Shiga toxin in enterohemorrhagic E.coli: regulation and novel anti-virulence strategies
- Department of Microbiology, University of Texas Southwestern Medical Center, Dallas, TX, USA
Enterohemorrhagic Escherichia coli (EHEC) are responsible for major outbreaks of bloody diarrhea and hemolytic uremic syndrome (HUS) throughout the world. The mortality associated with EHEC infections stems from the production and release of a potent Shiga toxin (Stx) by these bacteria. Stx induces cell death in endothelial cells, primarily in the urinary tract, causing HUS. Stx was first described in Shigella dysenteriae serotype I by Kiyoshi Shiga and was discovered later in EHEC. Multiple environmental cues regulate the expression of Stx, including temperature, growth phase, antibiotics, reactive oxygen species (ROS), and quorum sensing. Currently, there is no effective treatment or prophylaxis for HUS. Because antibiotics trigger Stx production and their use to treat EHEC infections is controversial, alternative therapeutic strategies have become the focus of intense research. One such strategy explores quorum sensing inhibitors as therapeutics. These inhibitors target quorum sensing regulation of Stx expression without interfering with bacterial growth, leading to the hypothesis that these inhibitors impose less selective pressure for bacteria to develop drug resistance. In this review, we discuss factors that regulate Stx production in EHEC, as well as novel strategies to prevent and/or minimize the development of HUS in infected subjects.
Shiga Toxin: An Introduction
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 is a deadly human pathogen that causes gastrointestinal (GI) infections worldwide. Annually in the United States, EHEC is responsible for an estimated 73,000 illnesses, 1800–3600 hospitalizations and from 61 to 541 deaths (Rangel et al., 2005) (www.cdc.gov). EHEC is a foodborne pathogen, and outbreaks occur through ingestion of contaminated food or water including: contaminated ground beef, steak, salami (Bell et al., 1994; Jay et al., 2004; Greig and Ravel, 2009), dairy products (raw milk, cheese, butter, cookie dough) (Jaeger and Acheson, 2000; Neil et al., 2009) and vegetables (spinach, lettuce, sprouts) (Rangel et al., 2005; Grant et al., 2006; Wendel et al., 2009; Sodha et al., 2011). A remarkable feature of EHEC infection is its low infectious dose; it is estimated that 50–100 colony forming units (CFUs) of EHEC is sufficient to cause disease in healthy individuals (Tilden et al., 1996).
Although EHEC is a human pathogen, this bacterium resides as a commensal in the GI tract of cattle (Savageau, 1983; Naylor et al., 2003). Fecal shedding from cattle represents an important source of EHEC contamination and transmission (Arthur et al., 2011). Many studies have focused on the eradication of EHEC from the GI tract of ruminants to reduce the infection rate of this enteric pathogen (Rivas et al., 2010; Vilte et al., 2011).
EHEC implements two major virulence strategies: production of Shiga toxin (Stx) and formation of attaching and effacing (AE) lesions on enterocytes (Kaper et al., 2004). EHEC contains a pathogenicity island termed the locus of enterocyte effacement (LEE), which is crucial for the development of AE lesions (McDaniel et al., 1995). The LEE encodes a master regulator for its own expression, ler (Mellies et al., 1999); a type 3 secretion (T3S) apparatus (Jarvis et al., 1995), translocator proteins EspA (Knutton et al., 1998) and EspB (Kenny and Finlay, 1995; Abe et al., 1997), an adhesin Intimin (Jerse et al., 1990) and its own receptor, the translocated intimin receptor (Tir) (Kenny et al., 1997). AE lesions are characterized by extensive actin remodeling of the host cell cytoskeleton, leading to effacement of the microvilli and formation of a pedestal-like structure beneath the bacteria (Tzipori et al., 1992; Deng et al., 2001; Campellone et al., 2004; Garmendia et al., 2004; Vingadassalom et al., 2010). It should be highlighted that the initial findings of AE lesion formation described in the references above (Jerse et al., 1990, 1995; Kenny and Finlay, 1995; Abe et al., 1997; Knutton et al., 1998; Mellies et al., 1999) were discovered in enteropathogenic E. coli (EPEC), however, the proteins Ler, Intimin, Tir, EspA, and EspB are conserved between EHEC and EPEC.
EHEC infection leads to a characteristic bloody diarrhea, which self-resolves in most cases. However, in approximately 5–7% of the cases, the patient develops a complication known as hemolytic uremic syndrome (HUS) (Karmali, 2004; Noris and Remuzzi, 2005; Tarr et al., 2005). HUS is the most serious sequelae of EHEC infection, and its incidence is higher in children and the elderly (Brandt et al., 1990; Gould et al., 2009). The disease is characterized by thrombocytopenia, hemolytic anemia and acute renal failure. In children infected by EHEC, HUS often develops soon after the onset of diarrhea (Wong et al., 2000). HUS is a life-threatening disease, contributing to a 5% mortality rate. The development of HUS is a direct result of the release of Stx, a very potent toxin encoded in a bacteriophage in the EHEC chromosome (Kaper et al., 2004). Stx also contributes to the development of hemorrhagic colitis by lysing endothelial cells from the intestinal mucosa (Tarr et al., 2005).
The current treatment for EHEC infection is supportive, due to the lack of specific therapies to clear the infection or to prevent its sequelae (Michael et al., 2009). Several drugs commonly used in the clinic to treat bacterial infections are not advised for treatment of EHEC-infected patients, such as antibiotics, antimotility agents, narcotics, and non-steroidal anti-inflammatory medicines (Tarr et al., 2005). The increasing number of outbreaks and HUS cases highlight the urgent need for new treatments.
In this review, we will focus on the mechanism by which Stx causes the development of HUS and how Stx expression is regulated within EHEC. We will discuss novel strategies that are currently being developed to treat EHEC infection and to ameliorate the severity of HUS.
Shiga Toxin and HUS
The first outbreak in the United States of EHEC O157:H7 was reported in 1982 and it was linked to the consumption of contaminated hamburger meat from McDonald's in Michigan. EHEC O157:H7 was a rare serotype prior to the 1982 outbreak (Riley et al., 1983). At present, however, O157:H7 is the major serotype responsible for outbreaks of hemorrhagic colitis throughout the world. EHEC O157:H7 encodes for Stx in a bacteriophage in the EHEC chromosome, and release of Stx by EHEC is the predominant cause of mortality in EHEC infections. A potent cytotoxin, Stx is produced by the enteric pathogens Shigella dysenteriae serotype I and a collective group of E. coli strains called Stx-producing E. coli (STEC). Stx from S. dysenteriae serotype I was first identified by Kiyoshi Shiga (Fraser et al., 2004). In 1983, it was first reported that EHEC could also produce Stx (O'Brien et al., 1983a), providing the link between Stx production and the development of HUS (Karmali et al., 1983). Initially, Stx was called Verotoxin due to its cytotoxicity against Vero cells in culture. Upon the discovery that Verotoxin could be neutralized by an antitoxin against purified Stx from Shigella, the name Stx came into use and is the more common name to date (O'Brien et al., 1983b). Stx and Verotoxin are used interchangeably, but for the purpose of this review, only Stx and the STEC strain EHEC will be discussed (Table 1).

Table 1. Stx family and variants.
Since 1982, EHEC has been recognized as a major enteric pathogen. An increase in the number of recent outbreaks is attributed to the emergence of more virulent strains (Kulasekara et al., 2009). In 1999, the largest reported outbreak to date occurred in the United States, as a result of a contaminated water well; 781 people were infected, with 9% hospitalized and 2% developing HUS (www.cdc.gov). In a 2006 outbreak caused by contaminated spinach, 29% of the patients developed HUS, indicating a rise in the incidence of HUS from previous outbreaks that reported an incidence of 15–20% (Grant et al., 2006). Genomic analysis of an isolate from the spinach outbreak revealed the presence of two stx-encoding genes, stx2 and stx2c (Neupane et al., 2011).
The highest number of HUS cases reported to date occurred in a 2011 German outbreak, suggesting that STEC has increased its virulence over time. Genomic analyses of this isolate determined that the infective strain was actually an enteroaggregative E.coli (EAEC) that had acquired a lambdoid-like phage encoding stx2 (Rasko et al., 2011). EAEC forms a biofilm in the intestine, a trait that makes treatment challenging (Sheikh et al., 2001). The combined ability to form a biofilm with the production of Stx resulted in a highly virulent STEC strain that caused 34 deaths and 908 HUS cases in 2011. The causative agent of the 2011 German outbreak was identified as a non-LEE Stx-producing E.coli serotype O104:H4, EAEC (Buchholz et al., 2011).
Mechanism of Action of Shiga Toxin
Stx inhibits protein synthesis and induces apoptosis. Stx is an AB5 toxin, constituted by a catalytic subunit A bound non-covalently to a pentamer of B subunits (Stein et al., 1992; Fraser et al., 1994). The A subunit has N-glycosidase activity that cleaves an adenosine residue from 28S ribosomal RNA of the 60S ribosomal subunit (Endo et al., 1988). As a result, it inhibits protein synthesis, causing cell death by apoptosis. The five B subunits form a structure that binds the globotriaosylceramide (Gb3) receptor on the surface of eukaryotic cells (Jacewicz et al., 1986; Lindberg et al., 1987). Gb3 is expressed by Paneth cells in the intestinal mucosa and by kidney epithelial cells [reviewed in (Tarr et al., 2005)] (Figure 1). Stx enters systemic circulation through absorption by the epithelium (Acheson et al., 1996), enabling its access to the kidneys. The damage of the GI epithelium caused by EHEC likely aids in Stx systemic absorption (Mukherjee et al., 2002). Upon receptor binding, Stx is endocytosed by the eukaryotic cell (Romer et al., 2007), bypasses the late endocytic pathway and undergo retrograde transport from the trans-Golgi network to the endoplasmic reticulum (ER) where it encounters its target (Sandvig et al., 1992; Mallard et al., 1998). These processes have been reviewed in detail elsewhere (Johannes and Romer, 2010).
