Development of a multiplex PCR assay for detection of Shiga toxin-producing Escherichia coli, enterohemorrhagic E. coli, and enteropathogenic E. coli strains
- 1 Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX, USA
- 2 Servicio Fisiopatogenia, Departamento de Bacteriología, Instituto Nacional de Enfermedades Infecciosas, Administración Nacional de Laboratorios e Institutos de Salud “Dr. Carlos G. Malbrán,”, Buenos Aires, Argentina
- 3 School of Science and Computer Engineering, University of Houston – Clear Lake, Houston, TX, USA
- 4 Department of Pathology, Sealy Center for Vaccine Development, University of Texas Medical Branch, Galveston, TX, USA
Escherichia coli O157:H7 and other pathogenic E. coli strains are enteric pathogens associated with food safety threats and which remain a significant cause of morbidity and mortality worldwide. In the current study, we investigated whether enterohemorrhagic E. coli (EHEC), Shiga toxin-producing E. coli (STEC), and enteropathogenic E. coli (EPEC) strains can be rapidly and specifically differentiated with multiplex PCR (mPCR) utilizing selected biomarkers associated with each strain’s respective virulence genotype. Primers were designed to amplify multiple intimin (eae) and long polar fimbriae (lpfA) variants, the bundle-forming pilus gene bfpA, and the Shiga toxin-encoding genes stx1 and stx2. We demonstrated consistent amplification of genes specific to the prototype EHEC O157:H7 EDL933 (lpfA1-3, lpfA2-2, stx1, stx2, and eae-γ) and EPEC O127:H6 E2348/69 (eae-α, lpfA1-1, and bfpA) strains using the optimized mPCR protocol with purified genomic DNA (gDNA). A screen of gDNA from isolates in a diarrheagenic E. coli collection revealed that the mPCR assay was successful in predicting the correct pathotype of EPEC and EHEC clones grouped in the distinctive phylogenetic disease clusters EPEC1 and EHEC1, and was able to differentiate EHEC1 from EHEC2 clusters. The assay detection threshold was 2 × 104 CFU per PCR reaction for EHEC and EPEC. mPCR was also used to screen Argentinean clinical samples from hemolytic uremic syndrome and diarrheal patients, resulting in 91% sensitivity and 84% specificity when compared to established molecular diagnostic procedures. In conclusion, our mPCR methodology permitted differentiation of EPEC, STEC and EHEC strains from other pathogenic E. coli; therefore, the assay becomes an additional tool for rapid diagnosis of these organisms.
Introduction
Rapid diagnosis of pathogenic E. coli strains is an increasingly important issue to address in public health. Infections with Shiga toxin-producing E. coli (STEC) and among those enterohemorrhagic E. coli (EHEC), can result in abdominal cramping and diarrhea (with or without blood). A small percentage of patients can progress to a more severe and often fatal condition called hemolytic uremic syndrome (HUS). STEC/EHEC strains are found in industrialized nations as well as developing countries and typical cases in the U.S. are associated with food-borne contamination. Enteropathogenic E. coli (EPEC) is frequently associated with outbreaks of infantile diarrhea in developing nations (Orskov et al., 1990), and is a contributor to diarrheagenic illnesses in human populations around the world (Ochoa et al., 2008).
Shiga toxin-producing E. coli/EHEC and EPEC strains encode a number of virulence factors in a chromosomally located pathogenicity island termed the locus for enterocyte effacement (LEE; McDaniel et al., 1995). Intimate adhesion of STEC/EHEC and EPEC to enterocytes is mediated in part by LEE-encoded intimin gene (eae), resulting in the formation of an attaching and effacing (A/E) lesion on the surface of the intestinal cells. In addition to virulence factors encoded in the LEE pathogenicity island, EHEC and EPEC possess one or more of the chromosomally encoded long polar fimbriae (lpf) loci. Together with intimin, Lpf is the only other well-characterized colonization factor of EHEC O157:H7 (Torres et al., 2002, 2004, 2007). Our group conducted an extensive study involving A/E-producing bacterial collections from Europe and South America and demonstrated a correlation between lpf genes and different genetic variants of the intimin genes in A/E-producing E. coli (AEEC; Torres et al., 2009). The lpf genes are also widely distributed throughout pathogenic and some commensal populations of E. coli and can be categorized into distinct allelic variants (Galli et al., 2010; Gomes et al., 2011). Additionally, other groups have also reported the potential use of intimin for diagnostics based on the correlation of intimin type and lineage of STEC/EHEC and EPEC strains (Tarr and Whittam, 2002; Zhang et al., 2002; Jores et al., 2003). Due to the broad distribution of lpf and eae genes in AEEC and their association with these pathogenic E. coli strains, the lpf and eae subtypes could be used to genetically identify and distinguish diverse STEC/EHEC and EPEC serogroups (Torres et al., 2009). From a clinical point of view, the inclusion of primers for stx genes is critical, as the progression to HUS is strongly influenced by the presence of Shiga toxin (Friedrich et al., 2002; Brooks et al., 2005; Hedican et al., 2009). Further, the bfpA gene can be used as marker to detect typical (bfp+) and atypical (bfp−) EPEC strains (Nataro and Kaper, 1998).
Current proposed approaches for the specific detection of EHEC strains in clinical samples or food matrices are focused on the detection of genes present in a limited number of serotypes. For example, the inclusion of the E. coli O157:H7 O-antigen marker rfbEO157 limits the detection of strains to O157 serogroups (Bai et al., 2010; Gordillo et al., 2011). Analysis of multiple O-group genes improves the detection capabilities of an assay, but does not remove the constraint of detecting only known serogroups (Madic et al., 2011). Similar constraints are present when targeting the H7 fliC flagellar antigen (Madic et al., 2010; Gordillo et al., 2011) or when employing O157 strain-specific methodologies (Ooka et al., 2009). A recent study utilized genes encoding intimin and Shiga toxin to detect EHEC and EPEC strains, yet the assay was not designed to specifically detect O157 strains (Pavlovic et al., 2010).
In developing countries, enteric pathogen identification is frequently time consuming and incomplete, resulting in potential misdiagnoses or mistreatments. Therefore, a rapid, specific assay designed to identify EHEC/STEC and EPEC strains in a public health setting would be advantageous to help ensure that a timely and proper response is initiated. Furthermore, assays like this one would accelerate the diagnosis and significantly reduce mortality in endemic areas. Specific identification of highly pathogenic EHEC would also be critical in the event of food-borne illness outbreaks or agroterrorism.
Therefore, our study addresses the aforementioned constraints on the detection of these diarrheagenic E. coli (DEC) categories by using genes that do not encode serogroup-specific antigens, yet can distinguish O157:H7 strains, as well as unknown serogroups. We examined the hypothesis that re-emerging and outbreak-associated E. coli strains can be rapidly, specifically, and easily distinguished using multiplex PCR (mPCR) amplification of specific biomarkers associated with each strain’s respective virulence genotype. The results of our mPCR assay indicate that this approach can provide a rapid method for detection of pathogenic E. coli strains. We demonstrated that lpfA subtypes could distinguish between EHEC and EPEC groups and most importantly, inclusion of lpfA variants permitted detection of EHEC O157:H7 in 100% of the cases, further supporting the importance of lpfA in molecular diagnostics approaches.
