Bloodstream form pre-adaptation to the tsetse fly in Trypanosoma brucei
 Eva Rico†
Eva Rico†  Balazs Szoor
Balazs Szoor Keith R. Matthews
Keith R. Matthews- Centre for Immunity, Infection and Evolution, School of Biological Sciences, University of Edinburgh, Edinburgh, UK
African trypanosomes are sustained in the bloodstream of their mammalian hosts by their extreme capacity for antigenic variation. However, for life cycle progression, trypanosomes also must generate transmission stages called stumpy forms that are pre-adapted to survive when taken up during the bloodmeal of the disease vector, tsetse flies. These stumpy forms are rather different to the proliferative slender forms that maintain the bloodstream parasitaemia. Firstly, they are non proliferative and morphologically distinct, secondly, they show particular sensitivity to environmental cues that signal entry to the tsetse fly and, thirdly, they are relatively robust such that they survive the changes in temperature, pH and proteolytic environment encountered within the tsetse midgut. These characteristics require regulated changes in gene expression to pre-adapt the parasite and the use of environmental sensing mechanisms, both of which allow the rapid initiation of differentiation to tsetse midgut procyclic forms upon transmission. Interestingly, the generation of stumpy forms is also regulated and periodic in the mammalian blood, this being governed by a density-sensing mechanism whereby a parasite-derived signal drives cell cycle arrest and cellular development both to optimize transmission and to prevent uncontrolled parasite multiplication overwhelming the host. In this review we detail recent developments in our understanding of the molecular mechanisms that underpin the production of stumpy forms in the mammalian bloodstream and their signal perception pathways both in the mammalian bloodstream and upon entry into the tsetse fly. These discoveries are discussed in the context of conserved eukaryotic signaling and differentiation mechanisms. Further, their potential to act as targets for therapeutic strategies that disrupt parasite development either in the mammalian bloodstream or upon their transmission to tsetse flies is also discussed.
Introduction
“This species …..is characterised by showing two distinct forms-the long and slender, and the short and stumpy. These are not sharply divided from one another, but are interconnected by intermediate forms, so as to form an unbroken series, or curve, from the shortest to the longest.”
“It has now been shown that the short forms are definitely capable of surviving in the transmitting host, but that does not in itself exclude the intermediate and long forms from a similar development. This apparently does not occur.”
“The shorter forms are those destined to carry on the cycle in the transmitting host.”
The presence of distinct morphological forms in the bloodstream of mammalian hosts infected with African trypanosomes was recognized very early in the research into these parasites and the link between this transition and disease transmission proposed soon after (Robertson, 1912). However, the connection between the different forms in the blood, termed “slender” and “stumpy” at their morphological extremes, and parasite transmission potential remained controversial for nearly a century thereafter. This was contributed to by the fact that the serial laboratory passage of trypanosomes through rodent hosts prior to the development of cryopreservation methods in the late 1950–1960s selected against the development of stumpy forms. This was because stumpy forms are irreversibly arrested in their cell cycle (Shapiro et al., 1984) and require uptake by tsetse flies in order to recommence cell division as midgut procyclic forms. The resulting laboratory-adapted “monomorphic” forms are similar in appearance to the slender-intermediate forms in natural infections and can differentiate to procyclic forms in culture if exposed to high concentrations of citrate/cis-aconitate (CCA; Brun and Schonenberger, 1981; Roditi et al., 1989). This complicated the interpretation of studies analysing the relative importance of stumpy forms in transmission and it is only over the last 15 years that the particular characteristics of stumpy forms that adapt them for transmission have been recognized [reviewed in MacGregor et al. (2012)]. This has been enabled by methods for the culture and transfection of pleomorphic trypanosomes (i.e., those capable of stumpy formation) (Reuner et al., 1997; Engstler and Boshart, 2004) and the availability of tools to analyse development of trypanosomes in the bloodstream quantitatively and in molecular detail (Reuner et al., 1997; Dean et al., 2009; MacGregor et al., 2011). All of these studies have reemphasized that stumpy forms fulfil a crucial role in the life-cycle of the trypanosomes, assisting both infection chronicity and transmission potential. Both of these components are linked, since the development of arrested stumpy forms in preparation for transmission also limits the ascending parasitaemia, prolonging host survival (Figure 1). This provides an elegant situation whereby the parasite creates an infection dynamic that balances parasite survival through immune evasion, host survival through restricted parasite virulence and an adaptation for transmission and establishment in the tsetse fly midgut (Lythgoe et al., 2007; Gjini et al., 2010; MacGregor et al., 2011, 2012). This infection dynamic is established by the ability of the trypanosomes to monitor their density in the mammalian bloodstream using a system in some ways analogous to the quorum sensing (QS) systems employed by bacteria. However, whilst those systems are often exploited by microbes to share resources or to co-operate to invade or colonize their current environment (Diggle et al., 2007), trypanosomes are also exploiting environmental information to prepare for their colonization of a new environment.
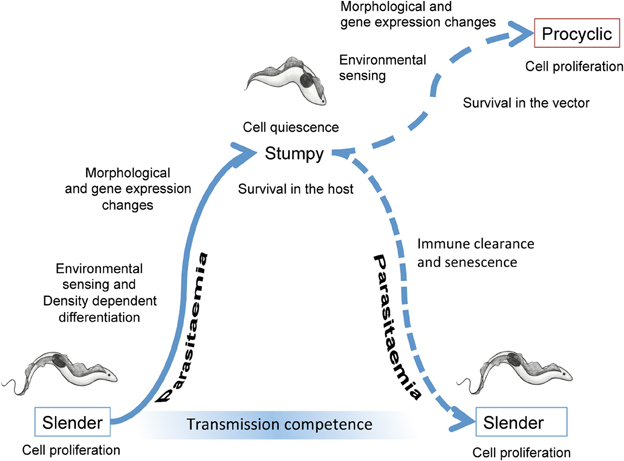
Figure 1. Events accompanying the development of trypanosomes in their mammalian host and upon transmission to tsetse flies. In the bloodstream, slender forms proliferate to increase the parasitaemia. Cell density is perceived via the release and detection of stumpy induction factor, this leading to changes in the morphology and gene expression of the parasites as they undergo development to stumpy forms. The stumpy forms are adapted to short term survival in the blood and transmission to tsetse flies. When taken up in a bloodmeal, stumpy forms perceive their new environment and then undergo morphological, metabolic and gene expression changes as they differentiate to procyclic forms. These colonize the tsetse midgut where, after further development and migration to the tsetse salivary glands, they become infective to new mammalian hosts.
In this review we detail the several mechanisms and processes that trypanosomes exploit to ensure their capacity for transmission through the development of stumpy forms. We firstly discuss the recent analyses of gene expression and protein changes that accompany the transition from proliferative slender forms to arrested stumpy forms. Secondly, we describe how the parasite regulates its production of transmission stages by density sensing mechanisms in the mammalian bloodstream. Thirdly, we detail how the parasite surface is adapted for environmental sensing and survival and for transmission of the parasite and, fourthly, how entry into the tsetse midgut is perceived by the parasite and the signal transduced to initiate the development to procyclic forms. Finally, we discuss how the developmental transitions that the trypanosome undergoes in preparation for, or during, transmission can be targeted in therapeutic approaches that act both to limit virulence and to prevent disease spread.
Gene Expression Regulation During the Slender to Stumpy Transition as a Pre-Adaptation for Differentiation
Overlaid onto the organization of the African trypanosome genome into multi-gene transcription units (Clayton, 2002) is its developmental regulation, with large scale changes in gene expression being observed during the parasite life cycle in order to adapt to the different conditions encountered in the mammal and insect environments. Recently, three different studies analysed the expression profile of mRNAs between bloodstream forms and procyclic forms utilizing pleomorphic slender forms, in vivo generated stumpy forms (differentiated from slender forms) and cells undergoing synchronous differentiation from stumpy to procyclic forms (Jensen et al., 2009; Kabani et al., 2009; Nilsson et al., 2010). Despite the different approaches used by the three laboratories [i.e., microarrays (Jensen et al., 2009; Kabani et al., 2009) vs. spliced leader trapping (Nilsson et al., 2010) or biological samples obtained from immunocompromised mice (Kabani et al., 2009; Nilsson et al., 2010) vs. rats (Jensen et al., 2009)], generally complementary results were achieved showing that, contrary to what had been previously suggested, a quite high percentage of T. brucei genes are differentially expressed between developmental forms in the blood and the tsetse midgut [25–40% of all genes, according to Jensen et al. (2009); Nilsson et al. (2010)]. Within these studies a number of group comparisons generated outcomes relevant to the developmental or environmental regulation of gene expression in the parasites. For example, Jensen et al. observed, surprisingly, that cultured bloodstream forms and in vivo derived slender forms showed similar expression profiles despite their predicted biological differences, whereas the distinctions between logarithmic and stationary procyclic forms were more pronounced (Jensen et al., 2009). Moreover, from all three studies it was clear that the stumpy forms constitute a pre-adaptation required for transmission, with these cells having already undergone many changes in expression when compared to slender forms.
Overall, the most highly regulated transcripts between slender and stumpy forms are those involved in metabolism, cell structure, protein transport, proteolysis and translation, as well as many transcripts with currently unknown function. As expected, slender forms express significantly more ESAGs and GRESAGs (both usually, but not always, found in telomeric and subtelomeric regions together with the VSGs) than stumpy forms (Kabani et al., 2009), with the exception of several ESAG9s that are known to be up-regulated in stumpy forms and have been suggested to be involved in interaction with the external environment (Barnwell et al., 2010). Consistent with slender forms being proliferative and stumpy forms being cell-cycle arrested quiescent forms, histones, DNA replication/repair and translation-related transcripts were found to be up-regulated in slender compared to stumpy forms (Kabani et al., 2009). Other transcripts showing higher expression in the proliferative forms included those associated with the cytoskeleton, especially those related to the flagellum, as well as transcripts involved in metabolism, predominantly those which are localized to the glycosome or are involved in glucose uptake and glycolysis (i.e., glucose transporter THT1 and PGKC). Contrasting with this, procyclins and PAD (Proteins Associated with Differentiation) family members involved in CCA-mediated differentiation, (Dean et al., 2009) are up-regulated in stumpy forms as is MSP-B, associated with the VSG cleavage necessary for the change of surface coat upon differentiation to the insect form (Gruszynski et al., 2006). Further, transcripts involved in cytoskeletal remodeling and membrane protein and lipid biosynthesis are increased in stumpy forms, perhaps contributing to the more robust characteristics of the transmissible forms (Nolan et al., 2000) and their need for rapid membrane rearrangement upon entry into the tsetse fly. It has also been known for some time that lysosome activity is enhanced in stumpy forms (Brickman and Balber, 1994), so the observed up-regulation of a chloride channel transcript in these forms (Kabani et al., 2009) could be explained by its role in the maintenance of lysosomal pH. Other transcripts elevated in stumpy forms are RNA helicases related to RNA processing and two zinc-finger proteins that have been demonstrated to be important in differentiation control (Hendriks et al., 2001; Hendriks and Matthews, 2005; Paterou et al., 2006) and that are apparently involved in the regulation of transmission stage transcripts (Walrad et al., 2012). Finally, metabolic enzymes, such as trypanosome alternative oxidase and fructose-2, 6-biphosphatase, as well as components of the cytochrome oxidase complex and other mithochondrial genes such as ATP synthase subunits and NADH dehydrogenase subunits are up-regulated in stumpy forms (Jensen et al., 2009; Kabani et al., 2009; Nilsson et al., 2010). All of these observations confirm the pre-adaptation of stumpy forms to the more complex metabolic requirements of life in the tsetse fly, which involves a well developed mitochondrion with functional Krebs cycle enzymes, respiratory chain and oxidative phosphorylation enzymes and an increased reliance on the oxidation of amino acids like proline. More recently, an analysis of the transcriptome of slender, intermediate and stumpy forms (Capewell et al., 2013) has shown that many of the changes observed between slender and stumpy forms are already present in the intermediate forms, highlighting that the preparation for transmission occurs early in the development of stumpy forms in the bloodstream, long before the dramatic environmental changes that accompany entry into the tsetse midgut are encountered. This developmental programming is therefore, a response to the parasites own density-sensing signal, and not a predominently environmental response, such as could be generated by the thermal stress or glucose depletion that occurs upon tsetse uptake.