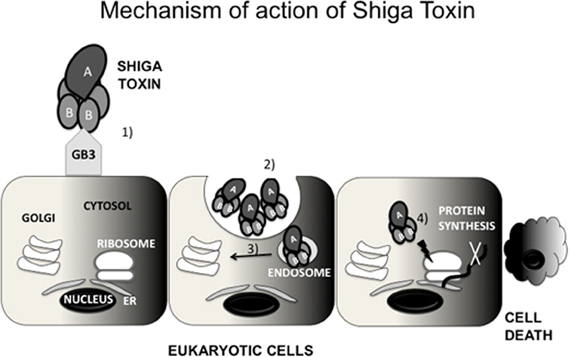
Figure 1. Mechanism of action of Shiga toxin (Stx). Stx is constituted by a pentamer of B subunits bound to a catalytic A subunit. The B subunits bind to globotriaosylceramide (Gb3) expressed by some eukaryotic cells (1) Stx is internalized by endocytosis (2) Subsequently, Stx undergoes retrograde transport to the trans-Golgi network (TGN) (3) and then to the endoplasmic reticulum (ER) (4) In the ER, Stx encounters its target, the ribosome, inactivating it (4) As a consquence, Stx inhibits protein synthesis, causing cell death by apoptosis.
The Stx family has two main groups, Stx1 and Stx2. The Stx1 family consists of the variants Stx1, Stx1c, and Stx1d. The Stx2 family is composed of the variants Stx2c, Stx2c2, Stx2d, Stx2dactivatable, Stx2e, and Stx2f (Muthing et al., 2009). The Stx2 variants differ in their biological activity, immunological reactivity, and the receptor to which they bind. These binding properties allow the Stx variants to target different cell types (Fraser et al., 2004).
Although Stx1 and Stx2 share enzymatic activity and structural features, they are immunogically distinct. Stx1 is nearly identical to Stx from Shigella, with a difference in only a single amino acid in the catalytic A subunit. Conversely, Stx2 shares only 55% amino acid sequence similarity to Stx1 (Fraser et al., 2004). Stx2 is more potent than Stx1 in humans (Nishikawa et al., 2000), and it is commonly associated with hemorrhagic colitis and HUS (Nataro and Kaper, 1998; Boerlin et al., 1999). The two subgroups of Stx can be found in different combinations in EHEC isolates. Epidemiological studies suggest that EHEC strains encoding Stx2 are more likely to cause severe disease than isolates that harbor only Stx1 or a combination of Stx1 and Stx2 (Ritchie et al., 2003).
There are conflicting reports concerning the effect of Stx on the generation of an inflammatory response. One study found that Stx and flagellin act in concert to activate expression of IL-8 and promote inflammation (Jandhyala et al., 2010). Matussek and colleagues reported an increased expression of IL-8 in the presence of Stx1 or Stx2 (Matussek et al., 2003). In contrast, Bellmeyer et al. (Bellmeyer et al., 2009) reported that while Stx activates IL-8, EHEC produces other factors that lead to suppression of the inflammatory response in intestinal epithelial cells.
Animal Models for EHEC Infection and Stx
Many animal models to study the pathogenesis of EHEC infection in vivo have been developed. Some of these models are more suitable to study colonization, while others are more informative to evaluate the effects of Stx. Nonetheless, no animal model currently available is able to mimic all aspects of EHEC-mediated disease, including infectious dose or development of hemorrhagic colitis and HUS (Mohawk and O'Brien, 2011). The animal models include: mice, rats, infant rabbits (Pai et al., 1986; Ritchie et al., 2003), gnotobiotic piglets, gnotobiotic calves, and primates.
A mouse model is usually the choice for in vivo studies, due to lower cost, easier maintenance, availability of reagents and knockout strains. The first mouse model for EHEC infection was developed by Wadolkowski and colleagues (Wadolkowski et al., 1990), and was based on the model developed by Myhal (Myhal et al., 1982), who studied colonization by non-pathogenic E.coli strains. In this model, the animals receive antibiotics in their drinking water prior to EHEC infection to reduce the microbial flora and diminish competition with EHEC. CD-1 mice receive antibiotics before an overnight fast and are subsequently infected with 1010 CFU of EHEC. Under these conditions, EHEC stably colonizes the small and large intestines for about a month. However, the mice do not develop diarrhea or any other hallmark GI symptoms of EHEC infection.
Because most of the mouse models developed to date require manipulation such as antibiotic treatment (Wadolkowski et al., 1990), dietary restrictions, or mitomycin C administration to improve Stx expression, Mohawk and colleagues developed a model that maintains an intact commensal flora (ICF) (Mohawk et al., 2010b). They infected BALB/c mice with different inoculums and assessed bacterial colonization by CFU recovery from intestinal segments and stool. A high infectious dose (109 CFU) resulted in the highest colonization rate and bacterial recovery compared to lower ones (105, 106, 107, and 108 CFU). The highest levels of Stx are detected in the large intestinal contents. Histopathology analysis of kidney sections shows mild kidney damage, suggesting Stx systemic effects. This study suggests that by using a high infectious dose, mice can be colonized by EHEC without alteration to their commensal gut microbiota. This model represents an advance in the use of mice to study EHEC colonization and persistence. However, a disadvantage of this particular model is the absence of hemolytic anemia and thrombocytopenia, making the ICF mice more attractive for colonization studies than evaluation of systemic effects resulting from EHEC infection. The reports using mice as a model for EHEC infection have to be interpreted with caution. Mice do not develop diarrhea, and AE lesions are not observed. Also, it is of note that the bacterial inoculum used in these experiments is many orders of magnitude higher than the estimated infectious dose of EHEC (50–100 CFU).
Gnotobiotic piglets are highly susceptible to EHEC infection. The gnotobiotic piglet model was developed by Tzipori in 1986 (Tzipori et al., 1986), and this model has been a useful tool to study Stx-mediated pathogenesis and the systemic effects of Stx. (Mukherjee et al., 2002). Upon infection by EHEC, these animals develop watery diarrhea, colitis and inflammation, and EHEC forms the characteristic AE lesions in their intestinal epithelium (Tzipori et al., 1986). Although the gnotobiotic piglets do not develop HUS, they demonstrate several signs of systemic disease including convulsions, tremor, ataxia, and brain hemorrhage. Formation of thrombotic microangiopathy, which causes acute kidney failure, was observed when gnotobiotic piglets were orally infected by 108–1010 CFU of EHEC (Gunzer et al., 2002). The gnotobiotic piglet model best represents the systemic effects of EHEC infection seen in humans, and it is an excellent model to study EHEC pathogenesis in vivo. However, a drawback of using this animal model is the requirement of a complex animal facility to perform the experiments.
The infant rabbit model is also used to study EHEC pathogenesis in vivo. Developed by Pai and colleagues in 1986 (Pai et al., 1986), 3-day-old infant rabbits are inoculated intragastrically with 108 CFU of EHEC. The rabbits develop diarrhea and colonic inflammation, and a portion succumb to death. In 2003, Ritchie and colleagues investigated the contribution of individual virulence factors on the development of disease by using EHEC isogenic mutants to infect infant rabbits (Ritchie et al., 2003). The infant rabbit model allowed the evaluation of Stx2, as well as the receptor and ligand crucial for AE lesion formation—Tir and Intimin, respectively, in the pathogenesis of EHEC infection, by inoculating 3-day-old New Zealand white rabbits with 5 × 108 CFU/90 g body weight of wild type EHEC or the EHEC isogenic mutants stx2, eae or tir. When the infant rabbits are infected with wild-type EHEC, the animals develop diarrhea and intestinal inflammation but not HUS. The absence of the Stx receptor Gb3 in the rabbit kidneys likely accounts for the lack of HUS in these animals (Zoja et al., 1992). Similarly to gnotobiotic piglets, infant rabbits infected with EHEC develop AE lesions on their intestinal epithelium (Ritchie et al., 2003). However, in the absence of Tir or Intimin, EHEC cannot colonize the intestine or cause disease, indicating the important role that AE lesion formation plays in pathogenesis. Stx2 promotes an inflammatory response at the intestinal mucosa but does not contribute to colonization, as shown by infection of rabbits with a stx2 mutant of EHEC or administration of purified Stx2. The infant rabbit model is useful to study the intestinal pathogenesis of EHEC infection, and it is advantageous to the gnotobiotic piglet model in its cost effectiveness.
The development of an animal model that would mimic the whole spectrum of EHEC disease would be invaluable to study preventive strategies and to test new therapies. While the infant rabbit model allows monitoring of hemorrhagic colitis and AE lesions without the requirement of complex animal facilities, the gnotobiotic piglet represents the best animal model available for investigation of systemic effects of EHEC infection. An animal model that mimics the human infectious dose (50–100 CFU) is not available yet. The remarkably low infectious dose is an important aspect of EHEC infection. Understanding the initial events that lead to colonization of the human intestine, especially the role of host factors, will provide insight into how EHEC can rapidly proliferate and establish disease in humans.
