cells (Wassenaar and Blaser, 1999; Poly and Guerry, 2008). In addition, individuals exposed to C. jejuni may develop postinfection sequelae including Reiter’s reactive arthritis or peripheral neuropathies including Miller–Fisher and Guillain–Barrè syndromes (Blaser and Engberg, 2008). Accumulating research activities over the last few years indicated that C. jejuni perturbs the normal absorptive capacity of the intestine by damaging epithelial cell functions either directly by cell invasion and/or the production of toxin(s), and indirectly by triggering inflammatory responses (Ketley, 1997; Wooldridge and Ketley, 1997).
Early reports of intestinal biopsies from patients and in vitro infection of cultured human intestinal epithelial cell lines have shown that C. jejuni is able to enter gut tissue cells (van Spreeuwel et al., 1985; Oelschlaeger et al., 1993; Wooldridge et al., 1996). Numerous studies indicated that C. jejuni encode a variety of adhesins including CadF, FlpA, JlpA, and PEB1 (Pei et al., 1998; Konkel et al., 2001; Poly and Guerry, 2008). For example, CadF and FlpA are well-characterized bacterial outer membrane proteins which bind fibronectin, an important extracellular matrix protein and bridging molecule to integrin receptors (Moser et al., 1997; Konkel et al., 2010). It has been postulated that host cell invasion by C. jejuni is one of the main reasons for tissue damage, and this process may proceed in a microtubule-dependent and/or actin-dependent fashion (Oelschlaeger et al., 1993; Hu and Kopecko, 1999; Biswas et al., 2004). Infection with C. jejuni triggers membrane ruffling in infected INT-407 intestinal epithelial cells followed by its entry exhibiting features of both the trigger and zipper mechanisms (Krause-Gruszczynska et al., 2007). Maximal adherence and invasion of INT-407 cells by C. jejuni requires CadF and is accompanied with increased levels of tyrosine phosphorylation of some host cell proteins (Biswas et al., 2004; Hu et al., 2006) such as the integrin-associated protein paxillin (Monteville et al., 2003), but the importance of these observations for the invasion process are unknown. Interestingly, CadF maybe also involved in the activation of the small Rho GTPases Rac1 and Cdc42, but the exact mechanism remained unclear (Krause-Gruszczynska et al., 2007). In addition, it has been shown that mutation of genes in the flagellar export system and ciaB (Campylobacter invasion antigen B), as well as deletion of kpsS and waaF genes, playing a role in the biosynthesis of capsular polysaccharide and lipooligosaccharide, respectively, resulted in reduced bacterial adhesion and invasion in vitro, indicating that these proteins could also play roles in host cell invasion (Karlyshev et al., 2000; Kanipes et al., 2004; Konkel et al., 2004; Guerry, 2007; Hu and Kopecko, 2008; Larson et al., 2008). However, some of these data are conflicting in the literature. For example, it is still unclear if the role of the flagellum during invasion is restricted to bacterial motility or secretion of bacterial factors into the medium; especially the role of secreted CiaB in invasion was called into question (Novik et al., 2010).
Pharmacological inhibitor studies have indicated that host heterotrimeric G proteins and certain protein kinases [EGF- and PDGF-receptor, phosphatidylinositol 3-kinase (PI3-K), and others] are also involved in epithelial cell entry of C. jejuni (Wooldridge et al., 1996; Hu et al., 2006; Watson and Galán, 2008). In addition, expression of dominant-negative mutants of caveolin-1 but not dynamin-II significantly decreased C. jejuni internalization suggesting that caveolin-1 or caveolae may also play a role in the uptake process (Watson and Galán, 2008). Once internalized in epithelial cells, C. jejuni co-localize with microtubules (Hu and Kopecko, 1999), they can survive for considerable time and consequently induce a cytotoxic response in vitro (Konkel et al., 1992; Day et al., 2000). The C. jejuni-containing intracellular vacuole deviates from the canonical endocytic pathway, and thus may avoid delivery into lysosomes and subsequent bacterial killing (Watson and Galán, 2008). Intracellular survival of C. jejuni may support the ability to evade host immune responses, causes relapse of the acute infection, and may establish long-term persistent infections (Lastovica, 1996; Day et al., 2000; Hofreuter et al., 2008). However, the molecular mechanism of early C. jejuni host cell invasion as well as the complex interplay of different bacterial and host factors at the pathogen–host cell interface is still not clear. Here we show that the activation of small Rho GTPase Rac1 is a major pathway in C. jejuni invasion. Using mouse knockout cell lines in Rac1 activation screens, invasion assays as well as scanning electron microscopy, we demonstrate a crucial role of fibronectin, integrin β1, focal adhesion kinase (FAK), and two guanine exchange factors, Tiam-1 (T-lymphoma invasion and metastasis gene-1) and DOCK180 (180-kDa dedicator of cytokinesis 1), in activating Rac1 and the induction of membrane ruffling during C. jejuni infection. Using C. jejuni knockout mutants we further investigated the potential role of the fibronectin-binding protein CadF and an intact flagellum in this process.
Materials and Methods
Bacterial Strains
The C. jejuni strains 81–176, 84–25, and F38011 were used in this study. The isogenic F38011ΔcadF, 81–176ΔcadF, 81–176ΔflaA/B, and 81–176ΔflhA mutants were kindly provided by Konkel et al. (1997) and Patricia Guerry (Goon et al., 2006), respectively. All C. jejuni strains were grown on Campylobacter blood-free selective Agar Base (Oxoid) containing Campylobacter growth supplement (Oxoid) or, when appropriate, on Mueller–Hinton (MH) agar amended with 50 μg/ml kanamycin or 30 μg/ml or chloramphenicol at 37°C under microaerobic conditions (generated by CampyGen, Oxoid) for 48 h.
Knockout Fibroblasts and Other Cell Lines
Human embryonic intestinal epithelial cells (INT-407), obtained from the American Type Culture Collection (ATCC CCL-6), were grown in Eagle’s minimum essential medium (MEM) containing L-glutamine and Earle’s salts (Gibco). Several mouse fibroblast knockout cell lines were cultured in RPMI1640 or DMEM medium, respectively, which were supplemented with 10% fetal calf serum (Gibco). Generation of the floxed FN+/+ mouse fibroblast cells and FN−/− knockout cells has been described (Nyberg et al., 2004; Schroeder et al., 2006). The FN−/− cells were grown in DMEM supplemented with 10% FCS or alternatively, in serum replacement medium for bacterial infection studies (Sigma Aldrich). Monolayers of GD25 mouse fibroblasts (integrin beta1−/−) or GD25 cells stably transfected with integrin β1A (GD25β1A) were grown in 10% fetal bovine serum (Wennerberg et al., 1996). Mouse knockout cells deficient in (FAK−/− cells) as well as stable expression of WT FAK in FAK−/− cells were described previously (Sieg et al., 1999). After reaching a confluency of about 70%, the cells were washed two times with phosphate-buffered saline (PBS), and then starved for 12 h.
Infection Studies
For the infection assays, the different cell lines were seeded to give 4 × 105 cells in 12-well tissue culture plates. The culture medium was replaced with fresh MEM without antibiotics 1 h before infection. Bacteria were suspended in (PBS, pH 7.4), added to the cells at a multiplicity of infection (MOI) of 100 and co-incubated with host cells for the indicated periods of time per experiment.
Inhibitor Studies
The pharmacological inhibitors methyl-beta cyclodextrin (MβCD, Sigma, 1–10 mM) and PF-573228 (Tocris; 10 μM) were added 30 min prior to infection and kept throughout the duration of the incubation period. All control cells were treated with the corresponding solvent for the same length of time. We have carefully checked the viability of cells in every experiment to exclude toxic effects resulting in loss of host cells from the monolayers. The experiments were repeated at least three times with similar results.
Plasmid DNA Transfection
The pcDNA3.1-FAK WT and corresponding Y397F, K454R, ΔPR1/ΔPR2, and Y925F mutants were kindly provided by the lab of D. Schlaepfer (Sieg et al., 1999). Transfection of plasmid constructs was performed using GeneJammer transfection reagent according to manufacturer’s instructions (Stratagene). After 48 h, transfected INT-407 cells were infected with C. jejuni strains. All FAK constructs were expressed as C-terminal HA-tag fusion proteins as described (Sieg et al., 1999). The efficiency of transfection was verified both by immunofluorescence microscopy and Western blotting using an α-HA antibody (NEB).
SiRNA Transfection
siRNA(s) directed against human DOCK180, Trio and control siRNA containing scrambled sequence were purchased from Santa Cruz. siRNAs targeted against human Rac1 and RhoA were synthesized as follows. Rac1 target sequence (5′-AAAACTTGCCTACTGATCAGT-3′), RhoA target sequence (5′-TACCTTATAGTTACTGTGTAA-3′) were utilized. For down-regulation of Tiam-1 both the siRNA from Santa Cruz and another one obtained from MWG-Biotech [Tiam-1 target sequence (5′-ACAGCTTCAGAAGCCTGAC-3′)], were used simultaneously. Transfection of siRNAs was performed using siRNA Transfection Reagent as recommended by the manufacturer (Santa Cruz).
