Comparative Whole-Genomic Analysis of an Ancient L2 Lineage Mycobacterium tuberculosis Reveals a Novel Phylogenetic Clade and Common Genetic Determinants of Hypervirulent Strains
 Rahim Rajwani1
Rahim Rajwani1  Wing Cheong Yam2
Wing Cheong Yam2  Ying Zhang3 Yu Kang4 Barry Kin Chung Wong5
Ying Zhang3 Yu Kang4 Barry Kin Chung Wong5  Kenneth Siu Sing Leung2
Kenneth Siu Sing Leung2  Kingsley King Gee Tam2
Kingsley King Gee Tam2  Ketema Tafess Tulu1 Li Zhu1
Ketema Tafess Tulu1 Li Zhu1  Gilman Kit Hang Siu1*
Gilman Kit Hang Siu1*- 1Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Hong Kong, Hong Kong
- 2Department of Microbiology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, Hong Kong
- 3Department of Molecular Microbiology and Immunology, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, United States
- 4CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China
- 5Department of Pathology, United Christian Hospital, Hong Kong, Hong Kong
Background: Development of improved therapeutics against tuberculosis (TB) is hindered by an inadequate understanding of the relationship between disease severity and genetic diversity of its causative agent, Mycobacterium tuberculosis. We previously isolated a hypervirulent M. tuberculosis strain H112 from an HIV-negative patient with an aggressive disease progression from pulmonary TB to tuberculous meningitis—the most severe manifestation of tuberculosis. Human macrophage challenge experiment demonstrated that the strain H112 exhibited significantly better intracellular survivability and induced lower level of TNF-α than the reference virulent strain H37Rv and other 123 clinical isolates.
Aim: The present study aimed to identify the potential genetic determinants of mycobacterial virulence that were common to strain H112 and hypervirulent M. tuberculosis strains of the same phylogenetic clade isolated in other global regions.
Methods: A low-virulent M. tuberculosis strain H54 which belonged to the same phylogenetic lineage (L2) as strain H112 was selected from a collection of 115 clinical isolates. Both H112 and H54 were whole-genome-sequenced using PacBio sequencing technology. A comparative genomics approach was adopted to identify mutations present in strain H112 but absent in strain H54. Subsequently, an extensive phylogenetic analysis was conducted by including all publically available M. tuberculosis genomes. Single-nucleotide-polymorphisms (SNPs) and structural variations (SVs) common to hypervirulent strains in the global collection of genomes were considered as potential genetic determinants of hypervirulence.
Results:Sequencing data revealed that both H112 and H54 were identified as members of the same sub-lineage L2.2.1. After excluding the lineage-related mutations shared between H112 and H54, we analyzed the phylogenetic relatedness of H112 with global collection of M. tuberculosis genomes (n = 4,338), and identified a novel phylogenetic clade in which four hypervirulent strains isolated from geographically diverse regions were clustered together. All hypervirulent strains in the clade shared 12 SNPs and 5 SVs with H112, including those affecting key virulence-associated loci, notably, a deleterious SNP (rv0178 p. D150E) within mce1 operon and an intergenic deletion (854259_ 854261delCC) in close-proximity to phoP.
Conclusion: The present study identified common genetic factors in a novel phylogenetic clade of hypervirulent M. tuberculosis. The causative role of these mutations in mycobacterial virulence should be validated in future study.
Introduction
Despite the continuous effort to combat tuberculosis (TB) in the past few decades, it remains to be a major health problem globally. In particular, the challenge from TB is worsened by the rapid emergence of multidrug resistant (MDR) and even extensively drug-resistant (XDR) TB cases. Development of new drugs and vaccines has proved difficult owing to our poor understanding of the pathogenesis of the causative agent, Mycobacterium tuberculosis.
M. tuberculosis is an intracellular pathogen that is able to modulate the host immune response and persist inside the macrophage, leading to a latent infection with limited replicative potential (Smith, 2003). However, previous studies demonstrated that some clinical strains have established virulence mechanisms that disrupt the delicate balance between replication and survival of its host. They multiply rapidly inside the host macrophage, followed by escaping from immune barriers in the lungs and spreading to other organs, causing more severe forms of the disease, such as tuberculous meningitis, even in immunocompetent individuals (Caminero et al., 2001; Alonso et al., 2011; Ribeiro et al., 2014).
In our previous study, virulence of 125 M. tuberculosis clinical strains was evaluated by measuring intracellular survival in peripheral-blood-monocyte-derived-macrophages (PBMDMs), as well as quantifying the release of pro-inflammatory cytokines (TNFα, IL-10, and IL-12) upon infection (Wong et al., 2007). Interestingly, one of the strains (H112) was shown to grow more rapidly and induce lower level of TNF-α than a large number of other strains, consistently in different batches of PBMDMs collected from multiple healthy donors (Wong et al., 2007). Moreover, clinical record revealed that H112 was isolated from the cerebrospinal fluid of an HIV-negative patient who had aggressive disease progression from pulmonary TB to tuberculous meningitis within 2 months. We hypothesized that the genome M. tuberculosis H112 encodes an altered set of virulence-factors that enables it to survive within the hostile macrophage environment more efficiently than other strains.