Materials and Methods
Strains
Enterohemorrhagic E. coli O157:H7 strain EDL933, EPEC O127:H6 strain E2348/69, E. coli K12 strain MG1655, E. coli HS, Salmonella enterica serovar Typhimurium 2157, Shigella flexneri M90T, adherent invasive E. coli O83:H1 NRG857c, enterotoxigenic E. coli H10407, enteroaggregative E. coli O42, and 78 isolates from the DEC Collection (Whittam et al., 1993) were grown in Luria–Bertani (LB) broth at 37°C with shaking. Genomic DNA (gDNA) was extracted from the cells using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA).
PCR
Single and mPCR reactions were carried out using REDTaq ReadyMix PCR Reaction Mix and REDTaq DNA Polymerase (Sigma, St. Louis, MO, USA) supplemented with the appropriate primers and template DNA. Oligonucleotide primer sequences were used from previously published work or manually designed to obtain amplicons of sufficiently different sizes to be resolved in the multiplex assay (Tables 1 and 2). Primer sets specifically designed in this study utilized sequences for eae-α (FM180568), eae-β (AF081186), eae-γ (AE005174), eae-δ (AJ875027), and lpfA1-1 (NC_011601). Single PCR reactions using REDTaq ReadyMix (Sigma, St. Louis, MO, USA) and 0.8 mM of each primer were performed under the following conditions: 94°C for 5 min; 30 cycles of 94°C for 30 s, 42°C for 45 s, 72°C for 35 s; 72°C for 10 min; hold at 10°C. Multiplex reactions using an additional 0.8 units of REDTaq DNA polymerase (1.4 units total per reaction – supplementation with extra polymerase permitted an increase in specificity and band intensity) were performed under the following conditions using the primer concentrations indicated in Table 1: 94°C for 5 min; 40 cycles of 94°C for 30 s, 59°C for 1 min 30 s, 72°C for 40 s; 72°C for 10 min; hold at 10°C. Products were analyzed on 1.5% agarose gels.
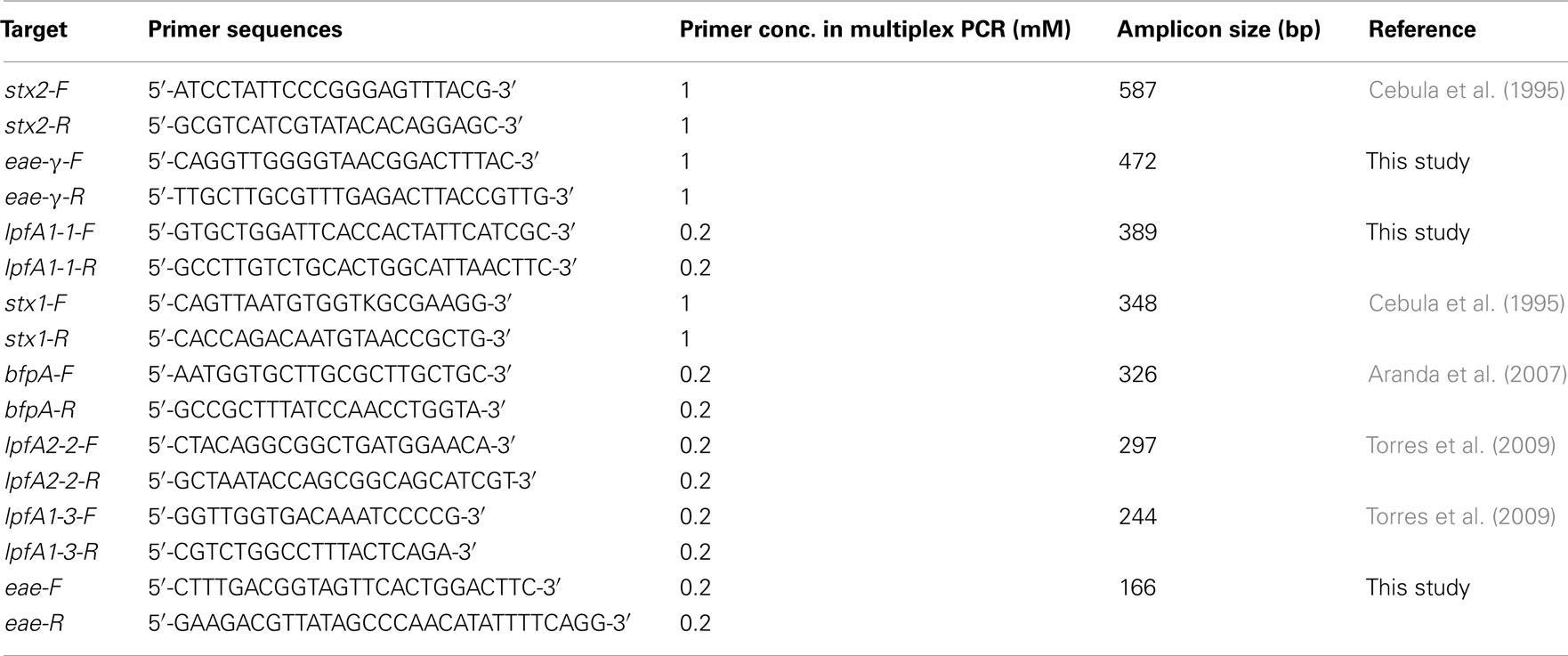
Table 1. Virulence-associated genes and primers used in this study.

Table 2. Gene profiles for differentiation of E. coli pathotypes.
Threshold of Multiplex PCR Assay Detection
The assay was performed by first re-suspending cells (EHEC O157:H7 EDL933 or EPEC O127:H6 E2348/69) freshly streaked onto LB agar to an estimated concentration of ∼4 × 109 cells/ml by monitoring the OD600. Ten-fold serial dilutions were made into sterile distilled water, which was then used as template directly in mPCR reactions.
Isolation and Molecular Characterization of Clinical Strains
One hundred fecal samples (43 HUS, 36 non-bloody diarrhea, and 21 bloody diarrhea cases) submitted to the National Reference Laboratory (NRL) in Buenos Aires, Argentina were studied. Fecal samples were plated either directly onto sorbitol MacConkey agar or after enrichment at 37°C for 4 h in trypticase soy broth with or without cefixime (50 ng/ml) and potassium tellurite (25 mg/ml). Confluent growth zones were first screened for stx1, stx2, and rfbO157 genes by mPCR (Leotta et al., 2005). A single PCR targeting the eae gene was performed (Karch et al., 1993; Karch and Bielaszewska, 2001) as well as testing of the eae variants (Ramachandran et al., 2003), if mPCR for the stx1, stx2, and rfbO157 genes was negative. Isolates with stx1, stx2, and/or eae genes were identified by standard biochemical tests, serotyped, and characterized by phenotypic and genotypic techniques (Rivas et al., 2011). For comparison purposes, the same DNA templates were screened by the mPCR developed in the present study using Platinum Taq DNA Polymerase (Invitrogen, Brazil). This study was carried out in strict accordance with the Guidelines of the National Institutes of Health and the Ministry of Health, Argentina. The protocol was approved by the Institutional Review Board of the University of Texas Medical Branch (IRB#11-081).