Gentamicin Protection Assay
Infection of eukaryotic cells at a density of 4 × 105 cells per well was carried out as described above. After infection, the cells were washed three times with 1 ml of pre-warmed MEM medium per well to remove non-adherent bacteria. To determine the colony-forming units (CFU) corresponding to intracellular bacteria, the INT-407 cell monolayers were treated with 250 μg/ml gentamicin (Sigma) at 37°C for 2 h, washed three times with medium, and then incubated with 1 ml of 0.1% (w/v) saponin (Sigma) in PBS at 37°C for 15 min. The treated monolayers were resuspended thoroughly, diluted, and plated on MH agar. To determine the total CFU corresponding to host-associated bacteria, the infected monolayers were incubated with 1 ml of 0.1% (w/v) saponin in PBS at 37°C for 15 min without prior treatment with gentamicin. The resulting suspensions were diluted and plated as described above. For each strain, the level of bacterial adhesion and uptake was determined by calculating the number of CFU. All experiments were routinely performed in triplicates. In parallel control experiments, 250 μg/ml gentamicin killed all extracellular bacteria (data not shown).
Rac1-GTP Pull-Down Assay
Rac1 activation in infected cells was determined with the Rac1 activation assay kit (Cytoskeleton), based on a pull-down assay using the Cdc42–Rac1 interactive binding domain of PAK1 fused to glutathione S-transferase, GST–CRIB (Sander et al., 1998). Briefly, host cells were grown to 70% confluency and serum-starved overnight. Subsequently, cells were incubated in PBS (pH 7.4) as a control or infected with C. jejuni (MOI of 100) in a time course. Uninfected and infected host cells were washed with PBS, resuspended in the assay buffer of the kit, and detached from dishes with a cell scraper. For a positive and negative control, a portion of the uninfected cell lysate was mixed with GTPγ-S and GDP for 15 min, respectively. Cell lysates (treated with bacteria, GTPγ-S, GDP or untreated) were mixed with the PAK–RBD slurry (1 h, 4°C). Finally, the beads were collected by centrifugation and washed three times with assay buffer. Activated Rac1 was then visualized by immunoblotting as described below. To confirm equal amounts of protein for each sample, aliquots of the lysates from different time points were also analyzed by immunoblotting. The GTPase activities were quantitated as band intensities representing the relative amount of active Rac1-GTP using the Lumi-Imager F1 software program (Roche).
G-LISA Assay
Rac1 activation in infected cells was also determined by a second approach, the G-LISA™ Rac1-activation assay (Cytoskeleton). Host cells were grown to 70% confluency in tissue culture petri dishes and serum depleted overnight. The cells were infected with C. jejuni for the indicated times per experiment. Subsequently, cells were washed with PBS, resuspended in lysis buffer of the kit and harvested from the dishes with cell scraper. Total protein concentration in each lysate was determined by protein assay reagent of the kit. The G-LISA’s contains a Rac1-GTP-binding protein immobilized on provided microplates. Bound active Rac1 was detected with a specific antibody and luminescence. The luminescence signal was quantified by using a microplate reader (SpectraFluor Plus, Tecan).
Generation of the Polyclonal α-FlaGELLIN and α-MOMP Antibodies
Polyclonal antiserum (α-FlaA and α-MOMP) was raised according to standard protocols (Biogenes GmbH) by immunization of two rabbits with a conserved FlaA-derived peptide (corresponding to amino acids 93–106: KTKATQAAQDGQSL) or a MOMP-derived peptide (amino acids 400–413: NLDQGVNTNESADH), which were conjugated to Limulus polyphemus hemocyanin carrier protein. The resulting antisera were affinity-purified and the specificity against their antigens was confirmed by Western blotting.
Motility Assay
Motility phenotypes of strains were tested in MH media containing 0.4% agar. Bacterial cells were harvested from a 36 h culture on conventional agar plates and resuspended in PBS to obtain an optical density at 600 nm of 0.45 (approximately 1 × 109 cfu/ml). The bacteria were incubated for 30 min in the presence or absence of 20 μg/ml α-FlaA antibody or pre-immune serum as control. To ensure that equal amounts of antibody were present on the entire agar surface in the α-FlaA sample, 50 μl of the antibody solutions were plated onto the corresponding plates. Subsequently, 2 μl of a bacterial suspension of 2 × 108 cfu/ml (±antibody) were stabbed into motility agar. Plates were incubated at 37°C under microaerophilic conditions for 24 h, followed by measuring the diameter of the resulting swarms. The results were the mean of at least five separate measurements from three experiments.
SDS-page and Immunoblotting
Proteins from transfected and/or infected host cells were separated on 10–15% polyacrylamide gels and blotted onto polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore). Staining with primary antibodies against Rac1 (Upstate), FAK-PY-397 (Biomol), FAK-PY-925 (NEB), FAK, RhoA, fibronectin, β1 integrin, Tiam-1, DOCK180, or GAPDH (all Santa Cruz) was performed according to the manufacturer’s instructions. As a secondary antibody, horseradish peroxidase-conjugated α-mouse, α-rabbit, or α-goat IgG was used (DAKO). Immunoreactive bands were visualized by ECL plus Western Blotting Detection System (Amersham Biosciences). Relative FAK kinase activities were quantitated as band intensities of the FAK-PY397 signals related to its FAK control blot using the Lumi-Imager F1 software program (Roche).
FESEM (Field Emission Scanning Electron Microscopy)
Host cell monolayers grown on coverslips were infected with C. jejuni strains for either 4 or 6 h, then fixed in cacodylate buffer (0.1 M cacodylate, 0.01 M CaCl2, 0.01 M MgCl2, 0.09 M sucrose; pH 6.9) containing 5% formaldehyde and 2% glutaraldehyde, and subsequently washed several times with cacodylate buffer. Samples were dehydrated with a graded series of acetone (10, 30, 50, 70, 90, and 100%) on ice for 15 min for each step. Samples in the 100% acetone step were allowed to reach room temperature before another change of 100% acetone. Samples were then subjected to critical-point drying with liquid CO2 (CPD030, Bal-Tec, Liechtenstein). Dried samples were covered with a 10 nm thick gold film by sputter coating (SCD040, Bal-Tec, Liechtenstein) before examination in a field emission scanning electron microscope (Zeiss DSM-982-Gemini) using the Everhart Thornley SE detector and the inlens detector in a 50:50 ratio at an acceleration voltage of 5 kV.
Statistical Analysis
All data were evaluated using Student’s t-test with SigmaStat statistical software (version 2.0). Statistical significance was defined by *p ≤ 0.05 and **p ≤ 0.005. All error bars shown in figures and those quoted following the ± signs represent SD.
Results
Campylobacter jejuni Invasion is Time-Dependent and Requires Rac1, Fibronectin, Integrins and FAK
In our previous studies we have shown that small Rho GTPases such as Rac1 and Cdc42 are activated by C. jejuni, and inhibitors as well the expression of dominant-negative constructs have indicated that active Rac1 maybe one important host factor required for bacterial entry into host cells (Krause-Gruszczynska et al., 2007). Here we aimed to identify the signaling pathway leading to Rac1 activation. First, we confirmed that Rac1 is activated in infected non-phagocytic intestinal epithelial cells (INT-407) using a novel commercial assay, called G-LISA (see Materials and Methods). The results show that wild-type (WT) C. jejuni strains 81–176, 84–25, or F38011 induced the generation of active Rac1-GTP during the course of infection (Figure 1A and data not shown). To confirm that Rac1 is indeed necessary for C. jejuni invasion, we downregulated Rac1 expression by siRNA. Down-regulation of Rac1 expression by about 90% lead to a significant drop in the number of intracellular bacteria as determined by gentamicin protection assays (Figure 1B). As controls, a scrambled siRNA or siRNA against RhoA, another Rho GTPase member, did not suppress C. jejuni invasion (Figures 1B,C). These results indicate that penetration of C. jejuni into cultured cells specifically requires Rac1.
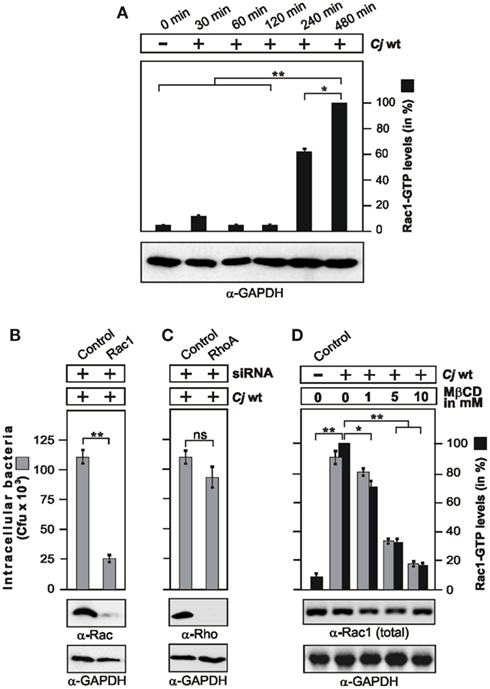
Figure 1. Campylobacter jejuni invasion is time-dependent, and requires Rac1 and lipid rafts. (A) Quantification of Rac1 activity during the course of infection. INT-407 cells were infected with WT C. jejuni 81–176 for indicated periods of time. The presence of active Rac1-GTP was quantified by G-LISA. One hundred percentage of activity corresponds to the highest amount of detected GTPase-GTP level. Similar amount of cells at every time point was confirmed by α-GAPDH Western blotting. (B,C) Effect of Rac1 and RhoA expression knockdown on C. jejuni invasion. INT-407 cells were transfected with Rac1-siRNA (B) or RhoA-siRNA (C), as well as a scrambled siRNA as control. After 48 h, cells were infected with C. jejuni for 6 h. Intracellular bacteria were quantified by gentamicin protection assays. Immunoblotting with α-Rac1 or α-RhoA antibodies confirmed down-regulation of the proteins. GAPDH expression levels were determined as control. (D) Effects of MβCD targeting lipid rafts on host cell internalization of C. jejuni. INT-407 monolayers were pre-incubated with the indicated concentrations of MβCD for 30 min, followed by 6 h infection with C. jejuni. Total intracellular C. jejuni were quantified by gentamicin protection assays. The presence of active Rac1-GTP was analyzed by CRIB–GST pull-down and quantified. One hundred percentage of activity corresponds to the highest amount of detected GTPase-GTP level (right lane). Similar quantities of total Rac1 and GAPDH were confirmed by Western blotting. *p < 0.05 and **p ≤ 0.005 were considered as statistically significant as compared to the control.
