Manipulation of Host Cholesterol by Obligate Intracellular Bacteria
 Dhritiman Samanta
Dhritiman Samanta Minal Mulye
Minal Mulye  Stacey D. Gilk
Stacey D. Gilk- Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, IN, USA
Cholesterol is a multifunctional lipid that plays important metabolic and structural roles in the eukaryotic cell. Despite having diverse lifestyles, the obligate intracellular bacterial pathogens Chlamydia, Coxiella, Anaplasma, Ehrlichia, and Rickettsia all target cholesterol during host cell colonization as a potential source of membrane, as well as a means to manipulate host cell signaling and trafficking. To promote host cell entry, these pathogens utilize cholesterol-rich microdomains known as lipid rafts, which serve as organizational and functional platforms for host signaling pathways involved in phagocytosis. Once a pathogen gains entrance to the intracellular space, it can manipulate host cholesterol trafficking pathways to access nutrient-rich vesicles or acquire membrane components for the bacteria or bacteria-containing vacuole. To acquire cholesterol, these pathogens specifically target host cholesterol metabolism, uptake, efflux, and storage. In this review, we examine the strategies obligate intracellular bacterial pathogens employ to manipulate cholesterol during host cell colonization. Understanding how obligate intracellular pathogens target and use host cholesterol provides critical insight into the host-pathogen relationship.
Introduction
To establish the intracellular niche, obligate intracellular pathogens must overcome numerous obstacles. First, the pathogen must recognize and bind to their target host cell. Second, they must penetrate the host cell plasma membrane, most often by tricking the host cell into engulfing the pathogen through phagocytosis. Once inside the target cell, the pathogen initially resides in a membrane-bound compartment called the phagosome that, under normal conditions, progressively acidifies to a mature phagolysosome. While some bacteria can thrive in this environment, others prevent fusion between the phagosome and lysosome, or escape the phagosome and replicate in the host cytosol. For vacuolar pathogens, the pathogen-containing vacuole must protect from the host innate immune the bacteria system, while at the same time allowing access to nutrients required for bacterial growth. In order to establish these highly specialized intracellular niche, pathogens manipulate host gene expression, metabolism, or trafficking pathways.
In contrast to facultative intracellular or extracellular pathogens, obligate intracellular pathogens rely on the host cell for a large percentage of their growth requirements. As a result, obligate intracellular pathogens have sophisticated mechanism to manipulate the host cell to obtain essential nutrients. One of the targeted host cell factors is cholesterol, a major lipid component of eukaryotic membranes that strongly influences membrane structure and function. Structurally, cholesterol affects membrane fluidity and permeability, with higher cholesterol levels increasing membrane rigidity. Cholesterol concentrates in membrane microdomains known as lipid rafts, which are specialized signaling platforms involved in signal transduction (Simons and Toomre, 2000). Further, intracellular cholesterol is a critical player in Golgi trafficking (Stüven et al., 2003), endocytic trafficking (van der Kant et al., 2013), and intra-organelle membrane contact sites (Eden et al., 2016). Pathogenic bacteria target cholesterol not only to gain entry to host cells but also to hijack host cell signaling pathways favorable for intracellular survival. This review discusses the role of cholesterol in host-pathogen interactions, from the perspective of obligate intracellular bacterial pathogens that reside in membrane-bound compartments (Chlamydia, Coxiella, Anaplasma, and Ehrlichia) or in the host cell cytoplasm (Rickettsia).
Intracellular Lifestyles
Chlamydia spp.
Chlamydia are obligate intracellular bacteria that cause trachoma (C. trachomatis serovars Ab, B, Ba, C), urogenital tract infections (C. trachomatis serovars D-K), lymphogranuloma venereum (C. trachomatis serovar L1, L2, L3) and pneumonia (C. pneumoniae and C. psittaci). Chlamydia have a biphasic life cycle, alternating between the infectious elementary body (EB) and the replicative reticulate body (RB). Following EB attachment to the host cell, the Chlamydia Type III secretion system (T3SS) injects proteins into the host cell cytosol, inducing actin rearrangement and bacterial uptake (Clifton et al., 2004). Once internalized, the bacteria block phagosome maturation and reside in a membrane-bound compartment known as the inclusion. The inclusion membrane contains both host and bacterial proteins and is non-fusogenic with endosomes and lysosomes, but intercepts nutrient-rich Golgi-derived vesicles and multivesicular bodies in the recycling pathway (Hackstadt et al., 1995, 1996; Heinzen et al., 1996; Beatty, 2006). Within the inclusion, EBs differentiate into RBs, which replicate and eventually differentiate back to EBs prior to host cell egress and reinfection. During this infectious cycle, Chlamydia T3SS effector proteins are translocated across the inclusion membrane and into the host cytosol, where they manipulate host pathways to divert nutrients such as amino acids, lipids, and iron to the inclusion (reviewed in Bastidas et al., 2013).
Coxiella burnetii
Coxiella burnetii is the causative agent of human Q fever, an aerosol-borne zoonotic disease characterized by flu-like symptoms during acute infection and endocarditis in chronically infected patients. Following phagocytosis of the environmentally stable C. burnetii small cell variant (SCV), the C. burnetii-containing phagosome matures through the endocytic pathway to a phagolysosome (Howe and Mallavia, 2000; Howe et al., 2003). The acidic pH of the phagolysosome activates C. burnetii metabolism and differentiation to the replicative form known as the large cell variant (LCV) (Hackstadt and Williams, 1981). At this point, fusion between the C. burnetii parasitophorous vacuole (PV) and host endosomes, lysosomes, and autophagosomes creates a large, phagolysosome-like vacuole that promotes bacterial replication (reviewed in Voth and Heinzen, 2007). C. burnetii actively manipulates host cell functions such as apoptosis and vesicular trafficking by secreting effector proteins into the cytoplasm through the Dot/Icm Type IV secretion system (T4SS) (reviewed in Moffatt et al., 2015).
Anaplasma phagocytophilum and Ehrlichia chaffeensis
Anaplasma phagocytophilum and Ehrlichia chaffeensis are tick-borne obligate intracellular bacterial pathogens belonging to the family Anaplasmataceae. A. phagocytophilum, the causative agent of human granulocytic anaplasmosis, infects human neutrophils and multiplies within vacuoles called inclusions or morulae. The A. phagocytophilum inclusion does not contain endosomal or lysosomal markers, although it interacts with the nutrient-rich endocytic and autophagic pathways (Webster et al., 1998; Mott et al., 1999; Niu et al., 2006). Like C. burnetii, the A. phagocytophilum T4SS effector proteins modulate host cell processes including autophagosome formation and SUMOylation (Niu et al., 2010; Al-Khedery et al., 2012; Beyer et al., 2015).
Ehrlichia chaffeensis exclusively infects human monocytes and macrophages and causes human monocytic ehrlichiosis. Similar to A. phagocytophilum inclusions, E. chaffeensis inclusions do not contain late endosomal markers and only weakly stain for lysosomal vacuolar ATPase (Barnewall et al., 1997). However, in contrast to A. phagocytophilum, E. chaffeensis inclusions are labeled with early endosomal markers, suggesting the E. chaffeensis resides in a vacuolar niche similar to an early endosome (Mott et al., 1999). In addition, the E. chaffeensis T4SS effector protein Etf-1 induces autophagy and redirects nutrients to the inclusion to support E. chaffeensis intracellular growth (Lin et al., 2016).
Rickettsia spp.
Rickettsia spp. are small coccobacilli and the causative agents of Rocky Mountain Spotted Fever (R. rickettsia), Mediterranean spotted fever (R. conorii), louse-borne typhus (R. prowazekii), and scrub typhus (Orientia tsutsugamushi). Most commonly transmitted to humans by arthropods, Rickettsia spp. infect and replicate in the endothelial cells of blood vessels and major organs. Following uptake by the host cell, rickettsial phospholipase degrades the phagosomal membrane, releasing the bacteria into the cytoplasm where they replicate freely (Walker et al., 2001). Several species of Rickettsia spread from cell to cell by reorganizing and mobilizing host cell actin (Heinzen et al., 1993), while O. tsutsugamushi escapes the host cell through a process similar to viral budding (Ogawa et al., 2014).
Role of Cholesterol in Bacterial Entry and Exit
Cholesterol-rich microdomains in the plasma membrane, known as lipid rafts, play a key role during pathogen attachment and entry into host cells. Lipid rafts are enriched in cholesterol, with 3- to 5-fold more cholesterol than the surrounding membrane (Pike, 2003). Lipid rafts also have higher concentrations of sphingolipids and signal transduction proteins such as integrins, kinases, phosphatases, and G protein-coupled receptors (Brown and Rose, 1992; Sargiacomo et al., 1993; Chun et al., 1994; Lisanti et al., 1994; Gorodinsky and Harris, 1995; Liu et al., 1996; Mineo et al., 1996; Bickel et al., 1997). Caveolae, a morphologically distinct subset of lipid rafts with a flask-shaped structure, contain cholesterol-binding caveolin proteins involved in clathrin-independent endocytosis (Chang et al., 1992; Stang et al., 1997; Orlandi and Fishman, 1998; Schubert et al., 2001; Lai, 2003). As lipid rafts are key components of cell signaling and endocytosis, bacterial pathogens often target lipid rafts during host cell entry. Hijacking raft-associated signaling proteins promotes internalization of intracellular bacteria and subsequent development of the intracellular niche.
Chlamydia spp.
The role of lipid rafts during Chlamydia host cell entry has been controversial. Early studies found that C. trachomatis serovar L2 and C. psittaci strain GPIC was associated with lipid rafts during infection of HeLa cells, and cholesterol depletion using methyl-beta-cyclodextrin (MβCD) inhibited bacterial entry in a dose-dependent manner (Jutras et al., 2003). Disrupting lipid rafts with the cholesterol-binding compounds filipin and nystatin impaired entry of C. pneumoniae, C. psittaci, and C. trachomatis serovars E, F, and K (but not A, B, and C) into HeLa cells (Stuart et al., 2003) and serovar K into J774 macrophages (Norkin et al., 2001). Further, although caveolin was not necessary for entry, these strains co-localized with the major caveolae protein caveolin-1 (Norkin et al., 2001; Stuart et al., 2003). Recent work suggests C. pneumoniae uptake by HL cells, but not attachment, also involves lipid rafts (Korhonen et al., 2012). Similarly, Gabel reported that MβCD cholesterol extraction inhibited entry of C. trachomatis serovars D, E, K, and L2 into HeLa cells (Gabel et al., 2004). However, disrupting lipid raft function with filipin, nystatin, or antibodies against cholera toxin B bound to lipid raft GM1 ganglioside had no effect on infection (Gabel et al., 2004). This study also did not find bacteria associated with lipid rafts or caveolin. Further, GPI-anchored proteins, which are enriched in lipid rafts, were not required for C. trachomatis entry into CHO cells (Jutras et al., 2003). In support of these findings, C. trachomatis serovar L2 entry into cholesterol-free DHCR24−/− mouse embryonic fibroblasts was identical to cholesterol supplemented cells (Gilk et al., 2013).