Regulation of Stx Expression
Phage Cycle and SOS Response
In EHEC O157:H7, Stx is encoded by two toxin-converting bacteriophages in the chromosome, 933W and 933J (O'Brien et al., 1984). The genes encoding stx are located in intact or partial genomes of prophages from the lambda family, which are integrated into the bacterial chromosome (Johansen et al., 2001). Activation of the phage lytic cycle leads to Stx production (Neely and Friedman, 1998), which is then released upon bacterial cell lysis.
The genes encoding Stx1 and Stx2 are located within the late phage genes, where expression of stxAB is under control of the phage cycle. Stx2 is only produced when the phage enters its lytic cycle (Tyler et al., 2004), while Stx1 is regulated by phage cycle and an iron-regulated promoter (Calderwood and Mekalanos, 1987; Wagner et al., 2002). The lambda phage remains quiescent due to the binding of the cI repressor to the right (OR) and left (OL) operator sites, in turn inhibiting the activity of the phage early promoters PR and PL (Waldor and Friedman, 2005). To enter its lytic cycle, the bacteriophage encoding stxAB takes advantage of the bacterial cell SOS response. Upon triggering of the SOS response, RecA is produced and activated (Muhldorfer et al., 1996). The activated form of RecA promotes cleavage of the cI repressor, de-repressing PR. Absence of cI leads to the expression of the anti-terminators N and Q. The protein Q then binds to PR' to activate the late phage genes, including stxAB. Bacteria undergo lysis and release Stx into the environment (Figure 2). A study of the promoters and regulatory regions of the 933W phage provides evidence that cI removal is required for Stx production by the bacterial cell. A 933W derivative encoding a non-cleavable repressor cannot produce Stx (Tyler et al., 2004).
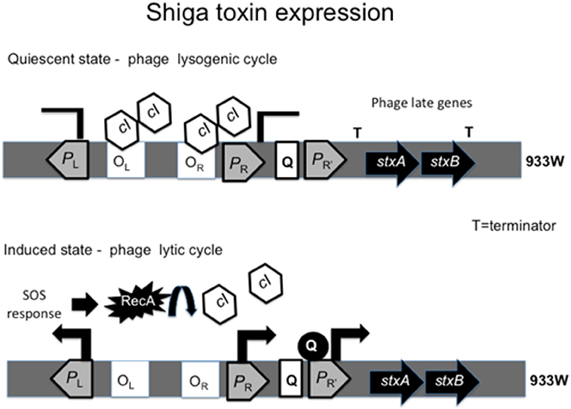
Figure 2. Regulation of Shiga toxin expression by the phage cycle. The cI repressor bound to operator sites OL and OR inhibitis transcription from the promoters PL and PR. The presence of terminator downtream of PR' inhibits transcription of stxAB and the phage remains quiescent. When SOS response is triggered, RecA cleaves cI, relieving PR repression and expression of antiterminators N and Q. Antiterminator Q binds PR' leading to transcription of stxAB genes with the late phage genes.
Regulation by Antibiotics
The use of antibiotics for the treatment of STEC infection is controversial due to its effect on induction of the phage lytic cycle (Zhang et al., 2000). Many studies have shown that antibiotics such as quinolones and mitomycin C increase the production of Stx (Zhang et al., 2000; McGannon et al., 2010). Specifically, mitomycin C is commonly used in research for prophage induction for its ability to interfere with DNA replication, triggering an SOS response (Los et al., 2010).
In a clinical study, Wong et al. followed the course of disease in 71 children who had diarrhea caused by EHEC O157:H7. Treatment of infected children with the antibiotic trimethoprim-sulfamethoxazole (SXT) led to a relative risk of 17.7% of developing HUS, while treatment based on beta-lactamic antibiotics contributed to a 13.4% relative risk (Wong et al., 2000). A recent study conducted by McGannon and colleagues supports the finding that the use of SXT and quinolones worsen a patient's outcome during EHEC infection (McGannon et al., 2010). These authors investigated the influence of antibiotics on Stx production: azithromicin (AZM), doxycycline (DOX), phosphomycin (FOS) and gentamycin (GEN) decreased Stx production, while ampicillin (AMP), SXT and ciprofloxacin (CIP) increased Stx production. The antibiotics ceftriaxone (CRO) and rifampin (RIF) did not affect Stx production. Their results confirm that antibiotics that target DNA synthesis trigger Stx production, and suggest that antibiotics that target protein synthesis and cell wall synthesis inhibit Stx production.
Quinolones are a class of antibiotics known to induce production of Stx, due to the activation of the SOS response in EHEC. A study conducted by Zhang et al. (Zhang et al., 2000) showed that administration of CIP and FOS caused a reduction in the bacterial burden of EHEC in the feces. However, treatment of infected mice with CIP resulted in increased levels of Stx in feces, as well as increased mortality. Conversely, treatment with FOS did not increase Stx production (Zhang et al., 2000). FOS is an inhibitor of cell wall synthesis, and it does not induce bacteriophages. This study contributes to a better understanding on the effects of different classes of antibiotics for treatment of EHEC infections.
Further in vivo investigations would help to determine whether Stx-inhibitory antibiotics are potentially beneficial to treat EHEC-infected individuals, yet it is still premature to endorse antibiotic treatment for EHEC infection.
Regulation by Other Mechanisms
Nitric oxide
Nitric oxide (NO) inhibits the synthesis of Stx by EHEC (Vareille et al., 2007). Vareille and colleagues reported that stx2 expression during stationary growth phase could be diminished by administration of the NO donor NOR4. The inhibitory effect of NO was evident when EHEC was grown in the presence of mitomycin C, a known inducer of Stx production. In addition, the use of the NO scavenger compound cPTIO could restore stx2 expression and Stx2 production in NO-treated EHEC cultures (Vareille et al., 2007). Interestingly, the inhibitory effect of NO was also seen at the phage particle levels. NOR4 reduced the number of 933W phage released after SOS-induction by mitomycin C. Treatment with NO dramatically reduces the expression of recA, therefore, the inhibitory effect of NO over Stx is due to interference with the bacterial SOS response.
Hydrogen peroxide (H2O2)
DNA-damaging agents such as mitomycin C and UV irradiation trigger Stx-expressing phages, yet these signals are unlikely to be encountered in vivo. Many investigators have focused on identifying agents present in the human intestine that might contribute to Stx production leading to HUS. Neutrophils and other cells produce NO and H2O2, that are known to damage DNA and potentially trigger an SOS response in bacteria (Imlay and Linn, 1987; Lobysheva et al., 1999).
Wagner and colleagues (Wagner et al., 2001) investigated the possible participation of H2O2 and neutrophils in Stx production and phage induction in EHEC. Exposure of EHEC to H2O2in vitro resulted in increased Stx production in a dose-dependent manner, as well as an increase in phage titers. Incubation of EHEC with human neutrophils increased Stx production, indicating that neutrophil-derived products might induce an SOS response leading to Stx release (Wagner et al., 2001). A study by Los and colleagues (Los et al., 2010) corroborated findings that H2O2 increases Stx production. An investigation of possible prophage triggers found that although high osmolarity did not affect prophage induction, the addition of millimolar concentrations of H2O2 resulted in phage production comparable to the amounts generated by addition of the antibiotic norphloxacin. Oxidative stress conditions during host colonization, such as reactive oxygen species (ROS) produced by commensal bacteria, might enhance Stx production in vivo, thereby contributing to EHEC pathogenesis.
Iron
The concentration of iron affects the production of Stx1; low-iron conditions lead to increased amounts of Stx1 (O'Brien et al., 1982). Regulation of stx1AB by iron has been demonstrated (Weinstein et al., 1988), but iron seems to have no impact on the expression of stx2AB genes (Muhldorfer et al., 1996). High levels of iron repress the activity of the stx1AB iron-dependent promoter, thereby reducing Stx1 production. Specifically, repression of stx1AB by iron is mediated by Fur (Calderwood and Mekalanos, 1987).
Environmental conditions
The conditions that govern the switch between lysogenic and lytic phage growth affect Stx production. During the lytic stage, a phage produces and releases Stx. The study of environmental cues and regulatory pathways that lead to expression of stx genes is fundamental to the development of novel strategies to prevent Stx production or counteract the effects of the toxin in order to avoid HUS.
Muhldorfer and colleagues (Muhldorfer et al., 1996) investigated the effects of different environmental conditions on the expression and production of Stx2. By using a translational reporter fusion for Stx2 synthesis, they demonstrated that the global regulatory protein H-NS regulates the expression of stx2 by modulating its promoter activity and the synthesis of Stx2 by modulation of the phage induction. Growth temperature has a moderate effect on Stx production and has been linked to H-NS regulation. The synthesis of Stx2 increased at 30°C but decreased at 42°C, in the hns-mutant strain. However, conditions such as pH, osmolarity, oxygen tension, acetates, iron levels, or carbon sources did not affect stx2 expression.
Stationary phase
Earlier studies suggest that stationary growth increases production of Stx. Konowalchuk et al. reported maximal cytotoxic activity when the bacterial cultures were shaken for 24 h (Konowalchuk et al., 1978), a condition where the bacterial cells enter stationary phase, and stxAB are induced (Bergholz et al., 2007) (Table 2).
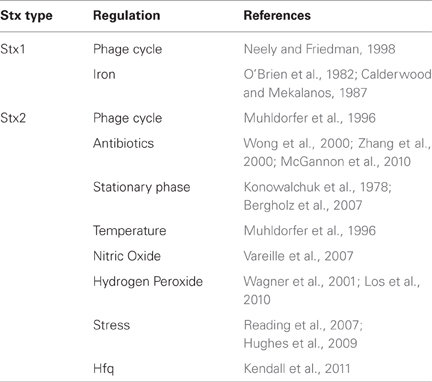
Table 2. Regulation of Stx expression.