Previous studies have shown that addition of methyl-beta cyclodextrin (MβCD), an agent that sequesters cholesterol, decreased the ability of C. jejuni to enter several cultured epithelial cell lines, suggesting that lipid rafts may be required for efficient host cell invasion (Watson and Galán, 2008). Thus, we investigated if intact lipid rafts may be required for C. jejuni-triggered Rac1 activation. Indeed, addition of MβCD blocked C. jejuni-induced Rac1 activation and internalization into INT-407 cells in a dose-dependent manner (Figure 1D), suggesting that a lipid raft-associated host receptor maybe targeted by C. jejuni to trigger downstream signaling leading to Rac1 activation. Since C. jejuni encodes CadF, an outer membrane protein binding to the extracellular matrix protein fibronectin (Konkel et al., 2001), we proposed that a classical fibronectin → integrin-β1 → FAK pathway could be involved in activating Rac1. To investigate this question, we utilized fibroblast cell lines derived from fibronectin−/−, integrin-β1−/− (so called GD25 cells) and FAK−/− knockout mice (Wennerberg et al., 1996; Sieg et al., 1999; Nyberg et al., 2004), which have the advantage that the cells are completely devoid of expressing the proteins of interest (Figures 2A–C). As controls, we infected in parallel experiments floxed fibronectin+/+ cells, GD25 cells re-expressing WT integrin-β1A (GD25β1A) and FAK−/− cells re-expressing WT FAK. Infection studies showed that WT C. jejuni can effectively enter the control cells, while the knockout cells exhibited significant deficiency for bacterial uptake (Figures 2A–C). In addition, transient transfection of FAK−/− knockout cells with HA-tagged WT FAK restored the capability of C. jejuni to enter these cells, while expression of FAK mutants that were either not capable of autophosphorylation (FAK Y397F), impaired kinase activity (FAK K454R), lacking several proline residues in two PxxP-motifs (FAKΔPR1/2) necessary for association with SH3-containing proteins such as p130CAS or Graf, or lacking the Grb2-binding site (FAK Y925F; Sieg et al., 1999; Hauck et al., 2002), significantly reduced the internalization of C. jejuni (p ≤ 0.001) by about 35–50% (Figure 2D). This indicates an important role of FAK signaling downstream of fibronectin and integrins in facilitating efficient uptake of C. jejuni.
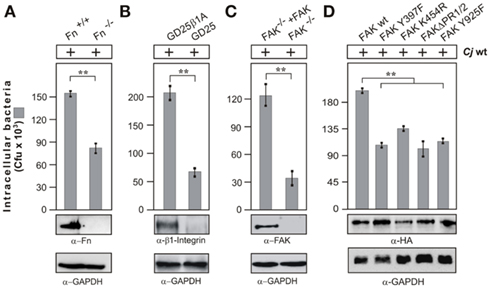
Figure 2. Importance of fibronectin, integrin β1, and FAK expression on C. jejuni invasion. The following cells lines were infected with WT C. jejuni 81–176 for 6 h. (A) Fibronectin-deficient cells (Fn−/−) and corresponding floxed WT cells (Fn+/+), (B) integrin β1-deficient cells (GD25) and GD25 stably re-expressing WT integrin β1A (GD25-β1A) cells, and (C) FAK-deficient cells (FAK−/−) and FAK−/− cells stably re-expressing WT FAK. (D) FAK−/− cells transiently transfected with the indicated HA-tagged FAK constructs. Intracellular Campylobacter cells were quantified by gentamicin protection assays. **p ≤ 0.005 was considered as statistically significant. Fibronectin, integrin β1, and FAK expression was analyzed by immunoblotting. Expression of the individual FAK constructs was verified using α-HA antibody. GAPDH expression levels were determined as loading control.
Campylobacter jejuni Induces Profound Membrane Ruffling and Invasion in WT Cells but Not in Fibronectin, Integrin, and FAK Knockout Cells
Next we infected the WT fibroblasts and their corresponding fibronectin−/−, GD25, and FAK−/− knockout cell lines and analyzed the interaction of C. jejuni with the surface of host cells by high resolution FESEM. First, we confirmed that the WT C. jejuni strains had single bipolar flagella (Figure 3A, white arrows). After infection, the micrographs revealed that the bacteria were able to attach to the host cell surface, followed by cellular invasion which was observed predominantly after 4–6 h of infection (Figure 3B). Interestingly, similar to our earlier observations with infected INT-407 cells (Krause-Gruszczynska et al., 2007), we found that C. jejuni entered the WT fibroblast cells in a very specific fashion, first with its leading flagellar tip followed by the opposite flagellar end (Figure 3B, yellow arrows). Tight engulfment of the bacteria and membrane ruffles (red arrows), filopodia-like structures (blue arrows) as well as the appearance of elongated microspikes (green arrowheads) were also regularly observed. In contrast, infection of fibronectin−/−, GD25 and FAK−/− knockout cell lines also revealed bound bacteria at the surface of the cells with short microspikes present, but no membrane ruffles or invading C. jejuni could be detected (Figure 3C). The predominant observation of membrane ruffling in WT cells confirms the typical occurrence of Rac1 GTPase activation followed by dynamic membrane rearrangements during the C. jejuni invasion process, and this depends on the expression of three crucial host cell factors, fibronectin, integrin β1, and FAK.
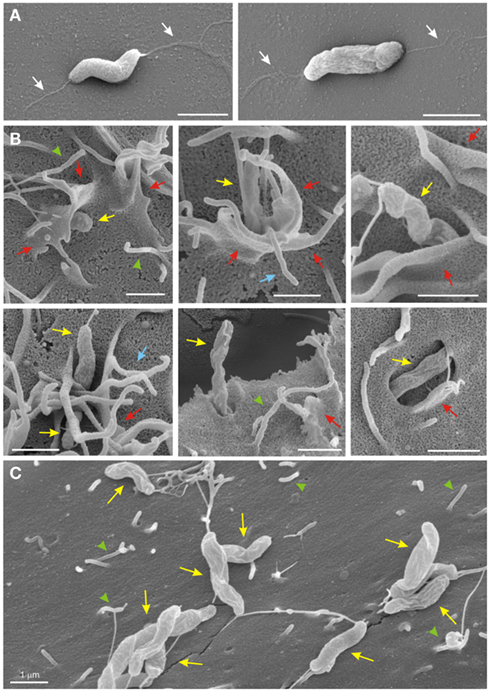
Figure 3. High resolution field emission scanning electron microscopy of C. jejuni invasion. (A) C. jejuni strains have single bipolar flagella (white arrows). (B) C. jejuni 81–176 infected for 4–6 h were able to induce their entry into the WT fibroblast target cells and were regularly associated with membrane ruffles (red arrows), filopodia-like structures (blue arrows) as well as elongated microspikes (green arrowheads). Invading bacteria were also marked (yellow arrows). (C) Infection of GD25 knockout cells with WT C. jejuni 81–176 for 6 h also revealed bacterial attachment, but membrane dynamics or invasion were not induced. Similar results were observed for the infected Fibronectin-deficient and FAK-deficient cells (data not shown).
WT but Not CadF Mutant C. jejuni Induce Profound FAK Activation
Focal adhesion kinase is an intracellular non-receptor tyrosine kinase and important modulator of integrin-dependent focal cell contacts, thereby orchestrating well-known integrin-initiated outside-in signaling events (Sieg et al., 1999; Hauck et al., 2002). FAK localizes with β1-integrins and becomes activated by autophosphorylation at position Y-397, and downstream signaling can be transmitted through subsequent phosphorylation of FAK at Y-925. The next experiment was therefore to examine whether C. jejuni can stimulate the autophosphorylation of FAK. For this purpose, FAK WT cells were infected with WT C. jejuni for different time periods and cell lysates were analyzed with an antibody specific for FAK phosphorylated at position Y-397. Quantification data show that C. jejuni-induced FAK autophosphorylation profoundly, and this correlated with increasing amounts of intracellular bacteria over time (Figure 4A). In addition, C. jejuni-induced the phosphorylation of FAK at Y-925 in a time-dependent manner (Figure 4B). To investigate the potential contribution of the CadF protein for FAK phosphorylation events, the effect of an isogenic ΔcadF mutant was examined under identical conditions. The results show that FAK autophosphorylation was widely impaired in infections with the ΔcadF mutant (Figures 4A,B). These findings suggest that expression of CadF maybe involved in C. jejuni-induced FAK kinase activity.