The discrepancies between these studies may be attributed to differences in bacterial isolates, host cell type, or pleotropic effects on membrane function by MβCD (Zidovetzki and Levitan, 2007). It is also possible that multiple, redundant entry pathways exist using different host cell receptors. Recent studies have shown that two C. pneumoniae proteins, adhesin CPn0473 and invasin Pmp21, bind to host epidermal growth factor receptor (EGFR) and trigger lipid raft-mediated uptake (Mölleken et al., 2013; Fechtner et al., 2016). C. trachomatis also adhered to the lipid raft protein EphrinA2 receptor (EphA2) (Chakraborty et al., 2012; Subbarayal et al., 2015), and the C. trachomatis invasin protein Ctad1 bound to β1 integrin in lipid rafts during internalization (Stallmann and Hegemann, 2016). Targeting multiple host cell lipid raft receptors may broaden host cell tropism and increase the potential for successful host cell colonization.
C. burnetii
During host cell entry, C. burnetii utilizes αVβ3 integrin (Capo et al., 1999), a transmembrane protein found in lipid rafts (Triantafilou and Triantafilou, 2003). In cholesterol-free DHCR24−/− fibroblasts, C. burnetii internalization decreased almost 90% compared to DHCR24−/− fibroblasts supplemented with exogenous cholesterol (Gilk et al., 2013). Blocking αVβ3 with antibodies or the αVβ3 ligand vitronectin further reduced internalization in DHCR24−/− fibroblasts, reinforcing the significance of lipid rafts in C. burnetii internalization. Targeting the αVβ3 pathway may be a strategy employed by C. burnetii to evade the host immune response. αVβ3 normally functions during macrophage phagocytosis of apoptotic cells (Wu et al., 2005), a process that suppresses the immune response by inhibiting macrophage production of interleukin (IL)-1β, IL-8, tumor necrosis factor (TNF)- α, leukotriene C4, and thromboxane B2 (Fadok et al., 1998). Thus, the αVβ3-dependent pathway may enable C. burnetii to enter host cells without eliciting an inflammatory response.
A. phagocytophilum and E. chaffeensis
A. phagocytophilum and E. chaffeensis have also been reported to utilize lipid rafts or caveolae for host cell entry (Lin and Rikihisa, 2003b). Similar to Chlamydia spp., cholesterol depletion using MβCD, or lipid raft disruption with nystatin or NBD-cholesterol, blocked infection by both A. phagocytophilum and E. chaffeensis (Lin and Rikihisa, 2003b). Removing host surface GPI-anchored proteins with phosphoinositide phospholipase C (PI-PLC) also inhibited entry of both bacterial species, suggesting lipid raft-associated GPI-anchored proteins are necessary for infection. Depletion of plasma membrane caveolae with cholera toxin B reduced A. phagocytophilum and E. chaffeensis entry by 90%, and both GM1 and caveolin-1 localized to early inclusions (Lin and Rikihisa, 2003b). These data suggest that A. phagocytophilum and E. chaffeensis utilize caveolae-mediated endocytosis for host cell entry.
Rickettsia spp.
Lipid rafts are involved in both host cell entry and exit of different Rickettsia spp. MβCD extraction of plasma membrane cholesterol blocked R. conorii uptake (Martinez et al., 2005). Further, the R. conorii outer membrane protein OmpB bound the host protein Ku70, a subunit of DNA-dependent protein kinase (DNA-PK) which is found in the nucleus and plasma membrane lipid rafts (Koike, 2002; Lucero et al., 2003; Martinez et al., 2005). Ku70 co-localized with R. conorii attached to Vero or HeLa cells, and an antibody directed against the extracellular N-terminus of Ku70 decreased R. conorii entry (Martinez et al., 2005). Thus, R. conorii OmpB facilitates host cell entry by binding to the lipid raft-associated host protein Ku70. In addition to OmpB, R. conorii OmpA bound the lipid raft integrins α2 and β1 on the cell surface (Upla et al., 2004; Hillman et al., 2013). While the signaling cascade triggered by Ku70 is not known, it may mimic integrin signaling and lead to actin-mediated phagocytosis (Martinez et al., 2005).
O. tsutsugamushi was found in lipid rafts of infected cells, suggesting the bacteria associate with lipid rafts (Kim et al., 2013). However, the inability of MβCD or filipin to impair O. tsutsugamushi host cell entry indicates that cholesterol or lipid rafts are not involved (Kim et al., 2013). While caveolin did not associate directly with the bacteria, the O. tsutsugamushi protein HtrA co-localized with caveolin during host cell exit, a process where the bacteria causes the plasma membrane to bulge out similar to viral budding (Kim et al., 2013). Thus, it appears that plasma membrane cholesterol is not involved in O. tsutsugamushi entry but plays a key role during bacterial egress from host cells.
Targeting lipid rafts is clearly an important strategy by obligate intracellular pathogens, with Chlamydia spp., C. burnetii, A. phagocytophilum, E. chaffeensis, and R. conorii all utilizing lipid rafts during host cell entry. Lipid rafts serve as signaling platforms to trigger host cell actin rearrangement and phagocytosis, thus allowing the bacteria to subvert host cell machinery to gain entry. Further, caveolin-mediated entry may facilitate development of the intracellular niche for vacuolar pathogens. For example, Chlamydia inclusion membrane caveolin may facilitate direct interception of Golgi-derived exocytic vesicles that are rich in sphingolipids and other nutrients essential for Chlamydia growth (Hackstadt et al., 1996; Van Ooij et al., 2000). While the presence of caveolin on the C. burnetii PV has not been explored, caveolin most likely plays an important role during host cell colonization by Chlamydia spp., A. phagocytophilum, and E. chaffeensis.
Cholesterol-rich Microdomains on Pathogen-containing Vacuoles
While the functions are less understood, cholesterol-rich microdomains are also found on the membrane of bacteria-containing vacuoles.
Chlamydia spp.
Filipin labeling of C. trachomatis-infected HeLa cells revealed cholesterol-rich microdomains on the inclusion membrane (Carabeo et al., 2003; Mital et al., 2010). These microdomains co-localized with four chlamydial inclusion membrane proteins (IncB, Inc101, Inc222, and Inc850) and active host Src-family kinases (SFKs) (Mital et al., 2010). SFKs were not required for Inc microdomain formation, leaving the relationship between SFKs and Inc proteins unclear (Mital and Hackstadt, 2011). It is not known whether the Incs are recruited to these inclusion microdomains, or if the Inc proteins themselves trigger microdomain formation. Inc222 and Inc850 stably interact with one another and may form a complex within the inclusion microdomains. While the functions of IncB, Inc101, and Inc222 are unknown, Inc850 may play a critical role during inclusion development by mediating microtubule-dependent trafficking of the nascent chlamydial inclusion to the perinuclear microtubule organizing center (MTOC) (Campbell et al., 1989a,b; Grieshaber et al., 2003). Inc850 was found to bind dynein light chain, DYNLT1, thus facilitating interactions between the inclusion and the host microtubule network that are required for trafficking to the MTOC (Mital et al., 2010, 2015). Inclusion trafficking to the MTOC is thought to promote homotypic fusion between inclusions, which serves as a potential mechanism to share nutrients or exchange genetic material (Richards et al., 2013). Chlamydia clinical strains that cannot undergo homotypic fusion cause less severe disease with fewer recoverable bacteria, suggesting this is an important virulence factor (Geisler et al., 2001). With the recent development of genetic tools in Chlamydia, the role of Inc850 in inclusion trafficking and virulence can now be tested.
In addition to the Incs, the host SFKs Fyn and Src localized to cholesterol-rich microdomains of C. trachomatis and C. pneumoniae inclusions but not the rodent species C. caviae and C. muridarum (Mital et al., 2010; Mital and Hackstadt, 2011). The SFKs are non-receptor membrane-associated tyrosine kinases that regulate microtubule-dependent trafficking by phosphorylating tubulin and binding to dynein-associated proteins (Macurek et al., 2008; Colello et al., 2010; Levi and Shalgi, 2010). SFK activity was necessary for dynein-dependent trafficking of the nascent C. trachomatis inclusion to the MTOC (Mital et al., 2010). In addition, Fyn was involved in sphingomyelin acquisition by Chlamydia, most likely through microtubule-dependent trafficking (Mital and Hackstadt, 2011). Chlamydia species that do not recruit SFKs to their inclusion membranes do not traffic to the MTOC but have increased inclusion development and bacterial growth in SFK-deficient cells, suggesting SFKs restrict growth of some Chlamydia species (Mital et al., 2010; Mital and Hackstadt, 2011).
C. burnetii
The C. burnetii PV membrane is sterol-rich and contains the lipid raft proteins flotillin-1 and flotillin-2 (Howe and Heinzen, 2006). It is not known, however, if there are organized microdomains on the PV membrane, or if these proteins play a role during C. burnetii infection. Like C. trachomatis, multiple C. burnetii PVs in a host cell will fuse, though there is no evidence for microtubule-dependent trafficking of the PV and the role of homotypic fusion in pathogenesis is not known.
Cholesterol-rich microdomains have been found on the pathogen-containing vacuoles of both C. trachomatis and C. burnetii, though the function has only been explored in C. trachomatis. Cholesterol or other sterols are also enriched in the A. phagocytophilum inclusion (Xiong et al., 2009). In addition to mediating microtubule-dependent trafficking as in the case of C. trachomatis inclusion, cholesterol may play a structural role in the membranes of pathogen-containing vacuoles. For example, high cholesterol membranes are more rigid and may create a stronger physical barrier between the pathogen and host cell defenses. Further, cholesterol regulates proteins involved in endosomal trafficking and fusion, and could facilitate recruitment of nutrient-rich endosomes to support bacterial growth.
Cholesterol Trafficking to Pathogen-containing Vacuoles
Endogenous cholesterol is synthesized in the endoplasmic reticulum (ER) and trafficked to the plasma membrane before distribution throughout the cell. The major source of exogenous cholesterol is through receptor-mediated uptake of cholesterol bound to low density lipoprotein (LDL). LDL particles are internalized by clathrin-mediated endocytosis and transported through the endocytic pathway to lysosomes, where cholesterol esters are hydrolyzed to free cholesterol for cellular use. Regardless of the source, cholesterol is transported throughout the cell by both vesicular and non-vesicular (e.g., cholesterol transport proteins and membrane contact sites) trafficking pathways.
Chlamydia spp.
While the bacteria lack the machinery to synthesize cholesterol, it is found in the C. trachomatis membrane in addition to inclusion microdomains (Wylie et al., 1997; Hatch and McClarty, 1998; Stephens et al., 1998; Carabeo et al., 2003). Chlamydia appears to actively acquire host cholesterol, as inhibiting bacterial protein synthesis with chloramphenicol drastically decreased inclusion cholesterol levels (Carabeo et al., 2003). This suggests that bacterial proteins, possibly secreted into the host cytoplasm, directly manipulate host cholesterol trafficking pathways. Both de novo synthesized and exogenous LDL-derived cholesterol trafficked to C. trachomatis inclusion through a microtubule-dependent process and involved transit through the Golgi apparatus before delivery to the inclusion membrane (Carabeo et al., 2003). However, Golgi-dependent cholesterol trafficking was not essential for Chlamydia replication, and cholesterol-rich multivesicular bodies (MVBs) also deliver cholesterol to the inclusion (Carabeo et al., 2003; Beatty, 2006, 2008). The MVB pathway may be essential, as disruption of the MVB trafficking decreased cholesterol in the C. trachomatis inclusion, delayed inclusion maturation, and reduced bacterial growth (Beatty, 2006, 2008). Interestingly, C. trachomatis inclusion formation and bacterial growth was unaffected in cholesterol-free DHCR24−/− mouse embryonic fibroblasts, suggesting cholesterol precursors may be sufficient for C. trachomatis infection (Gilk et al., 2013).