As well as during the transition from slender to stumpy forms, transcriptome changes accompany the synchronous differentiation from stumpy forms to procyclic forms (Kabani et al., 2009; Queiroz et al., 2009). For example the transcripts for ESAG2, ESAG11 and the glucose transporter THT1 are rapidly and progressively down-regulated, whilst EP2 and EP3 procyclin transcripts are up-regulated, preparing the surface coat for the insect stage. Re-entry into DNA synthesis around 8–10 h through differentiation (Matthews and Gull, 1994) is preceded by the induction of histone mRNAs 6 h after exposure to CCA. The activation of translation between 1 and 6 h (Capewell et al., 2013) is also accompanied by an enrichment of mRNAs for different nucleolar proteins and ribosomal components (re-entry into proliferation occurs at 8–19 h into the differentiation programme). The metabolic adaptation to the procyclic form is also confirmed by down-regulation of bloodstream form enriched phosphoglycerate kinase C mRNA and the up regulation of the procyclic form enriched PGKB (Gibson et al., 1988), whilst mitochondrial activation is accompanied by the up-regulation of the components of the cytochrome-oxidase complex soon after the initiation of the differentiation process (Kabani et al., 2009).
From all these studies, it seems clear that changes during the life cycle are accompanied by significant changes in gene expression at the mRNA level, but what is happening to the corresponding proteome? Here the changes appear qualitatively and quantitaviely different, with protein levels also showing a greater amplitude of change during development. A recent study by Gunasekera et al. used a combination of SILAC (Stable Isotope Labelling of Amino acids in Cell culture) and mass spectrometry to determine the differences in protein abundance between slender, stumpy and procyclic forms (Gunasekera et al., 2012). The study was able to detect 30% of the total proteome and, although there was some bias against very small proteins, proteins containing more than one transmembrane domain or very basic proteins, achieved good coverage across the genome. An interesting feature of this study was that around 30% (depending on expression parameters) of genes showed relatively low correlation between RNA and protein levels, even for some transcripts with a robust expression profile. This contrasts with a similar SILAC analysis of bloodstream and procyclic forms carried out by Urbaniak et al. (2012), that found a good correlation with transcriptome profiles (albeit with a greater dynamic range in the proteome studies). Nonetheless, analysis of the three life cycle stages (slender, stumpy, procyclic) by Gunasekera et al. showed major protein changes during the transition from slender to stumpy forms, with more than 500 proteins being increased in stumpy forms. Of these, proteins showing ≥2-fold increase in stumpy forms belonged mainly to the GO (Gene Ontology) terms: plasma membrane, peroxisome, nucleus, microtubule, lysosome, glycosome, and mitochondrion. Supporting the identified changes, this study showed good correlation with previous data for the levels of the bloodstream form enriched alternative terminal oxidase (TAO) (Chaudhuri et al., 1995) and for the stumpy-specific PAD1 transporter family involved in parasite differentiation (Dean et al., 2009). Moreover, the assembly of oxidative phosphorylation complexes III and IV was suggested as one of the final activation steps for energy production in the insect form, being present only in procyclic forms, whilst complexes I, II, and V were already present in stumpy forms. Finally, and supporting previous data obtained with the splice leader trapping approach by the same group (Nilsson et al., 2010), the study showed that one gene can have different protein products differing in their N-terminus, highlighting alternative splicing as a mechanism to create protein diversity.
These proteomic studies are generally in good agreement with a more recent analysis by Digital Serial Analysis of Gene Expression (Digital-SAGE) of the transcripts enriched in the polysomal fraction of slender and stumpy forms (Capewell et al., 2013). This analysis was designed to identify transcripts that might escape the overall translational repression characteristic of stumpy forms, and pointed to mitochondrial, ribonuclear complexes and macromolecular biosynthetic related transcripts being amongst those more elevated during development to stumpy forms. Moreover, the global translational quiescence of stumpy forms was supported by the depletion of mRNAs encoding ribosomal proteins in the stumpy polysomal material. However, only a few of the polysome-enriched stumpy transcripts showed an in vitro differentiation phenotype when analysed by genome wide RNAi analysis (Alsford et al., 2011) indicating a role distinct from the process of differentiation per se, with an alternative potential role in cell viability or development in the tsetse fly midgut.
Since gene expression in African trypanosomes is regulated at multiple levels (mRNA stability, translation and protein stability), it is important to understand the mechanisms involved in the control of these processes. In trypanosomatids, the developmental regulation of mRNA abundance has been primarily linked to the presence of regulatory sequences and structures located in the 3′untranslated region (3′UTR) (Clayton and Shapira, 2007) but that can also be present in the 5′UTR (Pasion et al., 1996; Mahmood et al., 2001). As mentioned earlier, alternative splicing has been also identified as a further potential regulatory step, with a total of 676 genes being observed to have changes in major splice site usage between developmental forms (Nilsson et al., 2010). These regulatory signals within untranslated regions are then acted upon by RNA binding proteins that operate to positively or negatively regulate the mRNA transcript's expression. Indeed, a large number of potential RNA binding proteins are encoded in the trypanosome genome (De Gaudenzi et al., 2005; Kramer et al., 2010; Kramer and Carrington, 2011). With regards to the control of developmental gene expression, procyclin genes are the best characterized, being regulated during early development in the tsetse midgut (Hehl et al., 1994; Furger et al., 1997; Walrad et al., 2009). These genes have similar, but distinct, 3′UTRs (Vassella et al., 2001a) and their differential expression is controlled by trans-acting proteins that include the small CCCH protein ZFP3, which specifically associates with EP1 and GPEET mRNA, rather than EP2 and EP3 mRNA, and promotes gene expression through association with the translational apparatus (Paterou et al., 2006; Walrad et al., 2009). Interestingly, a global analysis of the other mRNAs that are also stabilized by ZFP3 showed enrichment for mRNAs that were predominantly abundant in stumpy forms (Walrad et al., 2012), emphasing the importance of this protein in transmission events.
As highlighted above, there is a global decrease in protein synthesis during the transition from the slender to stumpy form (Brecht and Parsons, 1998; Capewell et al., 2013) and yet a number of genes escape this global repression of expression to be upregulated in stumpy forms. In a search for regulatory regions controlling transmission-stage gene expression Kabani et al. (2009) identified, using an oligonucleotide frequency scoring algorithm, two related hexanucleotide sequences (TCTTAC and TTCTTA) that were over-represented in the 3′UTR of transcripts predicted to be enriched in stumpy forms. Although no functional analysis of these motifs has been performed, the first of these showed a positional bias 150–200 nt downstream from the gene stop codon. To more functionally define regulatory regions, two recent studies have focused on the detailed identification of sequences and structures involved in the control of two validated stumpy-specific transcripts, encoding PAD1 and an ESAG9 copy. Utilizing CAT reporter assays in pleomorphic trypanosomes, it was demonstrated that both PAD1 and ESAG9 contain regulatory regions in their 3′UTRs that repress expression in the slender life stage and alleviate repression in the stumpy form (MacGregor and Matthews, 2012; Monk et al., 2013). For PAD1, the distal end of the 3′UTR was found to contain an mRNA silencing element that represses both mRNA and protein expression. However, specific deletion of this region did not alleviate protein expression silencing, suggesting the additional involvement of proximal regions within the 3′UTR. This was confirmed by the independent integration of the PAD1 proximal or distal 3′UTR regions into the 3′UTR of a constitutively expressed gene, which resulted in no repression at the mRNA level for the reporter gene. Nevertheless, repression was observed at the protein level, suggesting that protein control may be more stringent, consistent with the fact that PAD mRNA is elevated early in intermediate forms (MacGregor et al., 2011, 2012) whilst protein expression is only found in fully differentiated stumpy forms (Dean et al., 2009). In the ESAG9 3′UTR a regulatory region of just 34 nt was defined as a bifunctional element responsible for both positive and negative regulation depending on the developmental stage of the parasite (Monk et al., 2013). This element regulated mRNA levels and, in the context of the complete ESAG9 3′UTR, also protein levels. The simplest model for this regulation implicated a negative regulator binding this element in slender forms and repressing gene expression, whilst in stumpy forms this would be replaced by a positive regulator alleviating the repression of gene expression. Unlike PAD1, insertion of this short regulatory element into the 3′UTR of a constitutively expressed gene had a strong effect on mRNA only, suggesting that other sequences within the ESAG9 3′UTR might also contribute to the regulation of translation, for example by stabilizing the association of positive translational regulators. Hence, whilst no similarity in the sequence elements was found between the ESAG9 and PAD1 3′UTRs, in both cases stringent regulation at both the mRNA and protein level was observed to control their exclusive expression in the stumpy form. This stringency of regulation is likely to be important for ensuring the appropriate timing of the expression of genes involved in cell-cycle arrest or transmission during programmed development from the slender to stumpy form.
Signaling Mechanisms that Govern Stumpy Formation in the Mammalian Bloodstream
The progression of trypanosome infections in the mammalian host follows a wave-like pattern, at least early on, with the actively dividing slender forms dominating the ascending phase leading up to the peak, followed by G1/G0 arrest and differentiation to the stumpy forms. While the interplay of host immune factors and the antigenic variation of the parasite play a major role in the waves of parasitaemia, the slender to stumpy transition is an important additional contributor (Lythgoe et al., 2007; Gjini et al., 2010; MacGregor et al., 2011), primarily dictated by parasite density (Reuner et al., 1997). Indeed, pleomorphs reach a threshold parasitaemia before they undergo differentiation (Seed and Sechelski, 1989), and in vitro this is linked to the parasite number regardless of the culture seeding density (Reuner et al., 1997). Since monomorphic strains continue to grow to higher cell densities under similar growth conditions as the arrested pleomorphs a limitation in medium components seems unlikely and, indeed, nutrient replacement fails to restore the growth of dense pleomorph cultures (Hesse et al., 1995). Instead, early tentative evidence for a secreted factor controlling parasite growth was provided by the observation that plasma from infected animals at peak parasitaemia could inhibit trypanosome proliferation (Seed and Sechelski, 1989), whereas in vitro studies provided further support for the accumulation of a parasite-derived product able to induce stumpy formation (Reuner et al., 1997; Vassella et al., 1997). This density dependent transition from long slender forms to short stumpy forms is reminiscent of the QS mechanisms observed in several prokaryotes and essentially comprises three main components- the signaling molecule(s), the receptor(s) that perceive the cue(s) and the downstream modulators that relay the signal, triggering a series of changes that ultimately bring about differentiation.