The current study aims to investigate the genomic uniqueness of hypervirulent strain H112 by a two-step comparative genomic approach. The first step focuses on masking lineage-related genetic polymorphisms by a comparison with a genetically-related less-virulent strain, H54. The second step concentrates on identifying a subset of H112-specific mutations that are commonly found in hypervirulent strains reported elsewhere.
Materials and Methods
Ethical Approval and Biological Safety
This study has been approved by the Institutional Review Board of The Hong Kong Polytechnic University (Ref. number: RSA15096). All experiment involving viable M. tuberculosis culture were handled in biosafety level 3 (BSL-3) laboratory with an approved protocol (HSE(HKU)_20160104).
M. tuberculosis Strains
In our previous study, 125 M. tuberculosis clinical isolates collected in Hong Kong and Shanghai, China, between 2002 and 2004, were determined for the intra-macrophage survivabilities in PBMDMs and induction of pro-inflammatory cytokines by macrophages (Wong et al., 2007). A hypervirulent strain, H112, which demonstrated enhanced intracellular growth relative to 123 other strains was included in the present study. The strain was isolated from cerebrospinal fluid of a tuberculous meningitis patient who was a 51-year old male with no known comorbidity and was HIV-negative. The patient was first diagnosed as pulmonary TB in April 2004, but then failed to comply with anti-TB treatment course. The disease rapidly progressed into tuberculous meningitis 2 months later. The patient died in July 2004. In vitro drug susceptibility testing revealed that the strains were pan-susceptible to all first-line anti-TB drugs. Subsequently, another clinical isolate, H54, which belonged to the same phylogenetic lineage as the hypervirulent strain, was obtained from respiratory specimen of a newly diagnosed pulmonary TB patient. The patient was a 67-year old, HIV negative male admitted to the same clinical center in Hong Kong in October 2004. He was successfully cured upon the completion of a standard course of first-line anti-TB treatment. Strain H54 was used as comparator of the hypervirulent strain H112 in whole-genome sequence analysis in this study. M. tuberculosis reference strains H37Rv (ATCC 27294) and H37Ra (ATCC 25177) were purchased from American type tissue culture collection (ATCC) as virulent and avirulent control strains respectively for THP-1 cell challenge experiment.
MIRU-VNTR Typing
Frozen bacterial stocks were subcultured on Middlebrook 7H10 Agar to obtain isolated colonies. DNA was extracted from isolated colonies using Cobas Amplicor Respiratory kit (Roche Diagnostics, Germany) as previously described (Siu et al., 2011). Each of the 12 loci MIRU were amplified individually and resolved on 2.5% agarose gel electrophoresis to infer number of repeats at each locus based on amplicon size as described previously (Cowan et al., 2002). Phylogenetic analysis of the 12 loci MIRU was conducted using MIRU-VNTR plus (Weniger et al., 2010).
Spoligotyping
Prior to WGS, spoligotyping was performed according to standardized laboratory procedure described previously (Kamerbeek et al., 1997). The resulting spoligotypes were used to assign M. tuberculosis lineages to strains in TB-lineage (Shabbeer et al., 2012).
Infection of THP-1 Cells with Mycobacterium tuberculosis and Measurement of Growth Index
The THP-1 monocytic cell line (ATCC TIB-202) was cultured at density 3 × 108 cells per well into 24-well plate containing RMI1640 medium (GIBCO, USA) supplemented with 5% (v/v) fetal bovine serum (GIBCO, USA) at 37°C with 5% CO2. Viability of cells was assessed by staining with 0.4% (w/v) Trypan blue and differentiation was induced overnight by treatment with 20 nM phorbol 12-myristate 13-acetate (Sigma Aldrich, USA). The THP-1 cells were rested for 24 h by incubating in RMI1640 without PMA before infecting with mycobacterial strains. Differentiated THP-1 cells were then infected at a multiplicity of infection (MOI) of 1:1 overnight with M. tuberculosis strains pre-passaged through 25-guage needle. Extracellular bacteria were removed by washing three times with RMI1640 medium. The cells were lysed at day 0, 1, 3, and 6 using 0.1% (v/v) sodium dodecyl sulfate (SDS) and 10-fold diluted suspensions of intracellular bacteria were plated on Middlebrook 7H10 agar (DIFCO, USA) supplemented with 10% (v/v) oleic acid-albumin-dextrose-catalase (Becton Dickinson, USA). Colony forming units per ml were enumerated after 4 weeks. The experiment was performed in triplicate and mean CFU per ml was calculated. Growth index was determined as mean CFU per ml at day x divided by mean CFU per ml at day 0.
DNA Extraction
Genomic DNA for Pac-Bio sequencing was isolated as described previously (Benjak et al., 2015). Briefly, 10 ml of late-log phase culture of M. tuberculosis strain was prepared in 7H9 medium which was then pelleted and frozen at −80°C overnight. Pellet was resuspended into SET buffer (25% w/v sucrose, 50 mM EDTA, 50 mM Tris-HCL, pH:8.0) with the addition of lysozyme and incubated at 37°C overnight. This was followed by treatment with proteinase K and RNAase. DNA was purified once through Phenol-Chloroform-isoamyl alcohol (25:24:1) and secondly through chloroform-isoamyl alcohol (24:1) layer. Quality and integrity of DNA was checked through Qubit HS assay (Thermo Scientific, USA) and 0.8% agarose gel electrophoresis respectively.