In addition to Golgi-dependent and MVB trafficking, a third mechanism of cholesterol transport to the inclusion utilizes the high-density lipoprotein (HDL) biogenesis machinery involved in cholesterol efflux. HDL is formed when cholesterol and phospholipids are transported to extracellular ApoA-1 by the lipid binding proteins ATP-binding cassette transporters A1 and G1 (ABCA1, ABCG1), and CLA1. Intriguingly, while ABCA1, CLA1, and ApoA-1 localized to the inclusion membrane, both CLA1 and ApoA-1 were also found in discrete foci within the inclusion lumen (Cox et al., 2012). siRNA knockdown of ABCA1 or pharmaceutical inhibitors of ABCA1 and CLA1 transporter activity significantly reduced C. trachomatis growth (Cox et al., 2012). While the mechanism is not clear, it is possible that C. trachomatis diverts these proteins to the inclusion as a mechanism to obtain cholesterol.
C. burnetii
The C. burnetii PV is sterol-rich, although filipin labeling does not indicate that cholesterol or other sterols are present in the C. burnetii envelope (Howe and Heinzen, 2006). Despite encoding two unique eukaryote-like sterol reductase homologs (Seshadri et al., 2003; Beare et al., 2009; Gilk et al., 2013), C. burnetii does not appear to synthesize cholesterol and instead obtains cholesterol from the host cell. Cholesterol-rich MVBs fuse with the PV (Gilk et al., 2013) and both endogenous and LDL-derived cholesterol traffic to the PV through unknown pathways (Mulye et al., 2017). C. burnetii also interacts with host cholesterol trafficking by recruiting the host cholesterol-binding protein ORP1L (oxysterol binding protein related protein 1 long) to the PV in a T4SS-dependent manner (Justis et al., 2017). ORP1L is an endosome/lysosome-localized Rab7 effector protein that serves two conformation-dependent functions in host cells (Rocha et al., 2009). When bound to cholesterol, ORP1L takes on a compact conformation, allowing Rab7-RILP to interact with dynein motors and direct minus-end transport of endosomes along microtubules. Alternately, when not bound to cholesterol, ORP1L is in an extended conformation and binds to the VAP proteins on the ER, participating in endosome/lysosome-ER membrane contact sites (Rocha et al., 2009; van der Kant et al., 2013). Interestingly, while the C. burnetii PV membrane is sterol-rich, fluorescent co-localization and electron microscopy suggest that ORP1L participates in PV-ER contact sites and is likely not binding cholesterol on the PV membrane (Justis et al., 2017). ORP1L was required for optimal C. burnetii PV expansion possibly through ORP1L-dependent trafficking to the PV (Justis et al., 2017). ORP1L may also be involved in transfer of cholesterol from the PV to the ER, similar to the proposed role of ORP1L during adenovirus infection (Cianciola et al., 2013).
Inhibiting host cell cholesterol metabolism disrupted PV morphology and bacterial replication, demonstrating that cholesterol and other sterols play an important role in C. burnetii infection (Howe and Heinzen, 2006). Although cholesterol itself was not essential for C. burnetii infection of cholesterol-free DHCR24−/− fibroblasts (Gilk et al., 2013), increasing PV cholesterol through cholesterol supplementation or pharmacological inhibitors was bactericidal (Mulye et al., 2017). Intriguingly, C. burnetii death was due to increased PV acidification when PV cholesterol levels were elevated (Mulye et al., 2017). The proton pump vATPase plays a key role in lysosomal acidification and has been localized to the C. burnetii PV (Heinzen et al., 1996). vATPase activity is affected by lysosomal membrane cholesterol (Cox et al., 2007), leading to the possibility that cholesterol levels regulate PV pH through vATPase.
A. phagocytophilum
Cholesterol is an important component of the A. phagocytophilum and E. chaffeensis cell envelopes, providing physical integrity in the absence of typical bacterial lipid A and peptidoglycan (Lin and Rikihisa, 2003b). Cholesterol is also enriched on the A. phagocytophilum inclusion membrane and lumen (Xiong et al., 2009). Like Chlamydia and C. burnetii, A. phagocytophilum lacks genes for cholesterol biosynthesis and recruits host cholesterol by targeting cholesterol trafficking (Lin and Rikihisa, 2003a). A. phagocytophilum-infected cells have increased LDL uptake, resulting in 2-fold more cholesterol than uninfected cells (Xiong et al., 2009). LDL-derived cholesterol was trafficked to the A. phagocytophilum inclusion by the host cell protein NPC1 (Niemann-Pick disease, type C1), a cholesterol-binding membrane protein required for cholesterol transfer from endosomes and lysosomes to the ER (Karten et al., 2009). Within 24 h of infection, A. phagocytophilum recruited NPC1 to the inclusion membrane via NPC1-positive, LAMP1/2-negative vesicles (Xiong and Rikihisa, 2012). Bacterial protein synthesis was required for NPC1 recruitment, and bacterial growth was significantly impaired in NPC1-deficient cells (Xiong and Rikihisa, 2012). Furthermore, vesicle-associated membrane protein 4 (VAMP4) and syntaxin 16, which participate in LDL-cholesterol transport from NPC1 vesicles to the Golgi network, were also recruited to the A. phagocytophilum inclusion (Xiong and Rikihisa, 2012). Thus, A. phagocytophilum hijacks NPC1-mediated LDL trafficking as a method to divert cholesterol to the bacterial inclusion for incorporation into the bacterial envelope and inclusion membrane.
Given that cholesterol plays important roles in both the bacteria and vacuole membranes, it is no surprise that obligate intracellular pathogens have devised multiple ways to recruit host cell cholesterol. Cholesterol binding proteins, which serve to move cholesterol between membranes, localize to the vacuoles of both C. trachomatis and C. burnetii. Cholesterol-rich MVBs also fuse with the C. trachomatis and C. burnetii vacuoles, and LDL trafficking of internalized extracellular cholesterol is important for C. trachomatis, C. burnetii, and A. phagocytophilum. In the case of A. phagocytophilum, the bacteria target a specific subset of LDL-positive vesicles, while the underlying mechanism of LDL trafficking to the C. trachomatis inclusion is not known. Finally, the promiscuous fusogenicity of the C. burnetii PV may account for LDL delivery to this vacuole. Most likely, other undiscovered pathways also deliver cholesterol to the bacteria, all serving to provide this important resource to the intracellular niche.
Bacterial Modification of Cholesterol
While not possessing the full biosynthetic machinery to generate cholesterol de novo, both C. trachomatis and C. burnetii express cholesterol-modifying enzymes which may modify host cell cholesterol or cholesterol precursors.
C. trachomatis
The C. trachomatis gene CT149 is a putative carboxylic esterase containing a cholesterol recognition consensus sequence and two GXSXG cholesterol esterase motifs (Peters et al., 2012). Further, recombinant CT149 exhibited cholesterol esterase activity in vitro, and CT149 ectopic expression in HeLa cells decreased cholesterol ester levels, with a corresponding increase in free cholesterol (Peters et al., 2012). Antibodies generated against CT149 localized this protein to the bacteria and not the host cytoplasm, suggesting CT149 functions inside the inclusion (Peters et al., 2012). Interestingly, lipid droplets (LDs), which store cholesterol esters, were observed inside the chlamydial inclusion (Cocchiaro et al., 2008), presenting the possibility that CT149 liberates cholesterol from LDs for use by the bacteria.
C. burnetii
C. burnetii expresses two eukaryotic-like sterol reductase enzymes, CBU1158 and CBU1206 (Seshadri et al., 2003; Beare et al., 2009). CBU1206 has homology to Δ24 sterol reductases, which function in the final step of mammalian cholesterol or yeast ergosterol synthesis (Beare et al., 2009). Heterologous expression of CBU1206 in Saccharomyces cerevisae functionally complements deletion of the yeast Δ24 sterol reductase erg4, indicating CBU1206 can modify sterols (Gilk et al., 2010). However, if CBU1206 modifies cholesterol in C. burnetii or host cells is unknown. Further, CBU1206 contains nine predicted transmembrane domains and is most likely in the C. burnetii cell envelope, which raises interesting questions about when and where CBU1206 might modify host sterols (Gilk et al., 2010). The enzymatic capability for the putative C. burnetii Δ7 sterol reductase CBU1158 remains unexplored. Determining the importance of the C. burnetii putative sterol reductases during infection, along with their substrate specificity and location of action, will be critical to understanding the role of cholesterol and other sterols during C. burnetii infection.
Manipulating Cholesterol Homeostasis
Eukaryotic cells tightly regulate cholesterol levels by balancing metabolism (biosynthesis and breakdown), uptake, efflux, and storage. De novo biosynthesis occurs in the ER, with the conversion of HMG-CoA to mevalonate by HMG-CoA reductase (HMGR) being the rate-limiting step. Cellular cholesterol levels can also be increased through uptake of cholesterol bound to LDL via the LDL receptor (LDLR). Both biosynthesis and uptake are regulated at the expression level by sterol regulatory element-binding protein (SREBP) or liver X receptor (LXR) transcription factors, which increase transcription of HMGR and LDLR under low cholesterol conditions. When cellular cholesterol levels are high, cholesterol can be transported out of the cell (i.e., effluxed), broken down into bile acids or steroids, or esterified and stored in LDs. Chlamydia spp., C. burnetii, and A. phagocytophilum all target host cholesterol homeostasis, particularly at the level of gene transcription.
Cholesterol Metabolism and Uptake
Chlamydia spp.
C. pneumoniae has been linked to atherosclerosis, a disease that results from dyslipidemia, or increased levels of circulating lipids. In mice, peripheral blood monocytes can spread C. pneumoniae from the lung to the liver, a key site of lipid metabolism (Moazed et al., 1998; Marangoni et al., 2006). A recent in vivo study found that C. pneumoniae-infected mice had decreased hepatic bile acid levels and increased serum cholesterol levels, as compared to uninfected or C. trachomatis-infected mice (Marangoni et al., 2015). These changes have been linked to Cyp7a1 (cholesterol 7α-hydroxylase), a host enzyme that catalyzes cholesterol breakdown into bile acids. Along with two transcription factors involved in regulating Cyp7a1 expression, lxr-α and srebp1c, cyp7a1was downregulated in C. pneumoniae-infected liver cells (Marangoni et al., 2015). A separate study reported a dose-dependent decrease in cyp7a1 promoter activity in C. pneumoniae-infected human hepatocytes (Michelini et al., 2012). Together, these data suggest that through Cyp7a1, C. pneumoniae downregulates cholesterol catabolism in liver cells. C. pneumoniae also altered uptake of serum cholesterol in the mouse liver, by downregulating ldlr expression and upregulating expression of idol (inducible degrader of the LDLR) (Marangoni et al., 2015). By decreasing cholesterol catabolism in the liver, while also decreasing cholesterol uptake, C. pneumoniae infection may lead to increased circulating cholesterol and promote atherosclerosis.