In the case of African trypanosomes, the cells sense the increase in their numbers and consequently cease cell division. This ensures the parasite numbers do not overwhelm the host, as can occur when infections are initiated with differentiation-incompetent monomorphic lines (Turner et al., 1995). The development of a plate-based technique for growing pleomorphic strains in vitro (Vassella and Boshart, 1996), led to the discovery that a soluble, low molecular weight factor, termed Stumpy Induction Factor (SIF) was able to provide the signal for differentiation (Reuner et al., 1997; Vassella et al., 1997), ruling out a major contribution from the host and highlighting the signal as a parasite-intrinsic entity. Although monomorphs are incapable of differentiation, they do produce SIF (Vassella et al., 1997), and are therefore “signal-blind,” a necessary requirement for their selection in a background population of parasites producing SIF (MacGregor et al., 2012).
In terms of intracellular signaling, the addition of parasite-conditioned media (containing SIF) causes an increase in cAMP levels in pleomorphs (Vassella et al., 1997). This is consistent with the 2–3-fold increase in intracellular cAMP observed when early log phase slender parasites reach peak parasitaemia in rodents, which is followed by a decrease in the cAMP levels upon appearance of intermediate and stumpy forms (Mancini and Patton, 1981). This suggested cAMP might act as a second messenger conveying the SIF signal. Moreover, further support for a role of cAMP in development was provided by the observation that a cell permeable analog of cAMP (8-pCPT-2′-O-Me-cAMP) promoted G1/G0 arrest in pleomorphs on plates (Vassella et al., 1997), whereas procyclic cells were quite insensitive to high concentrations (up to 1 mM or more) of various cell-permeable cAMP derivatives (Oberholzer et al., 2007). However, unlike Plasmodium falciparum, where differentiation to transmissible gametocytes is mediated by cGMP (Hopp et al., 2012), the slender to stumpy transition in trypanosomes now appears not to be a conventional cyclic nucleotide response but is restricted to cell permeable cAMP analogs (Vassella et al., 1997). Furthermore, the hydrolysis resistant analog of cAMP (Sp-8-pCPT-2′-O-Me-cAMPS) is not anti-proliferative for monomorphic slender forms whereas the degradation products of cAMP, namely AMP and adenosine, have a more potent effect, indicating that these molecules are likely to be the key players in the signaling cascade rather than cyclic nucleotides (Laxman et al., 2006) (Figure 2). Supporting this, knockdown of the T.brucei PDEB family, which causes an increase in the levels of intracellular cAMP, failed to promote differentiation (Oberholzer et al., 2007) despite the ability of etazolate, a phosphodiesterase (PDE) inhibitor to induce this transition (Vassella et al., 1997), a discrepancy possibly due to the proposed lack of specificity of etazolate for trypanosomal PDEs (Laxman et al., 2006). Further evidence for cAMP not being the direct modulator of differentiation was recently provided by the observation that the levels of TbTOR4 protein (whose loss of function triggers the appearance of stumpy-like forms, see below) decreased when parasites were exposed to 8-pCPT-2′-O-Me-cAMP or 8-pCPT-2′-O-Me-AMP but not Sp-8-pCPT-2′-O-Me-cAMPS (Barquilla et al., 2012). Combined this points to a non-canonical intracellular signaling pathway operating to regulate stumpy formation, that does not involve cAMP as a second messenger.
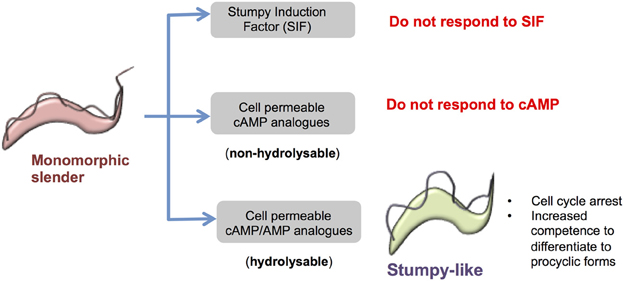
Figure 2. Response of monomorphic slender cells to cell permeable analogs of cAMP/AMP. Monomorphic slender cells have lost the capacity to generate stumpy forms in vivo, showing a reduced ability to respond to stumpy induction factor. However, when exposed to cell permeable analogs of cAMP or AMP these cells stop proliferation and express some, but not all, characteristics of stumpy forms, namely proliferation arrest, increased capacity for differentiation to procyclic forms and the increased expression of some mRNAs enriched in stumpy forms. This is not a cAMP-mediated response because hydrolysable but not non-hydrolysable cAMP analogs induce the transition and cell permeable AMP is considerably more potent than cell permeable cAMP. Hence the cell permeable cAMP is likely metabolized to cell permeable AMP to elicit the production of stumpy-like forms.
To date no component of a signaling pathway that has a direct role in promoting stumpy formation has been identified. However, genes whose loss of function triggers quiescence (and hence are differentiation inhibitors) have provided some molecular insight into the signaling cascade. For example, the deletion of a Zinc finger kinase (ZFK) in pleomorphs reduced growth and also increased the rate of the slender to stumpy transition, albeit only in vitro (Vassella et al., 2001b). Similarly, knock out of a mitogen activated protein kinase homolog, TbMAPK5, gave a similar phenotype, with the exception that stumpy formation in vivo was also accelerated (Domenicali Pfister et al., 2006). Whilst neither the ZFK nor MAPK knockouts could induce differentiation in monomorphs, deletion of the TOR kinase member, TbTOR4, stimulated monomorphs to generate stumpy-like forms (Barquilla et al., 2012), positioning this molecule downstream of, or on a distinct pathway to, the other inhibitors.
The evidence for the existence of the SIF signaling factor(s) itself remains circumstantial. It is believed to be a small (<500 Da), soluble and filterable factor, distinguishing it from other parasite communication systems, such as the secreted vesicular exosomes recently described for Plasmodium parasites (Mantel et al., 2013; Regev-Rudzki et al., 2013). Although the identity of SIF has eluded us to date, a recent intriguing finding on the ability of derivatives of the differentiation inducing factor (DIF) from Dictyostelium discoideum to reduce proliferation of T. cruzi parasites in vitro as well as in vivo (Nakajima-Shimada et al., 2013) suggests that there might be some conservation of eukaryotic QS processes. Until recently, one of the major obstacles to determining the molecular nature of SIF was the lack of a robust marker for stumpy formation that could enable high throughput screens for stumpy inducers. However, the discovery of PAD1 as a stumpy-specific marker (Dean et al., 2009) and the development of reporter lines using this marker (MacGregor and Matthews, 2012), now enables activity-guided assays for screening components of conditioned medium, assisting the purification of SIF.
Adaptations for Survival in the Mammalian Host by Stumpy form Trypanosomes and Signaling Events Upon Transmission
Surface Molecules
Once stumpy forms are generated from slender forms they are irreversibly arrested and have a limited lifespan in the bloodstream (Turner et al., 1995). Nonetheless, to maximize their potential for transmission, stumpy form trypanosomes exhibit a number of adaptations that both prolong their survival in the mammalian bloodstream (increasing their probability of uptake by tsetse flies) and promote their viability upon entering the tsetse midgut.
Perhaps the most obvious survival mechanism employed by trypanosomes in the mammalian blood is their expression of a variant surface glycoprotein, providing protection against the alternative pathway of complement activation (Ferrante and Allison, 1983) and, through antigen variation, antibody-mediated killing. This protection extends only to trypanosomes that have changed their surface antigen coat and is therefore, probably not available to non-proliferating stumpy forms. Nonetheless stumpy forms do show a particular adaptation allowing their preferential survival when exposed to increasing titres of antigen-specific antibody during a wave of infection. This process removes the bound antibodies from the surface of the parasite by using hydrodynamic flow forces (Engstler et al., 2007). Specifically, the flagellum of trypanosomes, attached along the cell body, propels the parasite anteriorly. This causes the movement of bound immune complexes by hydrodynamic drag forces backwards toward the posterior flagellar pocket. Using cell-surface VSG labeled with a membrane-impermeable blue-fluorescent dye, the majority of immunoglobulin bound to VSG was found to be transported to the lysosome, where the immunoglobulin was degraded, whereas the VSG was recycled to the cell surface. The directional movement of the immunoglobulin-VSG complex toward the posterior end of the cell and its subsequent rapid endocytosis provides a very efficient mechanism to clear immune complexes from the cell surface and is twice as rapid for stumpy forms as for slender forms (Engstler et al., 2007). Hence, this phenomenon could contribute to the greater resistance of stumpy forms to antibody-mediated killing than slender forms (McLintock et al., 1993), allowing the stumpy forms to predominate at peak parasitaemia and extending the period at which the infection is transmissible to tsetse flies.
Other molecules at the parasite surface and within the flagellar pocket show developmental regulation during development between slender and stumpy forms. Whilst ESAG9 is upregulated early during stumpy formation, as detailed earlier, the haptoglobin-haemoglobin receptor in the flagellar pocket is downregulated (Vanhollebeke et al., 2010). This molecule is involved in haptoglobin-haemoglobin uptake, which may no longer be needed in the quiescent stumpy form. However, it might also represent a protection mechanism for transmission stages against TLF1, since the haptoglobin haemoglobin receptor is the route for the uptake of the trypanosome lytic factor in human serum (Vanhollebeke et al., 2008). Since the resistance to TLF is mediated in Trypanosoma brucei rhodesiense by an expression site component, SRA (Xong et al., 1998), and expression site transcription is strongly down regulated in stumpy forms (Amiguet-Vercher et al., 2004), the downregulation of the haptoglobin-haemoglobin receptor in stumpy forms could prevent TLF1-mediated killing when SRA production is reduced. TLF2-mediated killing would remain possible, however, unless parasites were sequestered in the tissues or lymphatics, as has been suggested for metacyclic forms (Pays et al., 2006).
At the level of mRNA transcripts, the abundant invariant surface glycoproteins (ISGs) 65 and 75 [present at ~70,000 and 50,000 copies, respectively, (Ziegelbauer and Overath, 1992)] are also down regulated in stumpy forms with respect to slender forms (Nilsson et al., 2010). ISG proteins have type I transmembrane topology and intercalate with VSG on the surface (Leung et al., 2011). Although no identified function has been assigned for these surface proteins, ISG75 was recently identified in a genome-wide RNAi-screen to mediate the uptake of the trypanocidal drug suramin (Alsford et al., 2012). ISG65 and ISG75 have been found to be present at the surface and endosomal membranes and are turned over more rapidly than VSGs in a process involving ubiquitination of lysine residues in their cytoplasmic domains. Specifically, ISG65 has been shown to be ubiquitinated on residues K3 and K4 of the cytoplasmic tail, with mutation of these residues significantly increasing the protein half life (Chung et al., 2008). ISG75 is also ubiquitynated in T. brucei and the absence of cytoplasmic lysine residues renders the protein incapable of modification by ubiquitin and consequently stabilized (Leung et al., 2011).
As well as proteins at the cell surface membrane and flagellar pocket, it has been hypothesized that the parasite flagellum functions as a signaling organelle for integrating host-derived and parasite-derived signals and these could contribute to development both in the bloodstream and upon transmission to tsetse flies. For example, proteins involved in cyclic nucleotide signaling, Ca2+ signaling and phospho-signaling pathways are anchored to the T. brucei flagellum skeleton (Wu et al., 1994; Oberholzer et al., 2007; Smith et al., 2009). T. brucei calflagins Tb17, Tb24, Tb44 are all calcium sensors that associate with the flagellar membrane in a calcium-dependent and palmitoylation-dependent manner, and although their precise function its not clear, a role for these proteins in host survival and immune evasion has been described (Emmer et al., 2010). Monomorphic calflagin-deficient parasites exhibit a normal growth rate in vivo during the first week post-infection, but subsequently parasite numbers decrease to undetectable levels, followed by the emergence of a parasite population expressing alternative VSGs. This phenotype does not involve any failure in surface-bound antibody clearance or parasite motility, indicating that calflagins could promote parasite resistance to early host immunity. In a recent study, by performing flagellum purification, combined with affinity purification of surface exposed proteins and multidimensional protein identification technology (MudPIT), many novel and already identified proteins were found on the flagellum surface (Oberholzer et al., 2011). This included receptor-like proteins with a large extracellular domain, that could be suitable for binding host ligands, connected to an intracellular signaling module, for example a kinase domain, supporting the idea that the flagellum might be able to sense the environment and transmit signals inside the cell to assist their adaptation, differentiation or survival in the bloodstream.
