Identification of Conserved ABC Importers Necessary for Intracellular Survival of Legionella pneumophila in Multiple Hosts
 Amrita Lama
Amrita Lama  Rudd C. Johnson
Rudd C. Johnson Grace L. Rubenstein
Grace L. Rubenstein Eric D. Cambronne
Eric D. Cambronne- Department of Molecular Microbiology and Immunology, Oregon Health and Science University, Portland, OR, United States
It is established that the human pathogen Legionella pneumophila becomes significantly augmented for infection of macrophages after intracellular growth in amoebae when compared to like-strains cultivated in laboratory media. Based on this observation, we reasoned that the most critical virulence determinants of L.p. are expressed by responding to stimuli generated by the protozoan host specifically; a process we term “protozoan-priming.” We sought to identify L.p. virulence factors that were required for replication in amoebae in order to highlight the genes necessary for production of the most infectious form of the bacterium. Using a transposon mutagenesis screen, we successfully identified 12 insertions that produced bacteria severely attenuated for growth in amoebae, while retaining a functional Dot/Icm type IVb secretion system. Seven of these insertion mutants were found dispensable for growth in macrophages, revealing attractive therapeutic targets that reside upstream of the pathogen-human interface. Two candidates identified, lpg0730 and lpg0122 were required for survival and replication in amoebae and macrophage host cells. Both genes are conserved among numerous important human pathogenic bacteria that can persist or replicate in amoebae. Each gene encodes a component of an ATP binding cassette (ABC) transport complex of unknown function. We demonstrate the lpg0730 ortholog in Francisella tularensis subsp. novicida to be essential for colonization of both protozoan and mammalian host cells, highlighting conserved survival mechanisms employed by bacteria that utilize protozoa as an environmental reservoir for replication.
Introduction
Legionella pneumophila (L.p.) is a Gram-negative bacterium predominantly associated with freshwater environments. Free-living bacteria persist in water, yet replication is restricted to the confines of host protozoan cells (amoebae), where the bacterium is a facultative intracellular parasite (Fields, 1996; Abu Kwaik et al., 1998). It is an opportunistic human pathogen, where egress from host amoebae generates the most infectious form of the bacterium (Cirillo et al., 1994, 1999; Brieland et al., 1996). Aerosolization of contaminated water sources provides an invariant route of transmission, providing access to resident lung alveolar macrophages. Intracellular colonization and replication in host macrophages occurs in a similar fashion to the bacterial life-cycle in protozoa, manifesting a spectrum of pathologies collectively termed “legionellosis.” The range of presentation, which is largely owed to host immune-competence, includes self-limited flu-like illness (Pontiac fever) to a severe and often fatal pneumonia called Legionnaires' disease (Burillo et al., 2017).
In order to survive and replicate in eukaryotic cells, L.p. requires a specialized type IVb secretion system termed Dot/Icm (Marra et al., 1992; Berger and Isberg, 1993; Segal and Shuman, 1997). This multi-protein secretion apparatus functions to deliver up to 300 effector proteins into the host-cell (Zhu et al., 2011). Effectors collectively function in the subversion of host-cellular processes to facilitate establishment of a replication-permissive compartment, often termed Legionella-containing vacuole (LCV) (Roy, 2002). Strains defective for Dot/Icm-mediated transport of effectors are rendered largely avirulent (Roy et al., 1998). L.p. developed these intracellular survival strategies through close associations with protozoa in the environment. Strategies adopted for survival in protozoa therefore directly translated to efficient replication in the hostile environment encountered within the macrophage of the lung.
Importantly, evidence dating back over 20 years demonstrated that L.p. cultivated in amoebae prior to infection of macrophage cell lines or murine hosts were both hyper-invasive and hyper-virulent; upwards of 100-fold when compared to like-strains cultured in laboratory media; a phenomenon we term “protozoan-priming” (Figure 1) (Cirillo et al., 1994, 1999; Brieland et al., 1996, 1997a,b; Drennan et al., 2013). This effect has been experimentally ascribed to a transition from an intracellular replicative phase to a non-dividing transmissive form (Molofsky and Swanson, 2004). Similarly “transmissive” bacteria have been modeled in vitro by culturing L.p. to early stationary phase. Based on these early observations however, the discrepancy between the virulence properties associated with protozoan-primed vs. in vitro cultured bacteria could be exploited to reveal key determinants for L.p. dissemination.
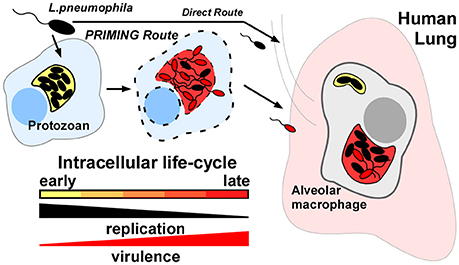
Figure 1. Protozoan-primed Legionella pneumophila are augmented for dissemination to human macrophages. L.p. strains that have completed an intracellular life-cycle in natural-host protozoa are up to 100x more infectious (Priming route) than like-strains cultivated in laboratory medium or persisting in freshwater (Direct route). Response to specific host-cellular cues must trigger expression of key virulence determinants for alveolar macrophage infection in the human lung.
We speculated that L.p. must respond to environmental stimuli provided exclusively by the protozoan host-cell in order to activate the expression of genes that contribute to the observed augmented infection phenotypes. With this notion in mind, we first sought to comprehensively identify genes necessary for intracellular survival in amoebae specifically, to potentially uncover therapeutic targets that would prevent generation of the highly virulent and most biologically relevant form of L.p. in the context of human infection. Additionally, these targets would have potential to reside upstream of the bacterial-human interface.
To this end, we conducted a genome-wide transposon mutagenesis screen in L.p. using intracellular survival and replication in the model protozoan Acanthamoeba castellanii (A.c.) as a primary evaluation criterion (Holden et al., 1984). Mutants that were attenuated or failed to replicate in A.c. (as determined using fluorescence microscopy) were next subjected to additional rounds of screening. Potential dot/icm insertions were selected-against based on sensitivity to growth on artificial media containing sodium chloride, and confirmed for a functional Dot/Icm transporter using an adenylate-cyclase reporter assay (Vogel et al., 1996; Cambronne and Roy, 2007). Host-cell specificity among several candidate insertions was demonstrated by examining intracellular replication in additional host cell types including murine and human macrophages.
Of particular interest were insertions in lpg0730 and lpg0122, each encoding a structural component of a distinct ATP binding cassette (ABC) transport complex (Theodoulou and Kerr, 2015). The assignment of substrate to each transport complex is unresolved. However, we found both loci conserved among several bacterial pathogens that can utilize protozoa as an intermediate reservoir for proliferation and transmission in the environment. Further, we demonstrated that disruption of the lpg0730 ortholog in Francisella tularensis subsp. novicida was essential for colonization of both protozoan and mammalian host-cells. Our data suggest that Lpg0730-containing ABC transport complexes therefore represent a conserved intracellular survival determinant that represents an attractive target for inhibiting proliferation in environmental host cells.
Results
Construction and Screening of L. pneumophila Mutant Library
We first generated a fluorescently-tractable isogenic L.p. strain harboring a single copy of gfpmut3 on the chromosome that would serve as a wild-type representation for mutagenesis (JR32::gfp) (Cormack et al., 1996). GFP production was driven by dual promoters in tandem. The isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible tac promoter was located immediately 5′ to the icmR promoter; active in early stationary phase (Neild and Roy, 2003). The gfp construct was inserted 3′ to the stop codon of the wipA effector locus (Figure 2A). This location was chosen due to the large 710 bp stretch of non-coding sequence 3′ to the monocistronic wipA. WipA, which was recently reported to harbor tyrosine phosphatase activity had been previously determined dispensable for intracellular survival of L.p. in both macrophage and amoebae (Ninio et al., 2005; Pinotsis and Waksman, 2017). We additionally constructed an isogenic ΔdotA::gfp strain for use as a negative control for intracellular replication (Roy et al., 1998). Both JR32::gfp and JR32ΔdotA::gfp produced GFP when cultured in vitro in the presence of IPTG (Figure 2B/insets). However, WT was the only strain capable of supporting intracellular replication, which could be detected using fluorescence microscopy (Figure 2B).