In contrast to liver cells, C. pneumoniae increased LDL uptake in infected human monocyte-derived macrophages (Kalayoglu and Byrne, 1998). This led to formation of foam cells, which are lipid-laden cells important in atherosclerosis progression. LDL uptake also increased approximately 2.5-fold in C. pneumoniae-infected human monocytes and human umbilical vein epithelial (HUVEC) cells (Yoshida et al., 2006; Evani and Ramasubramanian, 2016). In HUVEC cells, this was partially due to increased expression of cholesterol uptake receptors, including scavenger receptor A, LOX-1, and CD36, which all internalize oxidized low density lipoprotein (oxLDL) (Yoshida et al., 2006; Campbell et al., 2013; Sun et al., 2014). Interestingly, glycan from C. pneumoniae, but not C. trachomatis, bound and activated LOX-1 (Campbell et al., 2013). Recombinant C. pneumoniae Hsp60 also increased LOX-1 expression in endothelial cells of hypercholesterolemic rabbits, further suggesting that the bacteria actively interact with the LOX-1 pathway to increase LDL uptake (Lin et al., 2011).
Less is known about C. trachomatis manipulation of cholesterol metabolism. In human trophoblasts, C. trachomatis downregulated hmgr, leading to lower levels of cholesterol and the cholesterol-derived steroids estrogen and progesterone; this is hypothesized to impair trophoblast implantation and placentation during pregnancy (Azenabor et al., 2007). Although not the primary target organ, C. trachomatis has been found in mouse liver lesions and can cause perihepatitis in humans (Barteneva et al., 1996). Gene expression profiles in C. trachomatis-infected mouse livers reported upregulation of lxrα, lxrβ, and cyp7a1, suggesting cholesterol catabolism is elevated (Marangoni et al., 2015). C. trachomatis downregulated ldlr in human HepG2 hepatocellular cells, suggesting the bacteria decreases cholesterol uptake (Bashmakov et al., 2010). Cholesterol biosynthesis appears to be critical for C. trachomatis infection, as the HMGR inhibitor mevastatin reduced C. trachomatis growth (Bashmakov et al., 2010). The requirement for de novo host synthesized cholesterol would suggest sterol intermediates or cholesterol metabolites are important for C. trachomatis infection of liver cells.
C. burnetii
C. burnetii-infected Vero cells had a 73% increase in cellular cholesterol at 6 days post-infection, as compared to mock infected cells (Howe and Heinzen, 2006). Pharmaceutical inhibitors of either HMGR or LDL uptake blocked PV formation and bacterial growth, and both hmgr and ldlr were upregulated during C. burnetii infection of Vero cells (Howe and Heinzen, 2006). Intriguingly, this upregulation was not observed until 4 days post-infection, when the C. burnetii PV is large and the bacteria are in logarithmic growth. However, at 6 days post-infection the expression levels returned to the same level as mock-infected cells (Howe and Heinzen, 2006) indicating a temporal regulation of cholesterol biosynthesis and uptake. Supporting this hypothesis, C. burnetii PV size and growth were found most sensitive to cholesterol levels only during the first 2 days of infection (Mulye et al., 2017). As elevated cholesterol in the PV is bacteriolytic, it is possible that C. burnetii reduces PV cholesterol during early stages of infection and increases PV cholesterol later during PV maintenance. Given the role cholesterol plays in PV fusion with endosomes, as well as PV pH, C. burnetii most likely tightly regulates PV membrane cholesterol during the infectious cycle.
A. phagocytophilum
Infection of human premyelocytic leukemia cell line (HL-60) with A. phagocytophilum resulted in a 2-fold increase in total host cell cholesterol level (Xiong et al., 2009). Inhibitor studies and gene expression analyses revealed that A. phagocytophilum does not require de novo cholesterol synthesis but acquires cholesterol by upregulating LDLR at both the mRNA and the protein level (Xiong et al., 2009). Interestingly, A. phagocytophilum does not target SREBP, the primary transcription factor regulating ldlr expression. Instead, the 3′ end of the ldlr mRNA transcript is stabilized through an unknown mechanism (Xiong et al., 2009). However, the extracellular signal-regulated kinase (ERK) pathway appears to be involved, as ERK was upregulated during A. phagocytophilum infection, and inhibiting the upstream kinase MEK lowered ldlr expression levels and reduced bacterial infection (Xiong et al., 2009). How the bacterium targets this process still remains unknown, though it is clear that LDL uptake is essential for A. phagocytophilum pathogenesis.
Cholesterol Efflux
Excess cholesterol can be exported out of the cell by the ATP-binding cassette transporters ABCA1 and ABCG1. ABCG1 is found primarily in endosomes, while ABCA1 cycles between endosomes and the plasma membrane (Neufeld et al., 2001; Tarling and Edwards, 2011). While both ABCA1 and ABCG1 transfer cholesterol to a number of extracellular particles, ABCA1 promotes HDL assembly at the plasma membrane through binding ApoA-1, a main component of HDL (Phillips, 2013). By targeting host cholesterol efflux pathways, intracellular pathogens can further fine-tune host cholesterol to benefit bacterial growth.
Chlamydia spp.
C. pneumoniae infection decreased cholesterol efflux by downregulating expression of ABCA1 in multiple cell types including A549 lung epithelial cell lines (Korhonen et al., 2013), LDL-treated HUVEC cells (Sun et al., 2014), and THP-1 macrophage-derived foam cells (Zhao et al., 2014). ABCG1 was also downregulated in C. pneumoniae-infected HUVECs (Sun et al., 2014). Experimental measurement of cholesterol efflux to ApoA-1 showed a 50% decrease in C. pneumoniae-infected THP-1 macrophage-like foam cells compared to uninfected or heat-killed bacteria-infected cells (Zhao et al., 2014). Further, C. pneumoniae appeared to downregulate host cholesterol efflux by increasing microRNA miR-33 levels, which is produced from the SREBP intron and downregulates ABCA1 (Zhao et al., 2014). Upregulation of miR-33 was triggered by the innate immune pattern recognition receptor TLR2 (toll like receptor 2) which activates NF-kB-mediated upregulation of miR-33 upon bacterial recognition (Zhao et al., 2014). While viable organisms were required for this process, it is unknown if the bacteria are directly activating TLR2 or if this is strictly a host immune response. However, these data collectively indicate that C. pneumoniae targets efflux as a mechanism to further increase the levels of intracellular cholesterol.
C. burnetii
C. burnetii differentially regulated apoE and apoC gene expression in THP-1 macrophages (Ren et al., 2003; Mahapatra et al., 2010). In addition, a genome-wide RNA interference screen in HeLa cells revealed that siRNA depletion of apolipoproteins involved in lipid transport, including ApoA2, ApoC4, ApoL1, ApoL2, and ApoL5, affected the total number of C. burnetii PVs (McDonough et al., 2013). This suggests that cholesterol efflux may play an important role during C. burnetii infection, although the precise mechanisms and purpose are unknown.
Cholesterol Storage
Eukaryotic cells store excess cholesterol in lipid droplets (LD), specialized organelles comprised of a phospholipid monolayer surrounding a neutral lipid core of esterified cholesterol and triacylglycerols. Prior to packaging in ER-derived LDs, excess cholesterol is esterified by acyl CoA transferase (ACAT). LDs are coated by a special class of proteins called perilipins, which help prevent LD breakdown (Listenberger et al., 2007). LDs serve as an important source of lipids for membrane synthesis or energy metabolism, as well as immune modulators. Finally, LD accumulation leads to foam cell formation, a hallmark of atherosclerosis.
Chlamydia spp.
Atherosclerosis and foam cell formation play a significant role in C. pneumoniae pathogenesis. C. pneumoniae infection increased ACAT1 expression, and therefore esterified cholesterol, in THP-1 cells (Liu et al., 2010). Along with the decreased cholesterol efflux discussed earlier, this resulted in cholesterol accumulation within the host cell and promoted foam cell formation. The transcription factors LXR and the peroxisome proliferator-activated receptors PPARα and PPARγ, which regulate the expression of acat1 and other cholesterol homeostasis genes such as abca1 and abcg1, have been implicated in C. pneumoniae-induced foam cell formation (Chen et al., 2008; Naiki et al., 2008; Mei et al., 2009; Liu et al., 2010). For example, C. pneumoniae downregulated PPARα and PPARγ by targeting the c-Jun N-terminal kinase (JNK) branch of the MAP kinase pathway, leading to increased expression of acat1, abca1 and abcg1 and promoting foam cell formation (Mei et al., 2009; Liu et al., 2010). Treatment with PPARα and PPARγ agonists reversed this effect. Thus, C. pneumoniae induces foam cell formation by manipulating a signal transduction pathway that regulates both LD formation (ACAT1) as well as cholesterol efflux (ABCA1/G1).
During C. trachomatis infection, LDs were found inside the inclusion and the number of cytoplasmic LDs increased (Cocchiaro et al., 2008; Saka et al., 2015). LDs co-localized with the C. trachomatis inclusion protein IncA at the inclusion membrane and the lumen, suggesting that IncA participates in LD translocation into the inclusion (Cocchiaro et al., 2008). In addition, three C. trachomatis LD-associated proteins were identified: Lda1, Lda2, and Lda3 (Kumar et al., 2006). While the role of Lda1 and Lda2 have yet to be elucidated, Lda3 was localized to the inclusion, cytoplasmic LDs, and LDs within the inclusion (Cocchiaro et al., 2008). Lda3 overexpression decreased PLIN2 association with LDs, suggesting Lda3 replaces PLIN2 on LDs to promote LD translocation into the inclusion. While the role of LD translocation into the inclusion lumen is not clear, LDs could serve as a source of energy or membrane for intracellular C. trachomatis (Cocchiaro et al., 2008). As discussed earlier, the putative C. trachomatis cholesterol esterase (CT149) may hydrolyze cholesterol esters, freeing cholesterol for bacterial use (Peters et al., 2012). Further supporting the importance of LDs during C. trachomatis infection, inhibiting LD formation with either pharmaceutical inhibitors or gene knockouts significantly blocked C. trachomatis growth in epithelial cells and fibroblasts (Kumar et al., 2006; Peters and Byrne, 2015; Saka et al., 2015; Recuero-Checa et al., 2016). A recent proteomic analysis discovered that the LD proteome was altered during infection with C. trachomatis, with an enrichment of host lipid metabolism and biosynthesis proteins (Saka et al., 2015). Three additional bacterial inclusion proteins, Cap1, CTL0882, and IncG, were also found in the LDs. Together, these studies indicate that C. trachomatis actively manipulates LD formation, composition, and trafficking, potentially as a source of energy or lipids.