Single Molecule Real-Time (SMRT) Sequencing, de Novo Assembly, and Annotation
A total of 15 μg of genomic DNA (gDNA) from each M. tuberculosis strain was used to prepare 20-Kb libraries which were sequenced by P6-C4 chemistry using one SMRT cell per library (Pacific biosciences, USA). The library was loaded using MagBead One Cell per Well (OCPW version 1) protocol to capture data in 240 min movie time. De novo assembly from the resulting continuous long reads (CLR) was performed using Hierarchical Genome Assembly Process (HGAP.2) algorithm from SMRT portal (version 2.3.0) (Chin et al., 2013).
Phylogenetic Analysis and SNP Detection
Previously published assembled genomes (n = 22) from all eight M. tuberculosis complex (MTBC) lineages were downloaded from NCBI GenBank (Table S1). A multiple core-genome alignment was generated using Parsnp utility in Harvest Suite (Treangen et al., 2014). The alignment was then used to construct maximum-likelihood (ML) tree using generalized time reversible (GTR), proportion of invariable sites 0.0 and number of substitutions per categories as 4 in MEGA7 (Kumar et al., 2016). A multi-sample variant-call-file (VCF) was generated from core-genome alignment and SNPs present in H112 but absent in H54 were extracted using BCFtools (Li et al., 2009).
For detailed phylogenetic analysis, a total of 4,335 assembled genomes of M. tuberculosis in NCBI-Genbank (accessed on 8/12/2016) and un-assembled genome data-sets from previous studies (Zhang et al., 2013; Luo et al., 2015; Merker et al., 2015) were searched for lineage 2 strains harboring at least one of the H112-specific SNPs (i.e., SNPs present in H112 but absent in the comparator strain H54) (Table S2).
Subsequently, raw reads were aligned to the reference genome (Genbank accession NC_000962.3) using Bowtie2 and SNPs with depth greater than 5, allele frequency exactly 1, mapping quality greater than 20, and Phred-scaled variant quality in excess of 20 were called using SAMtools (Li et al., 2009). A list of all informative SNP positions was compiled and corresponding base calls for all samples were retrieved. SNP loci with missed call in any of the samples were discarded to obtain list of highly credible phylogenetically informative SNPs. Concatenated SNPs was used to construct ML tree using GTR model in MEGA7 as described above (Kumar et al., 2016).
As a quality-filtering step, nearly identical genomes (pair-wise SNP differences less than eight) or unusually long-branched on the phylogenetic tree (indicating sequencing-errors) were excluded. The final data-set comprised of 33 strains from 13 countries. Statistical support for clades was determined using bootstrap analysis with 100 pseudo-replicates. Trees were visualized in FigTree v1.4.3.
Furthermore, MSA was used to conduct principal component analysis (PCA) and compute pair-wise SNP distance within and between groups in JalView and MEGA7 respectively.
Predicting Impact of Non-synonymous SNPs
Deleterious impact of mutations on protein function was predicted using sorting intolerant from tolerant (SIFT 4G) (Kumar et al., 2009). Predictions with a score <0.05 were considered significant.
Detection of Structural Variations
Structural variations (SVs, ranging 1–10,000 bp) present in H112 but absent in H54 were identified using Assemblytics (Nattestad and Schatz, 2016). Briefly, genome assemblies were aligned against the reference genome M. tuberculosis H37Rv (NC_000962.3) using NUCmer with –minmatch 100 and –mincluster 500. The alignment file was used to call structural variations within and between alignments with at least 100 bp unique anchor sequence in Assemblytics (Nattestad and Schatz, 2016). SVs that do not overlap between H112 and H54 were identified using BEDtools (Quinlan and Hall, 2010). Presence of identified SVs in short-read sequencing data (used in detailed phylogenetic analysis) was verified by manual inspection of binary alignment map (BAM) files.
In addition, SVs mediated by IS6110 insertion were identified using IS-seeker (Adams et al., 2016). BLASTN was used to map complete or partial sequence of IS6110 element (Genbank accession X17348). Sequences flanking the mapped regions were retrieved and aligned to the reference genome (NC_000962.3) for annotation.
Genomic Data Availability
The genome sequencing data for strains sequenced in this study are deposited under NCBI Bio-Project accession number: PRJNA369711 (H112 accession CP019613; H54 accession CP019610).
Results
Selection of Control Strain to Mask Lineage-Specific Variability in Comparative Genomic Analysis
All available (n = 115/125;92%) isolates from previous study were cultivated from the frozen stocks and were subjected to DNA-fingerprinted based on polymorphisms within 12 MIRU loci. UPGMA tree-based analysis identified that hypervirulent strain H112 was genetically closest to H54 among other 113 strains. Genetic relationship between H112 and H54 was further confirmed by spoligotyping. Spoligotypes for both strains were found to be identical (000000000003771) and representative of lineage 2 (L2) (Figure 1). Together, both MIRU and spoligotype patterns indicated that H54 is genetically related to hypervirulent strain H112. Therefore, H54 was selected as a control strain to mask lineage-related genetic variation in subsequent comparative genomic analysis.