Regulation by Stress
Mammalian cells communicate with each other via hormones. Bacterial cells, although unicellular, can also communicate with each other through the production and detection of small molecules known as autoinducers (AIs) (Nealson et al., 1970). AIs are hormone-like compounds produced by commensal and pathogenic bacteria, and they are involved in the coordination of behavior and adaptive responses (Sperandio et al., 2003). This communication is known as quorum sensing, and it coordinates multiple phenotypes in bacteria (Fuqua et al., 1994).
Pathogenic bacteria use cell-to-cell communication to regulate the production of virulence factors. EHEC can also hijack the mammalian stress response hormones epinephrine (epi) and norepinephrine (NE) to regulate expression of its major pathogenic traits: AE lesion formation, flagellar motility, and Stx production (Sperandio et al., 2001). The communication between bacteria and its host is called inter-kingdom signaling (Pacheco and Sperandio, 2009).
Many quorum sensing systems are composed of two component systems (TCS) that incorporate a sensor histidine kinase (HK) and a response regulator (RR) to regulate signal transduction, including quorum sensing-mediated virulence regulation in EHEC (Clarke et al., 2006; Reading et al., 2007; Hughes et al., 2009). Upon sensing its signal, the HK autophosphorylates at a conserved histidine residue and subsequently transfers its phosphate to a conserved aspartate residue in the RR. Then, the RR undergoes conformational changes that allows it to bind DNA, promoting changes in gene expression (Stock et al., 2000).
EHEC encodes for the QseBC TCS (Sperandio et al., 2002), that senses bacterial-derived AI-3 and host-produced epi/NE (Hughes et al., 2009). The QseC sensor kinase is the first adrenergic sensor described in bacteria (Clarke et al., 2006), and it is the only sensor of AI-3 in EHEC. Hughes et al. (Hughes et al., 2009) reported that stx2 expression is diminished in the qseC mutant. Transcriptional analysis showed that QseC regulates stx2 expression through activation of recA. These findings demonstrate that the bacterial adrenergic receptor QseC participates in the generation of the SOS response in bacteria (Hughes et al., 2009). In the same study, it was demonstrated that the TCS QseEF (Reading et al., 2007) also regulates expression of stx2. QseEF expression is activated by QseBC, and QseC phosphorylates the non-cognate RR QseF. It was shown that QseC-mediated activation of stx2 expression occurs via QseF (Hughes et al., 2009). The QseC sensor is present in 25 species of Gram-negative bacteria (Rasko et al., 2008). Taken together, these results demonstrate that bacterial cell-to-cell signaling plays a major role in the regulation of stx expression and may represent a suitable target for novel anti-virulence therapies for EHEC.
The importance of global regulators in the modulation of stx2 gene expression is exemplified by a recent study performed by Kendall et al. (Kendall et al., 2011). It was demonstrated that the RNA chaperone Hfq regulates the expression of stx2. Hfq is an RNA chaperone that mediates post-transcriptional regulation through small RNAs (sRNA) (Vogel and Luisi, 2011). Deletion of hfq in EHEC 86–24 and EDL933 strains results in increased expression of stx2AB genes and increased production of Stx2 (Kendall et al., 2011). These data demonstrate that Hfq decreases stx2 expression and consequently plays an important role in the development of HUS upon EHEC infection.
Novel Therapies
The increasing knowledge on regulation of stx expression and its mechanism of action has contributed to the development of new strategies to counteract its effects and to prevent disease. To date, three major strategies seem promising: quorum sensing inhibitors to interfere with virulence and Stx production; vaccines and immunotherapies to prevent and treat infection by STEC; and toxin inhibitors to avoid receptor binding. We present and discuss these strategies in this section.
Inhibition of Quorum Sensing
Bacterial cells are able to sense their surroundings and regulate gene expression in an adaptive manner (Rasko and Sperandio, 2010). Environmental cues are used to fine-tune gene regulation and avoid energy waste. Pathogenic bacteria including EHEC can sense chemical signals present in the host environment to trigger the expression of virulence genes and ultimately promote colonization of the host (Hughes et al., 2009). Therefore, understanding the interaction between EHEC and its host is imperative to fight bacterial infection. The identification of mechanisms involved in the regulation of virulence gene expression allows the development of strategies to prevent that triggering event, diminishing the ability of the bacteria to colonize its host. By interfering with the sensing mechanisms harbored specifically by pathogenic bacteria, bacterial colonization of the host can be minimized or eliminated.
Many bacteria share the ability to sense epi/NE via the adrenergic sensor QseC (Sperandio et al., 2003). It has been reported that qseC knockouts in EHEC (Clarke et al., 2006), Salmonella typhimurium (Bearson and Bearson, 2008; Moreira et al., 2010) and Francisella tularensis (Weiss et al., 2007) are attenuated in animal models. Due to the important role of QseC in virulence regulation in these pathogens, Rasko et al. (Rasko et al., 2008) conducted a high-throughput screen (HTS) of a chemical library of small molecules in an attempt to identify an inhibitor of virulence regulation mediated by QseC. The HTS led to the discovery of the small molecule LED209 that targets QseC. LED209 presented little toxicity against bacterial and human cell lines and did not inhibit bacterial growth. In the same report, it was shown that LED209 counteracted the effects of epi on the expression of the LEE and stx2. Most importantly, LED209 did not trigger an SOS response in EHEC; in fact, the drug decreased stx2 expression. These results demonstrated that LED209 inhibits QseC-mediated virulence gene expression (Figure 3). The inhibitory effect of LED209 was also seen at the level of protein activity, since QseC autophosphorylation in the presence of epi was inhibited with the addition of LED209. The activity of LED209 was also tested against the QseC-encoding bacterial pathogens Salmonella typhimurium and Francisella tularensis, and it was shown to be effective in vivo against these pathogens (Rasko et al., 2008).
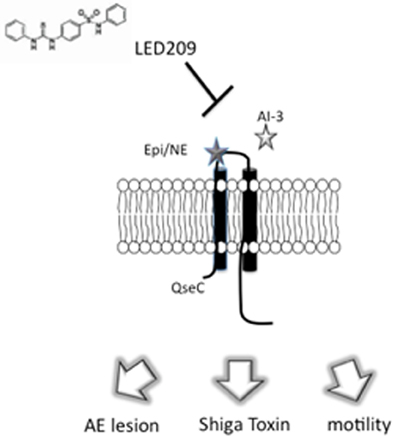
Figure 3. Inhibition of signal transduction by LED209. LED209 prevents autophosphorylation of the adrenergic sensor QseC, which is required to activate the expression of genes involved in colonization of the intestinal epitheliu.
Since quorum sensing inhibitors such as LED209 do not affect bacterial growth, it is thought that less selective pressure for resistance against the drug will occur. Also, quorum sensing inhibitors have the advantage of presenting selective toxicity, since the targeted sensory mechanisms are usually absent from eukaryotic cells. However, strategies involving reduction of virulence are not devoid of disadvantages. One of the drawbacks is the narrow spectrum of activity. The signaling systems might be pathogen-specific and not be suitable for use in the treatment of a diverse range of bacterial infections, in contrast to classic antibiotics. In addition, the presence of homologues of the target signal transduction systems in members of the microbiota might have an undesirable impact on the homeostasis of the GI tract. Lastly, the use of quorum sensing inhibitors has not been tested in clinical trials so no information exists about possible side effects in humans. Nonetheless, the multiple advantages of quorum sensing inhibitors make them attractive for treatment of bacterial infections. It appears that the future of treatment of EHEC-mediated disease might rely on the ability to design chemicals that directly target the bacterial receptors regulating Stx expression and production.
Pyocins
A study published by Ritchie et al. (Ritchie et al., 2011) explored the use of R-type pyocins to combat EHEC infection in vivo. R-type pyocins are bacteriocins produced by some Pseudomonas aeruginosa strains. These proteins form structures similar to tails of the bacteriophages from the Myoviridae family, which insert themselves into the bacterial cell envelope after contact with a specific cell receptor. R-type pyocins bind surface structures on the target cell and form pores at the cell membrane, causing membrane depolarization and cell death. The bactericidal activity of R-type pyocins toward EHEC was first investigated by Scholl et al. (Scholl et al., 2009), who constructed an EHEC O157-specific pyocin, AvR2-V10. These authors reported that AvR2-V10 could specifically degrade O157-LPS without inducing Stx production. Ritchie et al. (Ritchie et al., 2011) constructed a variant of AvR2-V10, named AvR2-V10.3, and tested it for antibacterial activity against EHEC in vivo using the infant rabbit model. Rabbits treated with AvR2-V10.3 after EHEC infection did not develop diarrhea and presented less intestinal inflammation. Also, administration of AvR2-V10.3 reduced intestinal colonization and bacterial shedding in stool. The pyocin treatment was also beneficial even when administrated after the onset of diarrhea. These studies suggest that R-type pyocins can be effective to prevent or treat symptoms caused by EHEC infection.
Vaccines and Immunotherapy
Other strategies under investigation for both the prevention and resolution of EHEC infection include vaccines, Stx receptor mimics and antibodies against Stx. Many immunization studies have focused on raising antibodies against purified subunits of the Stx.