C. burnetii
C. burnetii-containing foam cells have been found in heart valves of an infected patient (Brouqui et al., 1994), and LDs were observed in vitro in the C. burnetii PV lumen of infected human alveolar macrophages (Graham et al., 2013). LD formation may increase during infection, as the expression levels of the LD coat protein PLIN2 and fatty acid binding protein FABP4, which transfers fatty acids to the ER for packaging in LDs, were upregulated in infected THP-1 cells (Ren et al., 2003; Mahapatra et al., 2010). Furthermore, siRNA depletion of patatin-like phospholipase domain-containing protein 2 (PNPLA2), the phospholipase involved in LD breakdown, led to an increased number of C. burnetii PVs in HeLa cells (McDonough et al., 2013). A similar observation was made following depletion of the long chain fatty acyl-CoA ligase ACSL6, which is important in neutral lipid synthesis (McDonough et al., 2013; Teodoro et al., 2016). In addition, treatment of monkey kidney epithelial cells (Vero cells) with an LD-localized broad spectrum antiviral molecule ST699 inhibited C. burnetii intracellular growth (Sandoz et al., 2014). These data point to a role for LD homeostasis during infection, though it has not been determined if LDs are indeed targeted by C. burnetii or if C. burnetii growth is altered when LD formation is blocked.
A. phagocytophilum
A gene expression profiling study in A. phagocytophilum-infected HL-60 cells revealed increased expression of the major LD protein PLIN1 (de la Fuente et al., 2005; Manzano-Roman et al., 2008). Further studies revealed PLIN1 expression increased with bacterial replication, and siRNA knockdown of PLIN1 led to a 50% decrease in A. phagocytophilum replication (Manzano-Roman et al., 2008). As PLIN proteins are critical for LD formation, LDs most likely play an important role during A. phagocytophilim infection (Tansey et al., 2004; Brasaemle, 2007).
Rickettsia spp.
O. tsutsugamushi induced LD formation in mouse L-929 fibroblast cells (Ogawa et al., 2014). Although the difference in cholesterol ester levels was not determined, lipid composition analysis revealed O. tsutugamushi-induced LDs were enriched in triacylglycerols and could serve as an energy source for the bacteria. While these studies suggest that O. tsutsugamushi induce LD accumulation, their contribution to bacterial intracellular growth is yet to be determined.
Conclusion
For obligate intracellular bacteria, entry into the host cell and subsequent formation and maintenance of a vacuolar or cytoplasmic niche is essential for pathogen growth and survival. Due to cholesterol's multiple cellular functions, intracellular pathogens target cholesterol to obtain nutrients, membrane, or manipulate cellular signaling. This bacteria-host cholesterol interaction occurs at various stages of infection including host cell binding and internalization, niche formation, intracellular replication, and dissemination. The pathogens discussed in this review have appreciably different lifestyles with unique ways to manipulate host cell cholesterol (Figure 1).
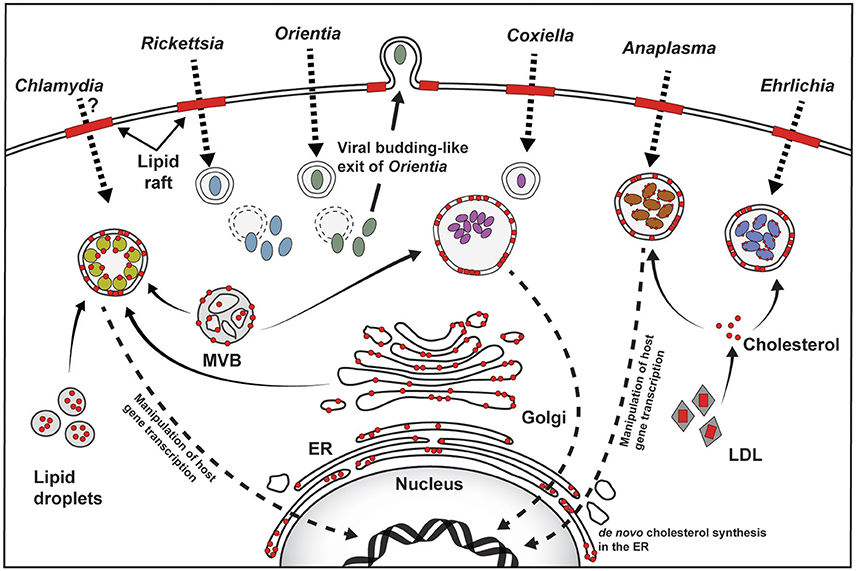
Figure 1. Overview of host cholesterol-pathogen interactions. For Rickettsia, Coxiella, Anaplasma, and Ehrlichia, attachment to the host cell plasma membrane and subsequent host cell entry involves cholesterol-rich lipid rafts (red bars). The role of lipid rafts for Chlamydia is unclear, and may be species- or host cell-specific, while Orientia utilizes lipid rafts for exit. Cholesterol (shown as red circles) is found in the pathogen-containing vacuoles, as well as the bacterial membrane of Chlamydia, Anaplasma, and Ehrlichia. Golgi-derived vesicles, multivesicular bodies (MVB), and lipid droplets traffic to the Chlamydia inclusion and serve as source of cholesterol. The source of cholesterol in the Coxiella vacuole is not clear, but may involve MVBs, while Anaplasma and Ehrlichia intercept LDL-derived cholesterol. Chlamydia, Coxiella, and Anaplasma target host gene expression to manipulate cholesterol homeostasis (Dotted arrow pointing to the nucleus).
Chlamydia spp. initially interact with cholesterol-rich lipid rafts during host cell entry, followed by targeting multiple host cholesterol trafficking pathways in order to establish the intracellular niche. Cholesterol is required in both the Chlamydia envelope and the inclusion membrane. While it has not been definitively demonstrated, cholesterol most likely plays a structural role in the bacterial envelope. Inclusion membrane cholesterol serves at least two purposes. First, inclusion cholesterol-rich microdomains contain both bacterial and host proteins involved in microtubule-dependent trafficking of the inclusion. Second, the cholesterol-binding protein caveolin may facilitate interactions between the inclusion and nutrient-rich endosomes and vesicles. In order to increase cholesterol availability, Chlamydia manipulates host cell cholesterol homeostasis. Both C. trachomatis and C. pneumoniae induce LD accumulation, which potentially provides lipids for bacterial growth. LDs appear particularly important to C. trachomatis, with bacterial proteins possibly facilitating translocation and breakdown of LDs in the inclusion lumen. C. pneumoniae reprograms cholesterol metabolism, increasing cholesterol levels that contribute to C. pneumoniae-mediated atherosclerosis and foam cell formation.
While Chlamydia spp. inclusions divert from the endocytic pathway, the C. burnetii niche is phagolysosome-like and highly fusogenic with host endosomes and autophagosomes. C. burnetii requires lipid rafts for entry into non-phagocytic cells, although the role of lipid rafts in macrophages has not been determined. Once inside the cell, the cholesterol requirement of C. burnetii appears complex and time-dependent. Due to the fusogenicity of the PV, it is likely that multiple sources of cholesterol traffic to the PV, though it is unknown how cholesterol trafficking or cholesterol levels change during PV development and maintenance. The bacteria are most sensitive to cholesterol during the initial stages of infection, before the PV is fully established. One possibility is that cholesterol regulates the C. burnetii T4SS, which secretes bacterial effector proteins necessary for PV expansion. Cholesterol also clearly influences PV pH, which is known to play an important role in C. burnetii metabolism. C. burnetii appears to have multiple mechanisms to manipulate host cholesterol levels, including targeting cholesterol efflux and storage. Further, C. burnetii may enzymatically modify cholesterol, or use membrane contact sites to transfer cholesterol from the PV to the host ER. Finally, our current knowledge of C. burnetii manipulation of host cholesterol is based on experiments with an avirulent strain, which contains truncated LPS compared to virulent bacteria. Studies with virulent bacteria will enable us to link host cholesterol to disease outcome during C. burnetii infection.
The importance of cholesterol during A. phagocytophilum, E. chaffeensis, and Rickettsia spp. infection is comparatively understudied. A. phagocytophilum and E. chaffeensis utilize lipid rafts during host cell entry. While A. phagocytophilum obtains cholesterol by hijacking the host LDL uptake and NPC1 trafficking, not much is known about E. chaffeensis and its effect on host cholesterol metabolism. Unlike the other discussed pathogens, Rickettsia spp. replicate freely in the cytoplasm and therefore have different cholesterol requirements than vacuolar pathogens. R. conorii targets cholesterol during entry, whereas O. tsutsugamushi requires it during egress from the host cell. Although O. tsutsugamushi infection increases LD accumulation, if and how these Rickettsia spp. target host cholesterol metabolism remains unknown.
Regardless of different bacterial life cycles and host pathways targeted, host cholesterol manipulation at varying stages of these intracellular life cycles seems to be the unifying theme (Table 1). Pathogen-mediated manipulation of host cell cholesterol metabolism still remains understudied, with a focus on gene expression analysis and little functional data. An additional limitation is a lack of in vivo data, which will be critical to fully understanding the role of cholesterol during infection. Finally, several questions still remain unanswered: (1) Is cholesterol manipulation cell type-dependent? (2) Can other sterols substitute for cholesterol? (3) What is the advantage of incorporating host cholesterol in the bacterial membrane? (4) What are the bacterial proteins responsible for manipulating host cholesterol? (5) Is disease outcome influenced by the patient cholesterol levels? Answering these and other questions will provide significant insight into the role of cholesterol during pathogenesis of obligate intracellular bacteria.
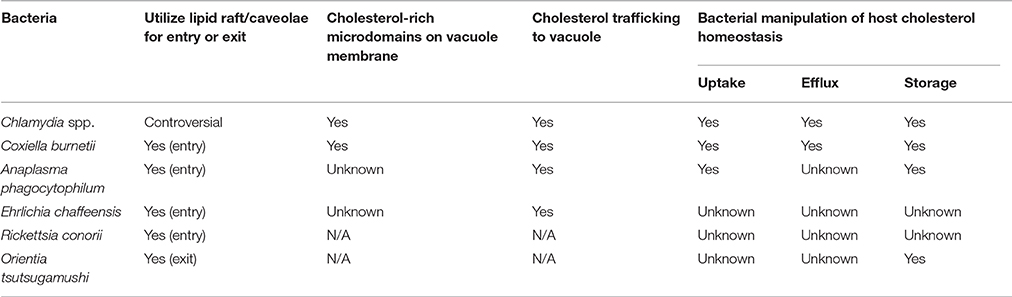
Table 1. Summary of manipulation of host cholesterol by obligate intracellular bacterial pathogens.
Author Contributions
All authors listed have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Seth Winfree for critical reading of this manuscript. This work was supported by the American Heart Association (14SDG18420034 to SG and 16POST27250157 to MM), the National Institutes of Health (AI121786 to SG and T32AI060519 to AJ), and the Showalter Trust (SG).
References
Al-Khedery, B., Lundgren, A. M., Stuen, S., Granquist, E. G., Munderloh, U. G., Nelson, C. M., et al. (2012). Structure of the type IV secretion system in different strains of Anaplasma phagocytophilum. BMC Genomics 13:678. doi: 10.1186/1471-2164-13-678
Azenabor, A. A., Kennedy, P., and Balistreri, S. (2007). Chlamydia trachomatis infection of human trophoblast alters estrogen and progesterone biosynthesis: an insight into role of infection in pregnancy sequelae. Int. J. Med. Sci. 4, 223–231. doi: 10.7150/ijms.4.223
Barnewall, R. E., Rikihisa, Y., and Lee, E. H. (1997). Ehrlichia chaffeensis inclusions are early endosomes which selectively accumulate transferrin receptor. Infect. Immun. 65, 1455–1461.