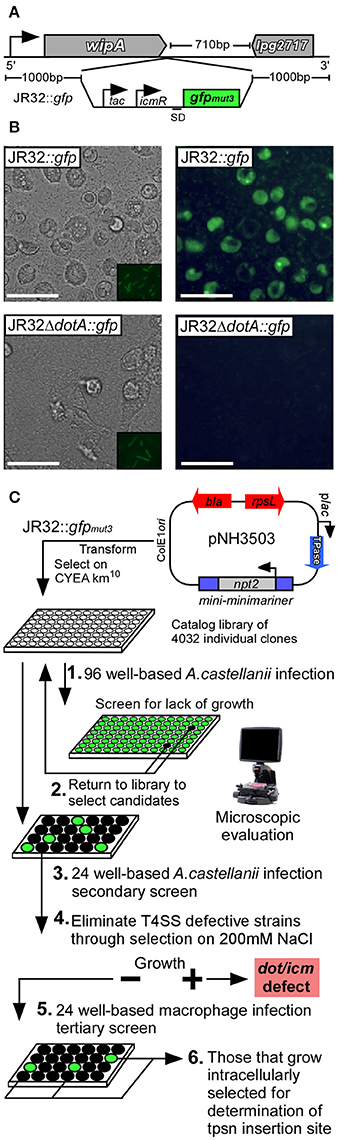
Figure 2. Schematic representation of genome-wide transposon mutagenesis of L. pneumophila and survival screen in multiple host-cell types. (A) gfpmut3 with tandem promoters (tac, icmR) was inserted on the L.p. JR32 chromosome 3′ to the wipA (lpg2718) locus using homologous recombination. (B) The JR32::gfp strain was used to generate a T4SS defective mutant through in-frame deletion of dotA, resulting in JR32ΔdotA::gfp. Both strains were cultured to post-exponential phase in the presence of IPTG (insets) and used to infect A.c. cells. Eighteen hours post-infection, light (left column) and fluorescence (right column) micrographs were captured to visualize replication-permissive vacuoles. Scale bar = 50 μm. (C) JR32::gfp was subjected to transposon mutagenesis using the plasmid pNH3503, where kanamycin resistant clones were transferred to 96-well culture plates. Each clone was cultured to post-exponential phase and used to infect A.c. cultures for 18 h (1). Individual wells were examined microscopically to evaluate abundance of fluorescent vacuoles. Mutants that lacked fluorescence were targeted for secondary infections of A.c. using controlled MOI (2–3). Mutants that passed secondary screen were subjected to cultivation on media supplemented with 150 mM NaCl in order to select against Dot/Icm-defective strains (4). Salt-sensitive mutants were subsequently used to infect CHO FcγRII-transgenic, J774.A1, and THP-1 monolayers to evaluate survival capacity using fluorescence microscopy (5). All remaining mutants were subjected to DNA sequencing to determine transposon insertion sites (6).
In order to provide unbiased coverage of the non-essential L.p. genome, we constructed a library of individual insertion mutants of L.p. strain JR32::gfp using a modified minimariner transposon (mini-minimariner) mutagenic strategy (Murata et al., 2006). For library construction, we used pNH3503 plasmid carrying the mini-minimariner transposon. This transposon targets TA di-nucleotides on the chromosome, integrating in a random fashion (Figure 2C) (Murata et al., 2006). Greater than 4,000 insertions were isolated using 20 individual rounds of mutagenesis, where 200–250 isolates were collected per round. PCR analysis of randomly selected mutants demonstrated the presence of transposon in every isolate examined (not shown). Individual mutants were cataloged in 96-well format for preservation (Figure 2C).
To screen the library, we first examined whether each mutant could replicate intracellularly in the model host protozoan A.c. (Holden et al., 1984). Individual mutants were used to infect A.c. in 96-well plate format for 18 h. Infected wells were examined via fluorescence microscopy. Mutants that were attenuated or unable to replicate intracellularly, as judged by reduction or absence of mature fluorescent vacuoles, were selected for further rounds of screening (Figure 2C).
A secondary screen of candidate mutants was performed in 24-well format under controlled multiplicity of infection. Successful candidates that were validated as attenuated or defective in intracellular replication were next tested for their capacity to grow under conditions of elevated sodium chloride concentration. WT L.p. cannot grow on media containing (150–200 mM) NaCl, a consequence of a functional Dot/Icm type IVb secretion system (Vogel et al., 1996). Conversely, L.p. mutations in several loci encoding structural components of the Dot/Icm transporter were shown to render L.p. permissive for growth on elevated [NaCl] (Figure 2C). This important selection criterion therefore allowed for identification of isolates that failed to divide in host cells yet presumably retained a functional Dot/Icm transporter. Candidates that satisfied the screening criteria were next used to infect a panel of mammalian host-cell types to evaluate specificity. Thirty-eight insertions were selected for further characterization, where 18 failed to replicate in A.c. and an additional 20 were attenuated by 50% or greater when compared to WT (as measured subjectively by total mature replication vacuoles per microscopic field).
Sequence Validation of Transposon Insertion Sites
Of the 38 isolates, 24 were successfully sequence validated, where transposon insertion sites were determined either by using the transposon insertion as a site for priming and subsequent amplification of purified genomic DNA or with “arbitrary” PCR (O'Toole and Kolter, 1998).
Twelve of the isolates were found in dot/icm-encoding loci, where insertions were limited to eight of the 27 genes that comprise the transport system (Table 1). These eight dot/icm genes therefore represented a functionally distinct class, as each failed to grow on elevated [NaCl]. An additional 12 insertions were located at the sites described in (Table 2). Each of these mutants were severely attenuated or failed to replicate in one of the four eukaryotic hosts examined. The remaining 14 mutants, displaying a range of intermediate phenotypes in A.c. infection were not sequenced but were cataloged. Infection phenotypes for these isolates are described in (Table S1).
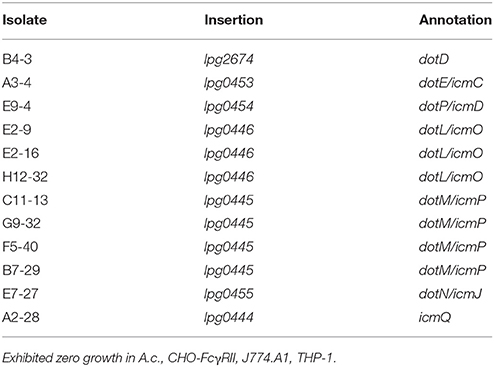
Table 1. NaCl-sensitive dot/icm insertions.

Table 2. Sequence-validated gene insertions identified in screen.
In addition to measuring survival and replication in A.c., each mutant was used to infect FcγRII transgenic Chinese Hamster Ovarian (CHO) cells (Nagai et al., 2005). Here bacteria were opsonized with polyclonal antisera directed against heat-killed WT L.p. prior to infection, which effectively stimulated phagocytosis and allowed for measure of survival in an artificial host system. Mutants were also used to infect the murine macrophage cell line J774.A1, or phorbol myristate acetate (PMA)-differentiated human THP-1 macrophages. Survival values indicated in Table 2 represent approximates of the total number of fluorescent vacuoles per microscopic field compared as a percentage to the total number of observed vacuoles in an infection using WT L.p. over the same time-course of infection.
Overall, the 24 mutants identified using the selection criteria could be divided into two classes: (I) dot/icm machinery mutants that retained salt-sensitivity, or (II) mutants that were attenuated for intracellular survival in one or multiple host cell types. We found some degree of host cell-specificity associated with survival among seven of the 12 class 2 insertions (Table 2). Additionally, 3 of 12 class II insertions were located in intergenic regions.
Of the insertions identified as a result of limited or complete failure to produce mature replication vacuoles in A.c., three interrupted dot/icm effector encoding genes. The product of sdhA has been implicated in vacuolar integrity and is necessary for survival in macrophages (Creasey and Isberg, 2012). The promoter regions of effector-encoding lem25 (lpg2422) and rvfA (lpg1797) were also interrupted (Huang et al., 2011). Complementation studies using plasmid-borne copies of lpg2422 or lpg2423 (encoded opposite direction from transposon insertion) in the H2-15 isolate failed to restore intracellular survival. Further the lpg1797 ORF alone, or in the context of native promoter failed to complement the A9-20 isolate. Therefore, the contribution of these transposon insertions to observed phenotypes remains unresolved.
Two additional insertions were found to interrupt genes encoding enzymes involved in amino acid metabolism. lpg2276 encodes a Glu/Leu/Phe/Val- family dehydrogenase, a NAD+ or NADP+-dependent enzyme that de-aminates the amino acid to a keto-acid form, which can be assimilated into the Kreb's cycle. The lpg1811 locus encodes aspartokinase-diaminopimelate decarboxylase, an enzyme important in lysine synthesis. The requirement for this gene was strictly limited to intracellular survival in the protozoan host.
Additional sequenced insertions included the proQm activator of ProP osmoprotectant transporter, which functions on ProP at a post-translational level. ProP responds to osmotic stress functioning as a zwitterion/proton symporter (Chaulk et al., 2011). Loss of proQm was found more detrimental for survival in the protozoan host. The D12-34 insertion was located between lspG and lspF, structural components of the type II secretion system in L.p. The Lsp secretion system has been previously implicated in intracellular survival of L.p. through secretion of multiple enzymes to the extracellular confines of the replication-permissive vacuolar compartment (Cianciotto, 2005). We also found hsp90 to be important for optimal survival in the protozoan cell, remaining dispensable in each of the metazoan cell types.
