Ectodomain Shedding by ADAM17: Its Role in Neutrophil Recruitment and the Impairment of This Process during Sepsis
 Hemant K. Mishra
Hemant K. Mishra Jing Ma
Jing Ma  Bruce Walcheck
Bruce Walcheck- Department of Veterinary and Biomedical Sciences, University of Minnesota, St. Paul, MN, USA
Neutrophils are specialized at killing bacteria and are recruited from the blood in a rapid and robust manner during infection. A cascade of adhesion events direct their attachment to the vascular endothelium and migration into the underlying tissue. A disintegrin and metalloproteinase 17 (ADAM17) functions in the cell membrane of neutrophils and endothelial cells by cleaving its substrates, typically in a cis manner, at an extracellular site proximal to the cell membrane. This process is referred to as ectodomain shedding and it results in the downregulation of various adhesion molecules and receptors, and the release of immune regulating factors. ADAM17 sheddase activity is induced upon cell activation and rapidly modulates intravascular adhesion events in response to diverse environmental stimuli. During sepsis, an excessive systemic inflammatory response against infection, neutrophil migration becomes severely impaired. This involves ADAM17 as indicated by increased levels of its cleaved substrates in the blood of septic patients, and that ADAM17 inactivation improves neutrophil recruitment and bacterial clearance in animal models of sepsis. Excessive ADAM17 sheddase activity during sepsis thus appears to undermine in a direct and indirect manner the necessary balance between intravascular adhesion and de-adhesion events that regulate neutrophil migration into sites of infection. This review provides an overview of ADAM17 function and regulation and its potential contribution to neutrophil dysfunction during sepsis.
Neutrophils
Neutrophils are the predominant leukocyte population in the blood of healthy individuals and serve a critical function in host protection and wound healing, as described by others in this research topic and in recent reviews (Kolaczkowska and Kubes, 2013; Mayadas et al., 2014). These innate immune cells are produced in the bone marrow and reside in the blood where they are poised for a rapid influx into sites of acute inflammation. Neutrophils are professional phagocytes that engulf bacteria and kill them through the release of lytic enzymes and reactive oxygen species. They can also impede the spread of extracellular pathogens through the production of neutrophil extracellular traps. Circulating neutrophils infiltrate sites of inflammation by an exquisitely orchestrated multistep adhesion cascade (Figure 1A) (Kolaczkowska and Kubes, 2013). The first step is their attachment to vascular endothelial cells (e.g., lining post-capillary venules) that have been activated by events in the underlying tissue. The loosely attached neutrophils are pushed along by the blood flow, causing them to roll, and survey the luminal surface of endothelial cells for chemokines that will promote their stimulation and more stable attachment and transmigration through the vascular wall. Neutrophil attachment and rolling is primarily mediated by selectin adhesion proteins (L-selectin on neutrophils and E- and P-selectin on activated endothelial cells) that recognize various mucin-like molecules, such as PSGL1. In addition to free-flowing neutrophils attaching directly to endothelial cells (referred to as 1° or direct attachment), they can also attach to other neutrophils that have already accumulated on the vascular endothelium (2° or indirect attachment) (Figure 1A). The latter process is mediated by L-selectin and PSGL1 (Walcheck et al., 1996b), and has been shown in vitro and in vivo to amplify neutrophil accumulation (Bargatze et al., 1994; Walcheck et al., 1996b; Sperandio et al., 2003; St. Hill et al., 2003). Indeed, neutrophil infiltration into inflamed tissues occurs in a prodigious manner and has been referred to as “swarming” based on in vivo imaging (Lämmermann, 2016).
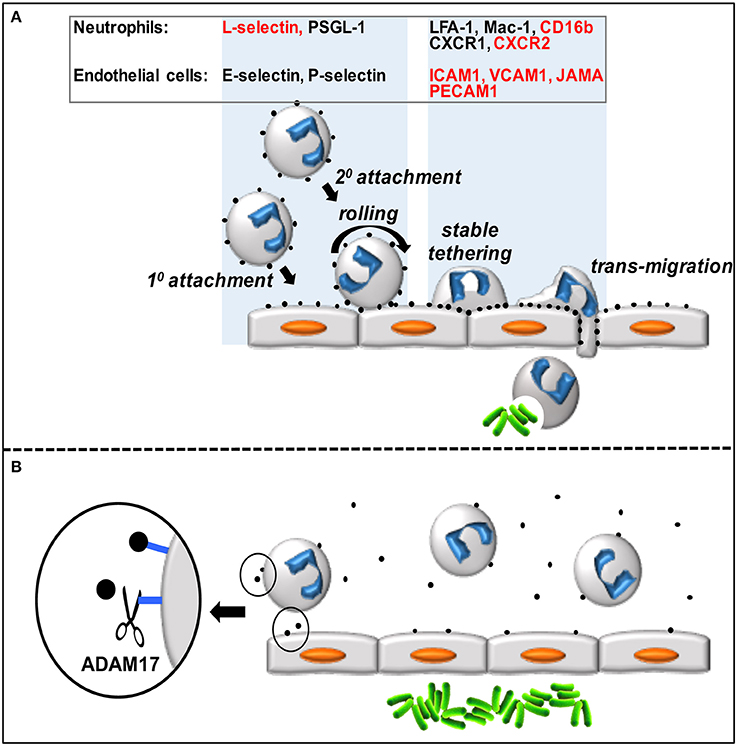
Figure 1. (A) Circulating neutrophils attach to and transmigrate through the vascular endothelium in a step-wise process. Neutrophils accumulate on the vascular endothelium by direct (1°) and indirect (2°) manners, roll and scan the endothelial cells for chemokines, which promotes stable tethers and eventual transmigration into the underlying tissue. Various neutrophil and endothelial cell adhesion molecules and receptors directly involved in this process (represented by black dots) are listed in the figure, and those that are ADAM17 substrates are indicated in red. (B) Over-activation of ADAM17 by inflammatory stimuli during sepsis may result in excessive ectodomain shedding by neutrophils and endothelial cells that in turn impairs neutrophil recruitment and bacterial (green rods) clearance.
Neutrophils attached to the vascular endothelium transition from rolling to firm adhesion upon their stimulation by chemokines, which induce a high affinity state by integrin adhesion proteins, such as LFA-1 and Mac-1. These integrins bind to the immunoglobulin superfamily members ICAM1 and ICAM2 that are upregulated in expression by activated endothelial cells. Neutrophil transmigration across the vascular wall also involves VCAM1, PECAM1, and JAMA. Upon entering the underlying tissue, neutrophils move in a directed manner, guided by a hierarchy of chemotactic factors, to the origin of pathogen and damaged cell-associated molecular patterns (PAMPs and DAMPs). The primary chemokine receptors expressed by human neutrophils involved in promoting their firm adhesion to the vascular wall and chemotaxis are CXCR1 (binds to CXCL6 and CXCL8) and CXCR2 (binds to CXCL1-3 and CXCL5-8) (Sadik et al., 2011). CXCR2 has been extensively examined in animal models as well (Stadtmann and Zarbock, 2012), and on mouse neutrophils this receptor binds to KC, MIP-2, and LIX (Cacalano et al., 1994; Goncalves and Appelberg, 2002; Sadik et al., 2011).
Neutrophil Dysfunction during Sepsis
Sepsis is a severe systemic inflammatory response to microbial pathogens (primarily bacterial and to a lesser degree fungal or viral), and is the primary cause of death from infection (Cohen et al., 2015). Since the early 1990s, this disorder was defined by using four categories; systemic inflammatory response syndrome, sepsis, severe sepsis, and septic shock. Due to increased scientific understanding of sepsis pathophysiology, the definition of the sepsis syndrome has been recently updated to just sepsis, defined as “life-threatening organ dysfunction due to a dysregulated host response to infection,” and septic shock; “a subset of sepsis where underlying circulatory and cellular/metabolic abnormalities are profound enough to substantially increase mortality” (Singer et al., 2016).