A strategy to reduce HUS development is the prevention of Stx exposure. Although considered a conservative approach in the field of infectious diseases, immunization of children against EHEC could prevent the occurrence of disease. Immunization represents a method to prevent HUS, especially in children, who succumb to acute kidney failure resulting from EHEC infection at a higher rate than adults. Many studies have focused on the development of a vaccine to prevent EHEC infection, while some studies have demonstrated the protective effect of a Stx vaccine in mouse or piglet models of infection. However, a vaccine against EHEC is still not commercially available.
Zhang et al. (Zhang et al., 2011) investigated if immunization with a combined subunit vaccine containing StxB and Tir fragments protected against EHEC infection. The authors constructed a multivalent protein constituted of Stx2B-Tir-Stx1B-Zot, where Zot is an antigen delivery tool that binds a receptor in the intestinal epithelium affecting mucosal permeability. Mice immunized intranasally with the multivalent protein had reduced colonization and reduced amounts of EHEC detected in their stool. Their results suggest that a multi-antigen vaccine that targets EHEC can reduce colonization in vivo. However, Stx levels or systemic effects of Stx were not assessed in this study.
Passive immunotherapy using antibodies against Stx for EHEC infection has been explored. Many antibodies have been developed, in an attempt to prevent the devastating pathogenesis of Stx. The idea behind the use of anti-Stx antibodies to prevent HUS relies on the time course of EHEC infection. After the onset of diarrhea, there is a window of time before the development of HUS when the administration of passive immunization might help to prevent the evolution of the disease. Mukherjee and colleagues (Mukherjee et al., 2002) developed a humanized monoclonal antibody (HuMAb) against Stx2. In this study, it was observed that the HuMAbs presented neutralization activity of 90% and contributed to mice survival in the presence of Stx injected intravenously.
Mohawk and colleagues (Mohawk et al., 2010a) investigated the ability of Stx-neutralizing antibodies to affect colonization by EHEC 86–24. This strain produces only Stx2. BALB/c mice harboring an ICF were first inoculated with polyclonal rabbit anti-Stx2 serum and then challenged with EHEC. The administration of serum had no impact on initial colonization, but it reduced the CFU detected after 3–5 days post-infection. The anti-Stx2 serum reduced mortality of mice and reduced weight loss at later stages in infection (Mohawk et al., 2010a). Vaccination of infected mice with a Stx2 toxoid resulted in decreased CFU detected in their feces, suggesting that active immunization leads to generation of Stx2-neutralizing antibodies in the intestine (Mohawk et al., 2010a). Although the results were positive, they have to be interpreted with caution. EHEC infection of mice does not mimic the human infection in many aspects. EHEC does not form AE lesions on mouse intestinal epithelial cells, which is a hallmark of the infection by this pathogen, nor do mice develop HUS.
Inhibitors of Toxin Receptors
Stx binds to the Gb3 receptor in endothelial cells (Lingwood et al., 1987), causing intense vascular damage and organ failure. Therefore, prevention of Stx binding to its receptor is a potential strategy to reduce Stx-mediated disease.
Many ligands that mimic the Stx receptor Gb3 have been designed and have shown modest effect of neutralizing toxin in vitro and in vivo (Kitov et al., 2000; Nishikawa et al., 2000). However, clinical trials using such compounds have not been effective (Trachtman et al., 2003). It is possible that the Stx-neutralizing molecules predominately target the less toxic Stx1 instead of the very potent Stx2 (Kulkarni et al., 2010). Therefore, new design strategies need account for the subtle variations between the two Stx forms.
Nishikawa and colleagues explored the neutralization of circulating Stx as a strategy to prevent HUS. Based on the interaction of the toxin with the trisaccharide moieties of its receptor Gb3, this group designed a series of carbosilane dendrimers carrying trisaccharide moieties at their termini that they termed SUPER TWIG. The rationale was that the compound would bind Stx in the blood, preventing its access to its receptor at the host cell. SUPER TWIG binds Stx with high affinity (Kd= 1.1 × 106− M) and inhibits incorporation of the toxin in vitro. Mice treated with SUPER TWIG were protected from EHEC infection upon challenge with a fatal dose. This group has optimized the SUPER TWIG structure and determined the binding site on Stx (Nishikawa et al., 2005), but more investigations on the mechanism of action are necessary before SUPER TWIG can be used as a therapeutic for STEC infections.
An effort to develop Stx2-specific ligands led to the identification of a novel ligand using an ELISA-based approach (Kale et al., 2008), and it served as the basis for the development of nanoparticles by the same research group (Kulkarni et al., 2010). Nanotechnology is a novel approach for the development of targeted drug delivery systems to treat several diseases including cancer and infectious diseases. Kulkarni et al. (Kale et al., 2008) developed glycan-encapsulated gold nanoparticles (GNP) to display ligands for Stx. These nanoparticles represent an efficient material for drug delivery, since it allows multivalent display of glycans in a similar way to the glycocalyx exposed at the cell surface. Analogs of the glycan Pk trisaccharide, which binds preferentially to Stx, were developed and used to coat GNP. Toxicity assays using Vero cells showed that the GNPs successfully neutralized Stx1 and Stx2, but not Stx2 variants Stx2c or Stx2d. Also, GNPs were not toxic toward Vero cells, based on a protein inhibition assay. These results indicated the therapeutic potential of ligand-coated nanoparticles, however, further studies on receptor specificity are needed to address the target of emerging variants of Stx.
A randomized clinical trial conducted by Trachtman et al. (Trachtman et al., 2003) assessed the effectiveness of a Stx inhibitor on the severity of HUS when administered in children presenting HUS associated with EHEC infection. One hundred and forty-five children were involved in the trial, and 100 of then received a daily dose of SYNSORB Pk, a drug developed by SYNSORB Biotech (Alberta, Canada), for seven days. SYNSORB Pk is constituted by silicone dioxide containing the trisaccharide moiety of Gb3 that mediates the Stx binding to the receptor. The rationale was that the drug would compete with the host cell receptor for Stx binding, preventing the interaction between Stx and Gb3, and consequently reducing the risk of HUS. Unfortunately, the drug did not reduce the severity of the disease. Possible explanations for the failure of this trial include that a low percentage of patients had detectable levels of free Stx in their stool or that EHEC attachment to the intestinal epithelium could affect drug access to Stx. Although the results were not positive, this study represents a major step toward determining a treatment of HUS.
Inhibitors of the Stx receptor Gb3 are also a target for HUS prevention. C-9 is a specific inhibitor of glucosylceramide (GL1) synthase (Zhao et al., 2007). Silberstein and colleagues (Silberstein et al., 2011) reported that C-9 was able to downregulate Gb3 expression and prevent Stx2 effects in a tissue culture model using human kidney cells. Further studies using a rat model showed that oral administration of C-9 for a period of six days protected animals from the effects of Stx2. It was observed that prophylactic and post-infection treatment with C-9 reduced the expression of Gb3 and diminished mortality in infected rats by 50%. Also, C-9 reduced renal tubular necrosis and goblet cell damage as well as prevented the onset of watery diarrhea in rats injected with Stx2. Gb3 inhibitors may represent a potential novel therapy to prevent Stx-mediated disease. Although C-9 counteracted many symptoms of Stx-mediated disease, more studies are needed to assess the possible side effects of downregulation of Gb3 in eukaryotic cells.
Conclusions
The increasing incidence of HUS caused by Stx raises the urgent need for alternative therapies for EHEC infection. Novel anti-virulence strategies include quorum sensing inhibitors, Stx-receptor analogs and pyocins. These therapies employ different mechanisms of action from classic antibiotics, and they are thought to cause less selective pressure for drug resistance than classic antibiotics. Although several alternative anti-Stx strategies have been described in the literature, only one drug reached a phase 1 clinical trial. Therefore, translational research is necessary to bring new drugs discovered in basic science studies to the clinic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, A., Kenny, B., Stein, M., and Finlay, B. B. (1997). Characterization of two virulence proteins secreted by rabbit enteropathogenic Escherichia coli, EspA and EspB, whose maximal expression is sensitive to host body temperature. Infect. Immun. 65, 3547–3555.
Acheson, D. W., Moore, R., De Breucker, S., Lincicome, L., Jacewicz, M., Skutelsky, E., and Keusch, G. T. (1996). Translocation of Shiga toxin across polarized intestinal cells in tissue culture. Infect. Immun. 64, 3294–3300.
Arthur, T. M., Nou, X. W., Kalchayanand, N., Bosilevac, J. M., Wheeler, T., and Koohmaraie, M. (2011). Survival of Escherichia coli O157, H7 on Cattle Hides. Appl. Environ. Microbiol. 77, 3002–3008.
Bearson, B. L., and Bearson, S. M. (2008). The role of the QseC quorum-sensing sensor kinase in colonization and norepinephrine-enhanced motility of Salmonella enterica serovar typhimurium. Microb. Pathog. 44, 271–278.
Bell, B. P., Goldoft, M., Griffin, P. M., Davis, M. A., Gordon, D. C., Tarr, P. I., Bartleson, C. A., Lewis, J. H., Barrett, T. J., Wells, J. G., Baron, R., and Kobayashi, J. (1994). A multistate outbreak of Escherichia coli O157, H7-associated bloody diarrhea and hemolytic uremic syndrome from hamburgers. JAMA 272, 1349–1353.
Bellmeyer, A., Cotton, C., Kanteti, R., Koutsouris, A., Viswanathan, V. K., and Hecht, G. (2009). Enterohemorrhagic Escherichia coli suppresses inflammatory response to cytokines and its own toxin. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G576–G581.
Bergholz, T. M., Wick, L. M., Qi, W., Riordan, J. T., Ouellette, L. M., and Whittam, T. S. (2007). Global transcriptional response of Escherichia coli O157, H7 to growth transitions in glucose minimal medium. BMC Microbiol. 7, 97.