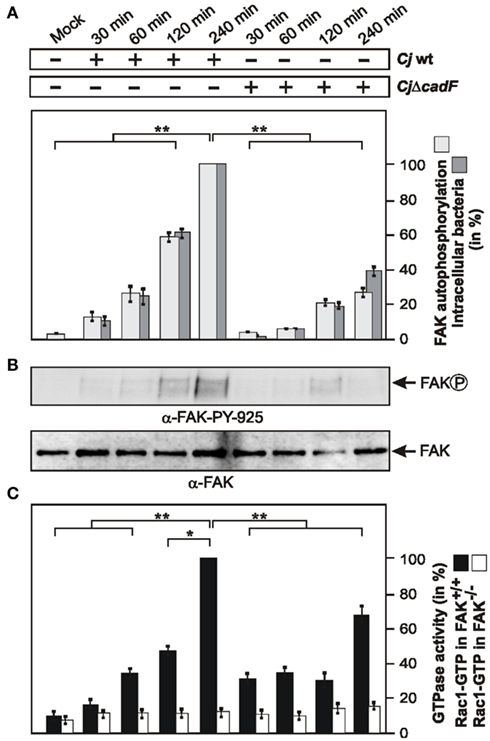
Figure 4. Importance of FAK for C. jejuni-induced Rac1 activation and role of the bacterial CadF protein. (A) FAK-positive fibroblasts were infected with WT C. jejuni F38011 or isogenic F38011ΔcadF for indicated periods of time. Quantification of FAK kinase activity during the course of infection. One hundred percentage of activity corresponds to the highest amount of detected FAK phosphorylated at Y-397 (lane 5). (B) Cell lysates were also analyzed by immunoblotting with α-FAK-PY-925. Total FAK expression levels were determined as control. (C) Quantification of Rac1 GTPase activity during the course of infection. FAK-positive and FAK-deficient cells were infected with C. jejuni WT F38011 or F38011ΔcadF mutant for the indicated periods of time. The presence of bound, active Rac1-GTP or was analyzed in CRIB–GST pull-down assays. One hundred percentage of activity corresponds to the highest amount of detected GTPase-GTP level. *p < 0.05 and **p ≤ 0.005 were considered as statistically significant as compared to the control.
Induction of Maximal Rac1-GTP Levels Requires the cadF Gene and is Strongly Impaired in FAK−/− Knockout Cells
Next we aimed to investigate if FAK is required for C. jejuni-induced Rac1 activation. For this purpose, FAK−/− knockout cells and FAK−/− cells re-expressing FAK were infected under the same conditions as shown in Figures 4A,B, followed by CRIB–GST pull-down assays. While growing levels of activated Rac1 were detected in FAK-positive cells over time, no detectable activation of Rac1 was found in FAK−/− cells during the entire course of infection (Figure 4C), indicating the involvement of FAK in signaling upstream of Rac1 activation during C. jejuni invasion. Furthermore, significantly reduced Rac1-GTP levels were observed in both FAK-positive and FAK−/− cells infected with the ΔcadF mutant (Figure 4C). These findings further support the view that cadF plays a role in signaling leading to FAK-mediated activation of Rac1. However, the ΔcadF mutant was still able to induce some GTPase activation in FAK-positive cells suggesting that other bacterial factor(s) are also implicated in this signaling (Figures 4A–C, right lanes).
The Guanine Exchange Factors Tiam-1 and DOCK180 are Required for Rac1 Activation and C. jejuni Invasion
Our next aim was to determine additional signaling components downstream of FAK and upstream of Rac1 activation during infection with C. jejuni. Cycling of Rho GTPases between the inactive and active forms is positively regulated by guanine nucleotide exchange factors (GEFs) and negatively regulated by GTPase activating proteins (GAPs). GEFs stimulate the exchange of GDP for GTP to generate the active form of a given GTPase, which is then capable of recognizing downstream targets (Schmidt and Hall, 2002; Hsia et al., 2003; Tomar and Schlaepfer, 2009). To identify which GEFs are involved in C. jejuni invasion, the expression of typical GEFs including DOCK180, Tiam-1, or Trio was downregulated using target-specific siRNA. While down-regulation of Tiam-1 and DOCK180 led to the significant reduction in C. jejuni internalization (Figures 5A,B), both down-regulation of Trio (Figure 5C) and transfection with non-targeting scrambled control siRNA sequence had no significant effect on C. jejuni uptake as quantified by gentamicin protection assays (Figures 5A–C). Importantly, the drop in invasion rates correlated with significantly reduced Rac1 GTP levels in CRIB-pull-down assays of Tiam-1 and DOCK180 siRNA-treated cells, but not in Trio siRNA-treated cells (Figures 5D–F). This suggests that two GEFs, Tiam-1 and DOCK180 (but not Trio), play a role in C. jejuni-induced Rac1 activation and invasion.
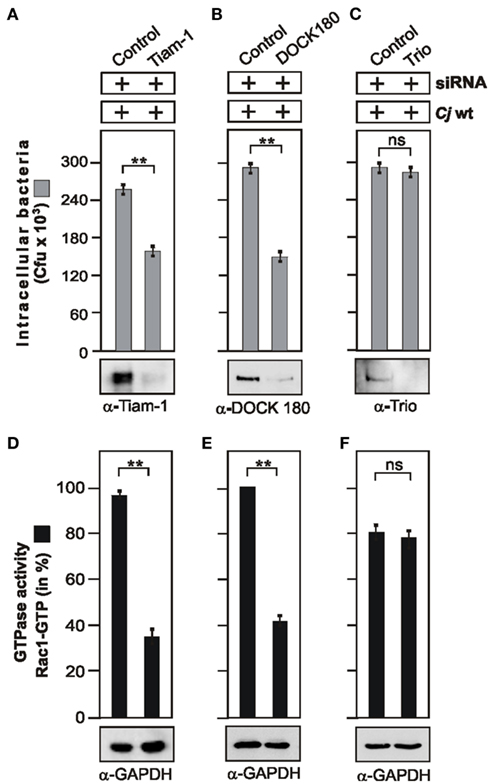
Figure 5. Importance of guanine exchange factors Tiam-1, DOCK180, and Trio for C. jejuni-induced Rac1 activation and bacterial invasion. INT-407 cells were transfected with siRNA against Tiam-1 (A), DOCK180 (B), or Trio (C), as well as a scrambled siRNA as control. After 48 h, cells were infected with WT C. jejuni 81–176 for 6 h. Intracellular bacteria were quantified by gentamicin protection assays. Immunoblotting with indicated antibodies confirmed successful knockdown of respective proteins. To detect Tiam-1 expression, immunoprecipitation with α-Tiam-1 antibody was performed. GAPDH expression levels were determined as control. (D–F) The presence of bound, active Rac1-GTP was analyzed in CRIB–GST pull-down assays for each indicated GEF. Quantification of Rac1 GTPase activity during the course of infection. One hundred percentage of activity corresponds to the highest amount of detected GTPase-GTP level. **p ≤ 0.005 was considered as statistically significant as compared to the control.
Tiam-1 and DOCK180 Act Cooperatively to Trigger Rac1 Activation and C. jejuni Invasion Downstream of FAK
We have noted that down-regulation of the individual GEFs Tiam-1 or DOCK180 did not lead to a complete blockade of Rac1 activity and C. jejuni uptake. We therefore proposed that both GEFs may act together in C. jejuni-infected cells. To investigate this question, Tiam-1 and DOCK180 expression was downregulated by siRNA, either alone or simultaneously, followed by G-LISA to determine Rac1 activity. The results show that simultaneous down-regulation of Tiam-1 and DOCK180 led to a profound block of C. jejuni-induced Rac1 activity (Figures 6A,B). A similar strong blockade of Rac1 levels was achieved in infected FAK−/− cells (Figure 4C) or by infection in the presence of the FAK kinase inhibitor PF-573228 (Figure 6C). As expected, simultaneous down-regulation of Tiam-1 and DOCK180 resulted not only in profound inhibition of Rac1 activity but also profound blockade of C. jejuni invasion (Figure 6D). These data suggest that one important pathway of C. jejuni host cell entry proceeds by activation of a FAK → Tiam-1/DOCK180 → Rac1 signaling cascade.
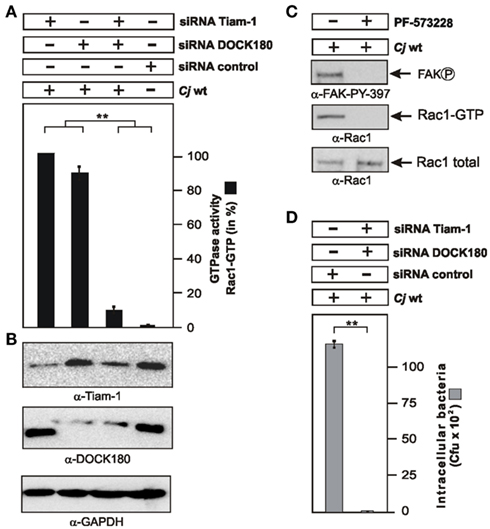
Figure 6. Importance of Tiam-1, DOCK180, and FAK activation on C. jejuni-induced Rac1 activation and bacterial invasion. (A) INT-407 cells were transfected for 48 h with siRNA for Tiam-1, DOCK180, or a scrambled siRNA as control. Quantification of Rac1 GTPase activity after infection with WT C. jejuni 81–176 for 6 h. The presence of bound, active Rac1-GTP or was analyzed by G-LISA. One hundred percentage of activity corresponds to the highest amount of detected GTPase-GTP level (lane 1). (B) Immunoblotting with the indicated antibodies confirmed knockdown of the proteins. GAPDH expression levels were determined as control. (C) Addition of FAK kinase inhibitor PF-573228 during a 6 h infection led to the disappearance of Rac1-GTP and FAK phosphorylated at Y-397. (D) Intracellular bacteria were quantified by gentamicin protection assays in the indicated siRNA-treated cells. **p ≤ 0.001 were considered as statistically significant as compared to the control.