Identification of Putative ABC Import Complexes
Of particular interest, was that four of the insertions interrupted putative ATP-binding cassette (ABC) transporter components, lpg0730 and lpg0122. (Table 2) (Theodoulou and Kerr, 2015). The lpg0730 locus is the final gene in a transcriptional unit that contains as many as 9 ORFs. It resides 3′ to the gene encoding phosphatidylglycerophosphate phosphatase (pgpA) involved in lipid metabolism and membrane homeostasis (Figure 3A) (Funk et al., 1992). Three unique transposon insertion sites were identified in the screen, highlighting the importance of this locus for survival in A.c. (Figure 3B). The 1,053 bp lpg0730 ORF encodes a 350 aa (38.7 kDa) protein with 9 putative transmembrane segments. It is annotated as a membrane permease of the UPF0118 superfamily, which encompasses many substrate-binding transporters. E. coli YdiK is the archetype of this family of proteins, whose function is not determined but it is a member of the purR regulon, responsive to purine concentration (Cho et al., 2011). These permeases share strong homology with a class of transmembrane domain (TMD) proteins found in ABC transport systems.
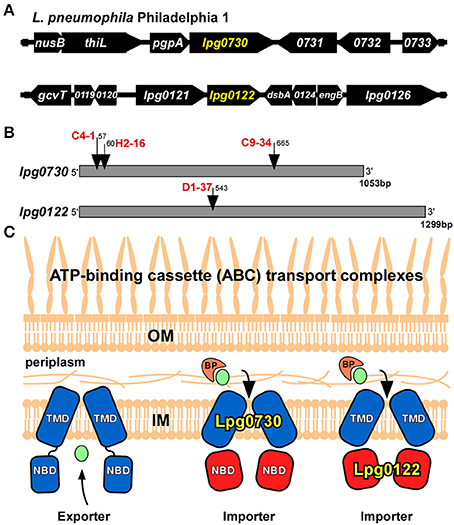
Figure 3. Transposon insertions found in ABC import complex components. (A) Location on the L.p. genome of lpg0730 and lpg0122. (B) Transposon insertion sites (nucleotide relative to +1) on the lpg0730 and lpg0122 ORF's for candidates identified in screen with clone designations in red. (C) Proposed location and function of Lpg0730 and Lpg0122 in the L.p. inner membrane. BP, binding protein (orange); TMD, transmembrane domain (blue); NBD, nucleotide binding domain (red); IM, inner membrane; OM, outer membrane, transported substrate depicted in green.
The lpg0122 locus is the second gene in a bi-cistronic transcriptional unit (Figure 3A). The D1-37 isolate was interrupted by the transposon at nucleotide position 543 of the 1,299 bp lpg0122 ORF (Figure 3B). The gene encodes a 432 aa (48.4 kDa) protein annotated as a nucleotide binding domain (NBD) component of an ABC transport system. Its sequence falls under the NtrD/SsuB transporter family that encompasses numerous ATP-binding cassette domains that are involved in the import of nitrates and sulfonates. Lpg0122 shares its N-terminal 252 aa with TauB, involved in taurine import (Pereira et al., 2015). The C-terminal region is annotated as an AAA-ATPase associated region. The product of lpg0121 is annotated as an ABC membrane permease (TMD).
We surmised lpg0730 and lpg0122 individually represented components of distinct ABC “importer” complexes, which could be extrapolated based on amino acid sequence and their arrangement on the L.p. genome. For ABC import complexes, the TMD and NBD components are generally distinct proteins, whereas in ABC exporters the TMD and NBD are each sub-domains of a single fusion protein (Figure 3C) (Theodoulou and Kerr, 2015). It is more likely that Lpg0122 (NBD) associates with the co-expressed Lpg0121 (TMD) than with Lpg0730. Furthermore, transposon interruptions of lpg0730 or lpg0122 exhibited different requirements for intracellular survival based on host cell type. Both Lpg0730 and Lpg0122 were determined to be highly conserved in Gram-negative and some Gram-positive bacteria when translated protein was used as template for homology search. This included several notable human pathogens that can use freshwater amoebae as an environmental intermediate. Multiple sequence alignment and phylogenetic assignment of Lpg0730, Lpg0122, and corresponding orthologous proteins are depicted in Figures S1, S2, respectively.
Intracellular survival relative to WT was grossly estimated visually for lpg0730::Tn and lpg0122::Tn as part of the screening process. In addition to A.c., where survival rates approximated zero and 10 percent respectively, polyclonal anti-legionella-opsonized strains were used to infect FcγRII-transgenic CHO monolayers to measure survival independent from internalization (Nagai et al., 2005). Here, both insertions generated identical survival phenotypes to those observed in A.c. When used to infect murine J774.A1 macrophages, lpg0730 and lpg0122 were found dispensable for intracellular survival, and performed identical to WT. These results suggested that each of these loci were critical for survival in particular host cell types (A.c., CHO FcγRII) while remaining completely dispensable in another (J774A.1). Curiously, although lpg0122::Tn also performed similar to WT during infection of human THP-1 macrophages, each of the three lpg0730::Tn mutants failed to replicate, suggesting a gradient of substrate concentration or availability among the macrophage hosts (Table 2).
Host Cell-Specific Activation of Candidate Genes
Some degree of host cell-specificity was observed for seven of 12 of the non-dot/icm insertion mutants. Differential genetic requirements for intracellular survival were most pronounced in lpg0730, lpg1811, and lpg2276 insertion mutants (Table 2). We sought to determine the expression profile of these loci in WT L.p. in the context of intracellular growth in multiple established host cell types. Two closely related Acanthamoeba species (A. castellanii, A. polyphaga), a more distantly related amoebae species (Hartmanella vermiformis), and J774.A1 macrophages were selected for the analyses. Total RNA was isolated from bacteria either immediately after exposure to host cells or 18 h post-infection (prior to host cell egress). Quantitative PCR was performed using primer sets to generate ~150 bp amplicons from lpg0730, lpg1811, lpg2276, and DNA gyrase B (gyrB), which was used for normalization. As depicted in Figure 4 each of the genes examined were highly activated in all three amoebae species while remaining inactivated or slightly repressed in J774.A1 over the time-course of infection. These results suggest that the micro-environments encountered by L.p. in various host phagosomes must be chemically diverse.
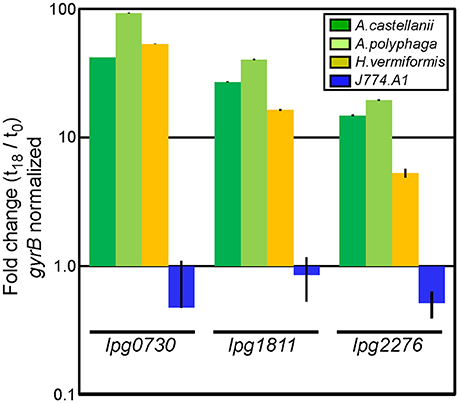
Figure 4. Genes identified in screen are exclusively activated during infection in protozoa. qPCR analysis using total bacterial RNA isolated immediately after exposure to, or 18 h post-infection of indicated host cells. Bars indicate fold-change of indicated transcripts (t18/t0) normalized to DNA gyrase B (gyrB). Infections were performed in triplicate (three biological replicates) with standard deviations indicated.
Characterization of lpg0730 and lpg0122 Mutant Strains
Our mutagenesis strategy selected only for non-essential genes. We therefore examined growth kinetics of the C4-1 (lpg0730) and D1-37 (lpg0122) insertion mutants through direct comparison to WT cultured in ACES-buffered yeast extract broth (AYE). As shown in Figures 5A,B the growth kinetics of all three strains were indistinguishable. Similar results were obtained through culture in defined synthetic media (not shown) (Warren and Miller, 1979). These results indicate that both lpg0730 and lpg0122 are dispensable for L. p. growth in vitro.