Adenylate Cyclase Signaling in the Mammalian Bloodstream
The T. brucei genome encodes more than 80 adenylate cyclases (ACs), many of which are at the cell surface (Alexandre et al., 1996; Bridges et al., 2008). The prototype gene of this family, termed ESAG4, is part of VSG expression sites and so bloodstream stage-specific, unlike the family of related genes, GRESAG4 (genes related to ESAG4) which are positioned outwith expression sites and constitutively transcribed throughout the life cycle of the parasite.
The ACs of T.brucei share a similar structure, with a large extracellular domain (900 amino acids) and a conserved intracellular catalytic domain (around 350 amino acids) separated by a single transmembrane domain; dimerization of the catalytic domain is required for activity. Although the basal cellular cyclic adenosine monophosphate (cAMP) concentration is extremely low, stress, such as acid treatment or osmotic lysis, activates ACs by up to two orders of magnitude (Rolin et al., 1996, 1998; Nolan et al., 2000). Recently, ESAG4 activity has been shown to be crucial to regulate processes involved in the early establishment of the parasitaemia in the host (Salmon et al., 2012b). By overexpressing a catalytically dead mutant (DNc) of ESAG4, total AC activity was reduced by 50% and transient growth retardation and cytokinesis abnormalities in culture were seen, subsequently compensated by an upregulation of GREG4.1 (Salmon et al., 2012a). In mice, however, the parasitaemia of DNc cells was considerably reduced, and the host survival time was extended. After analysing immune modulators affecting trypanosome infection such as tumor necrosis factor alpha (TNF-α), Interferon gamma (INF-γ), and Nitric oxide (NO), it was shown that TNF-α is crucially involved in controlling the early trypanosome parasitaemia and that the trypanosomal ACs inhibited TNF-α synthesis in liver myeloid cells by activating the myeloid cell PKA (Salmon et al., 2012b). Apparently, therefore, the trypanosome adenylyl cyclases inhibit the innate response of the host by shutting down TNF-α synthesis, helping the infection to establish.
As mentioned earlier, a peak in the activity of AC has also been linked to the transition from the slender form to the stumpy forms (Mancini and Patton, 1981; Vassella et al., 1997); slender cells increase their intracellular cAMP concentration until they reach peak parasitaemia, and then it decreases as the transition to intermediate and short stumpy forms commences. Interestingly, ESAG4 protein levels are reduced in the stumpy form by 93% compared to slender forms, although ablation by RNAi of the ESAG4+ESAG4L subfamily does not modify the slender to stumpy differentiation (Salmon et al., 2012a).
AC activity is also activated by incubation of slender forms at pH 5.5 (perhaps mimicking their activation upon macrophage uptake early in infection), but no similar effect is seen in stumpy forms (Nolan et al., 2000). This correlates with the greater tolerance to high extracellular H+ concentration of stumpy forms compared to slender forms, reflecting that stumpy cells are able to maintain a constant cytoplasmic pH. Nonetheless, during the differentiation from stumpy to procyclic forms a significant but transient increase in cyclase activity is observed at around 6 h into differentiation and after the initiation of cell proliferation (Rolin et al., 1998). However, since the first peak in AC activity occurs after the transformation into procyclics begins, it is unlikely that the rise in cAMP generated is involved in the early differentiation events in the tsetse.
Surface Molecules and Signaling Events as Stumpy forms Enter the tsetse Midgut
There are two known ways that VSG is released from the trypanosome cell surface during differentiation from stumpy to procyclic forms, glycophosphatidyl inositol (GPI) hydrolysis and endoproteolysis. Slender and stumpy trypanosomes contain an endogenous GPI phospholipase C (GPI-PLC) that is capable of hydrolyzing the GPI anchor found on VSG, but not procyclin. GPI-PLC is reported to be located exclusively in a linear array on the outside of the flagellar membrane, close to the flagellar attachment zone (Hanrahan et al., 2009). Deletion of both the GPI-PLC gene and a Zn2+-metalloprotease (TbMSP-B) gene prevents the differentiation of monomorphic bloodstream forms into procyclic forms, although the cells remain viable for several days (Grandgenett et al., 2007). In stumpy cells exposed to the differentiation triggers CCA, MSP-B mRNA and protein levels are upregulated, this occurring concurrently with the onset of proteolytic release of VSG (Gruszynski et al., 2006). In contrast, GPI-PLC mRNA levels quickly decrease after differentiation is induced. Protein levels decrease subsequently, although a dramatic loss of enzyme only occurs after the onset of cell division (most probably by cell dilution). Hence the combined action of GPI-PLC and MSP-B operate to enable the rapid and synchronous loss of VSG from differentiating stumpy form trypanosomes that occurs ~6 h after the initiation of differentiation (Gruszynski et al., 2006).
Two genes encoding surface carboxylate transporters, PAD1 and PAD2, have also been shown to be expressed at the surface of stumpy form T. brucei and important for the initiation of differentiation. These proteins have differential expression through the life cycle of the parasites, being absent in the replicative slender form and upregulated in the stumpy (PAD1 and PAD2) and procyclic form (PAD2) (Dean et al., 2009). By RNAi targeting both genes it was shown that they are not required for either stumpy formation or survival, but that they are required to perceive CCA at physiological concentrations to enable differentiation from stumpy into procyclic cells. PAD2 was also demonstrated to be released to the cell surface at 20°C, thereby demonstrating a redistribution of the transporter in response to cold shock. The expression profile and differential trafficking of the PAD proteins contributes to appropriate developmental responses in two ways (MacGregor and Matthews, 2010). Firstly, by being absent on slender forms but present on stumpy forms, sensitivity to the differentiation signals is restricted to the transmission-adapted stage. Second, the elevation of PAD2 at low temperature ensures that only those parasites that enter the arthropod midgut can detect and respond to the signal. This signal is then relayed via a phosphatase-signaling cascade to initiate the differentiation events that allow colonization of the tsetse midgut.
Signal Transduction of the Differentiation Signal Upon Entry into the tsetse Fly
Despite the detailed in silico analysis of the TriTryp kinome (Parsons et al., 2005) and phosphatome (Brenchley et al., 2007), a coherent assembly of environmental signaling pathways in trypanosomes is almost entirely missing. One exception is the protein phosphatase cascade that regulates differentiation from bloodstream stumpy forms to vector-adapted procyclic forms.
In stumpy forms, a tyrosine phosphatase (TbPTP1) (Szoor et al., 2006) prevents cells from differentiation until its activity is reduced in the presence of the developmental triggers CCA (Brun and Schonenberger, 1981), whose uptake is controlled by the carboxylate transporter PAD proteins (Dean et al., 2009), as detailed earlier. Recently, a D × D × T phosphatase, TbPIP39, was identified as a downstream regulator of this pathway and shown to be activated upon tyrosine-phosphorylation and, hence, negatively regulated by TbPTP1 (Figure 3). Moreover, the presence of TbPIP39 increased the activity of TbPTP1, reinforcing its own repression and thereby generating a potentially bistable regulatory switch (Szoor et al., 2010). Importantly, this self-repressive loop is interrupted and regulated by CCA, such that differentiation can be initiated. Specifically, in the presence of CCA, the activation of TbPTP1 by TbPIP39 is reduced, such that TbPIP39 becomes rapidly phosphorylated on tyrosine 278, the enzyme activated and differentiation is stimulated (Szoor et al., 2010). RNAi studies using stumpy form cells confirmed that reducing TbPIP39 levels inhibited differentiation, although delayed differentiation remained possible probably through the inability to completely eliminate TbPIP39 from the cells by RNAi. Localization studies showed that TbPIP39 is found in glycosomes, peroxisome-like organelles in trypanosomes, which are the site of glycolysis and several other metabolic activities (Michels et al., 2006). Supporting this, removal of the C-terminal glycosomal localization signal on TbPIP39 prevented its ability to promote differentiation, highlighting that the glycosome is the important site of action for the enzyme. Although the target(s) of TbPIP39 in the glycosome are unknown, as are the kinases that oppose its action, developmental effects mediated through metabolic perturbation are already well known, particularly with respect to their impact on surface protein expression (Morris et al., 2002; Vassella et al., 2004).
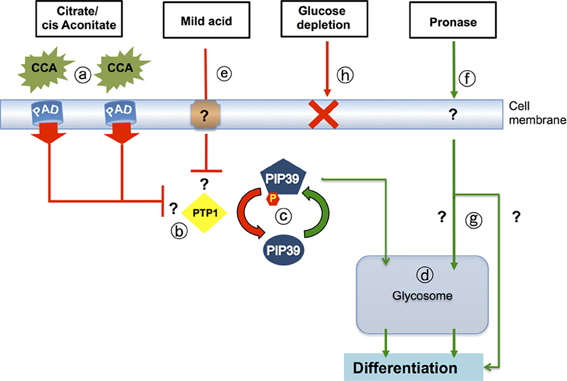
Figure 3. Signaling pathways that drive differentiation when trypanosomes enter the tsetse midgut. Citrate-cis aconitate (CCA) is transported by cell membrane PAD proteins (a), this being favored under cold shock conditions. CCA inhibits the activity of the differentiation inhibitor, TbPTP1 (b) a tyrosine phosphatase whose activity is normally enhanced by its substrate, TbPIP39. When TbPTP1 is inhibited, TbPIP39 becomes phosphorylated and more active (c). TbPIP39 can relocate to the glycosome (though this is not dependent upon its phosphorylation) whereupon it acts on unidentified substrates to promote differentiation (d). The same signaling pathway is stimulated by exposure of stumpy forms to mild acid conditions (pH 5.5 for 2 h) (e), which also generates phosphorylated TbPIP39. Exposure of stumpy forms to proteases at the cell surface (f) also stimulates differentiation, but this does not involve TbPTP1 or TbPIP39 and operates through an uncharacterized pathway (g) that may or may not involve the glycosome. It is also not necessary for differentiation in the midgut of tsetse. A reduction of environmental glucose (h) has also been suggested to initiate differentiation, but reducing medium glucose 20-fold, or using the transport inhibitor phloretin, does not cause the differentiation of stumpy forms to procyclic forms.
Although PAD proteins transport CCA and TbPTP1/TbPIP39 regulatory cross talk is controlled by the levels of these differentiation signals, trypanosomes have also been reported to be able to differentiate to procyclic forms in response to a number of other triggers. These include (i) mild acid treatment (Rolin et al., 1998), (ii) exposure of the parasite surface to limited protease digestion (Yabu and Takayanagi, 1988; Hunt et al., 1994; Sbicego et al., 1999) and (iii) a reduction of glucose, mediated either through the use of glucose-depleted media (Milne et al., 1998), or exposure to phloretin, an inhibitor of glucose uptake (Haanstra et al., 2011). With the identification of phosphorylated TbPIP39 as a signature of differentiation stimulated via TbPTP1, it has been possible to investigate, firstly, the efficiency of each of these reported stimuli on transmissible stumpy forms and, secondly, whether each signal is transduced through the same signaling pathway (Szoor et al., 2013).