The Surviving Sepsis Campaign (www.survivingsepsis.org) established standards for the diagnosis and management of sepsis, and this has led to decreases in early mortality (Dellinger and Vincent, 2005; Kumar et al., 2011). However, epidemiologic studies reveal that the incidence of sepsis is still on the rise, and this will likely continue as the general population ages, as immune compromising therapies for cancer and autoimmune disease become more prevalent, and as microbial antibiotic resistance increases. Remarkably, current estimates indicate that 1 million people with sepsis are hospitalized per year in the US and >30 million globally (Liu et al., 2014; Cohen et al., 2015). According to the Healthcare Cost and Utilization Project by the U.S. Department of Health & Human Services, Agency for Healthcare Research and Quality, sepsis is the most expensive condition treated in US hospitals (www.hcup-us.ahrq.gov).
Sepsis is initiated by the innate immune system's recognition and response to PAMPs and DAMPs. This response greatly affects immune homeostasis, with an acute phase that is both pro- and anti-inflammatory and a secondary phase in which the adaptive immune system is suppressed. The intensity and duration of these responses are associated with increased secondary infections and mortality (Gotts and Matthay, 2016). It is well–established in animal models subjected to sepsis and by clinical evidence in humans that circulating neutrophils become activated, which impairs their migration to sites of infection and causes them to sequester in the vascular beds of organs where they promote vascular occlusions and leakage, and tissue destruction (Alves-Filho et al., 2010b; Sônego et al., 2014; Lerman and Kim, 2015). These are key events that promote multiple organ failure and septic shock.
The multistep adhesion cascade by which circulating leukocytes infiltrate sites of inflammation requires rapid orchestration of adhesion and de-adhesion events. A critical mechanism that underpins this process is ectodomain shedding, which is the focus of this review. There is increasing evidence for aberrant regulation of ectodomain shedding during inflammatory disorders and its association with vascular dysfunction during sepsis (Gearing and Newman, 1993; Cowley et al., 1994; Muller Kobold et al., 1998; Zonneveld et al., 2014; Lerman and Kim, 2015).
Ectodomain Shedding
Ectodomain shedding is a proteolytic process in which cell surface proteins are cleaved at an extracellular location proximal to the cell membrane, resulting in the release of an intact ectodomain and the retention of a membrane-associated fragment (Weber and Saftig, 2012). Cleaved proteins include many type I and type II transmembrane proteins and some glycosylphosphatidylinositol (GPI)-linked proteins. Cell surface proteins that are shed have diverse functions and include adhesion molecules, cytokines, chemokines, growth factors, and their receptors (Scheller et al., 2011). The shedding process of these substrates regulates the density of cell surface receptors, the release of factors that serve as agonists, and the release of soluble receptors that can function as antagonists.
Ectodomain shedding primarily occurs by a disintegrin and metalloproteinases (ADAMs) and to a lesser degree by matrix metalloproteinases (MMPs), members of the adamalysin and matrixin subfamilies, respectively, of the metzincin metalloproteinase superfamily (Khokha et al., 2013). Metzincin derives its name from the conserved methionine amino acid adjacent to a zinc-binding motif in the catalytic region of the proteases. The ADAMs are type-1 transmembrane proteins with distinct modular domains consisting of, from N- to C-terminus, a metalloproteinase domain, disintegrin-like domain, cysteine-rich domain, an epidermal growth factor domain (note ADAM10 and 17 lack this domain), a transmembrane segment, and a cytoplasmic region (Figure 2) (Takeda, 2016). Twenty ADAMs have been identified in humans, excluding pseudogenes, and of these only 12 are proteolytically active (ADAM8, 9, 10, 12, 15, 17, 19, 20, 21, 28, 30, and 33) (Weber and Saftig, 2012; Hartmann et al., 2013; Takeda, 2016).
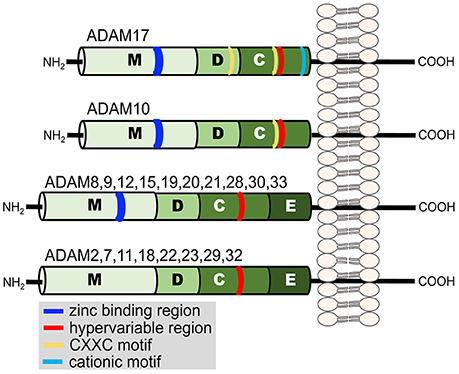
Figure 2. Illustration of the domain structure of the human ADAM family members. Each domain is indicated by a letter. Metalloproteinase (M), disintegrin-like (D), cysteine-rich (C), and epidermal growth factor (E). Additional regions of functional relevance discussed in the text are indicated in the key.
ADAM17 and ADAM10 are the most similar in terms of amino acid sequence and structure (Rosendahl et al., 1997; Maskos et al., 1998), and at this time they are the most widely studied. Though ADAM17 and ADAM10 are not redundant sheddases (Jones et al., 2016), there is some overlap in their substrate repertoire, which may serve a compensatory role and/or enable differential shedding of common substrates. Indeed, ADAM10 has been reported to function primarily in a constitutive manner, whereas ADAM17 is highly inducible, responding to various cellular stimuli (Le Gall et al., 2009), as described in more detail below. ADAM17's role in neutrophil effector functions has been broadly examined (Li et al., 2006; Walcheck et al., 2006; Bell et al., 2007; Chalaris et al., 2007; Horiuchi et al., 2007a; Schaff et al., 2008; Wang et al., 2009, 2010, 2011, 2013; Long et al., 2010, 2012; Arndt et al., 2011; Scheller et al., 2011; Tang et al., 2011; Mishra et al., 2015, 2016), and is discussed below.
ADAM17
Approximately 20 years ago, Roy Black's group and others provided direct evidence that ADAM17 converts transmembrane TNFα to its soluble form (Black et al., 1997; Moss et al., 1997). Soon afterwards this group also demonstrated through ADAM17 gene inactivation in mice that the sheddase had a much broader role than inflammation regulation and was essential for mammalian development due to EGFR ligand cleavage and EGFR signaling (Peschon et al., 1998). Global deletion of the Adam17 gene in mice is predominantly perinatal lethal (Peschon et al., 1998; Horiuchi et al., 2007a), though the degree of lethality depends on the background of the mouse strain (Li et al., 2007). However, mice expressing severely reduced levels of ADAM17, due to spontaneous or induced mutations of its gene, demonstrate significantly increased survival when compared to total inactivation of ADAM17 in mice (Brandl et al., 2010; Chalaris et al., 2010; Hassemer et al., 2010). ADAM17 deficiency has been reported in three humans so far. These patients suffered from severe inflammatory skin and bowel disease (Blaydon et al., 2011; Tsukerman et al., 2015). One patient remained alive at the time of the report and has “led a relatively normal life” (Blaydon et al., 2011).
A review by Scheller et al. in 2011 reported 76 putative substrates of ADAM17 (Scheller et al., 2011), which continues to increase, though only a handful of these substrates have been further verified in vivo. ADAM17 typically cleaves its substrates in a cis manner and an examination of the cleavage site of various ADAM17 substrates reveals no strict consensus sequence, consistent with the sheddase's promiscuous activity. Proteomic studies of ADAM17 cleavage site specificities have, however, revealed a high preference for alanine, leucine, and valine residues, and a low preference for a proline residue (Caescu et al., 2009; Thorp et al., 2011; Tucher et al., 2014). Indeed, a proline residue engineered into the cleavage site of the ADAM17 substrates CD16a, CD16b, and L-selectin completely abrogate their shedding (Zhao et al., 2001; Jing et al., 2015). Despite ADAM17's relaxed sequence specificity, the sheddase tends to require a cleavage region with an α-helical conformation and appropriate physical length (Kishimoto et al., 1995; Mezyk et al., 2003; Stawikowska et al., 2013). The specific site of cleavage may also depend on the type of membrane linkage by the substrate. For instance, the human IgG Fc receptor CD16a (FcγRIIIa) is a transmembrane protein and CD16b (FcγRIIIb) is GPI-linked to the plasma membrane. These substrates have identical cleavage regions, yet CD16a is cleaved at a single location (Lajoie et al., 2014; Jing et al., 2015), whereas CD16b is cleaved at three locations in close proximity (Galon et al., 1998; Jing et al., 2015).