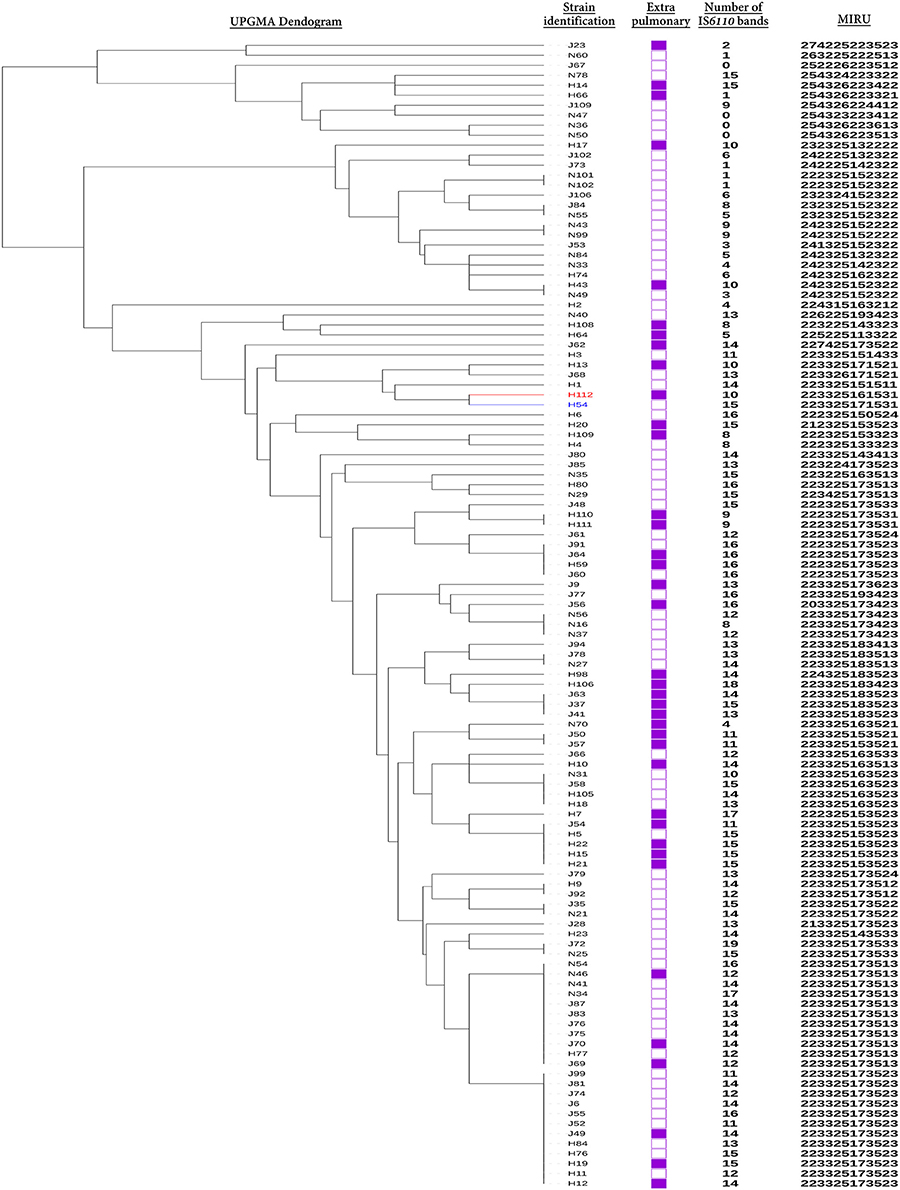
Figure 1. Selection of control strain. The relationship between Hypervirulent strain H112 (Red), control strain H54 (blue), and other 113 strains is indicated via dendogram. The dendogram was constructed using unweighted pair group method with arithmetic mean (UPGMA) algorithm based on MIRU. Additional information related to source of all strains that is extra-pulmonary (filled squares) or pulmonary TB (empty squares), number of IS6110-RFLP bands and MIRU patterns are also illustrated.
Reassessment of Intracellular Growth in Macrophage
H112 was demonstrated to have better survivability in PBMDMs in previous study (Wong et al., 2007). In present study, the ability of intracellular growth of H112 and H54 was reassessed and compared, as well as the reference strains in THP-1. The growth indices of M. tuberculosis strains inside THP-1 are shown in Figure 2. It was observed that shortly after infection (day 0–3), the bacterial counts of H112 increased, which indicated excellent adaptation to the harsh intracellular environment in human macrophage. On the contrary, the bacterial counts of H37Rv, H37Ra, and H54 remained constant or declined indicating reduced fitness of these strains accompanied by an early killing phase that macrophages attempted to eradicate the infection. This was followed by a second period (day 3–6) during which the cell number of H112 elevated further whereas the survival of H54 slightly declined. At day six after infection, both H112 and H37Rv showed 30–50% increase in cell numbers, confirming to their virulent phenotype, whereas H54 and H37Ra reduced in cell numbers by 12 and 92% respectively (Figure 2). Overall, the intracellular replication potential of H112 was found to be higher than H37Rv and clinical isolate of same major lineage, establishing itself as the hypervirulent strain.