Barteneva, N., Theodor, I., Peterson, E. M., and de la Maza, L. M. (1996). Role of neutrophils in controlling early stages of a Chlamydia trachomatis infection. Infect. Immun. 64, 4830–4833.
Bashmakov, Y. K., Zigangirova, N. A., Pashko, Y. P., Kapotina, L. N., and Petyaev, I. M. (2010). Chlamydia trachomatis growth inhibition and restoration of LDL-receptor level in HepG2 cells treated with mevastatin. Comp. Hepatol. 9:3. doi: 10.1186/1476-5926-9-3
Bastidas, R. J., Elwell, C. A., Engel, J. N., and Valdivia, R. H. (2013). Chlamydial intracellular survival strategies. Cold Spring Harb. Perspect. Med. 3:a010256. doi: 10.1101/cshperspect.a010256
Beare, P. A., Unsworth, N., Andoh, M., Voth, D. E., Omsland, A., Gilk, S. D., et al. (2009). Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect. Immun. 77, 642–656. doi: 10.1128/IAI.01141-08
Beatty, W. L. (2006). Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J. Cell Sci. 119, 350–359. doi: 10.1242/jcs.02733
Beatty, W. L. (2008). Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infect. Immun. 76, 2872–2881. doi: 10.1128/IAI.00129-08
Beyer, A. R., Truchan, H. K., May, L. J., Walker, N. J., Borjesson, D. L., and Carlyon, J. A. (2015). The Anaplasma phagocytophilum effector AmpA hijacks host cell SUMOylation. Cell. Microbiol. 17, 504–519. doi: 10.1111/cmi.12380
Bickel, P. E., Scherer, P. E., Schnitzer, J. E., Oh, P., Lisanti, M. P., and Lodish, H. F. (1997). Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem. 272, 13793–13802. doi: 10.1074/jbc.272.21.13793
Brasaemle, D. L. (2007). Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48, 2547–2559. doi: 10.1194/jlr.R700014-JLR200
Brouqui, P., Dumler, J. S., and Raoult, D. (1994). Immunohistologic demonstration of Coxiella burnetii in the valves of patients with Q fever endocarditis. Am. J. Med. 97, 451–458. doi: 10.1016/0002-9343(94)90325-5
Brown, D. A., and Rose, J. K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533–544. doi: 10.1016/0092-8674(92)90189-J
Campbell, L. A., Lee, A. W., Rosenfeld, M. E., and Kuo, C. C. (2013). Chlamydia pneumoniae induces expression of pro-atherogenic factors through activation of the lectin-like oxidized LDL receptor-1. Pathog. Dis. 69, 1–6. doi: 10.1111/2049-632X.12058
Campbell, S., Richmond, S. J., and Yates, P. (1989a). The development of Chlamydia trachomatis inclusions within the host eukaryotic cell during interphase and mitosis. J. Gen. Microbiol. 135, 1153–1165. doi: 10.1099/00221287-135-5-1153
Campbell, S., Richmond, S. J., and Yates, P. S. (1989b). The effect of Chlamydia trachomatis infection on the host cell cytoskeleton and membrane compartments. J. Gen. Microbiol. 135, 2379–2386. doi: 10.1099/00221287-135-9-2379
Capo, C., Lindberg, F. P., Meconi, S., Zaffran, Y., Tardei, G., Brown, E. J., et al. (1999). Subversion of monocyte functions by Coxiella burnetii: impairment of the cross-talk between alphavbeta3 integrin and CR3. J. Immunol. 163, 6078–6085.
Carabeo, R. A., Mead, D. J., and Hackstadt, T. (2003). Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc. Natl. Acad. Sci. U.S.A. 100, 6771–6776. doi: 10.1073/pnas.1131289100
Chakraborty, S., Veettil, M. V., Bottero, V., and Chandran, B. (2012). Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. U.S.A. 109, E1163–E1172. doi: 10.1073/pnas.1119592109
Chang, W. J., Rothberg, K. G., Kamen, B. A., and Anderson, R. G. (1992). Lowering the cholesterol content of MA104 cells inhibits receptor-mediated transport of folate. J. Cell Biol. 118, 63–69. doi: 10.1083/jcb.118.1.63
Chen, S., Sorrentino, R., Shimada, K., Bulut, Y., Doherty, T. M., Crother, T. R., et al. (2008). Chlamydia pneumoniae-induced foam cell formation requires MyD88-dependent and -independent signaling and is reciprocally modulated by liver X receptor activation. J. Immunol. 181, 7186–7193. doi: 10.4049/jimmunol.181.10.7186
Chun, M., Liyanage, U. K., Lisanti, M. P., and Lodish, H. F. (1994). Signal transduction of a G protein-coupled receptor in caveolae: colocalization of endothelin and its receptor with caveolin. Proc. Natl. Acad. Sci. U.S.A. 91, 11728–11732. doi: 10.1073/pnas.91.24.11728
Cianciola, N. L., Greene, D. J., Morton, R. E., and Carlin, C. R. (2013). Adenovirus RIDα uncovers a novel pathway requiring ORP1L for lipid droplet formation independent of NPC1. Mol. Biol. Cell 24, 3309–3325. doi: 10.1091/mbc.E12-10-0760
Clifton, D. R., Fields, K. A., Grieshaber, S. S., Dooley, C. A., Fischer, E. R., Mead, D. J., et al. (2004). A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. U.S.A. 101, 10166–10171. doi: 10.1073/pnas.0402829101
Cocchiaro, J. L., Kumar, Y., Fischer, E. R., Hackstadt, T., and Valdivia, R. H. (2008). Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. U.S.A. 105, 9379–9384. doi: 10.1073/pnas.0712241105
Colello, D., Reverte, C. G., Ward, R., Jones, C. W., Magidson, V., Khodjakov, A., et al. (2010). Androgen and Src signaling regulate centrosome activity. J. Cell Sci. 123, 2094–2102. doi: 10.1242/jcs.057505
Cox, B. E., Griffin, E. E., Ullery, J. C., and Jerome, W. G. (2007). Effects of cellular cholesterol loading on macrophage foam cell lysosome acidification. J. Lipid Res. 48, 1012–1021. doi: 10.1194/jlr.M600390-JLR200
Cox, J. V., Naher, N., Abdelrahman, Y. M., and Belland, R. J. (2012). Host HDL biogenesis machinery is recruited to the inclusion of Chlamydia trachomatis-infected cells and regulates chlamydial growth. Cell. Microbiol. 14, 1497–1512. doi: 10.1111/j.1462-5822.2012.01823.x
de la Fuente, J., Ayoubi, P., Blouin, E. F., Almazán, C., Naranjo, V., and Kocan, K. M. (2005). Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell. Microbiol. 7, 549–559. doi: 10.1111/j.1462-5822.2004.00485.x
Eden, E. R., Sanchez-Heras, E., Tsapara, A., Sobota, A., Levine, T. P., and Futter, C. E. (2016). Annexin A1 tethers membrane contact sites that mediate ER to endosome cholesterol transport. Dev. Cell 37, 473–483. doi: 10.1016/j.devcel.2016.05.005
Evani, S. J., and Ramasubramanian, A. K. (2016). Biophysical regulation of Chlamydia pneumoniae-infected monocyte recruitment to atherosclerotic foci. Sci. Rep. 6:19058. doi: 10.1038/srep19058
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 101, 890–898. doi: 10.1172/JCI1112
Fechtner, T., Galle, J. N., and Hegemann, J. H. (2016). The novel chlamydial adhesin CPn0473 mediates the lipid raft-dependent uptake of Chlamydia pneumoniae. Cell. Microbiol. 18, 1094–1105. doi: 10.1111/cmi.12569
Gabel, B. R., Elwell, C., van Ijzendoorn, S. C. D., and Engel, J. N. (2004). Lipid raft-mediated entry is not required for Chlamydia trachomatis infection of cultured epithelial. Cells 72, 7367–7373. doi: 10.1128/iai.72.12.7367-7373.2004
Geisler, W. M., Suchland, R. J., Rockey, D. D., and Stamm, W. E. (2001). Epidemiology and clinical manifestations of unique Chlamydia trachomatis isolates that occupy nonfusogenic inclusions. J. Infect. Dis. 184, 879–884. doi: 10.1086/323340
Gilk, S. D., Beare, P. A., and Heinzen, R. A. (2010). Coxiella burnetii expresses a functional Δ24 sterol reductase. J. Bacteriol. 192, 6154–6159. doi: 10.1128/JB.00818-10
Gilk, S. D., Cockrell, D. C., Luterbach, C., Hansen, B., Knodler, L. A., Ibarra, J. A., et al. (2013). Bacterial colonization of host cells in the absence of cholesterol. PLoS Pathog. 9:e1003107. doi: 10.1371/journal.ppat.1003107
Gorodinsky, A., and Harris, D. A. (1995). Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J. Cell Biol. 129, 619–627. doi: 10.1083/jcb.129.3.619
Graham, J. G., Macdonald, L. J., Hussain, S. K., Sharma, U. M., Kurten, R. C., and Voth, D. E. (2013). Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell. Microbiol. 15, 1012–1025. doi: 10.1111/cmi.12096
Grieshaber, S. S., Grieshaber, N. A., and Hackstadt, T. (2003). Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J. Cell Sci. 116, 3793–3802. doi: 10.1242/jcs.00695
Hackstadt, T., Rockey, D. D., Heinzen, R. A., and Scidmore, M. A. (1996). Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 15, 964–977.
Hackstadt, T., Scidmore, M. A., and Rockey, D. D. (1995). Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc. Natl. Acad. Sci. U.S.A. 92, 4877–4881. doi: 10.1073/pnas.92.11.4877
Hackstadt, T., and Williams, J. C. (1981). Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 78, 3240–3244. doi: 10.1073/pnas.78.5.3240
Hatch, G. M., and McClarty, G. (1998). Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect. Immun. 66, 3727–3735.
Heinzen, R. A., Scidmore, M. A., Rockey, D. D., and Hackstadt, T. (1996). Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect. Immun. 64, 796–809.
Heinzen, R. A., Hayes, S. F., Peacock, M. G., and Hackstadt, T. (1993). Directional actin polymerization associated with spotted fever group Rickettsia infection of vero cells. Infect Immun 61, 1926–1935.