Ectodomain shedding by ADAM17 is regulated in various manners, including gene expression, spatial redistribution of the sheddase and its substrates within the plasma membrane, proenzyme conversion, enzyme inhibition, and by allosteric control. The influences of these regulatory events differ per cell type, stimulus, and substrate. An interesting feature of ADAM17 compared to other ADAM family members is that its sheddase activity is greatly increased upon cell activation (Edwards et al., 2008; Gooz, 2010; Matthews et al., 2016). An example of the rate and efficiency of this process is demonstrated by L-selectin shedding. Resting neutrophils express from 50,000 to 100,000 L-selectin molecules on their surface and essentially all are cleaved within minutes of neutrophil activation (Kishimoto et al., 1989; Walcheck et al., 1996a). Heterogeneous stimuli induce ectodomain shedding in diverse cell types, and this is primarily mediated by serine and threonine kinase-dependent intracellular signaling pathways (Gechtman et al., 1999; Díaz-Rodríguez et al., 2002; Soond et al., 2005; Schwarz et al., 2014), including PKC and MAPKs in neutrophils (Fan and Derynck, 1999; Rizoli et al., 1999; Alexander et al., 2000; Wang et al., 2011). ADAM17 sheddase activity is also increased during neutrophil apoptosis (Walcheck et al., 2006; Chalaris et al., 2007; Wang et al., 2010, 2013), and this process required caspases and mitochondrial reactive oxygen species (Wang et al., 2011). An area of active debate is the proximal target(s) of the intracellular signals and how they affect ectodomain shedding by ADAM17.
Though numerous mechanisms by which ADAM17 sheddase activity is increased upon cell activation have been described, a predominant theme is that intracellular signaling induces changes in the intrinsic activity of ADAM17. Conformational changes in ADAM17 upon cell activation are apparent by the exposure of binding sites for small molecule inhibitors and antibodies (Le Gall et al., 2010; Willems et al., 2010). This may involve phosphorylation of ADAM17's cytoplasmic region, which occurs following cell activation by various stimuli (Díaz-Rodríguez et al., 2002; Soond et al., 2005; Schwarz et al., 2014). Such a means of induction, however, is confounded by several studies showing that the cytoplasmic region of ADAM17 is not required for its sheddase activity (Reddy et al., 2000; Wang et al., 2009; Le Gall et al., 2010; Schwarz et al., 2013). However, the cytoplasmic region of ADAM17 may participate in a negative regulatory process. Xu et al. reported that ADAM17 in resting cells forms dimers in the cell membrane that associate with tissue inhibitor of metalloproteinase 3 (TIMP3) (Xu et al., 2012), which forms a non-covalent complex with the catalytic region of ADAM17 and blocks its activity (Amour et al., 1998; Smookler et al., 2006; Wisniewska et al., 2008). The cytoplasmic region of ADAM17 has been shown to be critical for dimer formation, and cell activation and MAPK activity were associated with ADAM17 dimer conversion to monomers and TIMP3 dissociation (Xu et al., 2012). Other protein partners with ADAM17 include two inactive members of the Rhomboid family, iRhom 1 and 2, which control ADAM17 maturation and trafficking to the cell surface (Adrain et al., 2012; Mcilwain et al., 2012; Li et al., 2015). Interesting is that iRhom2 expression is restricted to hematopoietic cells, whereas iRhom1 is more widely expressed (Christova et al., 2013), but not in leukocytes (Issuree et al., 2013). The iRhoms have been proposed to also play a role in the induction of ADAM17 sheddase activity upon cell activation (Maretzky et al., 2013; Lorenzen et al., 2016). Intracellular stores of ADAM17 occur in certain cells (Doedens and Black, 2000; Schlöndorff et al., 2000), and through a process facilitated by the iRhoms, Lorenzen et al. reported that ADAM17 surface expression can rapidly increase upon overt cell activation with a phorbol ester (Lorenzen et al., 2016). However, the importance of rapid ADAM17 upregulation as a general inducer mechanism of ectodomain shedding is an area of debate since this was not observed with physiological stimuli (Walcheck et al., 2006; Lorenzen et al., 2016), or in various cells activated with phorbol esters (Doedens and Black, 2000; Doedens et al., 2003; Horiuchi et al., 2007b).
The disintegrin-like and cysteine-rich domains of ADAM17 also modulate its sheddase activity (Reddy et al., 2000; Gonzales et al., 2004; Wang et al., 2009; Willems et al., 2010; Düsterhöft et al., 2013; Sommer et al., 2016). These domains contain cysteine residues that provide strictly conserved disulfide bonds (Takeda, 2016). ADAM17 has two highly conserved cysteine-X-X-cysteine sequences (CXXC, where XX represents two other amino acids), one located in the disintegrin-like domain and the other in the cysteine-rich domain (Figure 2) (Wang et al., 2009). Site-directed mutagenesis revealed that these regions are critical for ADAM17 activity (Wang et al., 2009). Similarly, within the β-subunit of integrin adhesion proteins are cysteine-rich regions that contain CXXC sequences, and this motif has been reported to be an active site for the modification of allosteric disulfide bonds and rapid conformational switches (O'neill et al., 2000; Walsh et al., 2004; Xu et al., 2016). Interesting is that sulfhydryl-modifying agents are known to alter L-selectin shedding by human neutrophils. For instance, the reducing agent DTT inhibited L-selectin shedding, whereas the oxidizing agent H2O2 induced its shedding (Lynam et al., 1996; Bennett et al., 2000; Wang et al., 2009). ADAM17 sheddase activity can also be directly modified by redox agents in a cell free assay (Wang et al., 2009). These findings suggest that ADAM17 is an allosterically regulatable enzyme, which perhaps occurs by thiol isomerases (Wang et al., 2009; Willems et al., 2010; Düsterhöft et al., 2013). Another motif in ADAM17 that may regulate its conformation and enzymatic activity is a cluster of cationic amino acids located in the membrane proximal region of the cysteine-rich domain of the sheddase (Figure 2). Upon cell activation and apoptosis, cell surface exposure of negatively charged, membrane phosphatidylserine may interact with the cationic amino acids and in turn increase the proximity of ADAM17's catalytic region with certain substrates (Sommer et al., 2016).