Results
Multiplex PCR Analysis of Strains from a Diarrheagenic E. coli Collection
The present work describes the development of an mPCR assay for the rapid detection of specific categories of pathogenic E. coli. The assay is based on the use of pathotype-specific genes for the detection of medically relevant E. coli strains, and is able to specifically detect EHEC, typical and atypical EPEC, and STEC strains.
We demonstrated robust amplification of genes specific to EHEC O157:H7 strain EDL933 (lpfA1-3, lpfA2-2, stx1, stx2, and eae-γ) and EPEC O127:H6 strain E2348/69 (eae-α, lpfA1-1, and bfpA) using an optimized mPCR protocol with purified gDNA. Amplification of genes encoding virulence factors specific to pathogenic E. coli (Table 2) was first tested in single PCR reactions. One amplification product was observed in each case (data not shown). During optimization of the multiplex reaction, the addition of REDTaq DNA polymerase to the polymerase already present in the 1× solution of REDTaq ReadyMix permitted an increase in the annealing temperature (thereby increasing specificity) and robustness of the assay. Modification of primer concentration, total number of cycles, and primer annealing time were also optimized. To assess the efficacy of this assay on clinical isolates, gDNA from strains in the DEC collection (Whittam et al., 1993), representing a variety of serotypes from the EHEC (clonal groups EHEC1 and EHEC2) and EPEC (clonal groups EPEC1 and EPEC2), was screened using mPCR (Figure 1). Amplicon sizes from each DEC sample were compared to those in the control strains to compose a genotype for each representative DEC group member. We determined that mPCR analysis using gDNA from DEC isolates was successful in predicting the correct pathotype in 75.6% (59/78) of the total number of isolates; however the assay was able to predict the pathotype of all EHEC1 (DECs 3, 4, and 5) and EPEC1 (DECs 1 and 2) and distinguish between EHEC1 and EHEC2 (DECs 8, 9, and 10) pathogroups.
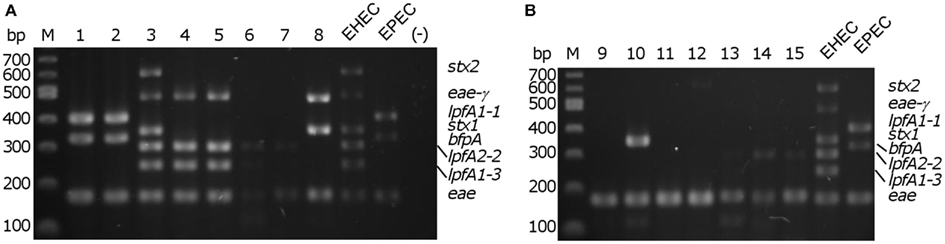
Figure 1. Multiplex PCR screening of representatives from each diarrheagenic E. coli collection (DEC) group. (A) DEC 1–8; (B) DEC 9–15, M – 100 bp DNA markers (NEB). EHEC/EPEC, positive controls; (−), no template control.
Analytical Sensitivity of Multiplex PCR Assay
To assess the specificity of the assay, the multiplex reaction setup was tested using gDNA from commensal E. coli strains and pathogenic enteric bacteria. Relatively low intensity amplicons slightly larger than the size of the eae product were observed in three of the seven strains tested (two E. coli strains [one commensal and one laboratory isolate] and Shigella flexneri; Figure 2). With the Salmonella strain used, we observed a product of approximately 300 bp, possibly due to amplification of an uncharacterized lpfA variant in that strain. A smaller, faint amplicon was also observed using the Salmonella strain; however, its size does not correspond to any of the amplicons expected in our assay. Analysis of the commensal E. coli HS revealed another faint, non-specific product not corresponding to the size of a target amplicon (Figure 2).
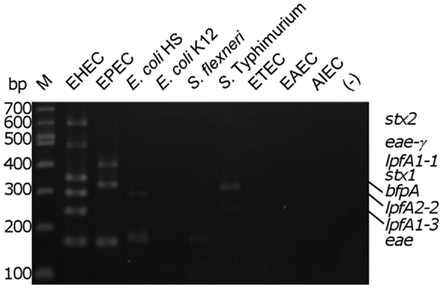
Figure 2. Determination of biological assay specificity. Multiplex PCR assay was performed using gDNA from pathogenic and commensal enteric bacteria. M, 100 bp DNA markers (NEB); EHEC, enterohemorrhagic E. coli; EPEC, enteropathogenic E. coli; E. coli HS and E. coli K12, human intestinal commensal and laboratory strains; S. flexneri, Shigella flexneri; S. typhimurium, Salmonella enterica serovar Typhimurium 2157; ETEC, enterotoxigenic E. coli H10407; EAEC, enteroaggregative E. coli O42; AIEC, adherent/invasive E. coli O83:H1; (−), no template control. The position of the amplicons in the EHEC and EPEC strains are indicated on the right of the figure.
Analytical Specificity of Multiplex PCR Assay
We then determined the in vitro threshold of detection with the EHEC and EPEC prototype strains. Colonies from freshly streaked LB agar plates were re-suspended in sterile distilled water to a concentration of ∼4 × 109 cells/mL. A 5 μl aliquot from each of the 10-fold serial dilutions were added to mPCR reactions to permit testing of a 10-fold range of template concentrations from 1 × 109 to 1 × 103 cells/mL in a 20 μl reaction. The threshold of detection for EHEC and EPEC in vitro was determined to be ∼2 × 104 CFU/reaction (corresponding to 1 × 106 cells/ml in each reaction), assessed by visibility of all six predicted amplicons for EHEC and all three amplicons for EPEC (Figure 3).
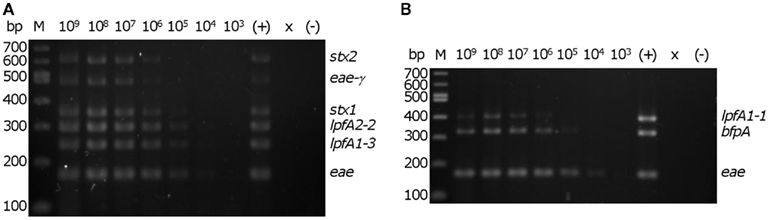
Figure 3. Determination of detection threshold. Serial dilutions of re-suspended EHEC EDL933 or EPEC E2348/69 colonies were used as template in the multiplex PCR reaction. M – 100 bp DNA markers (NEB). Number above each lane indicates the concentration (cells/ml) of bacteria per reaction. Consistent detection of (A) all of six EHEC EDL933-specific bands and (B) all three EPEC E2348/69-specific bands was observed using 2 × 104 cells in a 20 μl PCR reaction. ×, lane was not loaded; (−), no template control. The position of the amplicons in the EHEC and EPEC strains are indicated on the right of each panel.