Boerlin, P., McEwen, S. A., Boerlin-Petzold, F., Wilson, J. B., Johnson, R. P., and Gyles, C. L. (1999). Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 37, 497–503.
Brandt, M. L., O'Regan, S., Rousseau, E., and Yazbeck, S. (1990). Surgical complications of the hemolytic-uremic syndrome. J. Pediatr. Surg. 25, 1109–1112.
Buchholz, U., Bernard, H., Werber, D., Bohmer, M. M., Remschmidt, C., Wilking, H., Delere, Y., an der Heiden, M., Adlhoch, C., Dreesman, J., Ehlers, J., Ethelberg, S., Faber, M., Frank, C., Fricke, G., Greiner, M., Hohle, M., Ivarsson, S., Jark, U., Kirchner, M., Koch, J., Krause, G., Luber, P., Rosner, B., Stark, K., and Kuhne, M. (2011). German outbreak of Escherichia coli O104, H4 associated with sprouts. N. Engl. J. Med. 365, 1763–1770.
Calderwood, S. B., and Mekalanos, J. J. (1987). Iron regulation of Shiga-like toxin expression in Escherichia coli is mediated by the fur locus. J. Bacteriol. 169, 4759–4764.
Campellone, K. G., Robbins, D., and Leong, J. M. (2004). EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. cell 7, 217–228.
Clarke, M. B., Hughes, D. T., Zhu, C., Boedeker, E. C., and Sperandio, V. (2006). The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 103, 10420–10425.
Deng, W., Li, Y., Vallance, B. A., and Finlay, B. B. (2001). Locus of enterocyte effacement from Citrobacter rodentium: sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Infect. Immun. 69, 6323–6335.
Endo, Y., Tsurugi, K., Yutsudo, T., Takeda, Y., Ogasawara, T., and Igarashi, K. (1988). Site of action of a Vero toxin (VT2) from Escherichia coli O157, H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 171, 45–50.
Fraser, M. E., Chernaia, M. M., Kozlov, Y. V., and James, M. N. (1994). Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1, 59–64.
Fraser, M. E., Fujinaga, M., Cherney, M. M., Melton-Celsa, A. R., Twiddy, E. M., O'Brien, A. D., and James, M. N. (2004). Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157, H7. J. Biol. Chem. 279, 27511–27517.
Fuqua, W. C., Winans, S. C., and Greenberg, E. P. (1994). Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 176, 269–275.
Garmendia, J., Phillips, A. D., Carlier, M. F., Chong, Y., Schuller, S., Marches, O., Dahan, S., Oswald, E., Shaw, R. K., Knutton, S., and Frankel, G. (2004). TccP is an enterohaemorrhagic Escherichia coli O157, H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6, 1167–1183.
Gould, L. H., Demma, L., Jones, T. F., Hurd, S., Vugia, D. J., Smith, K., Shiferaw, B., Segler, S., Palmer, A., Zansky, S., and Griffin, P. M. (2009). Hemolytic uremic syndrome and death in persons with Escherichia coli O157, H7 infection, foodborne diseases active surveillance network sites, 2000–2006. Clin. Infect. Dis. 49, 1480–1485.
Grant, J., Wendelboe, A. M., Wendel, A., Jepson, B., Torres, P., Smelser, C., and Rolfs, R. T. (2008). Spinach-associated Escherichia coli O157, H7 outbreak, Utah and New Mexico (2006). Emerg. Infect. Dis. 14, 1633–1636.
Greig, J. D., and Ravel, A. (2009). Analysis of foodborne outbreak data reported internationally for source attribution. Int. J. Food Microbiol. 130, 77–87.
Gunzer, F., Hennig-Pauka, I., Waldmann, K. H., Sandhoff, R., Grone, H. J., Kreipe, H. H., Matussek, A., and Mengel, M. (2002). Gnotobiotic piglets develop thrombotic microangiopathy after oral infection with enterohemorrhagic Escherichia coli. Am. J. Clin. Pathol. 118, 364–375.
Hughes, D. T., Clarke, M. B., Yamamoto, K., Rasko, D. A., and Sperandio, V. (2009). The QseC adrenergic signaling cascade in Enterohemorrhagic E. coli (EHEC). PLoS Pathog. 5:e1000553. doi: 10.1371/journal.ppat.1000553
Imlay, J. A., and Linn, S. (1987). Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J. Bacteriol. 169, 2967–2976.
Jacewicz, M., Clausen, H., Nudelman, E., Donohue-Rolfe, A., and Keusch, G. T. (1986). Pathogenesis of shigella diarrhea. XI. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J. Exp. Med. 163, 1391–1404.
Jaeger, J. L., and Acheson, D. W. (2000). Shiga toxin-producing Escherichia coli. Curr. Infect. Dis. Rep. 2, 61–67.
Jandhyala, D. M., Rogers, T. J., Kane, A., Paton, A. W., Paton, J. C., and Thorpe, C. M. (2010). Shiga toxin 2 and flagellin from shiga-toxigenic Escherichia coli superinduce interleukin-8 through synergistic effects on host stress-activated protein kinase activation. Infect. Immun. 78, 2984–2994.
Jarvis, K. G., Giron, J. A., Jerse, A. E., McDaniel, T. K., Donnenberg, M. S., and Kaper, J. B. (1995). Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U.S.A. 92, 7996–8000.
Jay, M. T., Garrett, V., Mohle-Boetani, J. C., Barros, M., Farrar, J. A., Rios, R., Abbott, S., Sowadsky, R., Komatsu, K., Mandrell, R., Sobel, J., and Werner, S. B. (2004). A multistate outbreak of Escherichia coli O157, H7 infection linked to consumption of beef tacos at a fast-food restaurant chain. Clin. Infect. Dis. 39, 1–7.
Jerse, A. E., Yu, J., Tall, B. D., and Kaper, J. B. (1990). A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U.S.A. 87, 7839–7843.
Johannes, L., and Romer, W. (2010). Shiga toxins–from cell biology to biomedical applications. Nat. Rev. Microbiol. 8, 105–116.
Johansen, B. K., Wasteson, Y., Granum, P. E., and Brynestad, S. (2001). Mosaic structure of Shiga-toxin-2-encoding phages isolated from Escherichia coli O157, H7 indicates frequent gene exchange between lambdoid phage genomes. Microbiology 147, 1929–1936.
Kale, R. R., McGannon, C. M., Fuller-Schaefer, C., Hatch, D. M., Flagler, M. J., Gamage, S. D., Weiss, A. A., and Iyer, S. S. (2008). Differentiation between structurally homologous Shiga 1 and Shiga 2 toxins by using synthetic glycoconjugates. Angew. Chem. Int. Ed. Engl. 47, 1265–1268.
Kaper, J. B., Nataro, J. P., and Mobley, H. L. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140.
Karmali, M. A. (2004). Prospects for preventing serious systemic toxemic complications of Shiga toxin-producing Escherichia coli infections using Shiga toxin receptor analogues. J. Infect. Dis. 189, 355–359.
Karmali, M. A., Steele, B. T., Petric, M., and Lim, C. (1983). Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet 1, 619–620.
Kendall, M. M., Gruber, C. C., Rasko, D. A., Hughes, D. T., and Sperandio, V. (2011). Hfq virulence regulation in enterohemorrhagic Escherichia coli O157, H7 Strain 86–24. J. Bacteriol. 193, 6843–6851.
Kenny, B., DeVinney, R., Stein, M., Reinscheid, D. J., Frey, E. A., and Finlay, B. B. (1997). Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91, 511–520.
Kenny, B., and Finlay, B. B. (1995). Protein secretion by enteropathogenic Escherichia coli is essential for transducing signals to epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 92, 7991–7995.
Kitov, P. I., Sadowska, J. M., Mulvey, G., Armstrong, G. D., Ling, H., Pannu, N. S., Read, R. J., and Bundle, D. R. (2000). Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 403, 669–672.
Knutton, S., Rosenshine, I., Pallen, M. J., Nisan, I., Neves, B. C., Bain, C., Wolff, C., Dougan, G., and Frankel, G. (1998). A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J. 17, 2166–2176.
Konowalchuk, J., Dickie, N., Stavric, S., and Speirs, J. I. (1978). Comparative studies of five heat-labile toxic products of Escherichia coli. Infect. Immun. 22, 644–648.
Kulasekara, B. R., Jacobs, M., Zhou, Y., Wu, Z., Sims, E., Saenphimmachak, C., Rohmer, L., Ritchie, J. M., Radey, M., McKevitt, M., Freeman, T. L., Hayden, H., Haugen, E., Gillett, W., Fong, C., Chang, J., Beskhlebnaya, V., Waldor, M. K., Samadpour, M., Whittam, T. S., Kaul, R., Brittnacher, M., and Miller, S. I. (2009). Analysis of the genome of the Escherichia coli O157, H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect. Immun. 77, 3713–3721.
Kulkarni, A. A., Fuller, C., Korman, H., Weiss, A. A., and Iyer, S. S. (2010). Glycan encapsulated gold nanoparticles selectively inhibit shiga toxins 1 and 2. Bioconjug. Chem. 21, 1486–1493.
Lindberg, A. A., Brown, J. E., Stromberg, N., Westling-Ryd, M., Schultz, J. E., and Karlsson, K. A. (1987). Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 262, 1779–1785.
Lingwood, C. A., Law, H., Richardson, S., Petric, M., Brunton, J. L., De Grandis, S., and Karmali, M. (1987). Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 262, 8834–8839.