These studies revealed that CCA, mild acid and protease treatment of stumpy forms could all stimulate differentiation effectively, but that glucose depletion could not. This was a surprising finding, since glucose levels are believed to rapidly decrease in the tsetse blood meal. With respect to the signaling pathways used, monitoring the phosphorylation of TbPIP39 using a phospho-specific antibody revealed that CCA and mild acid both signal via the TbPTP1/TbPIP39 pathway, whereas protease treatment of the cells does not. Supporting the existence of distinct signaling pathways, ablation of TbPIP39 by RNAi reduced differentiation in response to CCA and mild acid but not protease treatment.
Since protease attack of the parasite surface is believed to occur in the tsetse midgut through the action of trypsin-like activities, the relevance of trypsin like activities in parasite differentiation in the tsetse fly has been recently investigated (Szoor et al., 2013). By feeding stumpy form parasites in the presence of Soybean trypsin inhibitor (STI), tsetse midgut trypsin activity could be almost completely ablated. Despite this, no difference in trypanosome differentiation was observed between control and treated tsetse flies, with equivalent levels of procyclic forms being generated in both populations. This highlighted that trypsin-like activities in the tsetse midgut are not solely responsible for the initiation of differentiation, although a contributory role for these activities remains possible. Supporting this, in vitro exposure of bloodstream trypanosomes to both CCA and protease treatment generates a more robust differentiation response than exposure to either trigger alone (Sbicego et al., 1999), suggesting that the two signals could act co-operatively to ensure effective parasite differentiation upon entry to the tsetse fly.
These findings support a central role for the CCA/PAD/TbPTP1/TbPIP39 signaling pathway in regulating differentiation both in vitro and in vivo, with protease attack of the parasite surface being dispensable for differentiation in the tsetse fly. Apparently, therefore, independent and redundant signaling pathways can operate to control parasite development.
Phenotypic Screening for Stumpy Formation Via Small Compound Screens
As well as providing fundamental biological insight into the developmental pathways operating in these evolutionarily ancient eukaryotes, analyses of the differentiation of trypanosomatids, particularly in the mammalian bloodstream, provides possible drug targets and approaches with the potential to limit the virulence or transmission of these important pathogens. The need for new, safe, efficacious drugs to target African trypanosomiasis is clear (Barrett et al., 2007; Simarro et al., 2012) and considerable efforts are currently underway to identify new therapeutics. The traditional route to identify such therapeutic agents has been to search for compounds that will target genes or pathways necessary for cell viability. Indeed, whole-cell based high-throughput screening for trypanostatic and trypanocidal agents is well established (Hesse et al., 1995; Mackey et al., 2006; Sykes and Avery, 2009; Jones et al., 2010; De Rycker et al., 2012; Sykes et al., 2012; Vodnala et al., 2013) and multiple classes of compounds with potential for development into new therapeutics are being pursued (Mackey et al., 2006; Jones et al., 2010; Sykes et al., 2012; Vodnala et al., 2013). However, phenotypic screens for genes or pathways involved in the establishment, maintenance or transmission of infection could provide an alternative or complementary route to the identification of novel therapeutics with anti-virulence or transmission-blocking potential. Indeed efforts have been made for other pathogens to develop high throughput phenotypic screens whereby small molecules are assessed for their ability to enhance or hinder a biological function, rather than cause direct cytotoxicity. This approach has the additional benefit that compounds perturbing defined cellular processes can provide invaluable tools for pathway dissection.
There are several examples of phenotypic screens that provide a template for dissecting virulence, transmission or other important pathways in pathogens. For example, screens targeting virulence factors in microbes have been used to identify compounds that prevent Type III (Kauppi et al., 2003) or Type IV (Paschos et al., 2011) secretion pathways in pathogenic gram-negative bacteria. Similarly, a compound that reduces adhesion of pathogenic fungi to mammalian epithelial cells was identified through screening for small molecules that block GPI-mannoprotein (e.g., fungal adhesins, where the GPI is removed and the protein linked to β-1,6-glucan) localization to the cell wall (Tsukahara et al., 2003). Such concepts have been also applied to protozoan parasites such as Toxoplasma gondii and Plasmodium falciparum. During infection, the malaria parasite P. falciparum uses multiple pathways for establishment, propagation, and maintenance of the parasitaemia and chemical genetic approaches have aided in the understanding of these processes. For example, a small screen of chemical protease inhibitors targeting the Falcipain-1 protease from cell lysate was then extended in whole cell assays to identify the role of this protease in red blood cell (RBC) invasion by merozoite forms (Greenbaum et al., 2002). This provided both increased understanding of the invasion process as well as a potential new antimalarial able to prevent merozoites from entering RBCs, thereby leaving them exposed to the host immune system. Additionally, protease inhibitors that prevent RBC rupture by P. falciparum have been identified through a high-throughput, whole-cell chemical screen, leading to the identification of two proteases, DPAP3 and PfSUB1, which function in release of the parasite from the host cell (Arastu-Kapur et al., 2008). As with Plasmodium, the parasite T.gondii must also invade host cells in order to establish and maintain an infection, requiring pathways for gliding motility, attachment to the host cell, secretion from the micronemes and invasion. Consequently, T.gondii invasion has been the target of multiple whole-cell high throughput small molecule screens (Carey et al., 2004; Hall et al., 2011; Kamau et al., 2012). These have successfully identified compounds targeting different parts of this pathway leading through follow up analysis to improved understanding of myosin motor activity during motility in one case (Heaslip et al., 2010) and to identification of the DJ-1 protein, required for microneme secretion and motility in another (Hall et al., 2011).
Clearly, the developmental biology of trypanosomes in the mammalian bloodstream offers similar possibilities for phenotypic screens. Without the slender form, the trypanosome parasitaemia could not be established or maintained whereas the stumpy form is necessary for cyclical transmission. As such, targeting either stage could eliminate the parasite or block its spread. For example, identification of compounds able to prevent slender cell proliferation or switching of the VSG coat would result in clearance of the parasites by the host immune system. Similarly, identification of compounds that drive proliferative slender forms to non-proliferative stumpy forms, or onward to procyclic forms, if irreversible and complete, would cause clearance of the parasitaemia altogether. Indeed, even if only partial induction of the population was achieved, this could lower the parasitaemia below the threshold needed for tsetse transmission, both reducing virulence and transmission potential possibly providing an adjunctive therapy for conventional trypanocidal drugs. In contrast, compounds able to prevent the slender to stumpy transition, perhaps through blocking of density sensing or operation of the stumpy induction pathway, would result in uncontrolled growth within the host. Whilst this would reduce transmission potential, it would significantly promote virulence and so would not be of therapeutic value.
In order to carry out a phenotypic chemical screen an assay must be designed that allows monitoring of the presence or absence of the phenotype in a simple, cheap and high-throughput manner, often through the use of a marker or reporter protein as a proxy for that phenotype. For developmental studies, molecular markers are known for the different life stages and reporter genes and assays are commonly utilized in the study of trypanosome molecular biology. For example, genetically modified reporter lines that combine a reporter gene with the stumpy-enriched PAD1 or ESAG9 3′UTR have been demonstrated to report on stumpy formation in vitro (MacGregor and Matthews, 2012; Monk et al., 2013) and such constructs can be modified in simple assays to screen for chemical inducers of this differentiation event (MacGregor and Matthews, unpublished data). Notwithstanding the caution that the monomorphic cells usually employed in high throughput screens may not reflect the phenotypic responses and drug sensitivities of the more field relevant pleomorphic lines, it is clear that screening for developmental defects in trypanosomes has potential for identifying novel anti-trypanosomal compounds and useful tool compounds for biological research.
Summary and Perspectives
The last few years have provided the first molecular insight into how trypanosomes monitor their environment and prepare for transmission as stumpy forms. Moreover, their environmental signaling pathways are now being uncovered in a coherent analysis that differs from the individual identification of molecules possible through earlier candidate gene analysis. This has been enabled through integration of information provided by high throughput transcriptome and proteomic analyses and through the capacity to extend out from identified nodes where key molecules have been identified using protein interaction studies. However, recently, the development of genome-wide RNAi selection approaches (Alsford et al., 2011, 2012) is providing a step change in the information that is available on molecular processes in trypanosomes. Although initially focused on understanding drug action, these screens have the potential to uncover the molecular components of developmental pathways or gene regulatory networks, particularly where the developmental expression of markers can be exploited to drive selectional screens. To date the application of these screens has been limited to monomorphic lines since their transfection efficiency is higher than the more biologically relevant pleomorphic lines that are crucial to understanding the developmental biology of trypanosomes in their mammalian host. However, as transfection efficiencies improve (MacGregor et al., 2013), RNAi screens will become tractable in pleomorphs and detailed analysis of slender and stumpy form biology will become possible. The application of increased HTS is also a powerful development for both anti-virulence therapies and for dissecting the biology of trypanosome development in its mammalian host. For example, the consequences of particular therapeutic or genetic perturbations can now be analyzed on a genome-wide scale through a combination of transcriptome, proteomic, and phosphoproteomic/post-translational modification analyses. The greatly reduced cost of genome sequencing also now means that different parasite strains can be economically analyzed, such that monomorphic laboratory-adapted lines can be compared with pleomorphic laboratory and field strains, assisting the identification of selected traits that render cells resistant to the stumpy formation signal.
What key aspects of the development of trypanosomes in the mammalian blood remain to be answered? One aspect is the nature of the signal that the trypanosome uses to regulate stumpy formation. Although a SIF or factors has been proposed, it remains the “Loch Ness monster” of trypanosome research, despite 15 years of effort. This failure could reflect several issues. Firstly, until recently, suitable reporter assays were not available to detect SIF activity, restricting the capacity to carry out bioassays. The development of PAD-based reporters to monitor stumpy formation (MacGregor and Matthews, 2012) provides the solution to this problem, applicable also to HTS for anti virulence compounds as detailed above. Secondly, SIF may not be a single entity, but a composite of several co-dependent signals that confound simple purification regimens. In bacterial QS, for example, multiple signals are often used to help the bacterial population perceive cell density in context- for example where there are low or high flow conditions that would change the “interpretation” of a single soluble signal (Diggle et al., 2007). Thirdly, the behavior of pleomorphic cells in culture does not fully reflect their behavior in mice, since parasites grown in liquid culture without methyl cellulose (unlike those on plates) do not form uniformly cell-cycle arrested stumpy forms despite their continued ability to readily undergo this transition in vivo. Finally, the evidence for an accumulated signal in stumpy formation remains circumstantial and indirect, such that a perception of depletion of a key environmental component is not yet formally excluded. In each case, the metabolomics-guided refinement of trypanosome culture media and the ability to detect subtle changes in media composition after parasite conditioning by small molecule profiling (Creek et al., 2013) provides one route to narrow down the entities that may control density sensing.
The response of the parasite to density sensing also needs to be dissected in detail. In particular, identification of the molecular components of the SIF reception pathway is tractable and offers the opportunity to generate a coherent picture of a key trypanosome environmental signaling pathway, providing detailed insight into how the parasite interacts with its environment. Upon entering the tsetse fly also, the environmental signaling pathways are being assembled, the link to regulation through the glycosomes (Szoor et al., 2010) providing an exciting area of metabolic control that could generate wide-ranging changes to the parasites' physiology, adapting them to life in the tsetse midgut.