Evaluation of Multiplex PCR Assay at NRL (Argentina)
Next, we tested the feasibility to implement our mPCR in a clinical setting, performing a trial study with samples received at the National Reference Laboratory (NRL), Argentina, and comparing the results to methods already optimized in that laboratory (Table 3). The clinical sensitivity and specificity of the assay was estimated to be 91% and 84%, respectively. Identification of at least one gene (eae, one of the stx genes, or rfbO157) was the basis for determining whether a given case was considered positive or negative.
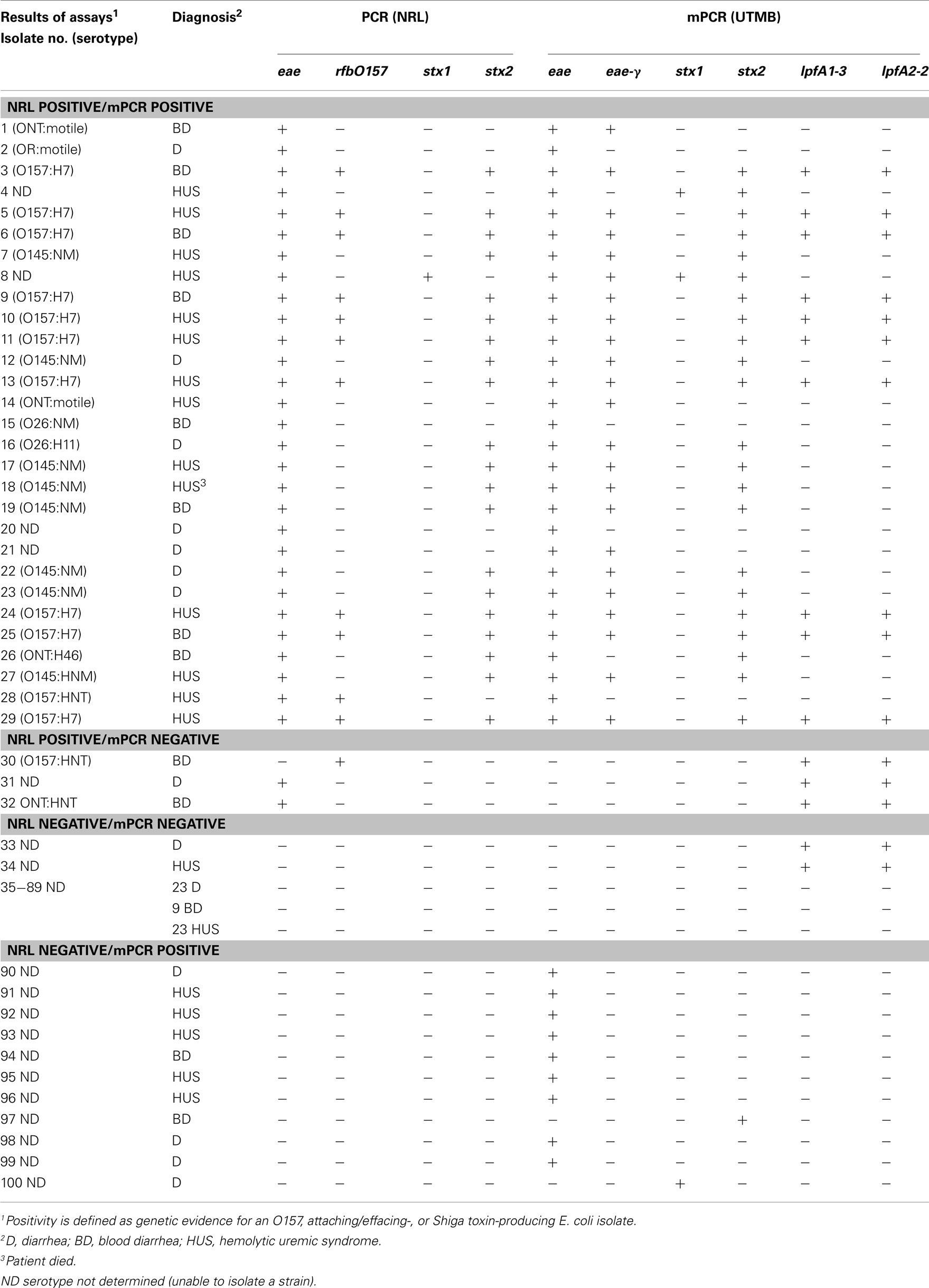
Table 3. Comparison of results between molecular diagnostic assays at NRL and the current proposed mPCR methodology.
Further analysis of non-O157 and non-O145 isolates revealed that although the mPCR assay typically identified eae-γ in these isolates, RFLP–PCR intimin typing (Ramachandran et al., 2003) at the NRL revealed eae-β or eae-Έ in the non-O157 and non-O145 cases (data not shown). These findings likely represent a false positive indication of the presence of eae-γ, as strains that possess eae-γ typically contain lpfA1-3 and lpfA2-2 as well, both of which are absent in the eae-β and eae-Έ strains.
In one positive case (Table 3, isolate 4), the mPCR assay was positive for both stx1 and stx2, while the stx-negative result at the NRL was confirmed using RFLP–PCR (Tyler et al., 1991; Zhang et al., 2002). In two other cases, the standard methodology revealed eae+ strains, whereas the mPCR assay indicated that the strains lacked intimin (Table 3, isolates 31 and 32). Interestingly, analysis of isolates 30–34 revealed that the mPCR produced amplicons for both lpfA1-3 and lpfA2-2only (Table 3). Despite indicating a positive result for two of the markers for O157:H7 strains, that serotype was not confirmed in those isolates (Table 3).
In the 11 cases where the NRL indicated negative results and the mPCR assay indicated positive results (Table 3, isolates 90–96, 97, 100), nine of them were considered false positives for eae, as they were further confirmed to be negative for eae subtypes by RFLP–PCR. Two strains identified by the described mPCR assay as either stx1+ (Table 3, isolate 100) or stx2+ (Table 3, isolate 97) were confirmed as stx-negative by RFLP–PCR.
While the assay can reliably detect EHEC, STEC, and typical or atypical EPEC strains, the repertoire of pathotype detection can be expanded by the inclusion of primers for lpfA1-2 and lpfA2-1. The LEE− negative STEC typically possess one or both of these lpfA subtypes (Galli et al., 2010; Gomes et al., 2011). Additionally, detection of lpfA1-2 and/or lpfA2-1 permits the differentiation of strains from the EHEC2 (DECs 8, 9, and 10) and EPEC2 (DECs 11 and 12) clonal groups (Figure 4, data not shown). The EHEC and EPEC pathotypes were identified after examining a number of diarrheagenic strains by multilocus enzyme electrophoresis and serotyping, and appear to represent distinct clonal lineages of pathogenic E. coli (Whittam and McGraw, 1996; Reid et al., 2000).