Regulation of Neutrophil Recruitment by ADAM17
Neutrophils and endothelial cells constitutively express ADAM17 on their cell surface (Walcheck et al., 2006; Koenen et al., 2009; Weskamp et al., 2010). In contrast to global ADAM17 inactivation, conditional ADAM17 knockout mice that lack ADAM17 in myeloid cells, all leukocytes, or endothelial cells are viable and lack any obvious developmental abnormalities (Horiuchi et al., 2007a; Long et al., 2010, 2012; Weskamp et al., 2010; Arndt et al., 2011; Dreymueller et al., 2012; Mishra et al., 2016). Interesting is that either conditional ADAM17 knockout mice or hematopoietic chimeric mice that lacked ADAM17 in leukocytes demonstrated accelerated neutrophil recruitment at sites of sterile inflammation as well as infection (Long et al., 2010, 2012; Arndt et al., 2011; Tang et al., 2011; Mishra et al., 2015, 2016). This was also observed in mice receiving short-term treatment with an ADAM17 inhibitor (Tang et al., 2011; Mishra et al., 2015), demonstrating that the neutrophil recruitment pattern was not a developmental effect. One mechanism accounting for the accelerated recruitment of neutrophils is the disruption of L-selectin shedding (Tang et al., 2011; Long et al., 2012), which enhanced neutrophil tethering to L-selectin ligands on the vascular endothelium (Tang et al., 2011). CXCR2 surface levels on mouse and human neutrophils are also regulated by ADAM17 (Mishra et al., 2015). It is well-described that this chemokine receptor undergoes a rapid downregulation in expression by internalization upon binding its chemokine ligands, which is a reversible process since the receptor can be recycled back to the cell surface to bind additional ligands (Stillie et al., 2009). CXCR2 is also downregulated following overt neutrophil activation by non-ligand stimuli, including various PAMPs (Khandaker et al., 1998, 1999; Doroshenko et al., 2002; Mishra et al., 2015). This process involves ADAM17 and does not result in a recycling pool of CXCR2 (Mishra et al., 2015). Relevant to human neutrophils is that CD16b, an ADAM17 substrate described above (Wang et al., 2013; Jing et al., 2015), is also known to facilitate neutrophil attachment and migration through the vascular wall at sites of inflammation (Tsuboi et al., 2008). Various adhesion molecules expressed by endothelial cells and platelets that regulate hemostasis, barrier function, and leukocyte transmigration are also substrates of ADAM17, including GPIbα, GPV, JAMA, ICAM1, PECAM1, and VCAM1 (Garton et al., 2003; Bergmeier et al., 2004; Brill et al., 2009; Koenen et al., 2009; Weskamp et al., 2010). Taken together, ADAM17 can regulate different aspects of the multi-step process by which circulating neutrophils infiltrate inflamed tissue sites.
ADAM17 Activity during Sepsis
Several lines of evidence from animal models and patients indicate aberrant ADAM17 activity during sepsis. Indeed, ADAM17 upregulation on the surface of circulating neutrophils was found to correlate with sepsis severity and patient outcome (Kermarrec et al., 2005). A recent study has also provided evidence that ADAM17 promoter polymorphism rs12692386 is a functional variant associated with the progression of sepsis severity (Shao et al., 2016). Patients with this polymorphism demonstrated an upregulation in ADAM17 expression and serum levels of several of its proinflammatory substrates (Shao et al., 2016). It has been reported that the plasma levels of several leukocyte- and endothelial cell-expressed, ADAM17 substrates are significantly elevated during sepsis, including L-selectin, ICAM-1, VCAM-1, CD16b, TNFα, IL-6R, TNFRI, and TNFRII, and some of these substrates demonstrated a positive correlation with disease severity (Ertel et al., 1994; Muller Kobold et al., 1998; Schulte et al., 2013; Zonneveld et al., 2014; Lerman and Kim, 2015). These adhesion proteins, receptors, and cell activating factors have a direct or indirect role in regulating neutrophil recruitment at sites of bacterial infection. Moreover, CXCR2 on the surface of circulating neutrophils is significantly downregulated during experimental sepsis and in human patients (Alves-Filho et al., 2009, 2010a).
Targeting leukocyte ADAM17 in animal models has been shown to greatly reduce damaging inflammation. For instance, ADAM17 inactivation in leukocytes significantly reduced tissue and plasma levels of proinflammatory factors and organ damage in localized and systemic endotoxemia models, in part, due to a marked reduction in TNFα levels and downstream effectors (Horiuchi et al., 2007a; Arndt et al., 2011). During E. coli infection, conditional ADAM17 knockout mice lacking ADAM17 in all leukocytes demonstrated a survival advantage and a marked reduction in bacterial levels at the site of infection (Long et al., 2010, 2012). In a model of polymicrobial sepsis, these conditional ADAM17 knockout mice also demonstrated enhanced survival, which corresponded with decreased bacteremia and levels of circulating proinflammatory cytokines, key determinants of sepsis severity (Mishra et al., 2016). Neutrophil recruitment at the site of infection was again found to be greatly increased in conditional ADAM17 knockout mice compared to control mice, and this likely accounted for the enhanced clearance of bacteria (Mishra et al., 2016).
Concluding Remarks
ADAM17 cleaves an assortment of type I and type II transmembrane proteins and GPI-anchored proteins at an extracellular site. Its sheddase activity is rapidly inducible and provides a mechanism for cells to respond very quickly to different environmental stimuli to reduce cell receptor densities. ADAM17 substrates on neutrophils and endothelial cells include L-selectin, CXCR2, CD16b, JAMA, ICAM1, PECAM1, and VCAM1, and the sheddase appears to function as a pivotal regulator of intravascular adhesion events (Figure 1A). It is well-established in animal models and by clinical evidence in humans that neutrophil recruitment at sites of infection is greatly impaired during the early stages of severe sepsis (Alves-Filho et al., 2010b; Sônego et al., 2014; Lerman and Kim, 2015). Sepsis may result in an over-induction of ADAM17 activity in neutrophils, endothelial cells, and other cells that in turn undermines the necessary balance between intravascular adhesion and de-adhesion events, and impairs neutrophil recruitment at the locus of infection (Figure 1B). Moreover, the ADAM17 substrate TNFα occurs at high levels in the blood during sepsis promoting neutrophil rigidity and the upregulation of integrin adhesion molecules, in turn causing occlusion of the microvasculature, ischemia, and tissue destruction through the release of cytotoxic factors (Brown et al., 2006; Alves-Filho et al., 2009; Lerman and Kim, 2015). Since there is not a strict consensus sequence at which ADAM17 cleaves, its fidelity may decrease during prolonged or excessive inflammation, resulting in more substrates and further cell dysfunction. In addition to aberrant ectodomain shedding during sepsis, various other mechanisms that underlie neutrophil dysfunction in the course of sepsis have been reported, as described in recent review articles (Sônego et al., 2014; Lerman and Kim, 2015; Zhang et al., 2016).
Despite years of active research, novel mechanistic insights about sepsis have not yet translated into effective host-directed drug treatments. Inflammation modulating research is shifting to therapeutic strategies to optimize the host's response to infection during sepsis. Therefore, it will be interesting to examine the targeting of ADAM17 as a host-directed therapeutic approach in patients. The potential benefits of ADAM17 inhibition on increasing neutrophil infiltration at sites of infection and reducing damaging inflammation may be exploited in clinical settings to reduce sepsis progression as well as its occurrence in high risk, general surgery patients. Of course, extrapolation of mouse model findings related to the effects of ADAM17 inactivation need to be confirmed in humans in which sepsis is a highly complex clinical syndrome. In addition, ADAM17-deficient mice are perinatal lethal (Peschon et al., 1998), mice expressing greatly reduced levels of ADAM17 demonstrate increased susceptibility to inflammatory diseases (Brandl et al., 2010; Chalaris et al., 2010), and loss-of-function mutations in ADAM17 cause inflammatory diseases in humans (Blaydon et al., 2011). In consideration of this, prolonged inhibition of ADAM17 could have detrimental consequences. However, pharmacological inhibitors of ADAM17 have advanced in specificity and progressed to clinical trials for cancer (for example, https://clinicaltrials.gov/ct2/show/record/NCT02141451), and have been reported to be well-tolerated (Friedman et al., 2009; Duffy et al., 2011). Thus, temporarily targeting ADAM17 for sepsis with highly specific inhibitors may not result in significant adverse effects. Moreover, it may be possible to selectively prevent the shedding of critical ADAM17 substrates that regulate leukocyte recruitment expressed by neutrophils, platelets, or endothelial cells by targeting their cleavage regions, which tend to vary between ADAM17 substrates, and in turn more precisely modulate leukocyte interactions with the vascular endothelium during sepsis.
Author Contributions
HM, JM, and BW: Substantial contributions to the conception, design, and drafting of the work; final approval of the version to be published; and agreement to be accountable for all aspects of the work.