Lobysheva, I. I., Stupakova, M. V., Mikoyan, V. D., Vasilieva, S. V., and Vanin, A. F. (1999). Induction of the SOS DNA repair response in Escherichia coli by nitric oxide donating agents: dinitrosyl iron complexes with thiol-containing ligands and S-nitrosothiols. FEBS Lett. 454, 177–180.
Los, J. M., Los, M., Wegrzyn, A., and Wegrzyn, G. (2010). Hydrogen peroxide-mediated induction of the Shiga toxin-converting lambdoid prophage ST2-8624 in Escherichia coli O157, H7. FEMS Immunol. Med. Microbiol. 58, 322–329.
Mallard, F., Antony, C., Tenza, D., Salamero, J., Goud, B., and Johannes, L. (1998). Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J. Cell Biol. 143, 973–990.
Matussek, A., Lauber, J., Bergau, A., Hansen, W., Rohde, M., Dittmar, K. E., Gunzer, M., Mengel, M., Gatzlaff, P., Hartmann, M., Buer, J., and Gunzer, F. (2003). Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 102, 1323–1332.
McDaniel, T. K., Jarvis, K. G., Donnenberg, M. S., and Kaper, J. B. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 92, 1664–1668.
McGannon, C. M., Fuller, C. A., and Weiss, A. A. (2010). Different classes of antibiotics differentially influence shiga toxin production. Antimicrob. Agents Chemother. 54, 3790–3798.
Mellies, J. L., Elliott, S. J., Sperandio, V., Donnenberg, M. S., and Kaper, J. B. (1999). The Per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 33, 296–306.
Michael, M., Elliott, E. J., Craig, J. C., Ridley, G., and Hodson, E. M. (2009). Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials. Am. J. Kidney Dis. 53, 259–272.
Mohawk, K. L., Melton-Celsa, A. R., Robinson, C. M., and O'Brien, A. D. (2010a). Neutralizing antibodies to Shiga toxin type 2 (Stx2) reduce colonization of mice by Stx2-expressing Escherichia coli O157, H7. Vaccine 28, 4777–4785.
Mohawk, K. L., Melton-Celsa, A. R., Zangari, T., Carroll, E. E., and O'Brien, A. D. (2010b). Pathogenesis of Escherichia coli O157, H7 strain 86-24 following oral infection of BALB/c mice with an intact commensal flora. Microb. Pathog. 48, 131–142.
Mohawk, K. L., and O'Brien, A. D. (2011). Mouse models of Escherichia coli O157, H7 infection and shiga toxin injection. J. Biomed. Biotechnol. 2011, 258185.
Moreira, C. G., Weinshenker, D., and Sperandio, V. (2010). QseC mediates Salmonella enterica serovar typhimurium virulence in vitro and in vivo. Infect. Immun. 78, 914–926.
Muhldorfer, I., Hacker, J., Keusch, G. T., Acheson, D. W., Tschape, H., Kane, A. V., Ritter, A., Olschlager, T., and Donohue-Rolfe, A. (1996). Regulation of the Shiga-like toxin II operon in Escherichia coli. Infect. Immun. 64, 495–502.
Mukherjee, J., Chios, K., Fishwild, D., Hudson, D., O'Donnell, S., Rich, S. M., Donohue-Rolfe, A., and Tzipori, S. (2002). Human Stx2-specific monoclonal antibodies prevent systemic complications of Escherichia coli O157, H7 infection. Infect. Immun. 70, 612–619.
Muthing, J., Schweppe, C. H., Karch, H., and Friedrich, A. W. (2009). Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb. Haemost. 101, 252–264.
Myhal, M. L., Laux, D. C., and Cohen, P. S. (1982). Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol. 1, 186–192.
Nataro, J. P., and Kaper, J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201.
Naylor, S. W., Low, J. C., Besser, T. E., Mahajan, A., Gunn, G. J., Pearce, M. C., McKendrick, I. J., Smith, D. G. E., and Gally, D. L. (2003). Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157, H7 in the bovine host. Infect. Immun. 71, 1505–1512.
Nealson, K. H., Platt, T., and Hastings, J. W. (1970). Cellular control of the synthesis and activity of the bacterial luminescent system. J. Bacteriol. 104, 313–322.
Neely, M. N., and Friedman, D. I. (1998). Functional and genetic analysis of regulatory regions of coliphage H-19B: location of shiga-like toxin and lysis genes suggest a role for phage functions in toxin release. Mol. Microbiol. 28, 1255–1267.
Neil, K. P., Biggerstaff, G., Macdonald, J. K., Trees, E., Medus, C., Musser, K. A., Stroika, S. G., Zink, D., and Sotir, M. J. (2012). A novel vehicle for transmission of Escherichia coli O157, H7 to humans: multistate outbreak of E. coli O157, H7 infections associated with consumption of ready-to-bake commercial prepackaged cookie dough–United States 2009. Clin. Infect. Dis. 54, 511–518.
Neupane, M., Abu-Ali, G. S., Mitra, A., Lacher, D. W., Manning, S. D., and Riordan, J. T. (2011). Shiga toxin 2 overexpression in Escherichia coli O157, H7 strains associated with severe human disease. Microb. Pathog. 51, 466–470.
Nishikawa, K., Matsuoka, K., Watanabe, M., Igai, K., Hino, K., Hatano, K., Yamada, A., Abe, N., Terunuma, D., Kuzuhara, H., and Natori, Y. (2005). Identification of the optimal structure required for a Shiga toxin neutralizer with oriented carbohydrates to function in the circulation. J. Infect. Dis. 191, 2097–2105.
Nishikawa, Y., Zhou, Z., Hase, A., Ogasawara, J., Cheasty, T., and Haruki, K. (2000). Relationship of genetic type of Shiga toxin to manifestation of bloody diarrhea due to enterohemorrhagic Escherichia coli serogroup O157 isolates in Osaka City, Japan. J. Clin. Microbiol. 38, 2440–2442.
O'Brien, A. D., LaVeck, G. D., Thompson, M. R., and Formal, S. B. (1982). Production of Shigella dysenteriae type 1-like cytotoxin by Escherichia coli. J. Infect. Dis. 146, 763–769.
O'Brien, A. D., Lively, T. A., Chang, T. W., and Gorbach, S. L. (1983a). Purification of Shigella dysenteriae 1 (Shiga)-like toxin from Escherichia coli 0157, H7 strain associated with haemorrhagic colitis. Lancet 2, 573.
O'Brien, A. O., Lively, T. A., Chen, M. E., Rothman, S. W., and Formal, S. B. (1983b). Escherichia coli O157, H7 strains associated with haemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (SHIGA) like cytotoxin. Lancet 1, 702.
O'Brien, A. D., Newland, J. W., Miller, S. F., Holmes, R. K., Smith, H. W., and Formal, S. B. (1984). Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 226, 694–696.
Pacheco, A. R., and Sperandio, V. (2009). Inter-kingdom signaling: chemical language between bacteria and host. Curr. Opin. Microbiol. 12, 192–198.
Pai, C. H., Kelly, J. K., and Meyers, G. L. (1986). Experimental infection of infant rabbits with verotoxin-producing Escherichia coli. Infect. Immun. 51, 16–23.
Rangel, J. M., Sparling, P. H., Crowe, C., Griffin, P. M., and Swerdlow, D. L. (2005). Epidemiology of Escherichia coli O157, H7 outbreaks, United States, 1982–2002. Emerging Infect. Dis. 11, 603–609.
Rasko, D. A., Moreira, C. G., Li de, R., Reading, N. C., Ritchie, J. M., Waldor, M. K., Williams, N., Taussig, R., Wei, S., Roth, M., Hughes, D. T., Huntley, J. F., Fina, M. W., Falck, J. R., and Sperandio, V. (2008). Targeting QseC signaling and virulence for antibiotic development. Science 321, 1078–1080.
Rasko, D. A., Webster, D. R., Sahl, J. W., Bashir, A., Boisen, N., Scheutz, F., Paxinos, E. E., Sebra, R., Chin, C. S., Iliopoulos, D., Klammer, A., Peluso, P., Lee, L., Kislyuk, A. O., Bullard, J., Kasarskis, A., Wang, S., Eid, J., Rank, D., Redman, J. C., Steyert, S. R., Frimodt-Moller, J., Struve, C., Petersen, A. M., Krogfelt, K. A., Nataro, J. P., Schadt, E. E., and Waldor, M. K. (2011). Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365, 709–717.
Rasko, D. A., and Sperandio, V. (2010). Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 9, 117–128.
Reading, N. C., Torres, A. G., Kendall, M. M., Hughes, D. T., Yamamoto, K., and Sperandio, V. (2007). A novel two-component signaling system that activates transcription of an enterohemorrhagic Escherichia coli effector involved in remodeling of host actin. J. Bacteriol. 189, 2468–2476.
Riley, L. W., Remis, R. S., Helgerson, S. D., McGee, H. B., Wells, J. G., Davis, B. R., Hebert, R. J., Olcott, E. S., Johnson, L. M., Hargrett, N. T., Blake, P. A., and Cohen, M. L. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685.
Ritchie, J. M., Greenwich, J. L., Davis, B. M., Bronson, R. T., Gebhart, D., Williams, S. R., Martin, D., Scholl, D., and Waldor, M. K. (2011). An Escherichia coli O157-specific engineered pyocin prevents and ameliorates infection by E. coli O157, H7 in an animal model of diarrheal disease. Antimicrob. Agents Chemother. 55, 5469–5474.