The Flagellum is also Involved in C. jejuni-Induced Rac1 Activation and Invasion
Since cadF is not the sole bacterial gene involved in C. jejuni-induced GTPase activation, the following aim was to search for other factor(s) playing a role in this signaling. The flagellar apparatus was reported to be one of the most intensively investigated pathogenicity determinant in C. jejuni (Konkel et al., 2004; Guerry, 2007). For this purpose, host cells were infected with WT strain 81–176 and its isogenic mutants ΔflaA/B, lacking the two major flagella subunits FlaA and FlaB (Goon et al., 2006), and ΔflhA, a key element involved in the coordinate regulation of late flagellar genes and other factors in C. jejuni (Carrillo et al., 2004). First, we confirmed the absence of flagella in both mutants (Figure 7A), followed by infection assays. As expected activated Rac1 was detected in FAK-positive cells between 2 and 4 h after infection with WT C. jejuni (Figures 7B,C). In contrast, no detectable Rac1 activation was found in cells infected with ΔflaA/B or ΔflhA mutants during the entire course of infection (Figures 7B,C), indicating an important role of these flagellar genes in activating Rac1 by C. jejuni, in addition to the contribution by cadF as shown above.
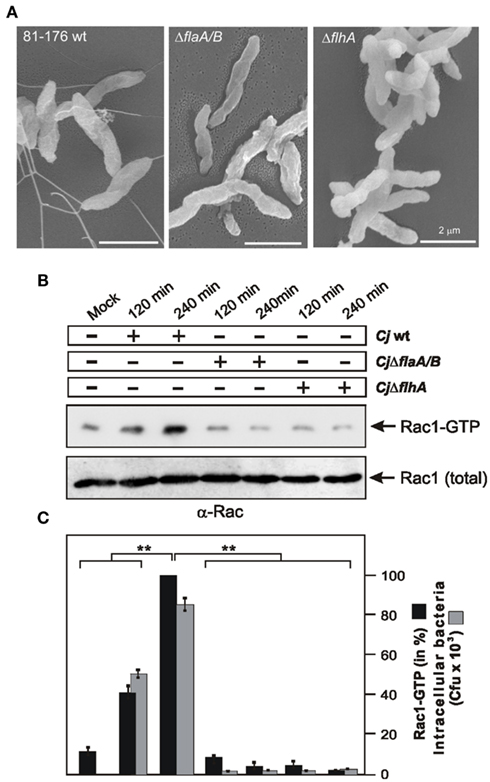
Figure 7. Importance of the flagellar apparatus for C. jejuni-induced activation of Rac1 and bacterial invasion. (A) High resolution field emission scanning electron microscopy of C. jejuni WT 81–176, 81–176ΔflaA/B, and 81–176ΔflhA mutants. (B) FAK-positive cells were infected with the indicated strains in a time course. The presence of bound, active Rac1-GTP was analyzed in CRIB–GST pull-down assays followed by Western blotting using α-Rac1 antibody. Similar quantities of individual GTPases at every time point were confirmed by Western blotting using equivalent volumes of cell lysates. (C) Quantification of Rac1-GTP levels during the course of infection. One hundred percentage of activity corresponds to the highest amount of detected GTPase-GTP level. The amount of intracellular bacteria was quantified by gentamicin protection assays.
There is some controversy in the literature of whether the C. jejuni flagellum-mediated bacterial motility is important for invasion or if the flagellum can secrete invasion-related bacterial factors in the supernatant (Konkel et al., 2004; Novik et al., 2010). To investigate if the flagellar effect on Rac1 is direct or indirect, we were searching for a condition in which the flagellar motility is not affected, but invasion can be impaired. We have generated an α-FlaA antibody against a conserved region at the N-terminus of FlaA, and this antibody recognizes C. jejuni FlaA proteins in Westernblots (Figure 8A). We then pre-incubated WT C. jejuni with the α-FlaA antibody or pre-immune serum as control followed by motility assays in soft agar (see Materials and Methods). Treatment of any of the used C. jejuni strains with the pre-immune serum revealed no significant differences in bacterial motility or invasion as compared to non-treated bacteria (data not shown). The results also showed that while presence of the α-FlaA antibody had a slight but no significant effect on motility of WT C. jejuni or ΔcadF mutant (Figures 8A,B), bacterial invasion was significantly impaired as determined by gentamicin protection assay (Figures 8C,D). This suggests that the flagellum of C. jejuni has a motility-independent activity leading to bacterial entry into host target cells.
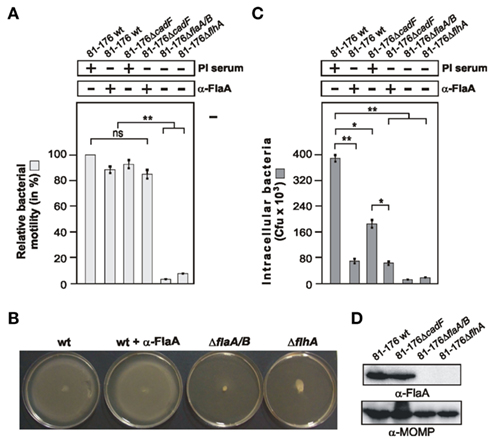
Figure 8. Different effects of an α-FlaA antibody on C. jejuni motility and host cell invasion. (A) Quantification of motility using the indicated WT and mutant strains in the presence of 20 μg/ml α-FlaA antibody or pre-immune (PI) serum as control. One hundred percentage of motility corresponds to the highest motility presented by C. jejuni WT strain 81–176. (B) Examples of motility phenotypes with indicated strains. (C) INT-407 cells were infected with the indicated WT and mutant strains for 6 h in presence or absence of 20 μg/ml α-FlaA antibody or PI serum as control. Intracellular Campylobacter cells were quantified by gentamicin protection assays. (D) The α-FlaA immunoblot shows that flagellin is expressed in WT and ΔcadF C. jejuni, but not in flagellar mutant bacteria. The α-MOMP immunoblot served as loading control. *p < 0.05 and **p ≤ 0.005 were considered as statistically significant as compared to the control.
Discussion
Host cell entry is an important process in the pathogenesis of many pathogenic bacteria and involves numerous steps such as bacterial binding at specific receptor sites, signaling to the host cell, re-programming of intracellular host signal transduction pathways, membrane and cytoskeletal rearrangements, and eventual engulfment of the bacterium, which commonly involves the activity of one or more small Rho GTPases. Rho family members, including Rac1, Cdc42, and RhoA, are small GTP-binding proteins that serve as guanine nucleotide-regulated switches which transmit external stimuli to modulate different cellular functions (Tran Van Nhieu et al., 1999; Schmidt and Hall, 2002; Cossart and Sansonetti, 2004; Rottner et al., 2005; Tomar and Schlaepfer, 2009). Host cell entry of C. jejuni is considered as one of the primary reasons for bacterial-caused tissue damage, however, the molecular mechanism of C. jejuni invasion is widely unclear. We have recently shown that C. jejuni infection of INT-407 cells is accompanied by time-dependent activation of small Rho GTPases, predominantly Rac1 (Krause-Gruszczynska et al., 2007). Using specific GTPase-modifying toxins, inhibitors and GTPase expression constructs we have also demonstrated that Rac1 activity is clearly involved in C. jejuni invasion. Here we aimed to identify the signaling pathway leading to C. jejuni-induced Rac1 activation. Using knockout cell lines of several host receptors (fibronectin−/−, GD25 integrin-β1−/−) and kinases (FAK−/−), siRNA transfection, scanning electron microscopy, Rho GTPase pull-downs, G-LISA, and gentamicin protection assays, we demonstrate that C. jejuni hijacks a major fibronectin → integrin beta1 → FAK → DOCK180/Tiam-1 signaling cascade (Hsia et al., 2003), which is crucial for triggering Rac1 GTPase activity followed by bacterial entry of host target cells. The major findings of this study are summarized in a proposed model (Figure 9) and are discussed below.
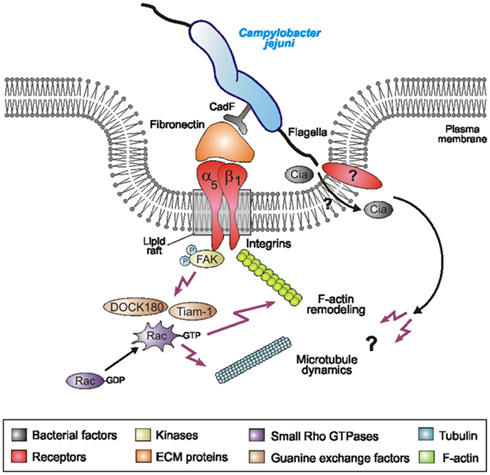
Figure 9. Proposed model for C. jejuni-induced signaling leading to Rac1 activation and bacterial invasion. C. jejuni adheres to host cells via the fibronectin-binding protein CadF which acts as a bridge engaging the integrin β1 receptor. Integrin occupancy and clustering in lipid rafts leads to recruitment and activation of the non-receptor tyrosine kinase FAK. Phosphorylation of FAK triggers a cascade of signals resulting in the formation of protein complexes leading to activation of GEFs, including DOCK180 and Tiam-1. Activated DOCK180 and Tiam-1 then induce the activation of Rac1. This potentially causes localized actin and/or microtubule rearrangements at the site of C. jejuni entry, resulting in bacterial uptake. In addition to C. jejuni cadF, some flagellar genes also appear to play a role. If the flagellum participates by sole bacterial motility, by translocating bacterial Cia effector proteins or targeting a host receptor directly is not yet clear.