Funding
Research in the authors' laboratory is funded by the National Institutes of Health, including the current grants HL128580 and AI107543.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adrain, C., Zettl, M., Christova, Y., Taylor, N., and Freeman, M. (2012). Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228. doi: 10.1126/science.1214400
Alexander, S. R., Kishimoto, T. K., and Walcheck, B. (2000). Effects of selective protein kinase C inhibitors on the proteolytic down-regulation of L-selectin from chemoattractant-activated neutrophils. J. Leukoc. Biol. 67, 415–422.
Alves-Filho, J. C., Freitas, A., Souto, F. O., Spiller, F., Paula-Neto, H., Silva, J. S., et al. (2009). Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc. Natl. Acad. Sci. U.S.A. 106, 4018–4023. doi: 10.1073/pnas.0900196106
Alves-Filho, J. C., Sônego, F., Souto, F. O., Freitas, A., Verri, W. A. Jr., Auxiliadora-Martins, M., et al. (2010a). Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16, 708–712. doi: 10.1038/nm.2156
Alves-Filho, J. C., Spiller, F., and Cunha, F. Q. (2010b). Neutrophil paralysis in sepsis. Shock 34(Suppl. 1), 15–21. doi: 10.1097/SHK.0b013e3181e7e61b
Amour, A., Slocombe, P. M., Webster, A., Butler, M., Knight, C. G., Smith, B. J., et al. (1998). TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 435, 39–44. doi: 10.1016/S0014-5793(98)01031-X
Arndt, P. G., Strahan, B., Wang, Y., Long, C., Horiuchi, K., and Walcheck, B. (2011). Leukocyte ADAM17 regulates acute pulmonary inflammation. PLoS ONE 6:e19938. doi: 10.1371/journal.pone.0019938
Bargatze, R. F., Kurk, S., Butcher, E. C., and Jutila, M. A. (1994). Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J. Exp. Med. 180, 1785–1792. doi: 10.1084/jem.180.5.1785
Bell, J. H., Herrera, A. H., Li, Y., and Walcheck, B. (2007). Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 82, 173–176. doi: 10.1189/jlb.0307193
Bennett, T. A., Edwards, B. S., Sklar, L. A., and Rogelj, S. (2000). Sulfhydryl regulation of L-selectin shedding: phenylarsine oxide promotes activation-independent L-selectin shedding from leukocytes. J. Immunol. 164, 4120–4129. doi: 10.4049/jimmunol.164.8.4120
Bergmeier, W., Piffath, C. L., Cheng, G., Dole, V. S., Zhang, Y., Von Andrian, U. H., et al. (2004). Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ. Res. 95, 677–683. doi: 10.1161/01.RES.0000143899.73453.11
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733. doi: 10.1038/385729a0
Blaydon, D. C., Biancheri, P., Di, W. L., Plagnol, V., Cabral, R. M., Brooke, M. A., et al. (2011). Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 365, 1502–1508. doi: 10.1056/NEJMoa1100721
Brandl, K., Sun, L., Neppl, C., Siggs, O. M., Le Gall, S. M., Tomisato, W., et al. (2010). MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 107, 19967–19972. doi: 10.1073/pnas.1014669107
Brill, A., Chauhan, A. K., Canault, M., Walsh, M. T., Bergmeier, W., and Wagner, D. D. (2009). Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 84, 137–144. doi: 10.1093/cvr/cvp176
Brown, K. A., Brain, S. D., Pearson, J. D., Edgeworth, J. D., Lewis, S. M., and Treacher, D. F. (2006). Neutrophils in development of multiple organ failure in sepsis. Lancet 368, 157–169. doi: 10.1016/S0140-6736(06)69005-3
Cacalano, G., Lee, J., Kikly, K., Ryan, A. M., Pitts-Meek, S., Hultgren, B., et al. (1994). Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265, 682–684. doi: 10.1126/science.8036519
Caescu, C. I., Jeschke, G. R., and Turk, B. E. (2009). Active-site determinants of substrate recognition by the metalloproteinases TACE and ADAM10. Biochem. J. 424, 79–88. doi: 10.1042/BJ20090549
Chalaris, A., Adam, N., Sina, C., Rosenstiel, P., Lehmann-Koch, J., Schirmacher, P., et al. (2010). Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 207, 1617–1624. doi: 10.1084/jem.20092366
Chalaris, A., Rabe, B., Paliga, K., Lange, H., Laskay, T., Fielding, C. A., et al. (2007). Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 110, 1748–1755. doi: 10.1182/blood-2007-01-067918
Christova, Y., Adrain, C., Bambrough, P., Ibrahim, A., and Freeman, M. (2013). Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 14, 884–890. doi: 10.1038/embor.2013.128
Cohen, J., Vincent, J. L., Adhikari, N. K. J., Machado, F. R., Angus, D. C., Calandra, T., et al. (2015). Sepsis: a roadmap for future research. Lancet Infect. Dis. 15, 581–614. doi: 10.1016/S1473-3099(15)70112-X
Cowley, H. C., Heney, D., Gearing, A. J., Hemingway, I., and Webster, N. R. (1994). Increased circulating adhesion molecule concentrations in patients with the systemic inflammatory response syndrome: a prospective cohort study. Crit. Care Med. 22, 651–657. doi: 10.1097/00003246-199404000-00022
Dellinger, R. P., and Vincent, J. L. (2005). The surviving sepsis campaign sepsis change bundles and clinical practice. Crit. Care 9, 653–654. doi: 10.1186/cc3952
Díaz-Rodríguez, E., Montero, J. C., Esparís-Ogando, A., Yuste, L., and Pandiella, A. (2002). Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol. Biol. Cell 13, 2031–2044. doi: 10.1091/mbc.01-11-0561
Doedens, J. R., and Black, R. A. (2000). Stimulation-induced down-regulation of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 275, 14598–14607. doi: 10.1074/jbc.275.19.14598
Doedens, J. R., Mahimkar, R. M., and Black, R. A. (2003). TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem. Biophys. Res. Commun. 308, 331–338. doi: 10.1016/S0006-291X(03)01381-0
Doroshenko, T., Chaly, Y., Savitskiy, V., Maslakova, O., Portyanko, A., Gorudko, I., et al. (2002). Phagocytosing neutrophils down-regulate the expression of chemokine receptors CXCR1 and CXCR2. Blood 100, 2668–2671. doi: 10.1182/blood.100.7.2668
Dreymueller, D., Martin, C., Kogel, T., Pruessmeyer, J., Hess, F. M., Horiuchi, K., et al. (2012). Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 4, 412–423. doi: 10.1002/emmm.201200217
Duffy, M. J., Mullooly, M., O'donovan, N., Sukor, S., Crown, J., Pierce, A., et al. (2011). The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin. Proteomics 8:9. doi: 10.1186/1559-0275-8-9
Düsterhöft, S., Jung, S., Hung, C. W., Tholey, A., Sonnichsen, F. D., Grötzinger, J., et al. (2013). Membrane-proximal domain of a disintegrin and metalloprotease-17 represents the putative molecular switch of its shedding activity operated by protein-disulfide isomerase. J. Am. Chem. Soc. 135, 5776–5781. doi: 10.1021/ja400340u
Edwards, D. R., Handsley, M. M., and Pennington, C. J. (2008). The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289. doi: 10.1016/j.mam.2008.08.001
Ertel, W., Scholl, F. A., Gallati, H., Bonaccio, M., Schildberg, F. W., and Trentz, O. (1994). Increased release of soluble tumor necrosis factor receptors into blood during clinical sepsis. Arch. Surg. 129, 1330–1336; discussion 1336–1337. doi: 10.1001/archsurg.1994.01420360120017
Fan, H., and Derynck, R. (1999). Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 18, 6962–6972. doi: 10.1093/emboj/18.24.6962
Friedman, S., Levy, R., Garrett, W., Doval, D., Bondarde, S., Sahoo, T., et al. (2009). Clinical benefit of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17, in combination with trastuzumab in metastatic HER2 positive breast cancer patients. Cancer Res. 69:5056. doi: 10.1158/0008-5472.SABCS-09-5056
Galon, J., Moldovan, I., Galinha, A., Provost-Marloie, M. A., Kaudewitz, H., Roman-Roman, S., et al. (1998). Identification of the cleavage site involved in production of plasma soluble Fc gamma receptor type III (CD16). Eur. J. Immunol. 28, 2101–2107. doi: 10.1002/(SICI)1521-4141(199807)28:07<2101::AID-IMMU2101>3.0.CO;2-W3.0.CO;2-W