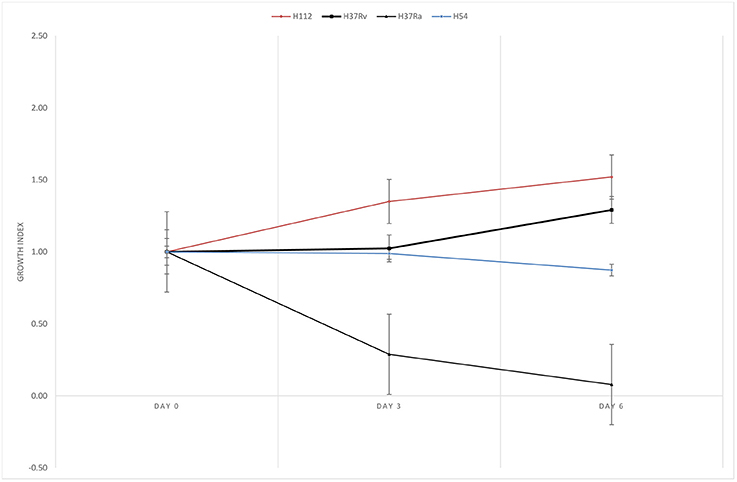
Figure 2. Reassessment of enhanced intracellular growth in THP-1 macrophage. Growth indexes showing multiplication of M. tuberculosis strains inside THP-1 macrophage. Higher growth index of hypervirulent strain H112 (red) relative to reference virulent strain H37Rv (black with squares), avirulent strain H37Ra (black with triangles), and control strains H54 (blue) were clearly demonstrated. Growth index was determined as mean CFU per ml at day 0, 3, 6 divided by mean CFU per ml at day 0, respectively. The lines represent mean of triplicate experiments.
Whole-Genome Sequencing and de Novo Assembly
The sequencing run yielded high-quality (quality score > 0.8) reads with sub-reads N-50 value greater than 10 Kb. Interestingly, fully closed circular genome assembled into single contig with an average sequencing depth of ~150X was obtained for both M. tuberculosis strains. Size of H112 and H54 genomes was nearly same (~4.4 megabases) (Table 1).
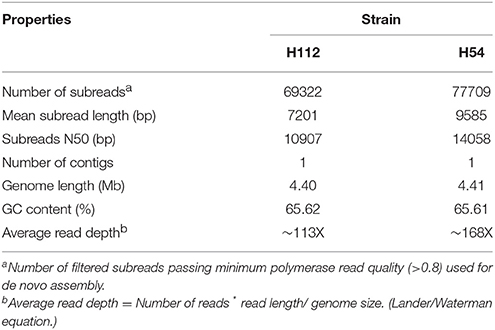
Table 1. Results of whole-genome sequencing and assembly.
Phylogenetic Placement Using WGS
In order to contextualize phylogenetic placement of H112 and H54 in the global phylogeny of Mycobacterium tuberculosis complex (MTBC), phylogenetic tree was reconstructed with the inclusion of representative genomes from all L2 sub-lineages (n = 8) and other MTBC major lineages (n = 14) (Figure 3). It was observed that H112 and H54 were represented by adjacent branches on the tree. Moreover, these branches were enclosed within a clade representative of L2 (Figure 3). According to SNP-based classification scheme for M. tuberculosis (Coll et al., 2014), H112 and H54 were identified as members of the same sub-lineage L2.2.1, which was also known as ancient L2. The sub-lineage assignment was also corroborated by absence of region-of-difference (RD)105, RD207, and RD181 in both strains H112 and H54. Overall, consistent with results obtained using conventional genotyping, H112 and H54 were found to be genetically-related at sub-lineage level.
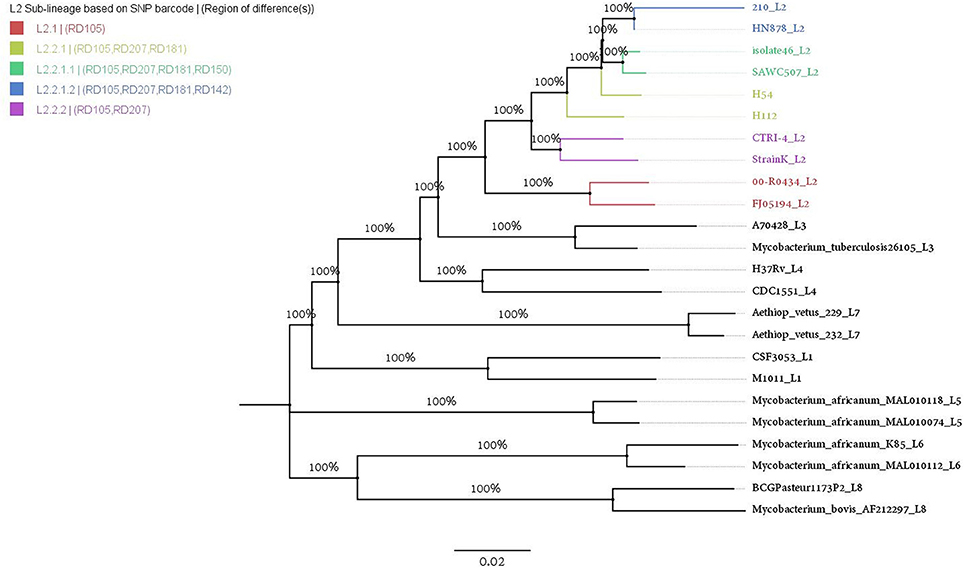
Figure 3. Phylogenetic placement of H112 and H54. Assignment of phylogenetic lineage to H112 and H54 is illustrated using ML-tree with representative strains from all eight major lineages (L1–L8) of M. tuberculosis complex (MTBC) and L2 sub-lineages. Branch labels indicate bootstrap values in percentage from 100 pseudo-replicates.