As these perception and response pathways become characterized and central regulators identified, therapeutic opportunities will certainly emerge. Parasite-specific essential proteins will provide novel drug targets, whereas the density-sensing mechanisms provide opportunities for perturbing signal transduction or manipulating the quorum-sensing signal. This represents an area of considerable interest in microbial therapeutics, since interfering with communication between microbes can generate imaginative therapeutic approaches whereby pathogen signals can be manipulated to block virulence under conditions where resistance is selected against (Brown et al., 2009). Clearly, the developmental events that underpin the pre-adaptation of trypanosomes for life in the tsetse fly provides a rich seam of exciting biology, molecular biology and therapeutic potential that will sustain trypanosome researchers, and excite the wider biological community, for many years to come.
Author Contributions
All authors contributed equally to this manuscript. The relevant sections were authored as follows: Gene expression (Eva Rico), Quorum sensing and signaling (Binny M. Mony), Adaptations at the parasite surface (Federico Rojas), Signaling in the tsetse fly (Balazs Szoor), Drug Screening (Paula MacGregor), Introductory and concluding sections and manuscript assembly (Keith R. Matthews).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the Matthews laboratory is funded by a Wellcome Trust Programme grant (088293MA) and project grant (092383MA) and by a Wellcome Trust strategic award (095831MA) to the Centre for Immunity, Infection, and Evolution. Federico Rojas is supported by the BBSRC “Silicon Trypanosome” initiative.
References
Alexandre, S., Paindavoine, P., Hanocq, Q.-J., Paturiaux, H.-F., Tebabi, P., and Pays, E. (1996). Families of adenylate cyclase genes in Trypanosoma brucei. Mol. Biochem. Parasitol. 77, 173–182. doi: 10.1016/0166-6851(96)02591-1
Alsford, S., Eckert, S., Baker, N., Glover, L., Sanchez-Flores, A., Leung, K. F., et al. (2012). High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 482, 232–236. doi: 10.1038/nature10771
Alsford, S., Turner, D. J., Obado, S. O., Sanchez-Flores, A., Glover, L., Berriman, M., et al. (2011). High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21, 915–924. doi: 10.1101/gr.115089.110
Amiguet-Vercher, A., Perez-Morga, D., Pays, A., Poelvoorde, P., Van Xong, H., Tebabi, P., et al. (2004). Loss of the mono-allelic control of the VSG expression sites during the development of Trypanosoma brucei in the bloodstream. Mol. Microbiol. 51, 1577–1588. doi: 10.1111/j.1365-2958.2003.03937.x
Arastu-Kapur, S., Ponder, E. L., Fonovic, U. P., Yeoh, S., Yuan, F., Fonovic, M., et al. (2008). Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat. Chem. Biol. 4, 203–213. doi: 10.1038/nchembio.70
Barnwell, E. M., van Deursen, F. J., Jeacock, L., Smith, K. A., Maizels, R. M., Acosta-Serrano, A., et al. (2010). Developmental regulation and extracellular release of a VSG expression-site-associated gene product from Trypanosoma brucei bloodstream forms. J. Cell. Sci. 123, 3401–3411. doi: 10.1242/jcs.068684
Barquilla, A., Saldivia, M., Diaz, R., Bart, J. M., Vidal, I., Calvo, E., et al. (2012). Third target of rapamycin complex negatively regulates development of quiescence in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 109, 14399–14404. doi: 10.1073/pnas.1210465109
Barrett, M. P., Boykin, D. W., Brun, R., and Tidwell, R. R. (2007). Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 152, 1155–1171. doi: 10.1038/sj.bjp.0707354
Brecht, M., and Parsons, M. (1998). Changes in polysome profiles accompany trypanosome development. Mol. Biochem. Parasitol. 97, 189–198. doi: 10.1016/S0166-6851(98)00151-0
Brenchley, R., Tariq, H., McElhinney, H., Szoor, B., Huxley-Jones, J., Stevens, R., et al. (2007). The TriTryp phosphatome: analysis of the protein phosphatase catalytic domains. BMC Genomics 8:434. doi: 10.1186/1471-2164-8-434
Brickman, M. J., and Balber, A. E. (1994). Trypanosoma brucei brucei and T. b. gambiense: stumpy bloodstream forms express more CB1 epitope in endosomes and lysosomes than slender forms. J. Eukaryot. Microbiol. 41, 533–536. doi: 10.1111/j.1550-7408.1994.tb01512.x
Bridges, D. J., Pitt, A. R., Hanrahan, O., Brennan, K., Voorheis, H. P., Herzyk, P., et al. (2008). Characterisation of the plasma membrane subproteome of bloodstream form Trypanosoma brucei. Proteomics 8, 83–99. doi: 10.1002/pmic.200700607
Brown, S. P., West, S. A., Diggle, S. P., and Griffin, A. S. (2009). Social evolution in micro-organisms and a Trojan horse approach to medical intervention strategies. Philos. Trans. R. Soc. Lond. B Biol. Soc. 364, 3157–3168. doi: 10.1098/rstb.2009.0055
Bruce, D., Harvey, D., Hamerton, A., Davey, J., and Bruce, L. (1912). The morphology of the Trypanosome causing disease in man in Nyasaland. Proc. R. Soc. Lond. B 85, 423–433. doi: 10.1098/rspb.1912.0068
Brun, R., and Schonenberger, M. (1981). Stimulating effect of citrate and cis-Aconitate on the transformation of Trypanosoma brucei bloodstream forms to procyclic forms in vitro. Z. Parasitenkd. 66, 17–24. doi: 10.1007/BF00941941
Capewell, P., Monk, S., Ivens, A., MacGregor, P., Fenn, K., Walrad, P., et al. (2013). Regulation of Trypanosoma brucei total and polysomal mRNA during development within its mammalian host. PLoS ONE 8:e67069. doi: 10.1371/journal.pone.0067069
Carey, K. L., Westwood, N. J., Mitchison, T. J., and Ward, G. E. (2004). A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc. Natl. Acad. Sci. U.S.A. 101, 7433–7438. doi: 10.1073/pnas.0307769101
Chaudhuri, M., Ajayi, W., Temple, S., and Hill, G. C. (1995). Identification and partial purification of a stage-specific 33 kDa mitochondrial protein as the alternative oxidase of the Trypanosoma brucei brucei bloodstream trypomastigotes. J. Eukaryot. Microbiol. 42, 467–472. doi: 10.1111/j.1550-7408.1995.tb05892.x
Chung, W. L., Leung, K. F., Carrington, M., and Field, M. C. (2008). Ubiquitylation is required for degradation of transmembrane surface proteins in trypanosomes. Traffic 9, 1681–1697. doi: 10.1111/j.1600-0854.2008.00785.x
Clayton, C. and Shapira, M. (2007). Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 156, 93–101. doi: 10.1016/j.molbiopara.2007.07.007
Clayton, C. E. (2002). NEW EMBO MEMBER'S REVIEW Life without transcriptional control? From fly to man and back again. EMBO J. 21, 1881–1888. doi: 10.1093/emboj/21.8.1881
Creek, D. J., Nijagal, B., Kim, D. H., Rojas, F., Matthews, K. R., and Barrett, M. P. (2013). Metabolomics guides rational development of a simplified cell culture medium for drug screening against Trypanosoma brucei. Antimicrob. Agents Chemother. 57, 2768–2779. doi: 10.1128/AAC.00044-13
Dean, S., Marchetti, R., Kirk, K., and Matthews, K. R. (2009). A surface transporter family conveys the trypanosome differentiation signal. Nature 459, 213–217. doi: 10.1038/nature07997
De Gaudenzi, J., Frasch, A. C., and Clayton, C. (2005). RNA-binding domain proteins in Kinetoplastids: a comparative analysis. Eukaryot. Cell 4, 2106–2114. doi: 10.1128/EC.4.12.2106-2114.2005
De Rycker, M., O'Neill, S., Joshi, D., Campbell, L., Gray, D. W., and Fairlamb, A. H. (2012). A static-cidal assay for Trypanosoma brucei to aid hit prioritisation for progression into drug discovery programmes. PLoS Negl. Trop. Dis. 6:e1932. doi: 10.1371/journal.pntd.0001932
Diggle, S. P., Griffin, A. S., Campbell, G. S., and West, S. A. (2007). Cooperation and conflict in quorum-sensing bacterial populations. Nature 450, 411–414. doi: 10.1038/nature06279
Domenicali Pfister, D., Burkard, G., Morand, S., Renggli, C. K., Roditi, I., and Vassella, E. (2006). A Mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryot. Cell. 5, 1126–1135. doi: 10.1128/EC.00094-06
Emmer, B. T., Daniels, M. D., Taylor, J. M., Epting, C. L., and Engman, D. M. (2010). Calflagin inhibition prolongs host survival and suppresses parasitemia in Trypanosoma brucei infection. Eukaryot. Cell 9, 934–942. doi: 10.1128/EC.00086-10
Engstler, M., and Boshart, M. (2004). Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 18, 2798–2811. doi: 10.1101/gad.323404
Engstler, M., Pfohl, P., Herminghaus, S., Boshart, M., Wiegerttjes, G., Heddergott, N., et al. (2007). Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell 131, 505–515. doi: 10.1016/j.cell.2007.08.046
Ferrante, A., and Allison, A. C. (1983). Alternative pathway activation of complement by African trypanosomes lacking a glycoprotein coat. Parasite Immunol. 5, 491–498. doi: 10.1111/j.1365-3024.1983.tb00763.x
Furger, A., Schurch, N., Kurath, U., and Roditi, I. (1997). Elements in the 3′ untranslated region of procyclin mRNA regulate expression in insect forms of Trypanosoma brucei by modulating RNA stability and translation. Mol. Cell. Biol. 17, 4372–4380.