Most of the recent studies investigating C. jejuni invasion utilized pharmacological inhibitors or dominant-negative constructs which may have side-effects, and thus only provide very limited clarity on host factors playing crucial roles in the bacterial entry process. Here we used for the first time a series of three knockout cell lines for infection assays. These cell lines have the great advantage that the respective genes of interest were completely deleted in the chromosomes. Thus, not even small traces of protein are expressed, allowing clear answers if certain genes are involved in a given response or not. In fact, C. jejuni has two reported fibronectin-binding proteins, CadF and FlpA (Moser et al., 1997; Konkel et al., 2010), and host cell entry of C. jejuni shown here was widely dependent on the expression of fibronectin, integrin β1 and FAK. Since integrin β1 is the natural receptor of fibronectin, our data suggest a cascade of signaling events in a fibronectin → integrin β1 → FAK dependent fashion. In addition, we found that Rac1-GTP levels induced by C. jejuni were significantly elevated in infected FAK-expressing but not in FAK-deficient cells, and Rac1 activation was confirmed conclusively by two independent approaches, CRIB-pull-down and G-LISA. In line with these observations, we observed membrane dynamics, ruffling, and engulfment of C. jejuni upon infection with WT control cells, but not in any of the infected knockout cell lines. Together, these new findings provide clear evidence that fibronectin, integrin β1, and FAK are major host factors playing not only a role in Rac1 activation but also host cell entry of the bacteria. By comparison, this scenario is very similar to that reported for Staphylococcus aureus, a Gram-positive pathogen expressing various fibronectin-binding proteins, because infected fibronectin−/− or FAK−/− cells were also severely impaired in their ability to internalize the latter bacteria (Agerer et al., 2005; Schroeder et al., 2006). Furthermore, integrin-mediated uptake of S. aureus depends on the integrity of membrane microdomains (Hoffmann et al., 2010), and this is in line with the finding that MβCD-treatment blocks internalization of C. jejuni. Our studies would suggest that S. aureus may also activate Rac1 by a similar sequence of events to trigger their uptake process.
By a strategy engaging fibronectin and integrin β1, C. jejuni seems to exploit the ability of this receptor complex to dynamically associate with the intracellular cytoskeleton and to generate the necessary pulling forces to promote bacterial uptake by the host cell. In non-infected healthy tissues, integrin β1-containing fibrillar cell adhesions are important for the organization of the extracellular matrix, as they co-align with fibronectin fibrils, and genetic elimination of integrin β1 in GD25 cells results in defects in the assembly of a fibrillar meshwork of extracellular fibronectin (Wennerberg et al., 1996; Danen and Yamada, 2001; Leiss et al., 2008). Cellular pulling forces generated via an integrin β1-mediated linkage to the actin-myosin network seem to be critical for fibronectin fibril formation, as force-triggered conformational changes are essential to expose cryptic oligomerization motifs within the fibronectin molecules (Sechler et al., 2001). Importantly, FAK has been shown to play a key role in the formation of a fibrillar fibronectin extracellular matrix. Cultured FAK−/− cells in vitro, as well as FAK−/− mouse embryos in vivo, fail to properly assemble fibronectin fibrils (Ilic et al., 2004; Leiss et al., 2008). In line with the fact that FAK−/− cells are unable to properly organize the extracellular fibronectin matrix, we found that these cells are deficient in the ability to internalize C. jejuni, suggesting that fibronectin/integrin-linkages to the actin-myosin network are disrupted and pulling forces are not provided. As one would expect from these results, C. jejuni profoundly stimulated FAK kinase activity during infection. The activation of FAK has also been described for other pathogens targeting integrins for bacterial invasion or other purposes, including S. aureus (Agerer et al., 2005), Yersinia pseudotuberculosis (Alrutz and Isberg, 1998; Eitel et al., 2005), and Helicobacter pylori (Kwok et al., 2007; Tegtmeyer et al., 2010), thus FAK seems a favorite target in bacterial pathogenesis.
The observation that FAK expression and activation is required for C. jejuni-triggered Rac1 activity and invasion, led us to investigate the involved signaling in more detail. In fact, C. jejuni-induced the phosphorylation of FAK at Y-397 and Y-925, and expression of FAK Y397F, K454R, Y925F, or ΔPR1/2 mutants in FAK−/− cells did not restore bacterial uptake as compared to WT FAK. A well-described GEF downstream of FAK is DOCK180 (Hauck et al., 2002; Hsia et al., 2003). The signaling cascade involves p130Cas, an adapter molecule binding to proline-rich residues in the carboxyl-terminal domain of FAK. p130Cas then associates with the adapter protein Crk and this complex activates DOCK180. In addition, it has been shown that expression of phospho-mimetic FAK-Y925E enhanced cell protrusions together with activation of the same DOCK180-dependent Rac1 signaling pathway, thus, phosphorylation of FAK at Y-925 is also involved this scenario (Deramaudt et al., 2011). These observations are in well agreement with our findings, suggesting that C. jejuni activates a classical FAK → p130CAS → Crk → DOCK180 → Rac1 signaling pathway. However, siRNA knockdown of DOCK180 expression was not sufficient to completely eliminate Rac1 activity and bacterial invasion. The other GEF required for C. jejuni-induced Rac1 activation was Tiam-1, but the molecular mechanism how FAK can target Tiam-1 is not yet clear. Interestingly, syndecan-2-mediated cell migration was diminished when cells were transfected with non-phosphorylatable FAK Y397F mutant or siRNA against Tiam-1, suggesting that a FAK → Tiam-1 → Rac1 signaling pathway is activated (Park et al., 2005). If FAK can stimulate Tiam-1 directly or via another factor, however, needs to be investigated in future studies.
The profound activation of Rac1 is a hallmark of numerous highly invasive pathogens such as Shigella flexneri and Salmonella enterica (Hardt et al., 1998; Tran Van Nhieu et al., 1999). These bacteria induce extensive membrane ruffling by the “trigger mechanism” requiring different Rho GTPases including Rac1, which are targeted directly through bacterial GEFs injected into the host cell by a type III secretion system (T3SS). Interestingly, C. jejuni does not encode a classical T3SS as reported above (Konkel et al., 1999; Hofreuter et al., 2006). This is in line with our observations that eukaryotic GEFs are required for C. jejuni-induced Rac1 activation, while homologs of well-known S. flexneri and S. enterica GEFs are absent in C. jejuni genomes. Our recent studies showed that C. jejuni pathogenicity factors such as plasmid pVir, cytolethal distending toxin (CDT), certain capsular genes and the adhesin PEB1 are not involved in C. jejuni-induced Rac1 activation nor invasion, and even some mutants including ΔkpsS or Δpeb1 even produced higher GTPase-GTP levels as compared to WT C. jejuni (Krause-Gruszczynska et al., 2007). Importantly, we found here that an isogenic ΔcadF mutant less efficiently induced activation of Rac1 as compared to WT bacteria. Although this is an indirect approach, this data suggests that the fibronectin-binding protein CadF, at least in part, could be involved in Rac1 activation. Thus, we propose that CadF does not only act as a canonical adhesin for bacterial binding to fibronectin, but can also stimulate integrins and FAK kinase activity, which subsequently activates downstream GEFs and Rac1, important in C. jejuni invasion. If the other fibronectin-binding protein FlpA may also play a role needs to be investigated in future studies. However, another proposed C. jejuni factor involved in Rac1 activation reported here is the flagellum, because ΔflaA/B or ΔflhA knockout mutants lacking FlaA expression and flagella induced only very little Rac1-GTP levels. The flagellar apparatus represents one of the most intensively investigated pathogenicity determinants in Campylobacter. FlaA/B proteins have been shown to be required for bacterial colonization in a number of animal models (Morooka et al., 1985; Wassenaar et al., 1993; Hendrixson and DiRita, 2004), and they play an active but yet unknown role in the invasion of epithelial cells (Wassenaar et al., 1991; Grant et al., 1993; Yao et al., 1994). The possible impact of the flagellum in invasion is controverse in the literature. One hypothesis is that the flagella, like their evolutionary related T3SS counterparts, can secrete invasion-triggering factors such as CiaB and others into the culture supernatant (Konkel et al., 1999, 2001, 2004). The other hypothesis is that flagella-mediated motility is the driving force to trigger C. jejuni invasion, but CiaB is not involved (Novik et al., 2010). We show here that flagella-driven motility is not significantly hampered by addition of an α-Flagellin antibody, but the invasive properties of C. jejuni were significantly inhibited. This suggests that motility and invasion are important, but not necessarily coupled processes. We propose that the flagellum, unlike its role in motility, may transport bacterial effectors into the medium or into the host cell, and/or the flagellum itself may bind a host cell receptor directly to trigger signaling involved in invasion (Figure 9). Future studies should investigate these different possibilities.