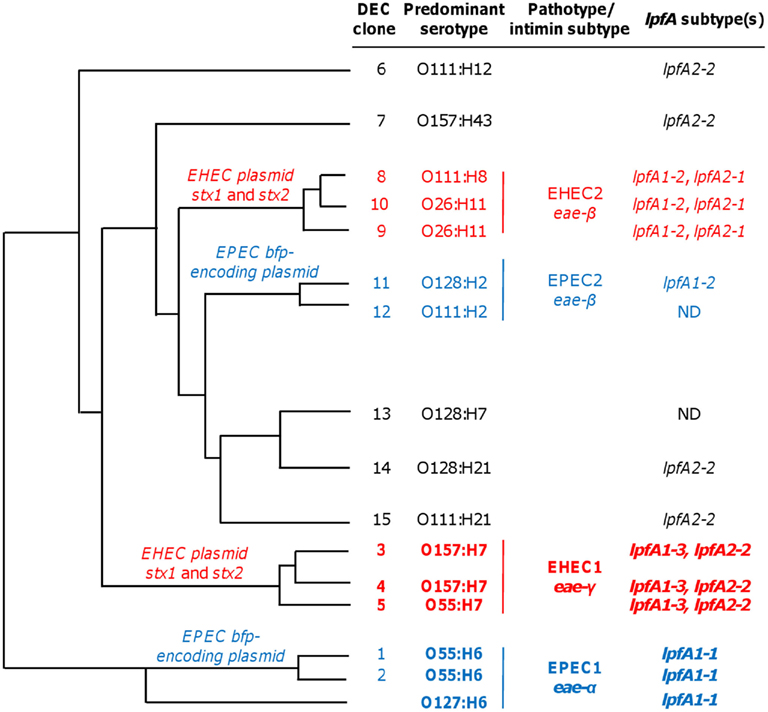
Figure 4. Phylogeny and lpfA subtype of isolates in diarrheagenic E. coli (DEC) collection. DEC clone numbers (Reid et al., 1999), corresponding predominant serotype of the given group, pathotype/intimin subtype, and information regarding lpfA subtype(s) for each group are indicated (Adapted from Torres, 1999 Ph.D. Dissertation). The lpfA1-2 and lpfA2-1 were previously identified in EHEC2 and EPEC2 isolates (Torres et al., 2009). ND, not determined (none of the three lpfA subtypes included in the multiplex PCR were detected, and lpfA subtypes were not studied further during the multiplex PCR project).
The unique ability of the multiplex assay to specifically detect EHEC and EPEC clonal groups is predominantly conferred by lpfA subtype analyses (Figure 4). Using DEC collection isolates (Whittam et al., 1993), clonal group EHEC1 (DECs 3, 4, and 5) were detected, in part, by the inclusion of primers amplifying EHEC O157:H7-specific lpfA subtypes 1-3 and 2-2. EPEC1 isolates (DECs 1 and 2) and EPEC O127:H6 were specifically identified by the amplification of lpfA1-1. The addition of lpfA subtyping was advantageous since lpfA subtypes differ among EHEC and EPEC clonal groups (Torres et al., 2009). As such, it permitted us to differentiate the more common EHEC1 and EPEC1 clonal groups from members of the EHEC2 and EPEC2 categories, respectively. Further specificity was conveyed by the amplification of pathotype-specific intimin subtypes: γ-intimin was used to detect EHEC1 isolates and α-intimin was used to detect EPEC1 isolates.
Discussion
A panel of eight genes was employed for the design of a sensitive and specific mPCR assay to facilitate detection of three pathotypes of E. coli that cause significant morbidity and mortality across the world – EHEC, STEC, and EPEC. The assay was also designed to be relatively low-cost, as compared to the financial burden of acquiring instrumentation and consumables to perform real-time PCR or multiplex bead-based assays. A collection of DEC strains (Whittam et al., 1993) was tested using the assay, and we demonstrated a relatively high degree of agreement between the mPCR results and strain information present in the DEC database. Evaluation of the specificity revealed no significant cross-reactivity of the primers with other E. coli pathotypes. The detection threshold of the assay was determined to be comparable to other PCR-based methods for detection of E. coli isolates (Aranda et al., 2007; Antikainen et al., 2009; Vidová et al., 2011). Of particular significance is the observation that defined combinations of lpfA subtypes permitted differentiation of EHEC and EPEC clonal groups.
Both EHEC and EPEC are AEEC strains, possessing the LEE-encoded gene products for development of the intestinal lesions (McDaniel and Kaper, 1997). The presence of the LEE-encoded adhesin intimin gene eae (Jerse and Kaper, 1991), is indicative of these strains, and; therefore, a first primer set was designed to amplify only eae-γ, previously demonstrated to be associated with the EHEC1 clonal group (Adu-Bobie et al., 1998; Reid et al., 1999). The second eae primer set was engineered to be more generic, amplifying the remaining major eae subtypes α, β, and δ (Adu-Bobie et al., 1998). Importantly, intimin α is associated with the EPEC1 clonal group, while intimin β is indicative of members of the EPEC2 clonal group (Adu-Bobie et al., 1998; Reid et al., 1999). Thus, the multiplex assay has the potential to detect a large number of AEEC strains.
In the multiplex strategy, the addition of primers for lpfA subtyping conferred the greatest increase in the ability of the assay to differentiate members of the EHEC and EPEC clonal groups. Lpf are elaborated appendages important for pathogenesis and adherence to cultured cells (Torres et al., 2002, 2004), persistence in animal models (Jordan et al., 2004; Torres et al., 2007), and tissue tropism in the human intestine (Fitzhenry et al., 2006). In addition, the combination of lpfA and eae subtyping can specifically detect EHEC O157:H7 (Torres et al., 2009), providing a distinct time advantage over more conventional culture- or immunoassay-based methodologies for the detection of O157:H7 strains.
Primer sets for both Shiga toxin 1 (stx1) and Shiga toxin 2 (stx2) were incorporated into the assay to facilitate identification of STEC strains, because early detection is critical for determining appropriate therapies for patients with suspected E. coli infections. Although there are a number of stx2 variants, we included primers based on the E. coli O157:H7 stx2 sequence due to the link between the development of HUS and the presence of stx2, particularly in O157:H7 strains (Friedrich et al., 2002; Brooks et al., 2005; Hedican et al., 2009). Conversely, the assay is capable of detecting non-O157 STEC, a group of under-diagnosed emerging pathogens (Coombes et al., 2011), and emerging LEE-negative STEC strains (Newton et al., 2009; Galli et al., 2010).