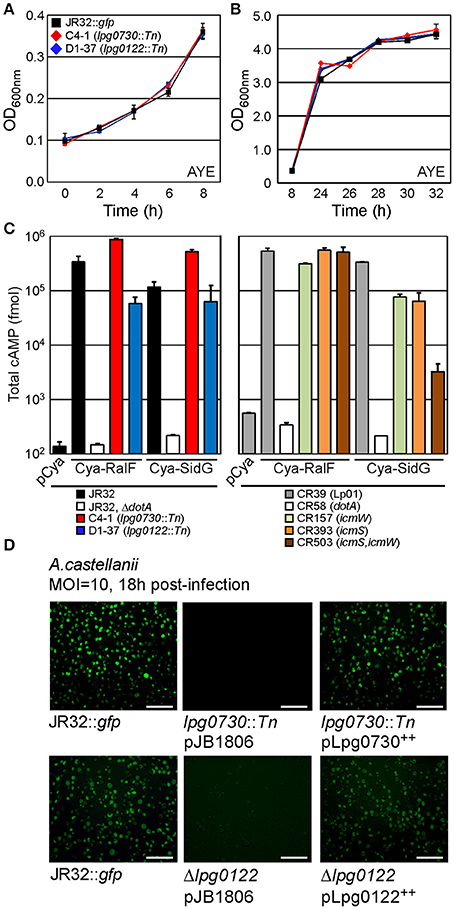
Figure 5. lpg0730 and lpg0122 are dispensable for replication in vitro and do not interfere with Dot/Icm-mediated translocation of effectors. (A) Indicated strains were suspended in AYE broth at an OD600 nm = 0.1 and cultivated at 37°C on shaker. Spectrophotometric measurements were taken at indicated times spanning lag to exponential phase transition. (B) As in (A) with measurements taken to cover late exponential to early stationary phase transition. (C) L.p. strains (indicated with colored bars) harboring plasmids producing adenylate cyclase (Cya) fusions to the L.p. effectors RalF or SidG were used to infect CHO FcγRII monolayers for 1 h. Cells were lysed after which total cAMP was extracted and quantified using immunosorbent assay. Bars indicate total cAMP produced per well (fmol). Experiments were performed in triplicate (three biological replicates) with standard deviations indicated. (D) lpg0730::Tn and Δlpg0122 were transformed with either empty vector (pJB1806) or plasmids that supported inducible expression of Lpg0730 or Lpg0122. Resulting strains were cultured to post-exponential phase in parallel with JR32::gfp and used to infect A.c. cultures for 18 h (MOI = 10). Representative fluorescence micrographs are shown for infected A.c. cells with each indicated bacterial strain. Fluorescent foci represent individual replication vacuoles. Scale bar = 100 μm.
It is possible that the severe intracellular growth attenuation observed for the C4-1 and D1-37 insertion mutants in A.c. was a result of detrimental effects on the type IV secretion pathway, which is essential for pathogenesis. We sought to determine whether interruptions in lpg0730 or lpg0122 generated strains that were defective for dot/icm-dependent translocation of effector proteins. Reporter proteins were utilized that consisted of the catalytic domain of the calmodulin-dependent adenylate cyclase (Cya) from Bordetella pertussis fused to the amino terminus of the established effectors RalF or SidG (Cambronne and Roy, 2007). The production of cyclic adenosine-monophosphate (cAMP) resulting from the translocation of a Cya-effector hybrid into CHO-FcγRII cells was used to measure productive translocation. As indicated in Figure 5C, both the Cya-RalF and Cya-SidG hybrids were translocated to the host cytosol with higher fidelity in the C4-1 (lpg0730) background than in the parent JR32 strain. Translocation of Cya-RalF in the D1-37 (lpg0122) strain was slightly attenuated when compared with JR32. However, there was no significant difference in translocation efficiency of Cya-SidG, an icmSW-dependent effector protein. Cya-SidG translocation was previously reported to be attenuated with deletions in either icmS or icmW, which form an adaptor complex in the L.p. cytoplasm that promotes delivery of a class of effectors to the substrate receptor complex (Figure 5C) (Cambronne and Roy, 2007). Even though SidG translocation is reduced in icmS or icmW mutants, both strains can form replication-permissive vacuoles with reduced kinetics in A.c. (Coers et al., 2000; Tilney et al., 2001). It is likely that a mutation in lpg0122 does not have a direct effect on type IV secretion, but may indirectly affect kinetics of vacuole maturation.
Both lpg0730 and lpg0122 were next targeted for deletion using homologous recombination in a tri-parental mating scheme. We successfully generated a marker-less deletion of the lpg0122 locus (Δlpg0122) on the JR32::gfp genetic background. Multiple attempts to generate a similar deletion of the lpg0730 locus, including production of variant recombination vectors, failed to generate the desired strain. We therefore continued our studies using the C4-1 transposon insertion.
We next performed genetic complementation in trans by cloning lpg0730 or lpg0122 into a low-copy plasmid, with expression guided by an IPTG-inducible tac promoter. Sequence validated plasmids were transformed either into the C4-1 (lpg0730) or Δlpg0122 strains, in parallel to empty vector (pJB1806). Parent JR32, C4-1, and Δlpg0122 harboring appropriate plasmids were cultured to early stationary phase and used to infect A.c. After 18 h of infection A.c. cultures were imaged using light and fluorescence microscopy. A productive infection with WT JR32::gfp can be visualized in Figure 5D. No vacuoles were visualized with the C4-1 (lpg0730) strain, and the lpg0122 deletion phenocopied the D1-37 isolate, with sparse vacuoles and low fluorescence. Production of Lpg0730 or Lpg0122 in trans fully restored A.c. infection fidelity to WT levels when evaluated microscopically (Figure 5D).
To quantitatively evaluate contribution of lpg0730 and lpg0122 to intracellular survival of L.p., lpg0730::Tn (C4-1) or Δlpg0122 were used to infect either A.c. or THP-1 macrophages over a 72 h time course. Parallel infections were performed with WT (JR32::gfp), dotA deletion (JR32::gfp, ΔdotA), or complement strains. The lpg0730::Tn strain failed to replicate in either A.c. (Figure 6A) or THP-1 (Figure 6C), and was less persistent than ΔdotA in both hosts. Production of Lpg0730 via low-copy plasmid restored survival and replication of lpg0730::Tn to WT levels in A.c. and THP-1 (Figures 6A,C). The lpg0122 deletion strain (Δlpg0122) was replication competent in both A.c. and THP-1 with significantly reduced kinetics. In A.c., maximal CFU counts were achieved 72 h post-infection and were nearly 100-fold lower than CFU's recovered in the WT infection (Figure 6B). Similar attenuation was observed over the infection time course in THP-1 (Figure 6D). Similar to Lpg0730, plasmid-derived Lpg0122 restored intracellular survival and replication to WT levels in both A.c. and THP-1 (Figures 6B,D). Overall both expression constructs complemented the growth attenuation observed in the two mutant strains.
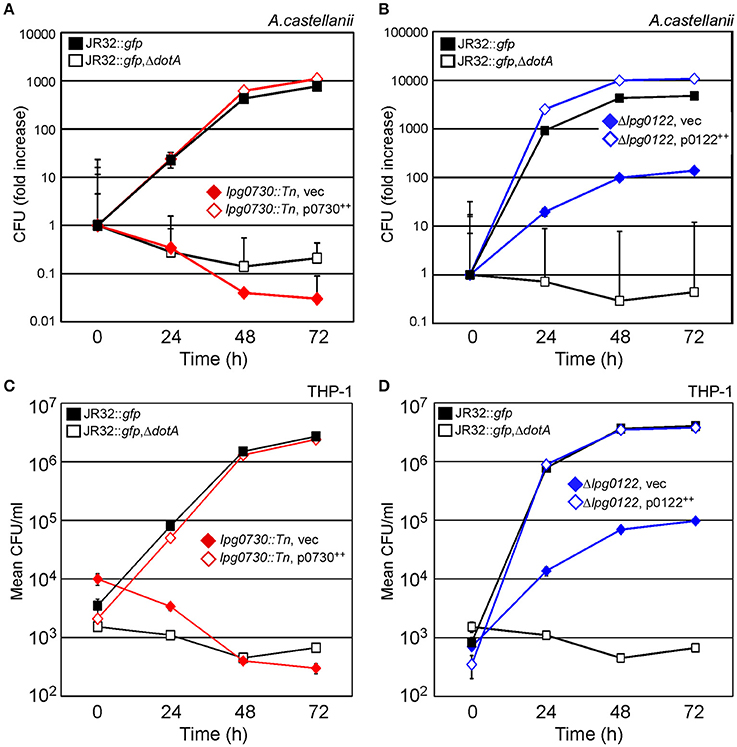
Figure 6. Intracellular survival and replication of lpg0730::Tn and Δlpg0122 in A.c. and THP-1 human macrophages is fully restored via complementation in trans. (A,B) Indicated strains were used to infect A.c. cultures (MOI = 1), where host cells were lysed at indicated times and total CFU were determined by colony count after serial dilution and cultivation on CYEA. Results are shown as fold increase over 72 h. Experiments were performed in triplicate (three biological replicates) with standard deviations indicated. (C,D) As in (A,B) with 48 h PMA-differentiated THP-1 monocytes substituted for A.c. Graphs indicate mean CFU/ml (three biological replicates) with standard deviations indicated.