Comparative Genomics between Hypervriulent Strain H112 and Control strain H54 within Phylogenetic Framework
Single Nucleotide Polymorphisms
There were 1,238 SNPs associated with hypervirulent strain H112. Interestingly, a vast majority (1,099/1,238; 88.7%) of H112-associated SNPs were common with H54 (Figure S1). SNPs shared between H112 and H54 constitute genetic variation related to lineage. For instance, SNPs accumulated as a member of L2.2.1 (85/1,099; 7.7%), L2.2 (111/1,099; 10.1%), L2 (112/1,099; 10.1%), and so on. There were 139 SNPs (139/1,238; 11.2%) present only in H112 (Table S3). Out of 139, 11 (11/139; 7.9%) were intergenic, 85 (85/139; 61.1%) were non-synonymous, 43 (43/139;30.9%) were synonymous and one (1/139; 0.7%) was non-sense SNP.
Structural Variations
Similarly, structural variations (SVs, i.e., insertions or deletions) in H112 and H54 were called relative to reference genome (NC_000962.3). Subsequently, SVs present in H112, but absent in H54 were identified (Table S4). There were 45 SVs present in H112 only. Out of 45, 31 (31/45;68.8%) were present within coding sequences, seven (7/45;15.5%) within repeat regions and another seven (7/45;15.5%) within intergenic regions.
Comparison of H112 with Global Circulating Strains
Phylogeny
For a more comprehensive comparison of H112 with global circulating strains, mutations present in H112, but absent in H54, were used to screen public genomes. Our search identified 33 strains collected from 13 countries that also harbored at least one of these mutations. WGS-based phylogeny clustered them into one large clade (ancestor clade) that could be further divided into two distinct clades (H112-clade and non-H112-clade) (Figure 4).

Figure 4. Extensive genomic comparisons between H112 and previously published genome sequences. (A) Maximum-likelihood phylogeny based on previously published genomes related to H112. Branch labels indicate bootstrap values on ratio scale 0–1. Scale bar indicates nucleotide substitutions per site. Strain identifications, country of isolation, and virulence is indicated adjacent to the corresponding tips. Country of isolation is mainland China unless otherwise indicated. For virulence column, filled circles represent high virulence and empty circle represent low virulence. No circle is drawn in case of unknown virulence. (B) Results of principal component analysis showing clear distinction between identified clades. Each dot represent a sequence used in the analysis. An ellipse is drawn to show distinction between H112 and non-H112 clade. (C) A Tukey boxplot showing comparison of pair-wise SNP differences within H112-clade, non-H112-clade and between them. Box covers 25 and 75th percentile of the data. Center line indicates median and cross indicates mean. Each circle represents a pairwise comparison in bell-swarm representation {n = 136 (non-H112-clade), 120 (H112-clade), 272 (Between-clades)}.
A small number of H112-specific SNPs (9/139; 6.4%), were shared among all members of ancestor clade. These included mutation in mce1 operon (yrbE1b p.A16T) and DNA-repair pathway (ogt p.R37L) (Table S3). No structural variation was common among all strains of ancestor clade (Table S4).
Notably, other than the nine SNPs common among ancestor clade, a group of 16 strains were found to share a total of 12 (12/139; 8.6%) H112-specific SNPs and five (5/45; 11.1%) H112-specific SVs, and were therefore classified as a distinct clade, which was named as H112-clade (Figure 4).
Other Highly Virulent Strains Related to H112
In addition to hypervirulent strain H112 described in this study, three other highly virulent strains clustered within H112-clade. These were M. tuberculosis GC1237 resulting in multiple outbreaks in Gran Canaria, Spain (Caminero et al., 2001; Alonso et al., 2011). M. tuberculosis Zt272 that resulted in increased bacterial load in lungs, decreased survival time and increased area of pneumonia relative to other strains in the intra-tracheally infected mice (Ribeiro et al., 2014). The M. tuberculosis Zaragoza strain caused outbreak in Zaragoza, Spain, and reached 18.7% of all isolates of M. tuberculosis in 2001 to 2004 (Jia et al., 2017; Rodríguez-Castillo et al., 2017). On the contrary, non-H112 clade strain included the streptomycin-dependent M. tuberculosis 18b strain, which has been reported to be severely attenuated in high-dose (>1,000 bacilli) aerosol infected mice model (Campos-Neto, 2016) although the virulence of other strains in this clade have not been phenotypically characterized.
Mutations Common between H112 and Other Highly Virulent Strains
Single nucleotide polymorphisms
There were 12 SNPs specific to the H112-clade: one (1/12;8.3%) intergenic, three (3/12;25%) synonymous, and eight (8/12;66.6%) non-synonymous SNPs (Table 2). Only one SNP (1/12;8.3%), Rv0178 (p. D150E), was predicted to have a deleterious impact on the protein function (SIFT score <0.05). Interestingly, Rv0178 is encoded within mammalian cell entry 1 (mce1) operon and has been implicated in modulating mycobacterial virulence.