Garton, K. J., Gough, P. J., Philalay, J., Wille, P. T., Blobel, C. P., Whitehead, R. H., et al. (2003). Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 278, 37459–37464. doi: 10.1074/jbc.M305877200
Gearing, A. J., and Newman, W. (1993). Circulating adhesion molecules in disease. Immunol. Today 14, 506–512. doi: 10.1016/0167-5699(93)90267-O
Gechtman, Z., Alonso, J. L., Raab, G., Ingber, D. E., and Klagsbrun, M. (1999). The shedding of membrane-anchored heparin-binding epidermal-like growth factor is regulated by the Raf/mitogen-activated protein kinase cascade and by cell adhesion and spreading. J. Biol. Chem. 274, 28828–28835. doi: 10.1074/jbc.274.40.28828
Goncalves, A. S., and Appelberg, R. (2002). The involvement of the chemokine receptor CXCR2 in neutrophil recruitment in LPS-induced inflammation and in Mycobacterium avium infection. Scand. J. Immunol. 55, 585–591. doi: 10.1046/j.1365-3083.2002.01097.x
Gonzales, P. E., Solomon, A., Miller, A. B., Leesnitzer, M. A., Sagi, I., and Milla, M. E. (2004). Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain. J. Biol. Chem. 279, 31638–31645. doi: 10.1074/jbc.M401311200
Gooz, M. (2010). ADAM-17: the enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 45, 146–169. doi: 10.3109/10409231003628015
Gotts, J. E., and Matthay, M. A. (2016). Sepsis: pathophysiology and clinical management. BMJ 353:i1585. doi: 10.1136/bmj.i1585
Hartmann, M., Herrlich, A., and Herrlich, P. (2013). Who decides when to cleave an ectodomain? Trends Biochem. Sci. 38, 111–120. doi: 10.1016/j.tibs.2012.12.002
Hassemer, E. L., Le Gall, S. M., Liegel, R., Mcnally, M., Chang, B., Zeiss, C. J., et al. (2010). The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics 185, 245–255. doi: 10.1534/genetics.109.113167
Horiuchi, K., Kimura, T., Miyamoto, T., Takaishi, H., Okada, Y., Toyama, Y., et al. (2007a). Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 179, 2686–2689. doi: 10.4049/jimmunol.179.5.2686
Horiuchi, K., Le Gall, S., Schulte, M., Yamaguchi, T., Reiss, K., Murphy, G., et al. (2007b). Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 18, 176–188. doi: 10.1091/mbc.E06-01-0014
Issuree, P. D., Maretzky, T., Mcilwain, D. R., Monette, S., Qing, X., Lang, P. A., et al. (2013). iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Invest. 123, 928–932. doi: 10.1172/jci66168
Jing, Y., Ni, Z., Wu, J., Higgins, L., Markowski, T. W., Kaufman, D. S., et al. (2015). Identification of an ADAM17 cleavage region in human CD16 (FcgammaRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS ONE 10:e0121788. doi: 10.1371/journal.pone.0121788
Jones, J. C., Rustagi, S., and Dempsey, P. J. (2016). ADAM Proteases and gastrointestinal function. Annu. Rev. Physiol. 78, 243–276. doi: 10.1146/annurev-physiol-021014-071720
Kermarrec, N., Selloum, S., Plantefeve, G., Chosidow, D., Paoletti, X., Lopez, A., et al. (2005). Regulation of peritoneal and systemic neutrophil-derived tumor necrosis factor-alpha release in patients with severe peritonitis: role of tumor necrosis factor-alpha converting enzyme cleavage. Crit. Care Med. 33, 1359–1364. doi: 10.1097/01.CCM.0000166359.47577.57
Khandaker, M. H., Mitchell, G., Xu, L., Andrews, J. D., Singh, R., Leung, H., et al. (1999). Metalloproteinases are involved in lipopolysaccharide- and tumor necrosis factor-alpha-mediated regulation of CXCR1 and CXCR2 chemokine receptor expression. Blood 93, 2173–2185.
Khandaker, M. H., Xu, L., Rahimpour, R., Mitchell, G., Devries, M. E., Pickering, J. G., et al. (1998). CXCR1 and CXCR2 are rapidly down-modulated by bacterial endotoxin through a unique agonist-independent, tyrosine kinase-dependent mechanism. J. Immunol. 161, 1930–1938.
Khokha, R., Murthy, A., and Weiss, A. (2013). Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 13, 649–665. doi: 10.1038/nri3499
Kishimoto, T. K., Jutila, M. A., Berg, E. L., and Butcher, E. C. (1989). Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science 245, 1238–1241. doi: 10.1126/science.2551036
Kishimoto, T. K., Kahn, J., Migaki, G., Mainolfi, E., Shirley, F., Ingraham, R., et al. (1995). Regulation of L-selectin expression by membrane proximal proteolysis. Agents Actions Suppl. 47, 121–134. doi: 10.1007/978-3-0348-7343-7_11
Koenen, R. R., Pruessmeyer, J., Soehnlein, O., Fraemohs, L., Zernecke, A., Schwarz, N., et al. (2009). Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood 113, 4799–4809. doi: 10.1182/blood-2008-04-152330
Kolaczkowska, E., and Kubes, P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175. doi: 10.1038/nri3399
Kumar, G., Kumar, N., Taneja, A., Kaleekal, T., Tarima, S., Mcginley, E., et al. (2011). Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest 140, 1223–1231. doi: 10.1378/chest.11-0352
Lajoie, L., Congy-Jolivet, N., Bolzec, A., Gouilleux-Gruart, V., Sicard, E., Sung, H. C., et al. (2014). ADAM17-mediated shedding of FcgammaRIIIA on human NK cells: identification of the cleavage site and relationship with activation. J. Immunol. 192, 741–751. doi: 10.4049/jimmunol.1301024
Lämmermann, T. (2016). In the eye of the neutrophil swarm-navigation signals that bring neutrophils together in inflamed and infected tissues. J. Leukoc. Biol. 100, 55–63. doi: 10.1189/jlb.1MR0915-403
Le Gall, S. M., Bobé, P., Reiss, K., Horiuchi, K., Niu, X. D., Lundell, D., et al. (2009). ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 20, 1785–1794. doi: 10.1091/mbc.E08-11-1135
Le Gall, S. M., Maretzky, T., Issuree, P. D., Niu, X. D., Reiss, K., Saftig, P., et al. (2010). ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 123, 3913–3922. doi: 10.1242/jcs.069997
Lerman, Y. V., and Kim, M. (2015). Neutrophil migration under normal and sepsis conditions. Cardiovasc. Hematol. Disord. Drug Targets 15, 19–28. doi: 10.2174/1871529X15666150108113236
Li, N., Boyd, K., Dempsey, P. J., and Vignali, D. A. (2007). Non-cell autonomous expression of TNF-alpha-converting enzyme ADAM17 is required for normal lymphocyte development. J. Immunol. 178, 4214–4221. doi: 10.4049/jimmunol.178.7.4214
Li, X., Maretzky, T., Weskamp, G., Monette, S., Qing, X., Issuree, P. D., et al. (2015). iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 6080–6085. doi: 10.1073/pnas.1505649112
Li, Y., Brazzell, J., Herrera, A., and Walcheck, B. (2006). ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood 108, 2275–2279. doi: 10.1182/blood-2006-02-005827
Liu, V., Escobar, G. J., Greene, J. D., Soule, J., Whippy, A., Angus, D. C., et al. (2014). Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 312, 90–92. doi: 10.1001/jama.2014.5804
Long, C., Hosseinkhani, M. R., Wang, Y., Sriramarao, P., and Walcheck, B. (2012). ADAM17 activation in circulating neutrophils following bacterial challenge impairs their recruitment. J. Leukoc. Biol. 92, 667–672. doi: 10.1189/jlb.0312112
Long, C., Wang, Y., Herrera, A. H., Horiuchi, K., and Walcheck, B. (2010). In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J. Leukoc. Biol. 87, 1097–1101. doi: 10.1189/jlb.1109763
Lorenzen, I., Lokau, J., Korpys, Y., Oldefest, M., Flynn, C. M., Kunzel, U., et al. (2016). Control of ADAM17 activity by regulation of its cellular localisation. Sci. Rep. 6:35067. doi: 10.1038/srep35067
Lynam, E. B., Rogelj, S., Edwards, B. S., and Sklar, L. A. (1996). Enhanced aggregation of human neutrophils by MnCl2 or DTT differentiates the roles of L-selectin and beta 2-integrins. J. Leukoc. Biol. 60, 356–364.