Conservation of lpg0730 and lpg0122 in Pathogens that Colonize Protozoa
In addition to Legionella species, many notable human pathogens are capable of colonizing protozoan cells. Mycobacterium, Salmonella, Staphylococcus, and many other Gram-negative and Gram-positive species can use amoebae as a nutrient rich environmental intermediate for replication and dissemination. We used the amino acid sequences of Lpg0730 and Lpg0122 to search for homologous and orthologous proteins in a subset of pathogenic species known to colonize amoebae use the BLASTP algorithm. To our surprise, both proteins were conserved in nearly every bacterial species examined. We next performed multiple sequence alignment and phylogenetic analysis using Clustal Omega. Of the species examined, Lpg0730, Lpg0122, and their respective homologs/orthologs separated into three clades from a common ancestral protein. The phylogenetic arrangement and multiple sequence alignments for Lpg0730 and Lpg0122 are shown in Figures S1, S2 respectively.
We sought to determine whether Lpg0730 homologs/orthologs were necessary for intracellular survival. We first constructed a C-terminal FLAG epitope-tagged version of Lpg0730 by cloning this construct into the same vector used for complementation. Using the same A.c. infection strategy employed in Figure 5D, WT (JR32::gfp), and dotA deletion (JR32::gfp, ΔdotA), and lpg0730::Tn (C4-1) pLpg0730FLAG were evaluated microscopically 18 h post-infection. Similar to expression of Lpg0730 alone appending the C-terminal FLAG epitope had no detrimental effect on intracellular survival and the fidelity of infection was restored to WT levels (Figure S3).
Lpg0730 is assigned by sequence to the PerM family of membrane permeases, which is exemplified by the YdiK protein in E. coli. Alignment of Lpg0730 and YdiK is shown in Figure S1. We sought to determine whether an Lpg0730 homolog from L. longbeachae (76% identity) or PerM orthologs from Salmonella enterica subsp. Typhimurium (25.2% identity) or Francisella tularemia subsp. novicida (22.6% identity) could complement the intracellular survival defect of the C4-1 insertion mutant. C-terminal FLAG epitope-tagged versions of each allele were cloned and transformed into the C4-1 strain. Each complementing strain was used for infection of A.c. as in Figure 5D. Expression of the L.longbeachae allele completely restored intracellular survival of L.p. C4-1, whereas both the Salmonella and Francisella alleles failed to restore survival of the mutant (not shown).
Although the Lpg0730 (PerM) ortholog in F.novicida could not restore survival of L.p., we had access to a sequence-cataloged transposon library of F.novicida U112 in the laboratory (Provided by F. Heffron). The library contained an insertion mutant in the perM locus (FTN0570). We used this strain and WT F.novicida U112 for infections of either A.c. or THP-1 macrophages. We determined that WT F.novicida persists in A.c. for over 24 h, while the FTN0570 failed to colonize amoebae (Figure 7A). The FTN0570 insertion mutant was also significantly attenuated for pathogenesis of THP-1 when compared to WT F.novicida over a 72 h time-course of infection (Figure 7B). These observations highlight the conservation of Lpg0730-like ABC import complexes and their requirement for optimal host cell colonization.
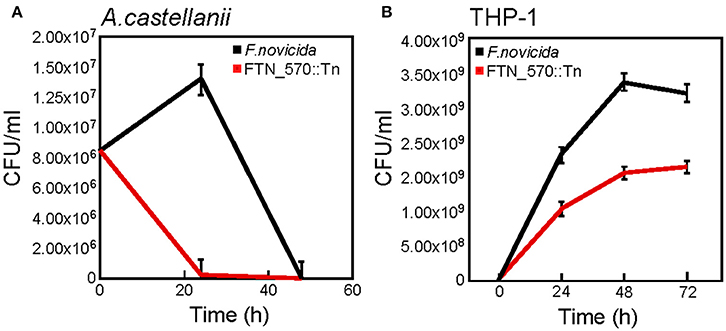
Figure 7. The lpg0730 ortholog FTN0570 from Francisella tularensis subsp. novicida is required for persistence/survival in A.c. and human THP-1 macrophages. (A) F.novicida WT (U112) or FTN0570 mutant (FTN0570::Tn) were used to infect A.c. cultures (MOI = 10), where host cells were lysed at indicated times and CFU/ml was determined by colony count after serial dilution and cultivation on supplemented TSA. (B) As in (A) except strains were used to infect differentiated THP-1 macrophages for indicated times prior to host cell lysis. Graphs indicate mean CFU/ml (three biological replicates) with standard deviations indicated.
Discussion
Details regarding physiological requirements of L.p. during the intracellular life-cycle are only beginning to be elucidated. Further, identification and characterization of L.p. factors required for intracellular survival even in the presence of a functional Dot/Icm transporter are even more limited. When we designed this screen, we anticipated that an abundance of candidates would be identified from the Dot/Icm effector protein catalog (Zhu et al., 2011). Just as dot/icm is required for virulence, nearly all effectors described to date are dispensable. We hypothesized that the extremely large catalog of effectors in L.p. (~10% of the genome) was owed to particular sets being required for colonization of a particular host. The taxonomically diverse host-range of L.p. could highlight specificity of particular subsets of effectors for survival in a particular host cell. We chose A. castellanii as host for the initial phase of the screen as a natural environmental host. A.c. transitions from trophozoite to cyst and reverts based upon nutrient availability (Lloyd, 2014).
Owing to our hypothesis, we indeed found a degree of host-cell specificity associated with several candidates identified in the screen, however we were surprised that only one effector, SdhA had a transposon insertion in its ORF (Laguna et al., 2006; Creasey and Isberg, 2012). Intergenic regions surrounding Lem25 and RvfA were also interrupted, but multiple complementation attempts were unsuccessful (Huang et al., 2011). Both of these mutants were more attenuated for survival in A.c. than in any of the metazoan host cells examined, so should be explored with greater scrutiny in the future. We also identified 12 insertions in dot/icm encoding genes. While none of these mutants supported translocation of Cya-RalF in the adenylate-cyclase reporter assay (not shown), it was notable that eight of the insertions were found in the DotLMN type IV coupling protein sub-complex (Sutherland et al., 2012). This provides additional evidence that separates the initial stages of substrate recognition (DotLMN) from a superstructure that renders L.p. sensitive to NaCl via cytoplasm accessible conduit.
Amino acid availability to L.p. while in a phagosome was revealed as a key physiological constraint, similar to early studies using defined chemical media. lpg2276 (Glu/Leu/Phe/Val dehydrogenase) was upregulated during intracellular replication in amoebae exclusively. Similarly lpg1811 (Aspartokinase-diaminopimelate decarboxylase), important in lysine synthesis was highly upregulated in amoebae. The gene was determined dispensable in all metazoan cell types, both here and in a separate study where the lpg1811 locus lost functionality over time when L.p. was continuously subjected to passage through a macrophage cell line (Ensminger et al., 2012). These results suggest that accessible lysine concentrations in the host must be distinct when comparing amoebae to macrophage. Other candidates identified in the screen can be directly correlated with particular environmental stressors encountered during colonization of protozoan cells. ProQm is a RNA-associated regulatory protein that governs activation of the ProP osmoprotectant transport system. Osmotic pressure changes are more likely to occur in the environmental host, especially in instances of desiccation upon encystment. HtpG (Hsp90) was found dispensable for survival in metazoan hosts, but was required for optimal infection of A.c., perhaps a result of elevated temperature used to accelerate A.c. infections.
The most striking result of the screen was the identification of three independent transposon insertions in the lpg0730 locus. Each of the insertions rendered L.p. completely attenuated for survival in A.c. and THP-1 host cells. lpg0730 and an additional candidate lpg0122 were both annotated to encode ABC import complex proteins (Theodoulou and Kerr, 2015). We selected these loci for further characterization as the identified substrates of the transporter families (YdiK and NtrD/SsrB, respectively) are variable and often ill-defined experimentally. Both lpg0730 and lpg0122 were demonstrated to be required for survival even in the presence of a functional Dot/Icm transporter, highlighting the importance of acquisition of particular substrates during colonization of the host cell.
Additionally, both Lpg0730 and Lpg0122 were conserved in multiple human pathogenic species that are capable of colonizing protozoa. Here we demonstrated the PerM ortholog in F. tularensis subsp. novicida was required for persistence in A.c. and optimal pathogenesis in THP-1 macrophages. This result supports the notion that in order to colonize amoebae, these distinct import complexes must be functional, and their activities take precedence over the contribution of other virulence mechanisms. Identification of the imported substrates will be of critical importance in order to develop methods to prevent acquisition of the particular nutrients supplied by the amoebae host.