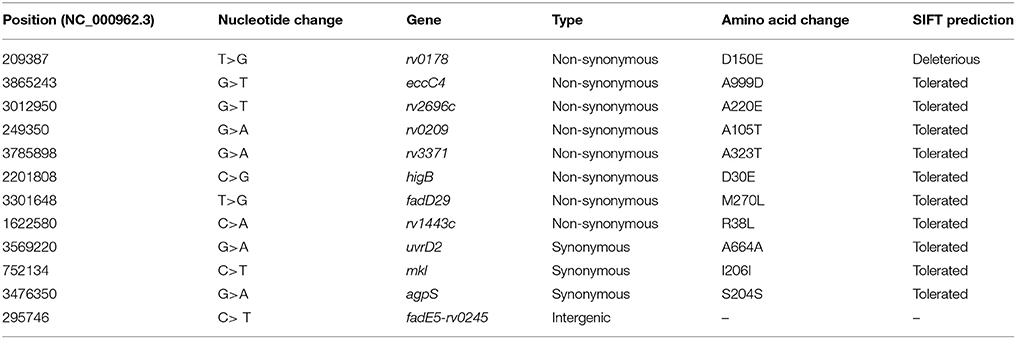
Table 2. Single nucleotide polymorphisms common to H112-clade.
Structural variations
Of the five SVs common among members of H112–clade, three (3/5;60%) were located within coding sequences, two (2/5;40%) were present within intergenic regions (Table 3). Among the coding sequences affected by SVs, two (rv2286c and rv0840c) were disrupted by IS6110 insertion events. In addition, there was a 9 bp deletion within a possible exported protein rv0633c. Both intergenic SVs were small deletions (i.e., less than three base-pairs). One of them was present between rv0759c and rv0760c, downstream of key virulence-associated operon phoPR. While, another one was in close proximity to IS6110 fragment, between rv2168c and rv2169c.
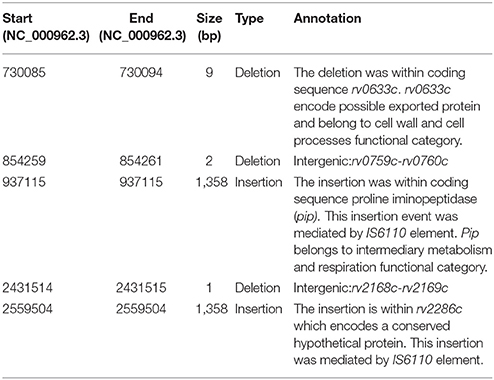
Table 3. Structural variation common to H112-clade.
Discussion
In this study, comparative whole genomic analysis between a hypervirulent M. tuberculosis clinical strain H112 and another clinical strain H54 of lower virulence from the same lineage was conducted to identify a set of mutations that are specific to the hypervirulent strain. Based on these mutations, we analyzed the phylogenetic relatedness of H112 with all available M. tuberculosis whole genomes, which led us to identify a novel phylogenetic cluster that encompassed H112 and the highly virulent strains reported elsewhere. All these strains shared a total of 12 SNPs and five SVs, which cannot be found in other strains with low-level virulence in L2 lineage. This is the first study to report the use of these genetic markers for successful clustering of highly virulent M. tuberculosis strains isolated from different parts of the world.
While several studies have suggested that modern phylogenetic groups of L2 (L2.2.1.1 and L2.2.1.2) are more virulent than ancient ones (L2.1, L2.2.1, L2.2.2) (Hanekom et al., 2010; Ribeiro et al., 2014), the current study has identified a highly virulent strain cluster within an ancient group (L2.2.1). In particular, our data point-toward existence of a highly virulent clade, H112-clade, comprising of at least three whole-genome sequenced hypervirulent strains, namely H112, GC1237, and Zt272. Another outbreak strain M. tuberculosis Zaragoza (MTZ) also harbors genetic markers of H112-clade, notably, IS6110 insertion within rv2286c and ogt (p.R36L) (López-Calleja et al., 2007), and thus could also be linked to H112-clade although whole-genome sequence for this strain has yet to be available. The enhanced virulence of these strains was previously demonstrated in individual studies and was shown to be independent of each other. In this study, through an extensive phylogenetic analysis, these strains were shown to be closely related to one another. The close relationship between these highly virulent strains suggested that genetic factors common to them are likely to contribute toward their hypervirulence, although the causative effect of these mutations should be further investigated in the future study.