Ritchie, J. M., Thorpe, C. M., Rogers, A. B., and Waldor, M. K. (2003). Critical roles for stx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inflammation in infant rabbits. Infect. Immun. 71, 7129–7139.
Rivas, L., Coffey, B., McAuliffe, O., McDonnell, M. J., Burgess, C. M., Coffey, A., Ross, R. P., and Duffy, G. (2010). In Vivo and Ex Vivo evaluations of bacteriophages e11/2 and e4/1c for use in the control of Escherichia coli O157, H7. Appl. Environ. Microb. 76, 7210–7216.
Romer, W., Berland, L., Chambon, V., Gaus, K., Windschiegl, B., Tenza, D., Aly, M. R., Fraisier, V., Florent, J. C., Perrais, D., Lamaze, C., Raposo, G., Steinem, C., Sens, P., Bassereau, P., and Johannes, L. (2007). Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 450, 670–675.
Sandvig, K., Garred, O., Prydz, K., Kozlov, J. V., Hansen, S. H., and van Deurs, B. (1992). Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 358, 510–512.
Savageau, M. A. (1983). Escherichia-coli habitats, cell-types, and molecular mechanisms of gene-control. Am. Nat. 122, 732–744.
Scholl, D., Cooley, M., Williams, S. R., Gebhart, D., Martin, D., Bates, A., and Mandrell, R. (2009). An engineered R-type pyocin is a highly specific and sensitive bactericidal agent for the food-borne pathogen Escherichia coli O157, H7. Antimicrob. Agents Chemother. 53, 3074–3080.
Sheikh, J., Hicks, S., Dall'Agnol, M., Phillips, A. D., and Nataro, J. P. (2001). Roles for Fis and YafK in biofilm formation by enteroaggregative Escherichia coli. Mol. Microbiol. 41, 983–997.
Silberstein, C., Lucero, M. S., Zotta, E., Copeland, D. P., Lingyun, L., Repetto, H. A., and Ibarra, C. (2011). A glucosylceramide synthase inhibitor protects rats against the cytotoxic effects of shiga toxin 2. Pediatr. Res. 69, 390–394.
Sodha, S. V., Lynch, M., Wannemuehler, K., Leeper, M., Malavet, M., Schaffzin, J., Chen, T., Langer, A., Glenshaw, M., Hoefer, D., Dumas, N., Lind, L., Iwamoto, M., Ayers, T., Nguyen, T., Biggerstaff, M., Olson, C., Sheth, A., and Braden, C. (2011). Multistate outbreak of Escherichia coli O157, H7 infections associated with a national fast-food chain 2006, a study incorporating epidemiological and food source traceback results. Epidemiol. Infect. 139, 309–316.
Sperandio, V., Torres, A. G., Giron, J. A., and Kaper, J. B. (2001). Quorum sensing is a global regulatory mechanism in enterohemorrhagic Escherichia coli O157, H7. J. Bacteriol. 183, 5187–5197.
Sperandio, V., Torres, A. G., Jarvis, B., Nataro, J. P., and Kaper, J. B. (2003). Bacteria-host communication: the language of hormones. Proc. Natl. Acad. Sci. U.S.A. 100, 8951–8956.
Sperandio, V., Torres, A. G., and Kaper, J. B. (2002). Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol. Microbiol. 43, 809–821.
Stein, P. E., Boodhoo, A., Tyrrell, G. J., Brunton, J. L., and Read, R. J. (1992). Crystal structure of the cell-binding B oligomer of verotoxin-1 from E. coli. Nature 355, 748–750.
Stock, A. M., Robinson, V. L., and Goudreau, P. N. (2000). Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215.
Tarr, P. I., Gordon, C. A., and Chandler, W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086.
Tilden, J. Jr., Young, W., McNamara, A. M., Custer, C., Boesel, B., Lambert-Fair, M. A., Majkowski, J., Vugia, D., Werner, S. B., Hollingsworth, J., and Morris, J. G. Jr. (1996). A new route of transmission for Escherichia coli: infection from dry fermented salami. Am. J. Public Health 86, 1142–1145.
Trachtman, H., Cnaan, A., Christen, E., Gibbs, K., Zhao, S., Acheson, D. W., Weiss, R., Kaskel, F. J., Spitzer, A., and Hirschman, G. H. (2003). Effect of an oral Shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children: a randomized controlled trial. JAMA 290, 1337–1344.
Tyler, J. S., Mills, M. J., and Friedman, D. I. (2004). The operator and early promoter region of the Shiga toxin type 2-encoding bacteriophage 933W and control of toxin expression. J. Bacteriol. 186, 7670–7679.
Tzipori, S., Montanaro, J., Robins-Browne, R. M., Vial, P., Gibson, R., and Levine, M. M. (1992). Studies with enteroaggregative Escherichia coli in the gnotobiotic piglet gastroenteritis model. Infect. Immun. 60, 5302–5306.
Tzipori, S., Wachsmuth, I. K., Chapman, C., Birden, R., Brittingham, J., Jackson, C., and Hogg, J. (1986). The pathogenesis of hemorrhagic colitis caused by Escherichia coli O157, H7 in gnotobiotic piglets. J. Infect. Dis. 154, 712–716.
Vareille, M., de Sablet, T., Hindre, T., Martin, C., and Gobert, A. P. (2007). Nitric oxide inhibits Shiga-toxin synthesis by enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 104, 10199–10204.
Vilte, D. A., Larzabal, M., Garbaccio, S., Gammella, M., Rabinovitz, B. C., Elizondo, A. M., Cantet, R. J. C., Delgado, F., Meikle, V., Cataldi, A., and Mercado, E. C. (2011). Reduced faecal shedding of Escherichia coli O157, H7 in cattle following systemic vaccination with gamma-intimin C(280) and EspB proteins. Vaccine 29, 3962–3968.
Vingadassalom, D., Campellone, K. G., Brady, M. J., Skehan, B., Battle, S. E., Robbins, D., Kapoor, A., Hecht, G., Snapper, S. B., and Leong, J. M. (2010). Enterohemorrhagic, E. coli requires N-WASP for efficient type III translocation but not for EspFU-mediated actin pedestal formation. PLoS Pathog. 6:e1001056. doi: 10.1371/journal.ppat.1001056
Vogel, J., and Luisi, B. F. (2011). Hfq and its constellation of RNA. Nat. Rev. Microbiol. 9, 578–589.
Wadolkowski, E. A., Burris, J. A., and O'Brien, A. D. (1990). Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157, H7. Infect. Immun. 58, 2438–2445.
Wagner, P. L., Acheson, D. W., and Waldor, M. K. (2001). Human neutrophils and their products induce Shiga toxin production by enterohemorrhagic Escherichia coli. Infect. Immun. 69, 1934–1937.
Wagner, P. L., Livny, J., Neely, M. N., Acheson, D. W., Friedman, D. I., and Waldor, M. K. (2002). Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol. Microbiol. 44, 957–970.
Waldor, M. K., and Friedman, D. I. (2005). Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 8, 459–465.
Weinstein, D. L., Holmes, R. K., and O'Brien, A. D. (1988). Effects of iron and temperature on Shiga-like toxin I production by Escherichia coli. Infect. Immun. 56, 106–111.
Weiss, D. S., Brotcke, A., Henry, T., Margolis, J. J., Chan, K., and Monack, D. M. (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 6037–6042.
Wendel, A. M., Johnson, D. H., Sharapov, U., Grant, J., Archer, J. R., Monson, T., Koschmann, C., and Davis, J. P. (2009). Multistate outbreak of Escherichia coli O157, H7 infection associated with consumption of packaged spinach, August-September 2006, the Wisconsin investigation. Clin. Infect. Dis. 48, 1079–1086.
Wong, C. S., Jelacic, S., Habeeb, R. L., Watkins, S. L., and Tarr, P. I. (2000). The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157, H7 infections. N. Engl. J. Med. 342, 1930–1936.
Zhang, X. H., He, K. W., Zhang, S. X., Lu, W. C., Zhao, P. D., Luan, X. T., Ye, Q., Wen, L. B., Li, B., Guo, R. L., Wang, X. M., Lv, L. X., Zhou, J. M., Yu, Z. Y., and Mao, A. H. (2011). Subcutaneous and intranasal immunization with Stx2B-Tir-Stx1B-Zot reduces colonization and shedding of Escherichia coli O157, H7 in mice. Vaccine 29, 3923–3929.
Zhang, X., McDaniel, A. D., Wolf, L. E., Keusch, G. T., Waldor, M. K., and Acheson, D. W. (2000). Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J. Infect. Dis. 181, 664–670.
Zhao, H., Przybylska, M., Wu, I. H., Zhang, J., Siegel, C., Komarnitsky, S., Yew, N. S., and Cheng, S. H. (2007). Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 56, 1210–1218.
Keywords: shiga toxin, enterohemorrhagic E. coli (EHEC), hemolytic uremic syndrome (HUS)
Citation: Pacheco AR and Sperandio V (2012) Shiga toxin in enterohemorrhagic E.coli: regulation and novel anti-virulence strategies. Front. Cell. Inf. Microbio. 2:81. doi: 10.3389/fcimb.2012.00081
Received: 13 January 2012; Accepted: 23 May 2012;
Published online: 07 June 2012.
Edited by:
Ken Bradley, University of California, USAReviewed by:
Elizabeth L. Hartland, The University of Melbourne, AustraliaMikhail A. Gavrilin, Ohio State University, USA
Copyright: © 2012 Pacheco and Sperandio. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Vanessa Sperandio, Department of Microbiology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390-9048, USA. e-mail: vanessa.sperandio@utsouthwestern.edu