Because the transmission of L.p. to humans is confined to dispersal via water droplet (aerosols), outbreaks of legionellosis are normally confined to a single source that has been compromised with infectious bacteria (Burillo et al., 2017). The barricade for controlling disease spread therefore resides predominantly upstream of the pathogen-human interface. A variety of methods are used both commercially and municipally to eliminate water-borne pathogens, including elevating temperature of contained water, lowering pH, or flushing water lines with heavy metals (Zgonc and Baideme, 2015). These treatment methods are often effective for elimination of L.p. and other bacteria. Contrarily, these sterilization scenarios can be insufficient to eradicate natural host amoebae; many of which can differentiate to an environmentally resistant cyst-form as part of their life-cycle (Lloyd, 2014).
Planktonic L.p. can be found in multiple physiological states in freshwater. Viable bacteria, which can be free-living or associated with polymicrobial biofilms, may be detected via culture on artificial media. Indeed L.p. has been demonstrated to remain infectious to human macrophage cell lines at low levels even after incubation at low temperature (10–15°C) in freshwater medium (Fraquil) for 6 months (Mendis et al., 2015). A second form, viable but not culturable (VBNC) could represent an adaptation of the bacterium to the nutrient-depleted environment that is freshwater. The VBNC state appears to be induced upon introduction of environmental stressors, especially temperature above 70°C (Ducret et al., 2015). VBNC L.p. were shown to be incapable of colonizing human airway epithelia or macrophage cell lines, but could be resuscitated when introduced to A. polyphaga. After completion of their intracellular life-cycle in amoebae, these protozoan-primed VBNC were now infectious to alveolar epithelial and macrophage cell lines (Epalle et al., 2014).
These results, along with the aforementioned studies comparing the infectivity of L.p. cultivated in laboratory medium compared to protozoan-primed bacteria, suggest that the state of the bacterium encountered at the human interface is most likely the transmissive form generated during egress from protozoa, and not the form generated through in vitro culture in rich medium. We felt it imperative to more thoroughly investigate the L.p.-protozoan interface in order to identify genetic factors required for colonization and replication of L.p. Because replication of L.p. in freshwater is restricted to the confines of protozoa, these genetic targets could be exploited to block proliferation of L.p. and other pathogens in amoebae, and if used in concert with current sterilization practices, could significantly reduce exposure of protozoan-primed L.p., and other bacteria to humans.
Materials and Methods
Bacterial Strains, Plasmids, Media, and Culture Conditions
All work performed in this study was approved by the OHSU Institutional Biosafety Committee (protocol #08-53). Bacterial strains, plasmids, and primers used in this study are summarized in Table S2. L.p. strains were cultured in ACES buffered yeast extract (AYE) or on AYE agar plates (0.2% activated charcoal) (CYEA) at 37°C. Media was supplemented with the following antibiotics where appropriate: chloramphenicol (Cm); (6.25 μg/ml), kanamycin (Kan); (20 μg/ml), streptomycin (Str); (50 μg/ml). Sucrose (5% w/v) was included in CYEA for counter-selection of recombinant plasmid pSR47S. Escherichia coli strains were cultured at 37°C in Luria Bertani (LB) medium supplemented with Cm (25 μg/ml), Kan (50 μg/ml), Str (50 μg/ml) where appropriate. F. novicida strains were cultured in Tryptic Soy Broth (TSB) supplemented with 0.1% cysteine at 37°C.
Cell Culture
Axenic A.c. strain Neff (ATCC 30010) and A. polyphaga strain (Puschkarew) Page CCAP 1501/3b (ATCC 30872) cells were propagated in supplemented PYG medium (ATCC 712) at room temperature (RT), harvested and diluted into fresh medium twice weekly. H. vermiformis strain CDC-19 (ATCC 50237) was propagated in SCGYEM medium (ATCC 1021) at 30°C, harvested and diluted into fresh medium weekly. Murine macrophage J774.A1 (ATCC TIB-67), human monocyte THP-1 (ATCC TIB-202) and FcγRII transgenic—Chinese Hamster ovary (CHO) cells (C. Roy laboratory) were maintained at 37°C with 5% CO2. Undifferentiated THP-1 cells were cultured in suspension in RPMI medium supplemented with 10% fetal bovine serum (FBS). Forty-eight hours prior to infection, cells were differentiated in 24 or 96-well tissue culture plates, using phorbol 12-myristate 13-acetate (PMA). CHO FcγRII and J774.A1 were propagated in αMEM and DMEM respectively, supplemented with 10% FBS.
In Vitro Growth Studies
L.p. strains were inoculated in triplicate into 10 ml AYE or minimal media to achieve an initial OD600 nm of 0.1 (Warren and Miller, 1979). Cultures were incubated on an orbital shaker (200 × rpm) at 37°C. One hundred microliters aliquots were collected from each culture at indicated times post-inoculation, suspended in 0.9 ml 1xPBS, and OD600 nm were calculated via spectrophotometer.
Transposon Mutagenesis
L.p. JR32::gfp was generated via single-copy allelic integration of the gfpmut3 locus on to the chromosome immediately 3′ to the wipA (lpg2718) locus. A region encompassing sequential tandem tac and icmR promoters located 5′ to the gfpmut3 ORF was amplified from the plasmid pECR350 using primers 551/552. Flanking 1 kb fragments located 5′ and 3′ to the wipA stop codon were amplified using 549/550 and 553/554 respectively. The three PCR products were mixed (1:1:1) and used as template for short overlap extension (SOE) PCR using the 549/554 primer set. The resulting ~2.8 kb PCR product was ligated into the pSR47S vector linearized with BamH1 using T4 DNA ligase (New England Biolabs). The resulting plasmid was designated pECL529. After sequence validation, pECL529 was transformed into E. coli DH5α λpir and introduced to JR32 via tri-parental mating scheme using E. coli DH5α, pRK600 as the helper strain. Transconjugants were generated through selection on CYEA Str, Kan. Colonies were isolated and plated on CYEA Str supplemented with 5% sucrose to force plasmid excision. Sucrose-resistant/kanamycin-sensitive isolates were examined using PCR to verify gfpmut3 insertion 3′ to the wipA locus. GFP positive isolates confirmed by fluorescence after growth in AYE broth supplemented with 1 mM IPTG. The resulting strain generated JR32::gfpmut3. The transposon-containing plasmid, pNH3503 was introduced into JR32::gfpmut3by electroporation. Twenty rounds of transformation and subsequent selection on CYEA Kan generated over 4,200 isolates which were randomly screened for the transposon insertion using the 573/574 primer set. Each individual insertion mutant was cultivated on CYEA in 96-well plates for 48 h at 37°C, re-suspended in stock solution (5% glycerol W/V, 2% peptone W/V), cataloged and stored at −80°C.
Intracellular Replication in Protozoa and Macrophages
Seventy-two hours quantitative intracellular growth assays were performed in a similar fashion to those previously described (Zamboni et al., 2003; Ninio et al., 2005). For the transposon mutagenesis screen, A.c. were suspended in supplemented ATCC 712 PYG media and cultured for 72 h, after which cells were harvested via centrifugation (400 × g) for 10 m. Media was aspirated and cells were suspended in infection media (3.4 mM Na citrate plus ATCC 712 salts) and seeded to tissue culture wells at 5 × 105 (0.5 ml) or 1.25 × 105 (0.125 ml) per well in 24 or 96-well format, respectively. Amoebae were allowed to adhere to plates overnight at RT. Host cells were infected with L.p. strains cultured to post-exponential phase in AYE broth (1 mM IPTG) at MOI = 10 (24-well) or ~10 (96-well). For the screening process, 96-well plates were floated on a 37°C water bath for 5 m and transferred to humidified incubator (37°C, 5%CO2) for 18 h. Intracellular growth was assessed qualitatively using light and fluorescence microscopy (AMG EVOSfl). J774.A1 were cultured to near-confluency, harvested by centrifugation, seeded (2.5 × 105/well) in 24-well plates and incubated overnight at 37°C, 5%CO2. One hour prior to infection, cells were washed 3x with 1x PBS, 0.5 ml fresh DMEM (10% FBS) was added to each well, and plates were returned to incubator. THP-1 monocytes were cultured for 3–5 days in RPMI (10% FBS) prior to harvest by centrifugation. Cells were resuspended in RPMI (10% FBS, 100 ng/ml PMA), seeded (2.5 × 105/well) in 24-well plates and incubated 48 h at 37°C, 5%CO2. One hour prior to infection, cells were washed 3x with 1x PBS, after which 0.5 ml RPMI (10% FBS) was added to each well. CHO FcγRII were cultivated in αMEM (10% FBS) and processed similar to J774.A1, with the exception being the inclusion of anti- heat-killed L.p. polyclonal antisera (Pacific Immunology) in the αMEM replacement media 1 h prior to infection. The A.c. and THP-1 infections with F. novicida were performed in a similar manner, using bacteria cultured to post-exponential phase in TSB 0.1% cysteine.
DNA Sequencing
Insertion mutants were sequenced either by PCR using purified genomic DNA as template for primer 780, or with “arbitrary” PCR modified from O'Toole and Kolter (1998). Briefly, the 1° PCR utilized purified gDNA as template for amplification with the 571/Arb1c primer set, followed by a 2° PCR using first PCR product as a template for amplification with the 780/781 primer set.