To further expand the detection capabilities, primers to amplify the bundle-forming pili subunit bfpA were incorporated into the assay. Because the bundle-forming pilus is a central virulence factor of EPEC strains, playing a putative role in initial attachment to enterocytes (Cleary et al., 2004) and microcolony formation (Hicks et al., 1998), the bfpA gene can be used as a marker for identification of typical EPEC (Nataro and Kaper, 1998) in conjunction with eae (Giron et al., 1993).
Our mPCR results with DEC collection isolates suggest that this approach will prove useful for rapid identification of these pathogenic E. coli strains. The incidence of non-specific amplification was low, and in many cases, the bands were faint compared to the intensities of the primer-specific products and outside the size range of the target amplicons (data not shown). The presence of non-specific amplicons is not anticipated to result in the misidentification of strains as DEC, and is not uncommon in mPCR approaches (Antikainen et al., 2009).
The diagnostic sensitivity and specificity was calculated to be 91% and 84%, respectively, when comparing to the “standard” methodology used by the NRL (Argentina) for routine detection of highly virulent STEC strains and our mPCR assay. These data strongly suggest that the mPCR approach described here is a relatively low-cost and feasible screening methodology for clinical fecal samples within 24 h of obtaining a specimen. Because Argentina possesses the highest incidence of post-enteric HUS in infants and children in the world, and O157:H7 and O145:NM are the most prevalent serotypes (Rivas et al., 2010, 2011), this new mPCR approach permits rapid identification of STEC strains involved in the majority of the cases (>70%) received at the NRL. Further, this method can also be used in areas where other STEC or EPEC strains are prevalent.
Assay specificity was determined by screening additional E. coli pathotypes, commensal E. coli, and a limited number of non-E. coli intestinal pathogens. This analysis revealed that the assay is highly specific; none of the unexpected amplification products correspond to the size of a predicted amplicon (Figure 2). Minor cross-reactivity of the broad-range intimin primers with a putative intimin gene in E. coli K12 (eaeH) and E. coli HS (EcHS_A0351) may account for the observation of a band migrating at approximately the same size as the specific intimin target observed using EHEC or EPEC. However, S. flexneri does not appear to possess a putative intimin sequence.
The threshold detection of our multiplex approach was assessed and the template concentrations at which all expected amplicons were clearly visible on the gel was set as the limit for detection of that pathotype. EHEC and EPEC were detectable at or above 2 × 104 CFU per reaction (Figure 3). Compared to other mPCR methodologies (Aranda et al., 2007; Antikainen et al., 2009; Vidová et al., 2011), the sensitivities determined here are slightly higher, perhaps owing to the selection of DNA polymerase. The REDTaq Ready Mix was chosen based on the premixed nature of the components, thereby reducing pipetting errors and increasing reproducibility, and its relatively low-cost, a factor critical for adoption of this assay in developing countries. The balance between assay cost and sensitivity can be adjusted based on the financial resources of the testing facility, suggesting that the purchase of more costly polymerases could increase assay sensitivity.
The observation that lpfA and eae subtypes are related to specific EHEC and EPEC clonal groups provides evidence of the lineage of pathogenic E. coli, but also permitted us to design an assay exploiting these phylogenetic relationships (Figure 4). In its current form, the mPCR assay can reliably distinguish strains in the EHEC1 and EPEC1 clonal groups, atypical EPEC, and differentiate O157 and non-O157 STEC strains. Interestingly, isolates from the EPEC2 group (DEC 11 and DEC 12) were negative for bfpA. This result likely reflects the specificity of the bfpA primer set for the bfpAα1 allele present in the O127:H6 prototype EPEC strain (Blank et al., 2000). Therefore, strategies for detection of EPEC2 strains should also include primers to amplify the β4 (DEC 11) and α2 (DEC 12) bfpA alleles (Blank et al., 2000). While not included in the same mPCR assay, detection of additional lpfA subtypes would permit identification of EHEC2 and EPEC2 strains, as well as afford detection of LEE-negative STEC (eae−, stx+, lpfA1-2+, and/or lpfA2-1+) and atypical EPEC (eae+, bfpA−, lpfA1-2+, and/or lpfA2-1+). Recent data also supports this notion, as Gomes et al. (2011) demonstrated that of the lpfA+ atypical EPEC strains tested, lpfA1-2 and lpfA2-1 were frequently present together in a given strain. Finally, the lpfA1-1 variant was used to identify typical EPEC strains; however, this allele was present in only 35% (16/46) of the isolates tested. Currently, we are exploring the possibility to incorporate alternative lpfA1 alleles identified in our initial screen (Torres et al., 2009) to increase the specificity of our assay.
In summary, we presented data supporting a mPCR approach that, with only eight virulence-associated genes, has the potential detect a wide range of pathogenic E. coli strains. The assay was tested with a collection of clinical isolates, resulting in a high degree of agreement between known strain information and the results of the mPCR. The specificity and sensitivity of the assay are such that diagnostic facilities in developing countries can easily incorporate this methodology into their workflow. The mPCR approach described here has potential for improving both diagnostics and epidemiological studies involving DEC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in the AGT laboratory was supported by NIH/NIAID grant 5-R01-AI079154. Douglas J. Botkin was supported by an NIH/NIAID T32 Postdoctoral Training Grant in Emerging and Re-emerging Infectious Diseases, 5-T32-AI007536-12. The authors would like to thank members of Servicio Fisiopatogenia and the Torres lab for their support and critical review of this project. The authors also thank Dr. Heidi Spratt for help with statistical analyses. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the RCE Programs Office, NIAID, or NIH.
References
Adu-Bobie, J., Frankel, G., Bain, C., Goncalves, A. G., Trabulsi, L. R., Douce, G., Knutton, S., and Dougan, G. (1998). Detection of intimins alpha, beta, gamma, and delta, four intimin derivatives expressed by attaching and effacing microbial pathogens. J. Clin. Microbiol. 36, 662–668.
Antikainen, J., Tarkka, E., Haukka, K., Siitonen, A., Vaara, M., and Kirveskari, J. (2009). New 16-plex PCR method for rapid detection of diarrheagenic Escherichia coli directly from stool samples. Eur. J. Clin. Microbiol. Infect. Dis. 28, 899–908.
Aranda, K. R., Fabbricotti, S. H., Fagundes-Neto, U., and Scaletsky, I. C. (2007). Single multiplex assay to identify simultaneously enteropathogenic, enteroaggregative, enterotoxigenic, enteroinvasive and Shiga toxin-producing Escherichia coli strains in Brazilian children. FEMS Microbiol. Lett. 267, 145–150.
Bai, J., Shi, X., and Nagaraja, T. G. (2010). A multiplex PCR procedure for the detection of six major virulence genes in Escherichia coli O157:H7. J. Microbiol. Methods 82, 85–89.
Blank, T. E., Zhong, H., Bell, A. L., Whittam, T. S., and Donnenberg, M. S. (2000). Molecular variation among type IV pilin (bfpA) genes from diverse enteropathogenic Escherichia coli strains. Infect. Immun. 68, 7028–7038.