Taken together, the work presented here demonstrates several lines of evidence for a clear role of fibronectin, integrin β1, FAK, DOCK180, Tiam-1, and Rac1 during host cell entry of three different isolates including the C. jejuni model strain 81–176. By comparison to other bacterial infection models, our electron microscopic studies of infected INT-407 and other cell lines reported here support the view that C. jejuni do not enter host cells by a robust trigger mechanism as known for Shigella and Salmonella, nor a classical “zipper mechanism” as described for Yersinia, but rather by a very specialized pathway sharing features of both mechanisms. Our electron microscopic pictures, showing that C. jejuni enter cells first with the leading flagellum (Figure 3B), may underline the important role of the flagellum in this context. Our findings suggest that the flagellum may act cooperatively with the CadF adhesin in order to establish initial host cell contact and trigger host cell signaling leading to Rac1 activation and invasion. Furthermore, there appears to be a separate, flagellum-mediated uptake mechanism, which is independent of CadF and which allows invasion in the absence of this adhesin. Together, these results add another important aspect to our understanding of the mechanism of C. jejuni pathogenesis. Since the exact pathway of C. jejuni invasion into host target cells is still not fully understood, it will be highly interesting to study in future the precise mechanism of how active Rac1 regulates actin rearrangements and/or microtubule dynamics involved in the bacterial entry process. It will be also interesting to investigate the role of another small Rho GTPase member, Cdc42, which is also activated by C. jejuni (Krause-Gruszczynska et al., 2007). Experiments are underway in our laboratory to investigate these signaling pathways in more detail.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ina Schleicher for excellent technical assistance, and Drs. Martin Blaser (New York University, USA), Michael Konkel (Pullman University, USA) and Patricia Guerry (Fayetteville State University, USA) for providing C. jejuni strains and mutants, respectively. We are also very grateful to Drs. David Schlaepfer (University of California, USA) and Staffan Johannsson (Uppsala University, Sweden) for providing FAK−/− and GD25 knockout cells as well as several plasmid constructs. The work of Steffen Backert is supported through a SFI grant (UCD 09/IN.1/B2609).
References
Agerer, F., Lux, S., Michel, A., Rohde, M., Ohlsen, K., and Hauck, C. R. (2005). Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalisation. J. Cell Sci. 118(Pt 10), 2189–2200.
Allos, B. M. (2001). Campylobacter jejuni infections: update on emerging issues and trends. Clin. Infect. Dis. 32, 1201–1206.
Alouf, J. E., and Popoff, M. R. (2005). The Comprehensive Sourcebook of Bacterial Protein Toxins, 3rd Edn. London: Academic Press.
Alrutz, M. A., and Isberg, R. R. (1998). Involvement of focal adhesion kinase in invasin-mediated uptake. Proc. Natl. Acad. Sci.U.S.A. 95, 13658–13663.
Biswas, D., Niwa, H., and Itoh, K. (2004). Infection with Campylobacter jejuni induces tyrosine-phosphorylated proteins into INT-407 cells. Microbiol. Immunol. 48, 221–228.
Blaser, M. J., and Engberg, J. (2008). “Clinical aspects of Campylobacter jejuni and campylobacter coli infections,” in Campylobacter, eds I. Nachamkin, C. M. Szymanski and M. J. Blaser (Washington, DC: American Society for Microbiology), 99–121.
Burns, D., Barbieri, J. T., Iglewski, B. H., and Rappuoli, R. (2003). Bacterial Protein Toxins. Washington, DC: American Society for Microbiology, 215–228.
Carrillo, C. D., Taboada, E., Nash, J. H., Lanthier, P., Kelly, J., Lau, P. C., Verhulp, R., Mykytczuk, O., Sy, J., Findlay, W. A., Amoako, K., Gomis, S., Willson, P., Austin, J. W., Potter, A., Babiuk, L., Allan, B., and Szymanski, C. M. (2004). Genome-wide expression analyses of Campylobacter jejuni NCTC11168 reveals coordinate regulation of motility and virulence by flhA. J. Biol. Chem. 279, 20327–20338.
Cossart, P., and Sansonetti, P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248.
Danen, E. H., and Yamada, K. M. (2001). Fibronectin, integrins, and growth control. J. Cell. Physiol. 189, 1–13.
Day, W.A. Jr., Sajecki, J. L., Pitts, T. M., and Joens, L. A. (2000). Role of catalase in Campylobacter jejuni intracellular survival. Infect. Immun. 68, 6337–6345.
Deramaudt, T. B., Dujardin, D., Hamadi, A., Noulet, F., Kolli, K., De Mey, J., Takeda, K., and Rondé, P. (2011). FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell 22, 964–975.
Eckmann, L., and Kagnoff, M. F. (2005). Intestinal mucosal responses to microbial infection. Springer Semin. Immunopathol. 27, 181–196.
Eitel, J., Heise, T., Thiesen, U., and Dersch, P. (2005). Cell invasion and IL-8 production pathways initiated by YadA of Yersinia pseudotuberculosis require common signalling molecules (FAK, c-Src, Ras) and distinct cell factors. Cell. Microbiol. 7, 63–77.
Fang, G., Araujo, V., and Guerrant, R. L. (1991). Enteric infections associated with exposure to animals or animal products. Infect. Dis. Clin. North Am. 5, 681–701.
Forsythe, S. J. (2000). “Food poisoning microorganisms,” in The Microbiology of Safe Food, ed. S. J. Forsythe (Abingdon: Blackwell Science publishers), 87–148.
Friedman, C. R., Neimann, J., Wegener, H. C., and Tauxe, R. V. (2000). “Epidemiology of Campylobacter jejuni infections in the United States and other industrialized nations,” in Campylobacter, eds I. Nachamkin and M. J. Blaser (Washington, DC: American Society for Microbiology), 121–138.
Goon, S., Ewing, C. P., Lorenzo, M., Pattarini, D., Majam, G., and Guerry, P. (2006). A sigma28-regulated nonflagella gene contributes to virulence of Campylobacter jejuni 81–176. Infect. Immun. 74, 769–772.
Grant, C. C. R., Konkel, M. E., Cieplak, W. Jr., and Tompkins, L. S. (1993). Role of flagella in adherence, internalization, and translocation of Campylobacter jejuni in nonpolarized and polarized epithelial cell cultures. Infect. Immun. 61, 1764–1771.
Hardt, W. D., Chen, L. M., Schuebel, K. E., Bustelo, X. R., and Galan, J. E. (1998). S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93, 815–826.
Hauck, C. R., Hsia, D. A., and Schlaepfer, D. D. (2002). The focal adhesion kinase – a regulator of cell migration and invasion. IUBMB Life 53, 115–119.
Hendrixson, D. R., and DiRita, V. J. (2004). Identification of Campylobacter jejuni genes involved in commensal colonization of the chick gastrointestinal tract. Mol. Microbiol. 52, 471–484.
Hoffmann, C., Berking, A., Agerer, F., Buntru, A., Neske, F., Chhatwal, G. S., Ohlsen, K., and Hauck, C. R. (2010). Caveolin limits membrane microdomain mobility and integrin-mediated uptake of fibronectin-binding pathogens. J. Cell Sci. 123(Pt 24), 4280–4291.
Hofreuter, D., Novik, V., and Galán, J. E. (2008). Metabolic diversity in Campylobacter jejuni enhances specific tissue colonization. Cell Host Microbe 4, 425–433.
Hofreuter, D., Tsai, J., and Watson, R. O. (2006). Unique features of a highly pathogenic Campylobacter jejuni strain. Infect. Immun. 74, 4694–4707.
Hsia, D. A., Mitra, S. K., Hauck, C. R., Streblow, D. N., Nelson, J. A., Ilic, D., Huang, S., Li, E., Nemerow, G. R., Leng, J., Spencer, K. S., Cheresh, D. A., and Schlaepfer, D. D. (2003). Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 160, 753–767.
Hu, L., and Kopecko, D. J. (1999). Campylobacter jejuni 81-176 associates with microtubules and dynein during invasion of human intestinal cells. Infect. Immun. 67, 4171–4182
Hu, L., and Kopecko, D. J. (2008). “Cell biology of human host cell entry by Campylobacter jejuni,” in Campylobacter, eds I. Nachamkin, C. M. Szymanski and M. J. Blaser (Washington, DC: American Society for Microbiology), 297–313.
Hu, L., McDaniel, J. P., and Kopecko, D. J. (2006). Signal transduction events involved in human epithelial cell invasion by Campylobacter jejuni 81–176. Microb. Pathog. 40, 91–100.
Ilic, D., Kovacic, B., Johkura, K., Schlaepfer, D. D., Tomasevic, N., Han, Q., Kim, J. B., Howerton, K., Baumbusch, C., and Ogiwara, N. (2004). FAK promotes organization of fibronectin matrix and fibrillar adhesions. J.Cell Sci. 117, 177–187.
Kanipes, M. I., Holder, L. C., Corcoran, A. T., Moran, A. P., and Guerry, P. (2004). A deep-rough mutant of Campylobacter jejuni 81–176 is noninvasive for intestinal epithelial cells. Infect. Immun. 72, 2452–2455.
Karlyshev, A. V., Linton, D., Gregson, N. A., Lastovica, A. J., and Wren, B. W. (2000). Genetic and biochemical evidence of a Campylobacter jejuni capsular polysaccharide that accounts for Penner serotype specificity. Mol. Microbiol. 35, 529–541.
Konkel, M. E., Garvis, S. D., Tipton, S., Anderson, D. E., and Cieplak, W. Jr. (1997). Identification and molecular cloning of a gene encoding a fibronectin binding protein (CadF) from Campylobacter jejuni. Mol. Microbiol. 24, 953–963.
Konkel, M. E., Hayes, S. F., Joens, L. A., and Cieplak, W. Jr. (1992). Characteristics of the internalization and intracellular survival of Campylobacter jejuni in human epithelial cell cultures. Microb. Pathog. 13, 357–370.
Konkel, M. E., Kim, B. J., Rivera-Amill, V., and Garvis, S. G. (1999). Bacterial secreted proteins are required for the internaliztion of Campylobacter jejuni into cultured mammalian cells. Mol. Microbiol. 32, 691–701.