Hillman, R. D. Jr., Baktash, Y. M., and Martinez, J. J. (2013). OmpA-mediated rickettsial adherence to and invasion of human endothelial cells is dependent upon interaction with α2β1 integrin. Cell. Microbiol. 15, 727–741. doi: 10.1111/cmi.12068
Howe, D., and Heinzen, R. A. (2006). Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell. Microbiol. 8, 496–507. doi: 10.1111/j.1462-5822.2005.00641.x
Howe, D., and Mallavia, L. P. (2000). Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect. Immun. 68, 3815–3821. doi: 10.1128/IAI.68.7.3815-3821.2000
Howe, D., Melnicákova, J., Barák, I., and Heinzen, R. A. (2003). Fusogenicity of the Coxiella burnetii parasitophorous vacuole. Ann. N.Y. Acad. Sci. 990, 556–562. doi: 10.1111/j.1749-6632.2003.tb07426.x
Justis, A. V., Hansen, B., Beare, P. A., King, K. B., Heinzen, R. A., and Gilk, S. D. (2017). Interactions between the Coxiella burnetii parasitophorous vacuole and the endoplasmic reticulum involve the host protein ORP1L. Cell. Microbiol. 19:e12637. doi: 10.1111/cmi.12637
Jutras, I., Abrami, L., and Dautry-Varsat, A. (2003). Entry of the Lymphogranuloma Venereum strain of Chlamydia trachomatis into host cells involves cholesterol-rich membrane domains. Infect. Immun. 71, 260–266. doi: 10.1128/IAI.71.1.260-266.2003
Kalayoglu, M. V., and Byrne, G. I. (1998). Induction of macrophage foam cell formation by Chlamydia pneumoniae. J. Infect. Dis. 177, 725–729. doi: 10.1086/514241
Karten, B., Peake, K. B., and Vance, J. E. (2009). Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1791, 659–670. doi: 10.1016/j.bbalip.2009.01.025
Kim, M. J., Kim, M. K., and Kang, J. S. (2013). Involvement of lipid rafts in the budding-like exit of Orientia tsutsugamushi. Microb. Pathog. 63, 37–43. doi: 10.1016/j.micpath.2013.06.002
Koike, M. (2002). Dimerization, translocation and localization of Ku70 and Ku80 proteins. J. Radiat. Res. 43, 223–236. doi: 10.1269/jrr.43.223
Korhonen, J. T., Olkkonen, V. M., Lahesmaa, R., and Puolakkainen, M. (2013). ABC-cassette transporter 1 (ABCA1) expression in epithelial cells in Chlamydia pneumoniae infection. Microb. Pathog. 61–62, 57–61. doi: 10.1016/j.micpath.2013.05.006
Korhonen, J. T., Puolakkainen, M., Haveri, A., Tammiruusu, A., Sarvas, M., and Lahesmaa, R. (2012). Chlamydia pneumoniae entry into epithelial cells by clathrin-independent endocytosis. Microb. Pathog. 52, 157–164. doi: 10.1016/j.micpath.2011.12.002
Kumar, Y., Cocchiaro, J., and Valdivia, R. H. (2006). The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr. Biol. 16, 1646–1651. doi: 10.1016/j.cub.2006.06.060
Lai, E. C. (2003). Lipid rafts make for slippery platforms. J. Cell Biol. 162, 365–370. doi: 10.1083/jcb.200307087
Levi, M., and Shalgi, R. (2010). The role of Fyn kinase in the release from metaphase in mammalian oocytes. Mol. Cell. Endocrinol. 314, 228–233. doi: 10.1016/j.mce.2009.08.027
Lin, F. Y., Lin, Y. W., Huang, C. Y., Chang, Y. J., Tsao, N. W., Chang, N. C., et al. (2011). GroEL1, a heat shock protein 60 of Chlamydia pneumoniae, induces lectin-like oxidized low-density lipoprotein receptor 1 expression in endothelial cells and enhances atherogenesis in hypercholesterolemic rabbits. J. Immunol. 186, 4405–4414. doi: 10.4049/jimmunol.1003116
Lin, M., Liu, H., Xiong, Q., Niu, H., Cheng, Z., Yamamoto, A., et al. (2016). Ehrlichia secretes Etf-1 to induce autophagy and capture nutrients for its growth through RAB5 and class III phosphatidylinositol 3-kinase. Autophagy 12, 2145–2166. doi: 10.1080/15548627.2016.1217369
Lin, M., and Rikihisa, Y. (2003a). Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect. Immun. 71, 5324–5331. doi: 10.1128/IAI.71.9.5324-5331.2003
Lin, M., and Rikihisa, Y. (2003b). Obligatory intracellular parasitism by Ehrlichia chaffeensis and Anaplasma phagocytophilum involves caveolae and glycosylphosphatidylinositol-anchored proteins. Cell. Microbiol. 5, 809–820. doi: 10.1046/j.1462-5822.2003.00322.x
Lisanti, M. P., Scherer, P. E., Vidugiriene, J., Tang, Z., Hermanowski-Vosatka, A., Tu, Y. H., et al. (1994). Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J. Cell Biol. 126, 111–126. doi: 10.1083/jcb.126.1.111
Listenberger, L. L., Ostermeyer-Fay, A. G., Goldberg, E. B., Brown, W. J., and Brown, D. A. (2007). Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J. Lipid Res. 48, 2751–2761. doi: 10.1194/jlr.M700359-JLR200
Liu, P., Ying, Y., Ko, Y. G., and Anderson, R. G. (1996). Localization of platelet-derived growth factor-stimulated phosphorylation cascade to caveolae. J. Biol. Chem. 271, 10299–10303. doi: 10.1074/jbc.271.17.10299
Liu, W., He, P., Cheng, B., Mei, C. L., Wang, Y. F., and Wan, J. J. (2010). Chlamydia pneumoniae disturbs cholesterol homeostasis in human THP-1 macrophages via JNK-PPAR? Dependent signal transduction pathways. Microbes Infect. 12, 1226–1235. doi: 10.1016/j.micinf.2010.09.004
Lucero, H., Gae, D., and Taccioli, G. E. (2003). Novel localization of the DNA-PK complex in lipid rafts. A putative role in the signal transduction pathway of the ionizing radiation response. J. Biol. Chem. 278, 22136–22143. doi: 10.1074/jbc.M301579200
Macurek, L., Dráberová, E., Richterová, V., Sulimenko, V., Sulimenko, T., Dráberová, L., et al. (2008). Regulation of microtubule nucleation from membranes by complexes of membrane-bound gamma-tubulin with Fyn kinase and phosphoinositide 3-kinase. Biochem. J. 416, 421–430. doi: 10.1042/BJ20080909
Mahapatra, S., Ayoubi, P., and Shaw, E. I. (2010). Coxiella burnetii Nine Mile II proteins modulate gene expression of monocytic host cells during infection. BMC Microbiol. 10:244. doi: 10.1186/1471-2180-10-244
Manzano-Roman, R., Almazán, C., Naranjo, V., Blouin, E. F., Kocan, K. M., and De La Fuente, J. (2008). Expression of perilipin in human promyelocytic cells in response to Anaplasma phagocytophilum infection results in modified lipid metabolism. J. Med. Microbiol. 57, 159–163. doi: 10.1099/jmm.0.47504-0
Marangoni, A., Donati, M., Cavrini, F., Aldini, R., Accardo, S., Sambri, V., et al. (2006). Chlamydia pneumoniae replicates in Kupffer cells in mouse model of liver infection. World J. Gastroenterol. 12, 6453–6457. doi: 10.3748/wjg.v12.i40.6453
Marangoni, A., Fiorino, E., Gilardi, F., Aldini, R., Scotti, E., Nardini, P., et al. (2015). Chlamydia pneumoniae acute liver infection affects hepatic cholesterol and triglyceride metabolism in mice. Atherosclerosis 241, 471–479. doi: 10.1016/j.atherosclerosis.2015.05.023
Martinez, J. J., Seveau, S., Veiga, E., Matsuyama, S., and Cossart, P. (2005). Ku70, a component of DNA-dependent protein kinase, is a mammalian receptor for Rickettsia conorii. Cell 123, 1013–1023. doi: 10.1016/j.cell.2005.08.046
McDonough, J. A., Newton, H. J., Klum, S., Swiss, R., Agaisse, H., and Roy, C. R. (2013). Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. MBio 4, 1–13. doi: 10.1128/mBio.00606-12
Mei, C., He, P., Cheng, B., Liu, W., Wang, Y. F., and Wan, J. J. (2009). Chlamydia pneumoniae induces macrophage-derived foam cell formation via PPAR alpha and PPAR gamma-dependent pathways. Cell Biol. Int. 33, 301–308. doi: 10.1016/j.cellbi.2008.12.002
Michelini, E., Donati, M., Aldini, R., Cevenini, L., Mezzanotte, L., Nardini, P., et al. (2012). Dual-color bioluminescent assay using infected HepG2 cells sheds new light on Chlamydia pneumoniae and human cytomegalovirus effects on human cholesterol 7α-hydroxylase (CYP7A1) transcription. Anal. Biochem. 430, 92–96. doi: 10.1016/j.ab.2012.08.003
Mineo, C., James, G. L., Smart, E. J., and Anderson, R. G. (1996). Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J. Biol. Chem. 271, 11930–11935. doi: 10.1074/jbc.271.20.11930
Mital, J., and Hackstadt, T. (2011). Diverse requirements for Src-family tyrosine kinases distinguish chlamydial species. mBio 2:e00031-11. doi: 10.1128/mBio.00031-11
Mital, J., Lutter, E. I., Barger, A. C., Dooley, C. A., and Hackstadt, T. (2015). Chlamydia trachomatis inclusion membrane protein CT850 interacts with the dynein light chain DYNLT1 (Tctex1). Biochem. Biophys. Res. Commun. 462, 165–170. doi: 10.1016/j.bbrc.2015.04.116
Mital, J., Miller, N. J., Fischer, E. R., and Hackstadt, T. (2010). Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell. Microbiol. 12, 1235–1249. doi: 10.1111/j.1462-5822.2010.01465.x
Moazed, T. C., Kuo, C. C., Grayston, J. T., and Campbell, L. A. (1998). Evidence of systemic dissemination of Chlamydia pneumoniae via macrophages in the mouse. J. Infect. Dis. 177, 1322–1325. doi: 10.1086/515280
Moffatt, J. H., Newton, P., and Newton, H. J. (2015). Coxiella burnetii: turning hostility into a home. Cell. Microbiol. 17, 621–631. doi: 10.1111/cmi.12432
Mölleken, K., Becker, E., and Hegemann, J. H. (2013). The Chlamydia pneumoniae invasin protein Pmp21 recruits the EGF receptor for host cell entry. PLoS Pathog. 9:e1003325. doi: 10.1371/journal.ppat.1003325
Mott, J., Barnewall, R. E., and Rikihisa, Y. (1999). Human granulocytic Ehrlichiosis agent and Ehrlichia chaffeensis reside in different cytoplasmic compartments in HL-60 cells. Infect. Immun. 67, 1368–1378.