Protein Sequence Alignment
L.p. reference amino acid sequences of polypeptides were derived from the Philadelphia 1 genome (NC_002942.5) National Center for Biotechnology Information (NCBI). Sequence alignments were performed using BLASTP with default parameters (NCBI/NLM). Phylogenetic analysis was performed by aligning L.p. proteins with individually selected bacterial species using BLASTP. Closest orthologous polypeptides from each species were aligned using Clustal Omega software (http://www.ebi.ac.uk/Tools/msa/clustalo/) European Molecular Biology Laboratory (EMBL).
Cya Translocation Assay
Translocation assays were performed as described previously (Nagai et al., 2005; Ninio et al., 2005). Briefly, monolayers containing 1 × 105 CHO FcγRII cells were infected with 3 × 106 opsonized L.p. (MOI = 30) expressing Cya hybrid proteins. After 1 h incubation at 37°C, 5% CO2, monolayers were washed with 1x PBS and lysed. Total cAMP was extracted and quantified using cAMP Biotrak Enzymeimmumoassay System (GE Healthcare).
Transcriptional Analyses
To analyze transcription of L.p. genes during intracellular growth in A. castellanii, A. polyphaga, H. vermiformis, and J774.A1, cells were seeded in appropriate media under conditions as suited for each, and challenged with L.p. at an MOI = 200. The infection was synchronized by centrifuging the cells at 400 × g for 10 m and bacterial RNA was extracted immediately (t0) and 18 h (t18) post-infection. For extraction, infection buffer was removed and monolayers were disrupted with a sterile cell scraper. One milliliter of PBS was added to each well and vigorously mixed with pipette. Suspensions were transferred to a 14 ml conical tube and centrifuged for 10 m at 3,000 rpm. Supernatants were aspirated and pellets were re-suspended in 10 ml ice cold sterile H2O (protozoan) or 10 ml ice cold sterile H2O, 0.1% Triton X-100 (macrophage) and incubated on ice for 10 m. Lysates were centrifuged for 10 m at 4,000 × rpm. Supernatants were aspirated and pellets were resuspended in 1 ml guanidine thiocyanate buffer (GTC) (4 M guanidine thiocyanate, 0.5% Na N-lauryl sarcosine, 25 mM Na citrate, pH 7.0, 1 M β-mercaptoethanol) and transferred to a microcentrifuge tube. Suspensions were passed through a 21 gauge needle 3x and centrifuged 10 m at 15,000 × rpm. After aspiration, pellets were washed with 1xPBS, 0.1% Tween-20 and centrifuged an additional 10 m at 15,000 × rpm. After aspiration, pellets were re-suspended in 100 μl of 10 mg/ml lysozyme (Fisher Bioreagents) in 10 mM Tris, pH 8.0. Samples were incubated 15 m at RT. 750 μl of 65°C Trizol reagent (Invitrogen) was added to each sample, mixed and centrifuged for 10 m at 15,000 × rpm. The aqueous phase was transferred to a microcentrifuge tube and mixed with 500 μl of 100% ethanol. Samples were finally transferred to an RNeasy column and isolated according to manufacturer instructions, except that the in-column DNAse treatment was increased to 1 h (Qiagen). Purified RNA samples were analyzed with a Nano-drop ND-1000 spectrophotometer and stored in aliquots at −80°C.
Five Hundred nanograms of total bacterial RNA isolated from intracellular WT L.p. was subjected to cDNA synthesis using a Takara BluePrint RT reagent kit with random hexameric priming (Clontech) according to manufacturer instructions. After cDNA synthesis was performed in a thermocycler, samples were diluted 1:15 and stored on ice prior to qRT-PCR analysis. Primer sets were designed to target specific transcripts on the L.p. genome (Figure 4, Table S2). qRT-PCR samples were prepared using BioRad IQ SYBR Green Supermix. Reactions for each primer set probe were performed in triplicate in a Bio-Rad clear 96-well multiplate on a Bio-Rad Opticon thermocycler. Results were analyzed using Step One Software, v2.2. Relative expression, ΔΔCt analysis, was performed using gyrB as a reference transcript.
Author Contributions
Conceptualization, methodology, funding acquisition, and project administration (EC). Investigation, data curation, validation, and formal analysis (AL, SD, RJ, GR, and EC). Visualization and original draft preparation (AL, SD, and EC). Review and Editing (EC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Drs. Hiroki Nagai and Joseph Vogel for plasmid constructs. Helen Wu, Gabrielle Ma, Taylor Toro, Ben Doron, and Dimitra Fellman contributed experimental data and technical assistance. This work was supported by grants AI123682, (National Institute of Allergy and Infectious Diseases) GM94623, (National Institute of General Medical Sciences) and by the OHSU Foundation (EC).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2017.00485/full#supplementary-material
Figure S1. Lpg0730 is conserved in multiple genera that colonize protozoa.(A) Clustal Omega (EMBL) phylogenetic cladogram depicting Lpg0730 (L.p.) and its relationship to homologous/orthologous polypeptide sequences found in the genera shown. (B) BLASTP amino acid sequence alignment (NCBI) of Lpg0730 (red) and homologous/orthologous polypeptide sequences found in the genera shown (multispecies). Nine representative transmembrane segments are shaded (gray). (C) BLASTP alignment of Lpg0730 and YdiK (E. coli). *indicate conserved residues. Transmembrane segments are shaded (gray). (D) BLASTP alignment of Lpg0730 and PerM (FTN0570) from F. tularensis subsp. novicida. *indicate conserved residues.
Figure S2. Lpg0122 is conserved in multiple genera that colonize protozoa.(A) Clustal Omega (EMBL) phylogenetic cladogram depicting Lpg0122 (L.p.) and its relationship to homologous/orthologous polypeptide sequences found in the genera shown. (B) BLASTP amino acid sequence alignment (NCBI) of Lpg0122 (blue) and homologous/orthologous polypeptide sequences found in the genera shown (multispecies). (C) BLASTP alignment of Lpg0730 and TauB (E. coli). *indicate conserved residues.
Figure S3. Complementation of lpg0730::Tn with Lpg0730FLAG. Cultured A.c. were infected with indicated strain/plasmid combinations for 18 h. Representative live images were captured using light and fluorescence microscopy. Scale bar = 200 μm.
References
Abu Kwaik, Y., Gao, L. Y., Stone, B. J., Venkataraman, C., and Harb, O. S. (1998). Invasion of protozoa by Legionella pneumophila and its role in bacterial ecology and pathogenesis. Appl. Environ. Microbiol. 64, 3127–3133.
Berger, K. H., and Isberg, R. R. (1993). Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x
Brieland, J. K., Fantone, J. C., Remick, D. G., LeGendre, M., McClain, M., and Engleberg, N. C. (1997a). The role of Legionella pneumophila-infected Hartmannella vermiformis as an infectious particle in a murine model of Legionnaire's disease. Infect. Immun. 65, 5330–5333.
Brieland, J., McClain, M., Heath, L., Chrisp, C., Huffnagle, G., LeGendre, M., et al. (1996). Coinoculation with Hartmannella vermiformis enhances replicative Legionella pneumophila lung infection in a murine model of Legionnaires' disease. Infect. Immun. 64, 2449–2456.
Brieland, J., McClain, M., LeGendre, M., and Engleberg, C. (1997b). Intrapulmonary Hartmannella vermiformis: a potential niche for Legionella pneumophila replication in a murine model of legionellosis. Infect. Immun. 65, 4892–4896.
Burillo, A., Pedro-Botet, M. L., and Bouza, E. (2017). Microbiology and epidemiology of legionnaire's disease. Infect. Dis. Clin. North Am. 31, 7–27. doi: 10.1016/j.idc.2016.10.002
Cambronne, E. D., and Roy, C. R. (2007). The Legionella pneumophila IcmSW complex interacts with multiple Dot/Icm effectors to facilitate type IV translocation. PLoS Pathog. 3:e188. doi: 10.1371/journal.ppat.0030188
Chaulk, S. G., Smith Frieday, M. N., Arthur, D. C., Culham, D. E., Edwards, R. A., Soo, P., et al. (2011). ProQ is an RNA chaperone that controls ProP levels in Escherichia coli. Biochemistry 50, 3095–3106. doi: 10.1021/bi101683a
Cho, B. K., Federowicz, S. A., Embree, M., Park, Y. S., Kim, D., and Palsson, B. O. (2011). The PurR regulon in Escherichia coli K-12 MG1655. Nucleic Acids Res. 39, 6456–6464. doi: 10.1093/nar/gkr307
Cianciotto, N. P. (2005). Type II secretion: a protein secretion system for all seasons. Trends Microbiol. 13, 581–588. doi: 10.1016/j.tim.2005.09.005
Cirillo, J. D., Cirillo, S. L., Yan, L., Bermudez, L. E., Falkow, S., and Tompkins, L. S. (1999). Intracellular growth in Acanthamoeba castellanii affects monocyte entry mechanisms and enhances virulence of Legionella pneumophila. Infect. Immun. 67, 4427–4434.