Maretzky, T., Mcilwain, D. R., Issuree, P. D., Li, X., Malapeira, J., Amin, S., et al. (2013). iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. U.S.A. 110, 11433–11438. doi: 10.1073/pnas.1302553110
Maskos, K., Fernandez-Catalan, C., Huber, R., Bourenkov, G. P., Bartunik, H., Ellestad, G. A., et al. (1998). Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc. Natl. Acad. Sci. U.S.A. 95, 3408–3412. doi: 10.1073/pnas.95.7.3408
Matthews, A. L., Noy, P. J., Reyat, J. S., and Tomlinson, M. G. (2016). Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: the emerging role of tetraspanins and rhomboids. Platelets. doi: 10.1080/09537104.2016.1184751. [Epub ahead of print].
Mayadas, T. N., Cullere, X., and Lowell, C. A. (2014). The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218. doi: 10.1146/annurev-pathol-020712-164023
Mcilwain, D. R., Lang, P. A., Maretzky, T., Hamada, K., Ohishi, K., Maney, S. K., et al. (2012). iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232. doi: 10.1126/science.1214448
Mezyk, R., Bzowska, M., and Bereta, J. (2003). Structure and functions of tumor necrosis factor-alpha converting enzyme. Acta Biochim. Pol. 50, 625–645.
Mishra, H. K., Johnson, T. J., Seelig, D. M., and Walcheck, B. (2016). Targeting ADAM17 in leukocytes increases neutrophil recruitment and reduces bacterial spread during polymicrobial sepsis. J. Leukoc. Biol. 100, 999–1004. doi: 10.1189/jlb.3VMAB1115-496RR
Mishra, H. K., Long, C., Bahaie, N. S., and Walcheck, B. (2015). Regulation of CXCR2 expression and function by a disintegrin and metalloprotease-17 (ADAM17). J. Leukoc. Biol. 97, 447–454. doi: 10.1189/jlb.3HI0714-340R
Moss, M. L., Jin, S. L., Milla, M. E., Bickett, D. M., Burkhart, W., Carter, H. L., et al. (1997). Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385, 733–736. doi: 10.1038/385733a0
Muller Kobold, A. C., Zijlstra, J. G., Koene, H. R., De Haas, M., Kallenberg, C. G., and Tervaert, J. W. (1998). Levels of soluble Fc gammaRIII correlate with disease severity in sepsis. Clin. Exp. Immunol. 114, 220–227. doi: 10.1046/j.1365-2249.1998.00727.x
O'neill, S., Robinson, A., Deering, A., Ryan, M., Fitzgerald, D. J., and Moran, N. (2000). The platelet integrin alpha IIbbeta 3 has an endogenous thiol isomerase activity. J. Biol. Chem. 275, 36984–36990. doi: 10.1074/jbc.M003279200
Peschon, J. J., Slack, J. L., Reddy, P., Stocking, K. L., Sunnarborg, S. W., Lee, D. C., et al. (1998). An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284. doi: 10.1126/science.282.5392.1281
Reddy, P., Slack, J. L., Davis, R., Cerretti, D. P., Kozlosky, C. J., Blanton, R. A., et al. (2000). Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 275, 14608–14614. doi: 10.1074/jbc.275.19.14608
Rizoli, S. B., Rotstein, O. D., and Kapus, A. (1999). Cell volume-dependent regulation of L-selectin shedding in neutrophils. A role for p38 mitogen-activated protein kinase. J. Biol. Chem. 274, 22072–22080. doi: 10.1074/jbc.274.31.22072
Rosendahl, M. S., Ko, S. C., Long, D. L., Brewer, M. T., Rosenzweig, B., Hedl, E., et al. (1997). Identification and characterization of a pro-tumor necrosis factor-α-processing enzyme from the ADAM family of zinc metalloproteases. J. Biol. Chem. 272, 24588–24593. doi: 10.1074/jbc.272.39.24588
Sadik, C. D., Kim, N. D., and Luster, A. D. (2011). Neutrophils cascading their way to inflammation. Trends Immunol. 32, 452–460. doi: 10.1016/j.it.2011.06.008
Schaff, U., Mattila, P. E., Simon, S. I., and Walcheck, B. (2008). Neutrophil adhesion to E-selectin under shear promotes the redistribution and co-clustering of ADAM17 and its proteolytic substrate L-selectin. J. Leukoc. Biol. 83, 99–105. doi: 10.1189/jlb.0507304
Scheller, J., Chalaris, A., Garbers, C., and Rose-John, S. (2011). ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32, 380–387. doi: 10.1016/j.it.2011.05.005
Schlöndorff, J., Becherer, J. D., and Blobel, C. P. (2000). Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 347 (Pt 1), 131–138. doi: 10.1042/bj3470131
Schulte, W., Bernhagen, J., and Bucala, R. (2013). Cytokines in sepsis: potent immunoregulators and potential therapeutic targets–an updated view. Mediators Inflamm. 2013:165974. doi: 10.1155/2013/165974
Schwarz, J., Broder, C., Helmstetter, A., Schmidt, S., Yan, I., Müller, M., et al. (2013). Short-term TNFα shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim. Biophys. Acta 1833, 3355–3367. doi: 10.1016/j.bbamcr.2013.10.005
Schwarz, J., Schmidt, S., Will, O., Koudelka, T., Köhler, K., Boss, M., et al. (2014). Polo-like kinase 2, a novel ADAM17 signaling component, regulates tumor necrosis factor alpha ectodomain shedding. J. Biol. Chem. 289, 3080–3093. doi: 10.1074/jbc.M113.536847
Shao, Y., He, J., Chen, F., Cai, Y., Zhao, J., Lin, Y., et al. (2016). Association study between promoter polymorphisms of ADAM17 and progression of sepsis. Cell. Physiol. Biochem. 39, 1247–1261. doi: 10.1159/000447830
Singer, M., Deutschman, C. S., Seymour, C. W., Shankar-Hari, M., Annane, D., Bauer, M., et al. (2016). The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810. doi: 10.1001/jama.2016.0287
Smookler, D. S., Mohammed, F. F., Kassiri, Z., Duncan, G. S., Mak, T. W., and Khokha, R. (2006). Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J. Immunol. 176, 721–725. doi: 10.4049/jimmunol.176.2.721
Sommer, A., Kordowski, F., Büch, J., Maretzky, T., Evers, A., Andrä, J., et al. (2016). Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 7:11523. doi: 10.1038/ncomms11523
Sônego, F., Alves-Filho, J. C., and Cunha, F. Q. (2014). Targeting neutrophils in sepsis. Expert Rev. Clin. Immunol. 10, 1019–1028. doi: 10.1586/1744666X.2014.922876
Soond, S. M., Everson, B., Riches, D. W., and Murphy, G. (2005). ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 118, 2371–2380. doi: 10.1242/jcs.02357
Sperandio, M., Smith, M. L., Forlow, S. B., Olson, T. S., Xia, L., Mcever, R. P., et al. (2003). P-selectin glycoprotein ligand-1 mediates L-selectin-dependent leukocyte rolling in venules. J. Exp. Med. 197, 1355–1363. doi: 10.1084/jem.20021854