Gibson, W. C., Swinkels, B. W., and Borst, P. (1988). Post-transcriptional control of the differential expression of phosphoglycerate kinase genes in Trypanosoma brucei. J. Mol. Biol. 201, 315–325. doi: 10.1016/0022-2836(88)90140-4
Gjini, E., Haydon, D. T., Barry, J. D., and Cobbold, C. A. (2010). Critical interplay between parasite differentiation, host immunity, and antigenic variation in trypanosome infections. Am. Nat. 176, 424–439. doi: 10.1086/656276
Grandgenett, P. M., Otsu, K., Wilson, H. R., Wilson, M. E., and Donelson, J. E. (2007). A function for a specific zinc metalloprotease of African trypanosomes. PLoS Pathog. 3:1432–1445. doi: 10.1371/journal.ppat.0030150
Greenbaum, D. C., Baruch, A., Grainger, M., Bozdech, Z., Medzihradszky, K. F., Engel, J., et al. (2002). A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science 298, 2002–2006. doi: 10.1126/science.1077426
Gruszynski, A. E., van Deursen, F. J., Albareda, M. C., Best, A., Chaudhary, K., Cliffe, L. J., et al. (2006). Regulation of surface coat exchange by differentiating African trypanosomes. Mol. Biochem. Parasitol. 147, 211–223. doi: 10.1016/j.molbiopara.2006.02.013
Gunasekera, K., Wuthrich, D., Braga-Lagache, S., Heller, M., and Ochsenreiter, T. (2012). Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genomics 13:556. doi: 10.1186/1471-2164-13-556
Haanstra, J. R., Kerkhoven, E. J., van Tuijl, A., Blits, M., Wurst, M., van Nuland, R., et al. (2011). A domino effect in drug action: from metabolic assault towards parasite differentiation. Mol. Microbiol. 79, 94–108. doi: 10.1111/j.1365-2958.2010.07435.x
Hall, C. I., Reese, M. L., Weerapana, E., Child, M. A., Bowyer, P. W., Albrow, V. E., et al. (2011). Chemical genetic screen identifies Toxoplasma DJ-1 as a regulator of parasite secretion, attachment, and invasion. Proc. Natl. Acad. Sci. U.S.A. 108, 10568–10573. doi: 10.1073/pnas.1105622108
Hanrahan, O., Webb, H., O'Byrne, R., Brabazon, E., Treumann, A., Sunter, J. D., et al. (2009). The glycosylphosphatidylinositol-PLC in Trypanosoma brucei forms a linear array on the exterior of the flagellar membrane before and after activation. PLoS Pathog. 5:e1000468. doi: 10.1371/journal.ppat.1000468
Heaslip, A. T., Leung, J. M., Carey, K. L., Catti, F., Warshaw, D. M., Westwood, N. J., et al. (2010). A small-molecule inhibitor of T. gondii motility induces the posttranslational modification of myosin light chain-1 and inhibits myosin motor activity. PLoS Pathog. 6:e1000720. doi: 10.1371/journal.ppat.1000720
Hehl, A., Vassella, E., Braun, R., and Roditi, I. (1994). A conserved stem-loop structure in the 3′ untranslated region of procyclin mRNAs regulates expression in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 91, 370–374. doi: 10.1073/pnas.91.1.370
Hendriks, E. F., and Matthews, K. R. (2005). Disruption of the developmental programme of Trypanosoma brucei by genetic ablation of TbZFP1, a differentiation-enriched CCCH protein. Mol Microbiol. 57, 706–716. doi: 10.1111/j.1365-2958.2005.04679.x
Hendriks, E. F., Robinson, D. R., Hinkins, M., and Matthews, K. R. (2001). A novel CCCH protein which modulates differentiation of Trypanosoma brucei to its procyclic form. EMBO J. 20, 6700–6711. doi: 10.1093/emboj/20.23.6700
Hesse, F., Selzer, P. M., Muhlstadt, K., and Duszenko, M. (1995). A novel cultivation technique for long-term maintenance of bloodstream form trypanosomes in vitro. Mol. Biochem. Parasitol. 70, 157–166. doi: 10.1016/0166-6851(95)00027-X
Hopp, C. S., Bowyer, P. W., and Baker, D. A. (2012). The role of cGMP signalling in regulating life cycle progression of Plasmodium. Microbes Infect. 14, 831–837. doi: 10.1016/j.micinf.2012.04.011
Hunt, M., Brun, R., and Kohler, P. (1994). Studies on compounds promoting the in vitro transformation of Trypanosoma brucei from bloodstream to procyclic forms. Parasitol. Res. 80, 600–606. doi: 10.1007/BF00933009
Jensen, B. C., Sivam, D., Kifer, C. T., Myler, P. J., and Parsons, M. (2009). Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics 10:482. doi: 10.1186/1471-2164-10-482
Jones, D. C., Hallyburton, I., Stojanovski, L., Read, K. D., Frearson, J. A., and Fairlamb, A. H. (2010). Identification of a kappa-opioid agonist as a potent and selective lead for drug development against human African trypanosomiasis. Biochem. Pharmacol. 80, 1478–1486. doi: 10.1016/j.bcp.2010.07.038
Kabani, S., Fenn, K., Ross, A., Ivens, A., Smith, T. K., Ghazal, P., et al. (2009). Genome-wide expression profiling of in vivo-derived bloodstream parasite stages and dynamic analysis of mRNA alterations during synchronous differentiation in Trypanosoma brucei. BMC Genomics 10:427. doi: 10.1186/1471-2164-10-427
Kamau, E. T., Srinivasan, A. R., Brown, M. J., Fair, M. G., Caraher, E. J., and Boyle, J. P. (2012). A focused small-molecule screen identifies 14 compounds with distinct effects on Toxoplasma gondii. Antimicrob. Agents Chemother. 56, 5581–5590. doi: 10.1128/AAC.00868-12
Kauppi, A. M., Nordfelth, R., Uvell, H., Wolf-Watz, H., and Elofsson, M. (2003). Targeting bacterial virulence: inhibitors of type III secretion in Yersinia. Chem. Biol. 10, 241–249. doi: 10.1016/S1074-5521(03)00046-2
Kramer, S., and Carrington, M. (2011). Trans-acting proteins regulating mRNA maturation, stability and translation in trypanosomatids. Trends Parasitol. 27, 23–30. doi: 10.1016/j.pt.2010.06.011
Kramer, S., Kimblin, N. C., and Carrington, M. (2010). Genome-wide in silico screen for CCCH-type zinc finger proteins of Trypanosoma brucei, Trypanosoma cruzi and Leishmania major. BMC Genomics 11:283. doi: 10.1186/1471-2164-11-283
Laxman, S., Riechers, A., Sadilek, M., Schwede, F., and Beavo, J. A. (2006). Hydrolysis products of cAMP analogs cause transformation of Trypanosoma brucei from slender to stumpy-like forms. Proc. Natl. Acad. Sci. U.S.A. 103, 19194–19199. doi: 10.1073/pnas.0608971103
Leung, K. F., Riley, F. S., Carrington, M., and Field, M. C. (2011). Ubiquitylation and developmental regulation of invariant surface protein expression in trypanosomes. Eukaryot. Cell 10, 916–931. doi: 10.1128/EC.05012-11
Lythgoe, K. A., Morrison, L. J., Read, A. F., and Barry, J. D. (2007). Parasite-intrinsic factors can explain ordered progression of trypanosome antigenic variation. Proc. Natl. Acad. Sci. U.S.A. 104, 8095–8100. doi: 10.1073/pnas.0606206104
MacGregor, P., and Matthews, K. R. (2010). New discoveries in the transmission biology of sleeping sickness parasites: applying the basics. J. Mol. Med. (Berl). 88, 865–871. doi: 10.1007/s00109-010-0637-y
MacGregor, P., and Matthews, K. R. (2012). Identification of the regulatory elements controlling the transmission stage-specific gene expression of PAD1 in Trypanosoma brucei. Nucleic Acids Res. 40, 7705–7717. doi: 10.1093/nar/gks533
MacGregor, P., Rojas, F., Dean, S., and Matthews, K. R. (2013). Stable transformation of pleomorphic bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 190, 60–62. doi: 10.1016/j.molbiopara.2013.06.007
MacGregor, P., Savill, N. J., Hall, D., and Matthews, K. R. (2011). Transmission stages dominate trypanosome within-host dynamics during chronic infections. Cell Host Microbe 9, 310–318. doi: 10.1016/j.chom.2011.03.013
MacGregor, P., Szoor, B., Savill, N. J., and Matthews, K. R. (2012). Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat. Rev. Microbiol. 10, 431–438. doi: 10.1038/nrmicro2779
Mackey, Z. B., Baca, A. M., Mallari, J. P., Apsel, B., Shelat, A., Hansell, E. J., et al. (2006). Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem. Biol. Drug Des. 67, 355–363. doi: 10.1111/j.1747-0285.2006.00389.x
Mahmood, R., Mittra, B., Hines, J. C., and Ray, D. S. (2001). Characterization of the Crithidia fasciculata mRNA cycling sequence binding proteins. Mol. Cell. Biol. 21, 4453–4459. doi: 10.1128/MCB.21.14.4453-4459.2001
Mancini, P. E., and Patton, C. L. (1981). Cyclic 3′, 5′-adenosine monophosphate levels during the developmental cycle of Trypanosoma brucei brucei in the rat. Mol. Biochem. Parasitol. 3, 19–31. doi: 10.1016/0166-6851(81)90074-8
Mantel, P. Y., Hoang, A. N., Goldowitz, I., Potashnikova, D., Hamza, B., Vorobjev, I., et al. (2013). Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe 13, 521–534. doi: 10.1016/j.chom.2013.04.009
Matthews, K. R., and Gull, K. (1994). Evidence for an interplay between cell cycle progression and the initiation of differentiation between life cycle forms of African trypanosomes. J. Cell Biol. 125, 1147–1156. doi: 10.1083/jcb.125.5.1147
McLintock, L. M., Turner, C. M., and Vickerman, K. (1993). Comparison of the effects of immune killing mechanisms on Trypanosoma brucei parasites of slender and stumpy morphology. Parasite Immunol. 15, 475–480. doi: 10.1111/j.1365-3024.1993.tb00633.x
Michels, P. A., Bringaud, F., Herman, M., and Hannaert, V. (2006). Metabolic functions of glycosomes in trypanosomatids. Biochim. Biophys. Acta 1763, 1463–1477. doi: 10.1016/j.bbamcr.2006.08.019
Milne, K. G., Prescott, A. R., and Ferguson, M. A. (1998). Transformation of monomorphic Trypanosoma brucei bloodstream form trypomastigotes into procyclic forms at 37 degrees C by removing glucose from the culture medium. Mol. Biochem. Parasitol. 94, 99–112. doi: 10.1016/S0166-6851(98)00055-3
Monk, S. L., Simmonds, P., and Matthews, K. R. (2013). A short bifunctional element operates to positively or negatively regulate ESAG9 expression in different developmental forms of Trypanosoma brucei. J. Cell Sci. 126, 2294–2304. doi: 10.1242/jcs.126011
Morris, J. C., Wang, Z., Drew, M. E., and Englund, P. T. (2002). Glycolysis modulates trypanosome glycoprotein expression as revealed by an RNAi library. EMBO J. 21, 4429–4438. doi: 10.1093/emboj/cdf474
Nakajima-Shimada, J., Hatabu, T., Hosoi, Y., Onizuka, Y., Kikuchi, H., Oshima, Y., et al. (2013). Derivatives of Dictyostelium discoideum differentiation-inducing factor-3 suppress the activities of Trypanosoma cruzi in vitro and in vivo. Biochem. Pharmacol. 85, 1603–1610. doi: 10.1016/j.bcp.2013.03.007
Nilsson, D., Gunasekera, K., Mani, J., Osteras, M., Farinelli, L., Baerlocher, L., et al. (2010). Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Pathog. 6:e1001037. doi: 10.1371/journal.ppat.1001037
Nolan, D. P., Rolin, S., Rodriguez, J. R., Van Den Abbeele, J., and Pays, E. (2000). Slender and stumpy bloodstream forms of Trypanosoma brucei display a differential response to extracellular acidic and proteolytic stress. Eur. J. Biochem. 267, 18–27. doi: 10.1046/j.1432-1327.2000.00935.x
Oberholzer, M., Langousis, G., Nguyen, H. T., Saada, E. A., Shimogawa, M. M., Jonsson, Z. O. et al. (2011). Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious Trypanosoma brucei. Mol. Cell. Proteomics 10, M111.010538. doi: 10.1074/mcp.M111.010538
Oberholzer, M., Marti, G., Baresic, M., Kunz, S., Hemphill, A., and Seebeck, T. (2007). The Trypanosoma brucei cAMP phosphodiesterases TbrPDEB1 and TbrPDEB2, flagellar enzymes that are essential for parasite virulence. FASEB J. 21, 720–731. doi: 10.1096/fj.06-6818com
Parsons, M., Worthey, E. A., Ward, P. N., and Mottram, J. C. (2005). Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6:127. doi: 10.1186/1471-2164-6-127
Paschos, A., den Hartigh, A., Smith, M. A., Atluri, V. L., Sivanesan, D., Tsolis, R. M., et al. (2011). An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect. Immun. 79, 1033–1043. doi: 10.1128/IAI.00993-10
Pasion, S. G., Hines, J. C., Ou, X., Mahmood, R., and Ray, D. S. (1996). Sequences within the 5′ untranslated region regulate the levels of a kinetoplast DNA topoisomerase mRNA during the cell cycle. Mol. Cell. Biol. 16, 6724–6735.