Mulye, M., Samanta, D., Winfree, S., Heinzen, R. A., and Gilk, S. D. (2017). Elevated cholesterol in the Coxiella burnetii intracellular niche is bacteriolytic. MBio 8, e02313-16. doi: 10.1128/mbio.02313-16
Naiki, Y., Sorrentino, R., Wong, M. H., Michelsen, K. S., Shimada, K., Chen, S., et al. (2008). TLR/MyD88 and liver X receptor alpha signaling pathways reciprocally control Chlamydia pneumoniae-induced acceleration of atherosclerosis. J. Immunol. 181, 7176–7185. doi: 10.4049/jimmunol.181.10.7176
Neufeld, E. B., Remaley, A. T., Demosky, S. J., Stonik, J. A., Cooney, A. M., Comly, M., et al. (2001). Cellular localization and trafficking of the human ABCA1 transporter. J. Biol. Chem. 276, 27584–27590. doi: 10.1074/jbc.M103264200
Niu, H., Kozjak-Pavlovic, V., Rudel, T., and Rikihisa, Y. (2010). Anaplasma phagocytophilum Ats-1 Is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog. 6:e1000774. doi: 10.1371/journal.ppat.1000774
Niu, H., Rikihisa, Y., Yamaguchi, M., and Ohashi, N. (2006). Differential expression of VirB9 and VirB6 during the life cycle of Anaplasma phagocytophilum in human leucocytes is associated with differential binding and avoidance of lysosome pathway. Cell. Microbiol. 8, 523–534. doi: 10.1111/j.1462-5822.2005.00643.x
Norkin, L. C., Wolfrom, S. A., and Stuart, E. S. (2001). Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Exp. Cell Res. 266, 229–238. doi: 10.1006/excr.2001.5202
Ogawa, M., Fukasawa, M., Satoh, M., Hanada, K., Saijo, M., Uchiyama, T., et al. (2014). The intracellular pathogen Orientia tsutsugamushi responsible for scrub typhus induces lipid droplet formation in mouse fibroblasts. Microbes Infect. 16, 962–966. doi: 10.1016/j.micinf.2014.09.004
Orlandi, P. A., and Fishman, P. H. (1998). Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 141, 905–915. doi: 10.1083/jcb.141.4.905
Peters, J., and Byrne, G. I. (2015). Chlamydia trachomatis growth depends on eukaryotic cholesterol esterification and is affected by Acyl-CoA:cholesterol acyltransferase inhibition. Pathog. Dis. 73:ftv028. doi: 10.1093/femspd/ftv028
Peters, J., Onguri, V., Nishimoto, S. K., Marion, T. N., and Byrne, G. I. (2012). The Chlamydia trachomatis CT149 protein exhibits esterase activity in vitro and catalyzes cholesteryl ester hydrolysis when expressed in HeLa cells. Microbes Infect. 14, 1196–1204. doi: 10.1016/j.micinf.2012.07.020
Phillips, M. C. (2013). New insights into the determination of HDL structure by apolipoproteins: thematic review series: high density lipoprotein structure, function, and metabolism. J. Lipid Res. 54, 2034–2048. doi: 10.1194/jlr.R034025
Pike, L. J. (2003). Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667. doi: 10.1194/jlr.R200021-JLR200
Recuero-Checa, M. A., Sharma, M., Lau, C., Watkins, P. A., Gaydos, C. A., and Dean, D. (2016). Chlamydia trachomatis growth and development requires the activity of host Long-chain Acyl-CoA Synthetases (ACSLs). Sci. Rep. 6:23148. doi: 10.1038/srep23148
Ren, Q., Robertson, S. J., Howe, D., Barrows, L. F., and Heinzen, R. A. (2003). Comparative DNA microarray analysis of host cell transcriptional responses to infection by Coxiella burnetii or Chlamydia trachomatis. Ann. N.Y. Acad. Sci. 990, 701–713. doi: 10.1111/j.1749-6632.2003.tb07447.x
Richards, T. S., Knowlton, A. E., and Grieshaber, S. S. (2013). Chlamydia trachomatis homotypic inclusion fusion is promoted by host microtubule trafficking. BMC Microbiol. 13:185. doi: 10.1186/1471-2180-13-185
Rocha, N., Kuijl, C., Van Der Kant, R., Janssen, L., Houben, D., Janssen, H., et al. (2009). Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150Glued and late endosome positioning. J. Cell Biol. 185, 1209–1225. doi: 10.1083/jcb.200811005
Saka, H. A., Thompson, J. W., Chen, Y. S., Dubois, L. G., Haas, J. T., Moseley, A., et al. (2015). Chlamydia trachomatis infection leads to defined alterations to the lipid droplet proteome in epithelial cells. PLoS ONE 10:e0124630. doi: 10.1371/journal.pone.0124630
Sandoz, K. M., Valiant, W. G., Eriksen, S. G., Hruby, D. E., Allen, R. D., and Rockey, D. D. (2014). The broad-spectrum antiviral compound ST-669 restricts chlamydial inclusion development and bacterial growth and localizes to host cell lipid droplets within treated cells. Antimicrob. Agents Chemother. 58, 3860–3866. doi: 10.1128/AAC.02064-13
Sargiacomo, M., Sudol, M., Tang, Z., and Lisanti, M. P. (1993). Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J. Cell Biol. 122, 789–807. doi: 10.1083/jcb.122.4.789
Schubert, W., Frank, P. G., Razani, B., Park, D. S., Chow, C. W., and Lisanti, M. P. (2001). Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J. Biol. Chem. 276, 48619–48622. doi: 10.1074/jbc.C100613200
Seshadri, R., Paulsen, I. T., Eisen, J. A., Read, T. D., Nelson, K. E., Nelson, W. C., et al. (2003). Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 100, 5455–5460. doi: 10.1073/pnas.0931379100
Simons, K., and Toomre, D. (2000). Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39. doi: 10.1038/35036052
Stallmann, S., and Hegemann, J. H. (2016). The Chlamydia trachomatis Ctad1 invasin exploits the human integrin β1 receptor for host cell entry. Cell. Microbiol. 18, 761–775. doi: 10.1111/cmi.12549
Stang, E., Kartenbeck, J., and Parton, R. G. (1997). Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol. Biol. Cell 8, 47–57. doi: 10.1091/mbc.8.1.47
Stephens, R. S., Kalman, S., Lammel, C., Fan, J., Marathe, R., Aravind, L., et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759. doi: 10.1126/science.282.5389.754
Stuart, E. S., Webley, W. C., and Norkin, L. C. (2003). Lipid rafts, caveolae, caveolin-1, and entry by Chlamydiae into host cells. Exp. Cell Res. 287, 67–78. doi: 10.1016/S0014-4827(03)00059-4
Stüven, E., Porat, A., Shimron, F., Fass, E., Kaloyanova, D., Brügger, B., et al. (2003). Intra-golgi protein transport depends on a cholesterol balance in the lipid membrane. J. Biol. Chem. 278, 53112–53122. doi: 10.1074/jbc.M300402200
Subbarayal, P., Karunakaran, K., Winkler, A., and Rother, M. (2015). EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 11:e1004846. doi: 10.1371/journal.ppat.1004846
Sun, S., Cheng, B., Wu, X., Wu, Q., Qi, B., Wu, J., et al. (2014). Chlamydia pneumoniae disrupts lipid metabolism in human umbilical vein endothelial cells. Mol. Med. Rep. 10, 1150–1156. doi: 10.3892/mmr.2014.2295
Tansey, J., Sztalryd, C., Hlavin, E., Kimmel, A., and Londos, C. (2004). The central role of perilipin a in lipid metabolism and adipocyte lipolysis. IUBMB Life 56, 379–385. doi: 10.1080/15216540400009968
Tarling, E. J., and Edwards, P. A. (2011). ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl. Acad. Sci. U.S.A. 108, 19719–19724. doi: 10.1073/pnas.1113021108
Teodoro, B. G., Sampaio, I. H., Bomfim, L. H. M., Queiroz, A. L., Silveira, L. R., Souza, A. O., et al. (2016). Long-Chain Acyl-CoA Synthetase 6 regulates lipid synthesis and mitochondrial oxidative capacity in human and rat skeletal muscle. J. Physiol. 595, 677–693. doi: 10.1113/JP272962
Triantafilou, K., and Triantafilou, M. (2003). Lipid raft microdomains: key sites for Coxsackievirus A9 infectious cycle. Virology 317, 128–135. doi: 10.1016/j.virol.2003.08.036
Upla, P., Marjomaki, V., Kankaanpaa, P., Ivaska, J., Hyypia, T., van der Goot, F. G. et al. (2004). Clustering Induces a Lateral Redistribution of α2β1 Integrin from Membrane Rafts to Caveolae and Subsequent Protein Kinase C-dependent Internalization. Mol. Biol. Cell 15, 625–636. doi: 10.1091/mbc.E03-08-0588
van der Kant, R., Fish, A., Janssen, L., Janssen, H., Krom, S., Ho, N., et al. (2013). Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 126, 3462–3474. doi: 10.1242/jcs.129270
Van Ooij, C., Kalman, L., Van Ijzendoorn, S., Nishijima, M., Hanada, K., Mostov, K., et al. (2000). Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cell. Microbiol. 2, 627–637. doi: 10.1046/j.1462-5822.2000.00077.x
Voth, D. E., and Heinzen, R. A. (2007). Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell. Microbiol. 9, 829–840. doi: 10.1111/j.1462-5822.2007.00901.x
Walker, D. H., Feng, H. M., and Popov, V. L. (2001). Rickettsial phospholipase a2 as a pathogenic mechanism in a model of cell injury by typhus and spotted fever group rickettsiae. Am. J. Trop. Med. Hyg. 65, 936–942. doi: 10.4269/ajtmh.2001.65.936
Webster, P., IJdo, J. W., Chicoine, L. M., and Fikrig, E. (1998). The agent of human granulocytic ehrlichiosis resides in an endosomal compartment. J. Clin. Invest. 101, 1932–1941. doi: 10.1172/JCI1544
Wu, Y., Singh, S., Georgescu, M. M., and Birge, R. B. (2005). A role for Mer tyrosine kinase in αvβ5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 118, 539–553. doi: 10.1242/jcs.01632
Wylie, J. L., Hatch, G. M., and McClarty, G. (1997). Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J. Bacteriol. 179, 7233–7242. doi: 10.1128/jb.179.23.7233-7242.1997
Xiong, Q., Lin, M., and Rikihisa, Y. (2009). Cholesterol-dependent Anaplasma phagocytophilum exploits the low-density lipoprotein uptake pathway. PLoS Pathog. 5:e1000329. doi: 10.1371/journal.ppat.1000329
Xiong, Q., and Rikihisa, Y. (2012). Subversion of NPC1 pathway of cholesterol transport by Anaplasma phagocytophilum. Cell. Microbiol. 14, 560–576. doi: 10.1111/j.1462-5822.2011.01742.x
Yoshida, T., Koide, N., Mori, I., Ito, H., and Yokochi, T. (2006). Chlamydia pneumoniae infection enhances lectin-like oxidized low-density lipoprotein receptor (LOX-1) expression on human endothelial cells. FEMS Microbiol. Lett. 260, 17–22. doi: 10.1111/j.1574-6968.2006.00286.x
Zhao, G., Mo, Z. C., Tang, S. L., Ouyang, X. P., He, P., Lv, Y., et al. (2014). Chlamydia pneumoniae negatively regulates ABCA1 expression via TLR2-Nuclear factor-kappa B and miR-33 pathways in THP-1 macrophage-derived foam cells. Atherosclerosis 235, 519–525. doi: 10.1016/j.atherosclerosis.2014.05.943
Keywords: cholesterol, lipid raft, Chlamydia, Coxiella, Rickettsia, Anaplasma, lipid droplet
Citation: Samanta D, Mulye M, Clemente TM, Justis AV and Gilk SD (2017) Manipulation of Host Cholesterol by Obligate Intracellular Bacteria. Front. Cell. Infect. Microbiol. 7:165. doi: 10.3389/fcimb.2017.00165
Received: 06 January 2017; Accepted: 18 April 2017;
Published: 05 May 2017.
Edited by:
Rey Carabeo, Washington State University, USAReviewed by:
Eric Ghigo, Centre National de la Recherche Scientifique, FranceChristopher Thompson, Imperial College London, UK
Copyright © 2017 Samanta, Mulye, Clemente, Justis and Gilk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stacey D. Gilk, sgilk@iupui.edu