St. Hill, C. A., Alexander, S. R., and Walcheck, B. (2003). Indirect capture augments leukocyte accumulation on P-selectin in flowing whole blood. J. Leukoc. Biol. 73, 464–471. doi: 10.1189/jlb.1002491
Stadtmann, A., and Zarbock, A. (2012). CXCR2: From Bench to Bedside. Front. Immunol. 3:263. doi: 10.3389/fimmu.2012.00263
Stawikowska, R., Cudic, M., Giulianotti, M., Houghten, R. A., Fields, G. B., and Minond, D. (2013). Activity of ADAM17 (a disintegrin and metalloprotease 17) is regulated by its noncatalytic domains and secondary structure of its substrates. J. Biol. Chem. 288, 22871–22879. doi: 10.1074/jbc.M113.462267
Stillie, R., Farooq, S. M., Gordon, J. R., and Stadnyk, A. W. (2009). The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 86, 529–543. doi: 10.1189/jlb.0208125
Takeda, S. (2016). ADAM and ADAMTS family proteins and snake venom metalloproteinases: a structural overview. Toxins 8:155. doi: 10.3390/toxins8050155
Tang, J., Zarbock, A., Gomez, I., Wilson, C. L., Lefort, C. T., Stadtmann, A., et al. (2011). Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood 118, 786–794. doi: 10.1182/blood-2010-11-321406
Thorp, E., Vaisar, T., Subramanian, M., Mautner, L., Blobel, C., and Tabas, I. (2011). Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 286, 33335–33344. doi: 10.1074/jbc.M111.263020
Tsuboi, N., Asano, K., Lauterbach, M., and Mayadas, T. N. (2008). Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity 28, 833–846. doi: 10.1016/j.immuni.2008.04.013
Tsukerman, P., Eisenstein, E. M., Chavkin, M., Schmiedel, D., Wong, E., Werner, M., et al. (2015). Cytokine secretion and NK cell activity in human ADAM17 deficiency. Oncotarget 6, 44151–44160. doi: 10.18632/oncotarget.6629
Tucher, J., Linke, D., Koudelka, T., Cassidy, L., Tredup, C., Wichert, R., et al. (2014). LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J. Proteome Res. 13, 2205–2214. doi: 10.1021/pr401135u
Walcheck, B., Herrera, A. H., St. Hill, C., Mattila, P. E., Whitney, A. R., and Deleo, F. R. (2006). ADAM17 activity during human neutrophil activation and apoptosis. Eur. J. Immunol. 36, 968–976. doi: 10.1002/eji.200535257
Walcheck, B., Kahn, J., Fisher, J. M., Wang, B. B., Fisk, R. S., Payan, D. G., et al. (1996a). Neutrophil rolling altered by inhibition of L-selectin shedding in vitro. Nature 380, 720–723. doi: 10.1038/380720a0
Walcheck, B., Moore, K. L., Mcever, R. P., and Kishimoto, T. K. (1996b). Neutrophil-neutrophil interactions under hydrodynamic shear stress involve L-selectin and PSGL-1. A mechanism that amplifies initial leukocyte accumulation of P-selectin in vitro. J. Clin. Invest. 98, 1081–1087. doi: 10.1172/JCI118888
Walsh, G. M., Sheehan, D., Kinsella, A., Moran, N., and O'neill, S. (2004). Redox modulation of integrin [correction of integin] alpha IIb beta 3 involves a novel allosteric regulation of its thiol isomerase activity. Biochemistry 43, 473–480. doi: 10.1021/bi0354536
Wang, Y., Herrera, A. H., Li, Y., Belani, K. K., and Walcheck, B. (2009). Regulation of mature ADAM17 by redox agents for L-selectin shedding. J. Immunol. 182, 2449–2457. doi: 10.4049/jimmunol.0802770
Wang, Y., Robertson, J. D., and Walcheck, B. (2011). Different signaling pathways stimulate a disintegrin and metalloprotease-17 (ADAM17) in neutrophils during apoptosis and activation. J. Biol. Chem. 286, 38980–38988. doi: 10.1074/jbc.M111.277087
Wang, Y., Wu, J., Newton, R., Bahaie, N. S., Long, C., and Walcheck, B. (2013). ADAM17 cleaves CD16b (FcgammaRIIIb) in human neutrophils. Biochim. Biophys. Acta 1833, 680–685. doi: 10.1016/j.bbamcr.2012.11.027
Wang, Y., Zhang, A. C., Ni, Z., Herrera, A., and Walcheck, B. (2010). ADAM17 activity and other mechanisms of soluble L-selectin production during death receptor-induced leukocyte apoptosis. J. Immunol. 184, 4447–4454. doi: 10.4049/jimmunol.0902925
Weber, S., and Saftig, P. (2012). Ectodomain shedding and ADAMs in development. Development 139, 3693–3709. doi: 10.1242/dev.076398
Weskamp, G., Mendelson, K., Swendeman, S., Le Gall, S., Ma, Y., Lyman, S., et al. (2010). Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ. Res. 106, 932–940. doi: 10.1161/CIRCRESAHA.109.207415
Willems, S. H., Tape, C. J., Stanley, P. L., Taylor, N. A., Mills, I. G., Neal, D. E., et al. (2010). Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem. J. 428, 439–450. doi: 10.1042/BJ20100179
Wisniewska, M., Goettig, P., Maskos, K., Belouski, E., Winters, D., Hecht, R., et al. (2008). Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J. Mol. Biol. 381, 1307–1319. doi: 10.1016/j.jmb.2008.06.088
Xu, P., Liu, J., Sakaki-Yumoto, M., and Derynck, R. (2012). TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 5:ra34. doi: 10.1126/scisignal.2002689
Xu, X. R., Carrim, N., Neves, M. A., Mckeown, T., Stratton, T. W., Coelho, R. M., et al. (2016). Platelets and platelet adhesion molecules: novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb. J. 14:29. doi: 10.1186/s12959-016-0100-6
Zhao, L., Shey, M., Farnsworth, M., and Dailey, M. O. (2001). Regulation of membrane metalloproteolytic cleavage of L-selectin (CD62l) by the epidermal growth factor domain. J. Biol. Chem. 276, 30631–30640. doi: 10.1074/jbc.M103748200
Zhang, F., Liu, A. L., Gao, S., Ma, S., and Guo, S. B. (2016). Neutrophil dysfunction in sepsis. Chinese Med. J. 129:2741. doi: 10.4103/0366-6999.193447
Keywords: adhesion, leukocyte, inflammation, infection, bacteria
Citation: Mishra HK, Ma J and Walcheck B (2017) Ectodomain Shedding by ADAM17: Its Role in Neutrophil Recruitment and the Impairment of This Process during Sepsis. Front. Cell. Infect. Microbiol. 7:138. doi: 10.3389/fcimb.2017.00138
Received: 30 January 2017; Accepted: 04 April 2017;
Published: 25 April 2017.
Edited by:
Silvia Mercedes Uriarte, University of Louisville, USAReviewed by:
Elizabeth Gardiner, Australian National University, AustraliaSusan M. Bueno, Pontifical Catholic University of Chile, Chile
Copyright © 2017 Mishra, Ma and Walcheck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruce Walcheck, walch003@umn.edu