Brooks, J. T., Sowers, E. G., Wells, J. G., Greene, K. D., Griffin, P. M., Hoekstra, R. M., and Strockbine, N. A. (2005). Non-O157 Shiga toxin-producing Escherichia coli infections in the United States, 1983–2002. J. Infect. Dis. 192, 1422–1429.
Cebula, T. A., Payne, W. L., and Feng, P. (1995). Simultaneous identification of strains of Escherichia coli serotype O157:H7 and their Shiga-like toxin type by mismatch amplification mutation assay-multiplex PCR. J. Clin. Microbiol. 33, 248–250.
Cleary, J., Lai, L. C., Shaw, R. K., Straatman-Iwanowska, A., Donnenberg, M. S., Frankel, G., and Knutton, S. (2004). Enteropathogenic Escherichia coli (EPEC) adhesion to intestinal epithelial cells: role of bundle-forming pili (BFP), EspA filaments and intimin. Microbiology 150, 527–538.
Coombes, B. K., Gilmour, M. W., and Goodman, C. D. (2011). The evolution of virulence in non-O157 Shiga toxin-producing Escherichia coli. Front. Microbiol. 2:90. doi: 10.3389/fmicb.2011.00090
Fitzhenry, R., Dahan, S., Torres, A. G., Chong, Y., Heuschkel, R., Murch, S. H., Thomson, M., Kaper, J. B., Frankel, G., and Phillips, A. D. (2006). Long polar fimbriae and tissue tropism in Escherichia coli O157:H7. Microbes Infect. 8, 1741–1749.
Friedrich, A. W., Bielaszewska, M., Zhang, W. L., Pulz, M., Kuczius, T., Ammon, A., and Karch, H. (2002). Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 185, 74–84.
Galli, L., Torres, A. G., and Rivas, M. (2010). Identification of the long polar fimbriae gene variants in the locus of enterocyte effacement-negative Shiga toxin-producing Escherichia coli strains isolated from humans and cattle in Argentina. FEMS Microbiol. Lett. 308, 123–129.
Giron, J. A., Donnenberg, M. S., Martin, W. C., Jarvis, K. G., and Kaper, J. B. (1993). Distribution of the bundle-forming pilus structural gene (bfpA) among enteropathogenic Escherichia coli. J. Infect. Dis. 168, 1037–1041.
Gomes, T. A., Hernandes, R. T., Torres, A. G., Salvador, F. A., Guth, B. E. C., Vaz, T. M., Irino, K., Silva, R. M., and Vieira, M. A. (2011). Adhesin-encoding genes from Shiga toxin-producing Escherichia coli are more prevalent in atypical than in typical enteropathogenic E. coli. J. Clin. Microbiol. 49, 3334–3337.
Gordillo, R., Cordoba, J. J., Andrade, M. J., Luque, M. I., and Rodriguez, M. (2011). Development of PCR assays for detection of Escherichia coli O157:H7 in meat products. Meat Sci. 88, 767–773.
Hedican, E. B., Medus, C., Besser, J. M., Juni, B. A., Koziol, B., Taylor, C., and Smith, K. E. (2009). Characteristics of O157 versus non-O157 Shiga toxin-producing Escherichia coli infections in Minnesota, 2000–2006. Clin. Infect. Dis. 49, 358–364.
Hicks, S., Frankel, G., Kaper, J. B., Dougan, G., and Phillips, A. D. (1998). Role of intimin and bundle-forming pili in enteropathogenic Escherichia coli adhesion to pediatric intestinal tissue in vitro. Infect. Immun. 66, 1570–1578.
Jerse, A. E., and Kaper, J. B. (1991). The eae gene of enteropathogenic Escherichia coli encodes a 94-kilodalton membrane protein, the expression of which is influenced by the EAF plasmid. Infect. Immun. 59, 4302–4309.
Jordan, D. M., Cornick, N., Torres, A. G., Dean-Nystrom, E. A., Kaper, J. B., and Moon, H. W. (2004). Long polar fimbriae contribute to colonization by Escherichia coli O157:H7 in vivo. Infect. Immun. 72, 6168–6171.
Jores, J., Zehmke, K., Eichberg, J., Rumer, L., and Wieler, L. H. (2003). Description of a novel intimin variant (type zeta) in the bovine O84:NM verotoxin-producing Escherichia coli strain 537/89 and the diagnostic value of intimin typing. Exp. Biol. Med. (Maywood) 228, 370–376.
Karch, H., Bohm, H., Schmidt, H., Gunzer, F., Aleksic, S., and Heesemann, J. (1993). Clonal structure and pathogenicity of Shiga-like toxin-producing, sorbitol-fermenting Escherichia coli O157:H7. J. Clin. Microbiol. 31, 1200–1205.
Karch, H., and Bielaszewska, M. (2001). Sorbitol-fermenting Shiga toxin-producing Escherichia coli O157:H(−) strains: epidemiology, phenotypic and molecular characteristics, and microbiological diagnosis. J. Clin. Microbiol. 39, 2043–2049.
Leotta, G. A., Chinen, I., Epszteyn, S., Miliwebsky, E., Melamed, I. C., Motter, M., Ferrer, M., Marey, E., and Rivas, M. (2005). Validation of a multiplex PCR for detection of Shiga toxin-producing Escherichia coli. Rev. Argent. Microbiol. 37, 1–10.
Madic, J., Peytavin de Garam, C., Vingadassalon, N., Oswald, E., Fach, P., Jamet, E., and Auvray, F. (2010). Simplex and multiplex real-time PCR assays for the detection of flagellar (H-antigen) fliC alleles and intimin (eae) variants associated with enterohaemorrhagic Escherichia coli (EHEC) serotypes O26:H11, O103:H2, O111:H8, O145:H28 and O157:H7. J. Appl. Microbiol. 109, 1696–1705.
Madic, J., Vingadassalon, N., de Garam, C. P., Marault, M., Scheutz, F., Brugere, H., Jamet, E., and Auvray, F. (2011). Detection of Shiga toxin-producing Escherichia coli serotypes O26:H11, O103:H2, O111:H8, O145:H28, and O157:H7 in raw-milk cheeses by using multiplex real-time PCR. Appl. Environ. Microbiol. 77, 2035–2041.
McDaniel, T. K., Jarvis, K. G., Donnenberg, M. S., and Kaper, J. B. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 92, 1664–1668.
McDaniel, T. K., and Kaper, J. B. (1997). A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 23, 399–407.
Nataro, J. P., and Kaper, J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201.
Newton, H. J., Sloan, J., Bulach, D. M., Seemann, T., Allison, C. C., Tauschek, M., Robins-Browne, R. M., Paton, J. C., Whittam, T. S., Paton, A. W., and Hartland, E. L. (2009). Shiga toxin-producing Escherichia coli strains negative for locus of enterocyte effacement. Emerging. Infect. Dis. 15, 372–380.