Cirillo, J. D., Falkow, S., and Tompkins, L. S. (1994). Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect. Immun. 62, 3254–3261.
Coers, J., Kagan, J. C., Matthews, M., Nagai, H., Zuckman, D. M., and Roy, C. R. (2000). Identification of Icm protein complexes that play distinct roles in the biogenesis of an organelle permissive for Legionella pneumophila intracellular growth. Mol. Microbiol. 38, 719–736. doi: 10.1046/j.1365-2958.2000.02176.x
Cormack, B. P., Valdivia, R. H., and Falkow, S. (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173, 33–38. doi: 10.1016/0378-1119(95)00685-0
Creasey, E. A., and Isberg, R. R. (2012). The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl. Acad. Sci. U.S.A. 109, 3481–3486. doi: 10.1073/pnas.1121286109
Drennan, S. L., Lama, A., Doron, B., and Cambronne, E. D. (2013). Tractable mammalian cell infections with protozoan-primed bacteria. J. Vis. Exp. doi: 10.3791/50300
Ducret, A., Chabalier, M., and Dukan, S. (2015). Characterization and resuscitation of ‘non-culturable’ cells of Legionella pneumophila. BMC Microbiol. 14:3. doi: 10.1186/1471-2180-14-3
Ensminger, A. W., Yassin, Y., Miron, A., and Isberg, R. R. (2012). Experimental evolution of Legionella pneumophila in mouse macrophages leads to strains with altered determinants of environmental survival. PLoS Pathog. 8:e1002731. doi: 10.1371/journal.ppat.1002731
Epalle, T., Girardot, F., Allegra, S., Maurice-Blanc, C., Garraud, O., and Riffard, S. (2014). Viable but not culturable forms of Legionella pneumophila generated after heat shock treatment are infectious for macrophage-like and alveolar epithelial cells after resuscitation on Acanthamoeba polyphaga. Microb. Ecol. 69, 215–224. doi: 10.1007/s00248-014-0470-x
Fields, B. S. (1996). The molecular ecology of legionellae. Trends Microbiol. 4, 286–290. doi: 10.1016/0966-842X(96)10041-X
Funk, C. R., Zimniak, L., and Dowhan, W. (1992). The pgpA and pgpB genes of Escherichia coli are not essential: evidence for a third phosphatidylglycerophosphate phosphatase. J. Bacteriol. 174, 205–213. doi: 10.1128/jb.174.1.205-213.1992
Holden, E. P., Winkler, H. H., Wood, D. O., and Leinbach, E. D. (1984). Intracellular growth of Legionella pneumophila within Acanthamoeba castellanii Neff. Infect. Immun. 45, 18–24.
Huang, L., Boyd, D., Amyot, W. M., Hempstead, A. D., Luo, Z. Q., O'Connor, T. J., et al. (2011). The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 13, 227–245. doi: 10.1111/j.1462-5822.2010.01531.x
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N., and Isberg, R. R. (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18745–18750. doi: 10.1073/pnas.0609012103
Lloyd, D. (2014). Encystment in Acanthamoeba castellanii: a review. Exp. Parasitol. 145(Suppl.), S20–S27. doi: 10.1016/j.exppara.2014.03.026
Marra, A., Blander, S. J., Horwitz, M. A., and Shuman, H. A. (1992). Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. U.S.A. 89, 9607–9611. doi: 10.1073/pnas.89.20.9607
Mendis, N., McBride, P., and Faucher, S. P. (2015). Short-term and long-term survival and virulence of Legionella pneumophila in the defined freshwater medium fraquil. PLoS ONE 10:e0139277. doi: 10.1371/journal.pone.0139277
Molofsky, A. B., and Swanson, M. S. (2004). Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53, 29–40. doi: 10.1111/j.1365-2958.2004.04129.x
Murata, T., Delprato, A., Ingmundson, A., Toomre, D. K., Lambright, D. G., and Roy, C. R. (2006). The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8, 971–977. doi: 10.1038/ncb1463
Nagai, H., Cambronne, E. D., Kagan, J. C., Amor, J. C., Kahn, R. A., and Roy, C. R. (2005). A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U.S.A. 102, 826–831. doi: 10.1073/pnas.0406239101
Neild, A. L., and Roy, C. R. (2003). Legionella reveal dendritic cell functions that facilitate selection of antigens for MHC class II presentation. Immunity 18, 813–823. doi: 10.1016/S1074-7613(03)00140-7
Ninio, S., Zuckman-Cholon, D. M., Cambronne, E. D., and Roy, C. R. (2005). The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol. Microbiol. 55, 912–926. doi: 10.1111/j.1365-2958.2004.04435.x
O'Toole, G. A., and Kolter, R. (1998). Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28, 449–461. doi: 10.1046/j.1365-2958.1998.00797.x
Pereira, C. T., Moutran, A., Fessel, M., and Balan, A. (2015). The sulfur/sulfonates transport systems in Xanthomonas citri pv. citri. BMC Genomics 16:524. doi: 10.1186/s12864-015-1736-5
Pinotsis, N., and Waksman, G. (2017). Structure of the WipA protein reveals a novel tyrosine protein phosphatase effector from Legionella pneumophila. J. Biol. Chem. 292, 9240–9251. doi: 10.1074/jbc.M117.781948
Roy, C. R. (2002). The Dot/lcm transporter of Legionella pneumophila: a bacterial conductor of vesicle trafficking that orchestrates the establishment of a replicative organelle in eukaryotic hosts. Int. J. Med. Microbiol. 291, 463–467. doi: 10.1078/1438-4221-00154
Roy, C. R., Berger, K. H., and Isberg, R. R. (1998). Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28, 663–674. doi: 10.1046/j.1365-2958.1998.00841.x
Segal, G., and Shuman, H. A. (1997). Characterization of a new region required for macrophage killing by Legionella pneumophila. Infect. Immun. 65, 5057–5066.
Sutherland, M. C., Nguyen, T. L., Tseng, V., and Vogel, J. P. (2012). The legionella IcmSW complex directly interacts with DotL to mediate translocation of adaptor-dependent substrates. PLoS Pathog. 8:e1002910. doi: 10.1371/journal.ppat.1002910
Theodoulou, F. L., and Kerr, I. D. (2015). ABC transporter research: going strong 40 years on. Biochem. Soc. Trans. 43, 1033–1040. doi: 10.1042/BST20150139
Tilney, L. G., Harb, O. S., Connelly, P. S., Robinson, C. G., and Roy, C. R. (2001). How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114(Pt 24), 4637–4650.
Vogel, J. P., Roy, C., and Isberg, R. R. (1996). Use of salt to isolate Legionella pneumophila mutants unable to replicate in macrophages. Ann. N. Y. Acad. Sci. 797, 271–272. doi: 10.1111/j.1749-6632.1996.tb52975.x
Warren, W. J., and Miller, R. D. (1979). Growth of Legionnaires disease bacterium (Legionella pneumophila) in chemically defined medium. J. Clin. Microbiol. 10, 50–55.
Zamboni, D. S., McGrath, S., Rabinovitch, M., and Roy, C. R. (2003). Coxiella burnetii express type IV secretion system proteins that function similarly to components of the Legionella pneumophila Dot/Icm system. Mol. Microbiol. 49, 965–976. doi: 10.1046/j.1365-2958.2003.03626.x
Zgonc, D., and Baideme, M. (2015). Distributed treatment systems. Water Environ. Res. 87, 1196–1207. doi: 10.2175/106143015X14338845155624
Keywords: Legionella, pathogenesis, transposon mutagenesis, Acanthamoeba castellanii, macrophage, Dot/Icm T4b secretion system, ABC transporter, Francisella
Citation: Lama A, Drennan SL, Johnson RC, Rubenstein GL and Cambronne ED (2017) Identification of Conserved ABC Importers Necessary for Intracellular Survival of Legionella pneumophila in Multiple Hosts. Front. Cell. Infect. Microbiol. 7:485. doi: 10.3389/fcimb.2017.00485
Received: 11 September 2017; Accepted: 13 November 2017;
Published: 30 November 2017.
Edited by:
Elizabeth L. Hartland, Hudson Institute of Medical Research, AustraliaReviewed by:
Julia Walochnik, Medical University of Vienna, AustriaSunny Shin, University of Pennsylvania, United States
Copyright © 2017 Lama, Drennan, Johnson, Rubenstein and Cambronne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric D. Cambronne, cambronn@ohsu.edu