Paterou, A., Walrad, P., Craddy, P., Fenn, K., and Matthews, K. (2006). Identification and stage-specific association with the translational apparatus of TbZFP3, a ccch protein that promotes trypanosome life cycle development. J. Biol. Chem. 281, 39002–39013. doi: 10.1074/jbc.M604280200
Pays, E., Vanhollebeke, B., Vanhamme, L., Paturiaux-Hanocq, F., Nolan, D. P., and Perez-Morga, D. (2006). The trypanolytic factor of human serum. Nat. Rev. Microbiol. 4, 477–486. doi: 10.1038/nrmicro1428
Queiroz, R., Benz, C., Fellenberg, K., Hoheisel, J. D., and Clayton, C. (2009). Transcriptome analysis of differentiating trypanosomes reveals the existence of multiple post-transcriptional regulons. BMC Genomics 10:495. doi: 10.1186/1471-2164-10-495
Regev-Rudzki, N., Wilson, D. W., Carvalho, T. G., Sisquella, X., Coleman, B. M., Rug, M., et al. (2013). Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 153, 1120–1133. doi: 10.1016/j.cell.2013.04.029
Reuner, B., Vassella, E., Yutzy, B., and Boshart, M. (1997). Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol. Biochem. Parasitol. 90, 269–280. doi: 10.1016/S0166-6851(97)00160-6
Robertson, M. (1912). Notes on the polymorphism of Trypanosoma gambiense in the blood and its relation to the exogenous cycvle in Glossina palpalis. Proc. R. Soc. Lond. B 85, 241–539. doi: 10.1098/rspb.1912.0048
Roditi, I., Schwarz, H., Pearson, T. W., Beecroft, R. P., Liu, M. K., Richardson, J. P. et al. (1989). Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J. Cell Biol. 108, 737–746. doi: 10.1083/jcb.108.2.737
Rolin, S., Hanocq, Q.-J., Paturiaux, H.-F., Nolan, D., Salmon, D., Webb, H., et al. (1996). Simultaneous but independent activation of adenylate cyclase and glycosylphosphatidylinositol-phospholipase C under stress conditions in Trypanosoma brucei. J. Biol. Chem. 271, 10844–10852. doi: 10.1074/jbc.271.18.10844
Rolin, S., Hancocq-Quertier, J., Paturiaux-Hanocq, F., Nolan, D. P., and Pays, E. (1998). Mild acid stress as a differentiation trigger in Trypanosoma brucei. Mol. Biochem. Parasitol. 93, 251–262. doi: 10.1016/S0166-6851(98)00046-2
Salmon, D., Bachmaier, S., Krumbholz, C., Kador, M., Gossmann, J. A., Uzureau, P., et al. (2012a). Cytokinesis of Trypanosoma brucei bloodstream forms depends on expression of adenylyl cyclases of the ESAG4 or ESAG4-like subfamily. Mol. Microbiol. 84, 225–242. doi: 10.1111/j.1365-2958.2012.08013.x
Salmon, D., Vanwalleghem, G., Morias, Y., Denoeud, J., Krumbholz, C., Lhomme, F., et al. (2012b). Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science 337, 463–466. doi: 10.1126/science.1222753
Sbicego, S., Vassella, E., Kurath, U., Blum, B., and Roditi, I. (1999). The use of transgenic Trypanosoma brucei to identify compounds inducing the differentiation of bloodstream forms to procyclic forms. Mol. Biochem. Parasitol. 104, 311–322. doi: 10.1016/S0166-6851(99)00157-7
Seed, J. R., and Sechelski, J. B. (1989). Nature of the trypanocidal factor in human serum. J. Parasitol. 75, 1003–1006. doi: 10.2307/3282889
Shapiro, S. Z., Naessen, J., Liesegang, B., Moloo, S. K., and Magondu, J. (1984). Analysis by flow cytometry of DNA synthesis during the life cycle of African trypanosomes. Acta Trop. 41, 313–323.
Simarro, P. P., Franco, J., Diarra, A., Postigo, J. A., and Jannin, J. (2012). Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology 139, 842–846. doi: 10.1017/S0031182012000169
Smith, T. K., Vasileva, N., Gluenz, E., Terry, S., Portman, N., Kramer, S., et al. (2009). Blocking variant surface glycoprotein synthesis in Trypanosoma brucei triggers a general arrest in translation initiation. PLoS ONE 4:e7532. doi: 10.1371/journal.pone.0007532
Sykes, M. L., and Avery, V. M. (2009). Development of an Alamar Blue viability assay in 384-well format for high throughput whole cell screening of Trypanosoma brucei brucei bloodstream form strain 427. Am. J. Trop. Med. Hyg. 81, 665–674. doi: 10.4269/ajtmh.2009.09-0015
Sykes, M. L., Baell, J. B., Kaiser, M., Chatelain, E., Moawad, S. R., Ganame, D., et al. (2012). Identification of compounds with anti-proliferative activity against Trypanosoma brucei brucei strain 427 by a whole cell viability based HTS campaign. PLoS Negl. Trop. Dis. 6:e1896. doi: 10.1371/journal.pntd.0001896
Szoor, B., Dyer, N., Ruberto, I., Acosta Serrano, A., and Matthews, K. R. (2013). Independent pathways can transduce the life-cycle differentiation signal in Trypanosoma brucei. PLoS Pathog 9:e1003689. doi: 10.1371/journal.ppat.1003689
Szoor, B., Ruberto, I., Burchmore, R., and Matthews, K. (2010). A novel phosphatase cascade reglates differentiation in trypanosomes via a glycosomal signaling pathway. Genes Dev. 24, 1306–1316. doi: 10.1101/gad.570310
Szoor, B., Wilson, J., McElhinney, H., Tabernero, L., and Matthews, K. R. (2006). Protein tyrosine phosphatase TbPTP1, a molecular switch controlling life cycle differentiation in trypanosomes. J. Cell Biol. 175, 293–303. doi: 10.1083/jcb.200605090
Tsukahara, K., Hata, K., Nakamoto, K., Sagane, K., Watanabe, N. A., Kuromitsu, J., et al. (2003). Medicinal genetics approach towards identifying the molecular target of a novel inhibitor of fungal cell wall assembly. Mol. Microbiol. 48, 1029–1042. doi: 10.1046/j.1365-2958.2003.03481.x
Turner, C. M. R., Aslam, N., and Dye, C. (1995). Replication, differentiation, growth and the virulence of Trypanosoma brucei infections. Parasitology 111, 289–300. doi: 10.1017/S0031182000081841
Urbaniak, M. D., Guther, M. L., and Ferguson, M. A. (2012). Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS ONE 7:e36619. doi: 10.1371/journal.pone.0036619
Vanhollebeke, B., De Muylder, G., Nielsen, M. J., Pays, A., Tebabi, P., Dieu, M., et al. (2008). A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science 320, 677–681. doi: 10.1126/science.1156296
Vanhollebeke, B., Uzureau, P., Monteyne, D., Perez-Morga, D., and Pays, E. (2010). Cellular and molecular remodeling of the endocytic pathway during differentiation of Trypanosoma brucei bloodstream forms. Eukaryot. Cell 9, 1272–1282. doi: 10.1128/EC.00076-10
Vassella, E., Acosta-Serrano, A., Studer, E., Lee, S. H., Englund, P. T., and Roditi, I. (2001a). Multiple procyclin isoforms are expressed differentially during the development of insect forms of Trypanosoma brucei. J. Mol. Biol. 312, 597–607. doi: 10.1006/jmbi.2001.5004
Vassella, E., Kramer, R., Turner, C. M., Wankell, M., Modes, C., van den Bogaard, M., et al. (2001b). Deletion of a novel protein kinase with PX and FYVE-related domains increases the rate of differentiation of Trypanosoma brucei. Mol Microbiol. 41, 33–46. doi: 10.1046/j.1365-2958.2001.02471.x
Vassella, E., and Boshart, M. (1996). High molecular mass agarose matrix supports growth of bloodstream forms of pleomorphic Trypanosoma brucei strains in axenic culture. Mol. Biochem. Parasitol. 82, 91–105. doi: 10.1016/0166-6851(96)02727-2
Vassella, E., Probst, M., Schneider, A., Studer, E., Renggli, C. K., and Roditi, I. (2004). Expression of a major surface protein of Trypanosoma brucei insect forms is controlled by the activity of mitochondrial enzymes. Mol. Biol. Cell. 15, 3986–3993. doi: 10.1091/mbc.E04-04-0341
Vassella, E., Reuner, B., Yutzy, B., and Boshart, M. (1997). Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell. Sci. 110(pt 21), 2661–2671.
Vodnala, S. K., Lundback, T., Sjoberg, B., Svensson, R., Rottenberg, M. E., and Hammarstrom, L. G. (2013). In vitro and in vivo activities of 2-aminopyrazines and 2-aminopyridines in experimental models of human African trypanosomiasis. Antimicrob. Agents Chemother. 57, 1012–1018. doi: 10.1128/AAC.01870-12
Walrad, P., Paterou, A., Acosta Serrano, A., and Matthews, K. R. (2009). Differential trypanosome surface coat regulation by a CCCH protein that co-associates with procyclin mRNA cis-elements. PLoS Pathog. 5:e1000317. doi: 10.1371/journal.ppat.1000317
Walrad, P. B., Capewell, P., Fenn, K., and Matthews, K. R. (2012). The post-transcriptional trans-acting regulator, TbZFP3, co-ordinates transmission-stage enriched mRNAs in Trypanosoma brucei. Nucleic Acids Res. 40, 2869–2883. doi: 10.1093/nar/gkr1106
Wu, Y., Deford, J., Benjamin, R., Lee, M. G., and Ruben, L. (1994). The gene family of EF-hand calcium-binding proteins from the flagellum of Trypanosoma brucei. Biochem. J. 304(pt 3), 833–841.
Xong, H. V., Vanhamme, L., Chamekh, M., Chimfwembe, C. E., Van Den Abbeele, J., Pays, A., et al. (1998). A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell 95, 839–846. doi: 10.1016/S0092-8674(00)81706-7
Yabu, Y., and Takayanagi, T. (1988). Trypsin-stimulated transformation of Trypanosoma brucei gambiense bloodstream forms to procyclic forms in vitro. Parasitol. Res. 74, 501–506. doi: 10.1007/BF00531625
Keywords: Trypanosoma, differentiation, slender, stumpy, signaling, gene expression, drug-screening
Citation: Rico E, Rojas F, Mony BM, Szoor B, MacGregor P and Matthews KR (2013) Bloodstream form pre-adaptation to the tsetse fly in Trypanosoma brucei. Front. Cell. Infect. Microbiol. 3:78. doi: 10.3389/fcimb.2013.00078
Received: 23 August 2013; Accepted: 24 October 2013;
Published online: 14 November 2013.
Edited by:
Jan Van Den Abbeele, Institute of Tropical Medicine, BelgiumCopyright © 2013 Rico, Rojas, Mony, Szoor, MacGregor and Matthews. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keith R. Matthews, Centre for Immunity, Infection and Evolution, School of Biological Sciences, University of Edinburgh, Kings Buildings, West Mains Road, Edinburgh EH9 3JT, UK e-mail: keith.matthews@ed.ac.uk
†These authors have contributed equally to this work.