Konkel, M. E., Klena, J. D., Rivera-Amill, V., Monteville, M. R., Biswas, D., Raphael, B., and Mickelson, J. (2004). Secretion of virulence proteins from Campylobacter jejuni is dependent on a functional flagellar export apparatus. J. Bacteriol. 186, 3296–3303.
Konkel, M. E., Larson, C. L., and Flanagan, R. C. (2010). Campylobacter jejuni FlpA binds fibronectin and is required for maximal host cell adherence. J. Bacteriol. 192, 68–76.
Konkel, M. E., Monteville, M. R., Rivera-Amill, V., and Joens, L. A. (2001). The pathogenesis of Campylobacter jejuni-mediated enteritis. Curr. Issues Intest. Microbiol. 2, 55–71.
Krause-Gruszczynska, M., Rohde, M., Hartig, R., Genth, H., Schmidt, G., Keo, T., Koenig, W., Miller, W. G., Konkel, M. E., and Backert, S. (2007). Role of the small Rho GTPases Rac1 and Cdc42 in host cell invasion of Campylobacter jejuni. Cell. Microbiol. 9, 2431–2444.
Kwok, T., Zabler, D., Urman, S., Rohde, M., Hartig, R., Wessler, S., Misselwitz, R., Berger, J., Sewald, N., König, W., and Backert, S. (2007). Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449, 862–866.
Larson, C. L., Christensen, J. E., Pacheco, S. A., Minnich, S. A., and Konkel, M. E. (2008). “Campylobacter jejuni secretes proteins via the flagellar type III secretion system that contribute to host cell invasion and gastroenteritis,” in Campylobacter, eds I. Nachamkin, C. M. Szymanski and M. J. Blaser (Washington, DC: American Society for Microbiology), 315–332.
Lastovica, A. J. (1996). “Campylobacter/Helicobacter bacteraemia in Cape Town, South Africa 1977–95,” in Campylobacters, Helicobacters and Related Organisms, eds D. G. Newell, J. M. Ketley, and R. A. Feldman (New York: Plenum Press), 475–479.
Leiss, M., Beckmann, K., Giros, A., Costell, M., and Faessler, R. (2008). The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr. Opin. Cell Biol. 20, 502–507.
Monteville, M. R., Yoon, J. E., and Konkel, M. E. (2003). Maximal adherence and invasion of INT 407 cells by Campylobacter jejuni requires the CadF outer-membrane protein and microfilament reorganization. Microbiology 149(Pt 1), 153–165.
Morooka, T., Umeda, A., and Amako, K. (1985). Motility as an intestinal colonization factor for Campylobacter jejuni. J. Gen. Microbiol. 131, 1973–1980.
Moser, I., Schroeder, W., and Salnikow, J. (1997). Campylobacter jejuni major outer membrane protein and a 59-kDa protein are involved in binding to fibronectin and INT-407 cell membranes. FEMS Microbiol. Lett. 157, 233–238.
Nachamkin, I., Szymanski, C. M., and Blaser, M. J. (2008). Campylobacter. Washington, DC: American Society for Microbiology, 163–198.
Novik, V., Hofreuter, D., and Galán, J. E. (2010). Identification of Campylobacter jejuni genes involved in its interaction with epithelial cells. Infect. Immun. 78, 3540–3553.
Nyberg, P., Sakai, T., Cho, K. H., Caparon, M. G., Fässler, R., and Björck, L. (2004). Interactions with fibronectin attenuate the virulence of Streptococcus pyogenes. EMBO J. 23, 2166–2174.
Oelschlaeger, T. A., Guerry, P., and Kopecko, D. J. (1993). Unusual microtubule-dependent endocytosis mechanisms triggered by Campylobacter jejuni and Citrobacter freundii. Proc. Natl. Acad. Sci. U.S.A. 90, 6884–6888.
Park, H., Han, I., Kwon, H. J., and Oh, E. S. (2005). Focal adhesion kinase regulates syndecan-2-mediated tumorigenic activity of HT1080 fibrosarcoma cells. Cancer Res. 65, 9899–9905.
Pei, Z., Burucoa, C., Grignon, B., Baqar, S., Huang, X. Z., Kopecko, D. J., Bourgeois, A. L., Fauchere, J. L., and Blaser, M. J. (1998). Mutation in the peb1A locus of Campylobacter jejuni reduces interactions with epithelial cells and intestinal colonization of mice. Infect. Immun. 66, 938–943.
Poly, F., and Guerry, P. (2008). Pathogenesis of Campylobacter. Curr. Opin. Gastroenterol. 24, 27–31.
Potturi-Venkata, L. P., Backert, S., Vieira, S. L., and Oyarzabal, O. A. (2007). Evaluation of logistic processing to reduce cross-contamination of commercial broiler carcasses with Campylobacter spp. J. Food Prot. 70, 2549–2554.
Rottner, K., Stradal, T. E., and Wehland, J. (2005). Bacteria-host-cell interactions at the plasma membrane: stories on actin cytoskeleton subversion. Dev. Cell 9, 3–17.
Salyers, A. A., and Whitt, D. D. (1994). Bacterial Pathogenesis. Washington, DC: American Society for Microbiology, 260–268.
Sander, E. E., van Delft, S., ten Klooster, J. P., Reid, T., van der Kammen, R. A., Michiels, F., and Collard, J. G. (1998). Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143, 1385–1398.
Schmidt, A., and Hall, A. (2002). Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609.
Schroeder, A., Schroeder, B., Roppenser, B., Linder, S., Sinha, B., Faessler, R., and Aepfelbacher, M. (2006). Mol. Biol. Cell 17, 5198–5210.
Sechler, J. L., Rao, H., Cumiskey, A. M., Vega-Colon, I., Smith, M. S., Murata, T., and Schwarzbauer, J. E. (2001). A novel fibronectin binding site required for fibronectin fibril growth during matrix assembly. J. Cell Biol. 154, 1081–1088.
Sougioultzis, S., and Pothoulakis, C. (2003). Bacterial infections: small intestine and colon. Curr. Opin. Gastroenterol. 19, 23–30.
Tam, C. C. (2001). Campylobacter reporting at its peak year of 1998: don’t count your chickens yet. Commun. Dis. Public Health 4, 194–199.
Tegtmeyer, N., Hartig, R., Delahay, R. M., Rohde, M., Brandt, S., Conradi, J., Takahashi, S., Smolka, A. J., Sewald, N., and Backert, S. (2010). A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J. Biol. Chem. 285, 23515–23526.
Thanassi, D. G., and Hultgren, S. J. (2000). Multiple pathways allow protein secretion across the bacterial outer membrane. Curr. Opin. Cell Biol. 12, 420–430.
Tomar, A., and Schlaepfer, D. D. (2009). Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 21, 676–683.
Tran Van Nhieu, G., Caron, E., Hall, A., and Sansonetti, P. J. (1999). IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 18, 3249–3262.
van Spreeuwel, J. P., Duursma, G. C., Meijer, C. J., Bax, R., Rosekrans, P. C., and Lindeman, J. (1985). Campylobacter colitis: histological immunohistochemical and ultrastructural findings. Gut 26, 945–951.
Wassenaar, T. M., and Blaser, M. J. (1999). Pathophysiology of Campylobacter jejuni infections of humans. Microbes Infect. 1, 1023–1033.
Wassenaar, T. M., Bleumink-Pluym, N. M. C., and van der Zeijst, B. A. M. (1991). Inactivation of Campylobacter jejuni flagellin genes by homologous recombination demonstrates that flaA but not flaB is required for invasion. EMBO J. 10, 2055–2061.
Wassenaar, T. M., van der Zeijst, B. A. M., Ayling, R., and Newell, D. G. (1993). Colonization of chicks by motility mutants of Campylobacter jejuni demonstrates the importance of flagellin A expression. J. Gen. Microbiol. 139, 1171–1175.
Watson, R. O., and Galán, J. E. (2008). Campylobacter jejuni survives within epithelial cells by avoiding delivery to lysosomes. PLoS Pathog. 4, e14. doi:10.1371/journal.ppat.0040014
Wennerberg, K., Lohikangas, L., Gullberg, D., Pfaff, M., Johansson, S., and Faessler, R. (1996). Beta 1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 132, 227–238.
Wooldridge, K. G., and Ketley, J. M. (1997). Campylobacter-host cell interactions. Trends Microbiol. 5, 96–102
Wooldridge, K. G., Williams, P. H., and Ketley, J. M. (1996). Host signal transduction and endocytosis of Campylobacter jejuni. Microb. Pathog. 21, 299–305.
Keywords: Rho family GTPases, molecular pathogenesis, cellular invasion, signaling, virulence
Citation: Boehm M, Krause-Gruszczynska M, Rohde M, Tegtmeyer N, Takahashi S, Oyarzabal OA and Backert S (2011) Major host factors involved in epithelial cell invasion of Campylobacter jejuni: role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1. Front. Cell. Inf. Microbio. 1:17. doi: 10.3389/fcimb.2011.00017
Received: 30 September 2011; Accepted: 24 November 2011;
Published online: 12 December 2011.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Stefan Bereswill, Charité-University Medicine Berlin, GermanyChristof R. Hauck, Universitaet Konstanz, Germany
Nick Dorrell, London School of Hygiene and Tropical Medicine, UK
Copyright: © 2011 Boehm, Krause-Gruszczynska, Rohde, Tegtmeyer, Takahashi, Oyarzabal and Backert. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Steffen Backert, School of Biomolecular and Biomedical Sciences, University College Dublin, Ardmore House, Belfield Campus, Dublin, Ireland. e-mail: steffen.backert@ucd.ie
† Manja Boehm and Malgorzata Krause-Gruszczynska have contributed equally to this work.