Ochoa, T. J., Barletta, F., Contreras, C., and Mercado, E. (2008). New insights into the epidemiology of enteropathogenic Escherichia coli infection. Trans. R. Soc. Trop. Med. Hyg. 102, 852–856.
Ooka, T., Terajima, J., Kusumoto, M., Iguchi, A., Kurokawa, K., Ogura, Y., Asadulghani, M., Nakayama, K., Murase, K., Ohnishi, M., Iyoda, S., Watanabe, H., and Hayashi, T. (2009). Development of a multiplex PCR-based rapid typing method for enterohemorrhagic Escherichia coli O157 strains. J. Clin. Microbiol. 47, 2888–2894.
Orskov, F., Whittam, T. S., Cravioto, A., and Orskov, I. (1990). Clonal relationships among classic enteropathogenic Escherichia coli (EPEC) belonging to different O groups. J. Infect. Dis. 162, 76–81.
Pavlovic, M., Huber, I., Skala, H., Konrad, R., Schmidt, H., Sing, A., and Busch, U. (2010). Development of a multiplex real-time polymerase chain reaction for simultaneous detection of enterohemorrhagic Escherichia coli and enteropathogenic Escherichia coli strains. Foodborne Pathog. Dis. 7, 801–808.
Ramachandran, V., Brett, K., Hornitzky, M. A., Dowton, M., Bettelheim, K. A., Walker, M. J., and Djordjevic, S. P. (2003). Distribution of intimin subtypes among Escherichia coli isolates from ruminant and human sources. J. Clin. Microbiol. 41, 5022–5032.
Reid, S. D., Betting, D. J., and Whittam, T. S. (1999). Molecular detection and identification of intimin alleles in pathogenic Escherichia coli by multiplex PCR. J. Clin. Microbiol. 37, 2719–2722.
Reid, S. D., Herbelin, C. J., Bumbaugh, A. C., Selander, R. K., and Whittam, T. S. (2000). Parallel evolution of virulence in pathogenic Escherichia coli. Nature 406, 64–67.
Rivas, M., Chinen, I., Miliwebsky, E., Galli, L., Repetto, H. A., and Masana, M. (2011). “Epidemiology of Argentinean Shiga toxin-producing Escherichia coli,” in Population Genetics of Bacteria: A Tribute to Thomas S. Whittam, eds S. T. Walk, and P. C. H. Feng (Washington, DC: ASM Press), 109–132.
Rivas, M., Padola, N. L., Lucchesi, P. M. A., and Massana, M. (2010). “Diarrheagenic Escherichia coli in Argentina,” in Pathogenic Escherichia coli in Latin America, ed. A. G. Torres (Oak Park, IL: Bentham Science Publishers Ltd.), 142–161.
Tarr, C. L., and Whittam, T. S. (2002). Molecular evolution of the intimin gene O111 clones of pathogenic Escherichia coli. J. Bacteriol. 184, 479–487.
Torres, A. G. (1999). Characterization of the Heme Transport System in Escherichia coli O157:H7, and Importance of Iron Uptake Systems in Virulence. University of Texas at Austin. PhD Dissertation.
Torres, A. G., Blanco, M., Valenzuela, P., Slater, T. M., Patel, S. D., Dahbi, G., Lopez, C., Barriga, X. F., Blanco, J. E., Gomes, T. A., Vidal, R., and Blanco, J. (2009). Genes related to long polar fimbriae of pathogenic Escherichia coli strains as reliable markers to identify virulent isolates. J. Clin. Microbiol. 47, 2442–2451.
Torres, A. G., Giron, J. A., Perna, N. T., Burland, V., Blattner, F. R., Avelino-Flores, F., and Kaper, J. B. (2002). Identification and characterization of lpfABCC′DE, a fimbrial operon of enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 70, 5416–5427.
Torres, A. G., Kanack, K. J., Tutt, C. B., Popov, V., and Kaper, J. B. (2004). Characterization of the second long polar (LP) fimbriae of Escherichia coli O157:H7 and distribution of LP fimbriae in other pathogenic E. coli strains. FEMS Microbiol. Lett. 238, 333–344.
Torres, A. G., Milflores-Flores, L., Garcia-Gallegos, J. G., Patel, S. D., Best, A., La Ragione, R. M., Martinez-Laguna, Y., and Woodward, M. J. (2007). Environmental regulation and colonization attributes of the long polar fimbriae (LPF) of Escherichia coli O157:H7. Int. J. Med. Microbiol. 297, 177–185.
Tyler, S. D., Johnson, W. M., Lior, H., Wang, G., and Rozee, K. R. (1991). Identification of verotoxin type 2 variant B subunit genes in Escherichia coli by the polymerase chain reaction and restriction fragment length polymorphism analysis. J. Clin. Microbiol. 29, 1339–1343.
Vidová, B., Tóthová, E., Blahut, L., Horváthová, V., and Godány, A. (2011). Multiplex PCR for detection of Escherichia coli O157:H7 in foods. Biologia 66, 401–405.
Whittam, T. S., and McGraw, E. A. (1996). Clonal analysis of EPEC serogroups. Rev. Microbiol. 27, 7–16.
Whittam, T. S., Wolfe, M. L., Wachsmuth, I. K., Orskov, F., Orskov, I., and Wilson, R. A. (1993). Clonal relationships among Escherichia coli strains that cause hemorrhagic colitis and infantile diarrhea. Infect. Immun. 61, 1619–1629.
Keywords: Shiga toxin-producing E. coli, enterohemorrhagic E. coli, enteropathogenic E. coli, E. coli O157, diagnostics
Citation: Botkin DJ, Galli L, Sankarapani V, Soler M, Rivas M and Torres AG (2012) Development of a multiplex PCR assay for detection of Shiga toxin-producing Escherichia coli, enterohemorrhagic E. coli, and enteropathogenic E. coli strains. Front. Cell. Inf. Microbio. 2:8. doi: 10.3389/fcimb.2012.00008
Received: 14 December 2011; Paper pending published: 16 January 2012;
Accepted: 28 January 2012; Published online: 14 February 2012.
Edited by:
Nora Lía Padola, Universidad Nacional del Centro de la Provincia de Buenos Aires, ArgentinaReviewed by:
Analía Inés Etcheverría, Universidad Nacional del Centro de la Provincia de Buenos Aires, ArgentinaRoberto Mauricio Vidal, Universidad De Chile, Chile
Copyright: © 2012 Botkin, Galli, Sankarapani, Soler, Rivas and Torres. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alfredo G. Torres, Department of Microbiology and Immunology, University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555, USA. e-mail: altorres@utmb.edu
†Present address: Douglas J. Botkin, NASA Johnson Space Center, Houston, TX, USA.; Vinoth Sankarapani, UT Health Medical School, Department of Neurosurgery, Houston, TX, USA.; Michael Soler, Christus Spohn Hospital Memorial Corpus Christi-Memorial, Corpus Christi, TX, USA.