WGS has emerged as a powerful tool to identify genetic determinants of mycobacterial virulence. Notably, comparative genomics approach has revealed possible associations between genetic polymorphisms and virulence diversity in M. tuberculosis (Jia et al., 2017; Rodríguez-Castillo et al., 2017). However, virulence of the M. tuberculosis strains used in most of these studies was largely unknown. In addition, no effort was made to eliminate lineage-related mutations, leading to identification of numerous genetic mutations that could be irrelevant to mycobacterial virulence. Conversely, in the present study, hypervirulence of strain H112 was well-defined by enhanced survivability in macrophage models of M. tuberculosis infection as well as severe clinical manifestation (TB meningitis) in a 51-year old immunocompetent patient. By genomic comparison with a less-virulent strain from same phylogenetic lineage, the hypervirulent strain-specific mutations were distinguished from the massive lineage-related mutations. These mutations were then used to screen for public genomes, and those which were commonly shared by hypervirulent M. tuberculosis strains isolated from different global regions were considered as potential genetic determinants of mycobacterial virulence. This analysis cascade helped us to pinpoint 12 SNPs out of 1,238 SNPs obtained immediately from WGS of H112.
The impact of the interested SNPs on protein function was further predicted by computational algorithm. Only one non-synonymous mutation, D150E, in rv0178 within mce1 operon was predicted to be deleterious. The mce1 operon forms a cluster of 13 genes encoding two Yrb-like permeases (YrbE1A and YrbE1B), six core mce proteins (Mce1A-F), fatty-acid-CoA ligase (FadD5), and four conserved hypothetical proteins (Rv0175-78) (Shimono, 2003). Deletion of mce1A or yrbE1B has been shown to abolish expression of other mce1 proteins as well (Shimono, 2003). Intravenous infection with mce1A or yrbE1B deleted strain of M. tuberculosis (analogous to complete absence of mce1 operon) was shown to result in increased bacillary load and poorly organized granulomas in mice (Shimono, 2003). It is interesting to speculate that deleterious mutation within mce1 operon, identified here, might be one of the contributing factors to increased virulence of H112-clade.
It was also observed that a 2 bp deletion in the intergenic region rv0759c-rv0760c, was present in all members of H112-clade. Both rv0759c and rv0760c encode conserved hypothetical proteins with unknown function. The intergenic deletion was located~400 bp downstream of phoPR operon. PhoPR is a two-component system that plays an important role in mycobacterial virulence. A deleterious SNP in phoP has been demonstrated as one of the reasons for avirulence of strain H37Ra (Lee et al., 2008). On the other hand, a promoter mutation linked with an increased phoP expression was found in an outbreak strain of M. bovis (Soto et al., 2004). It is well-known that downstream gene variants could also modulate the expression level of upstream gene, particularly, by altering the binding sites for transcriptional regulators. Interestingly, the intergenic region rv0759c-rv0760c has been identified as a binding target for an alternative sigma factor, Sigma F (Rodrigue et al., 2007), which is a regulator of phoP (Hümpel et al., 2010). We hypothesize that in H112-clade, deletion within regulatory region rv0759c-rv0760c, might confer hypervirulence by modulating expression of upstream gene phoP.
The current study was still limited in several aspects. First, the study was focused on only one hypervirulent strain and a control strain. However, both strains were selected from a cohort of more than one hundred strains after extensive genetic and virulence comparisons. The genome-sequence of hypervirulent strain H112 was also compared with other highly virulent strains to overcome limitations associated with sample size. Second, virulence of strains H112 and H54, was not determined in animal models of M. tuberculosis infection. However, an alternative model of M. tuberculosis infection, human macrophage, which is also well-established in the field, was used to assay virulence. The results were further interpreted in context of clinical background of strains. Third, the information regarding the virulence of M. tuberculosis other than the four hypervirulent strains in H112-clade were not available. The accuracy of using the interested mutations as epidemiological markers for tracing hypervirulent M. tuberculosis strains therefore required further investigation.
Most importantly, the functional impacts of H112-clade-specific mutations have not been experimentally validated. In the future study, their causative roles in mycobacterial virulence should be determined through genetic manipulation. For instance, the mutations could be introduced into the wildtype M. tuberculosis genome using allelic exchange approach (Gopinath et al., 2015), or alternatively, the wildtype genes could be transformed into H112 to compensate the respective mutated gene (Siu et al., 2014). The virulence of the genetically manipulated strains should be assessed in cell culture and mice models of M. tuberculosis infection.
In the present study, we identified a novel phylogenetic clade that encompassed highly virulent M. tuberculosis strains isolated from different geographic regions. The genetic mutations common to them may explain the mechanism underlying the enhanced virulence of these strains. The causative effect of these mutations on mycobacterial virulence should be experimentally validated in the future study.
Author Contributions
Conceived and designed the experiments: RR, WY, YZ, and GS. Performed the experiments: RR, BW, KL, KKT, KTT, LZ, and GS. Analyzed the data: RR, YZ, YK, and GS. Contributed reagents, materials, analysis tool: RR, WY, KTT, LZ, and GS. Wrote the paper: RR, WY, YZ, YK, KL, and GS.
Funding
This work was supported by Fund for ECS Project Rated 3.5 (Central Research Grant) from The Hong Kong Polytechnic University (G-YBK4).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sabrina Wai Chi TO for assistance with P3 Laboratory maintenance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2017.00539/full#supplementary-material
References
Adams, M. D., Bishop, B., and Wright, M. S. (2016). Quantitative assessment of insertion sequence impact on bacterial genome architecture. Microb. Genomics 2:e000062. doi: 10.1099/mgen.0.000062
Alonso, H., Aguilo, J. I., Samper, S., Caminero, J. A., Campos-Herrero, M. I., Gicquel, B., et al. (2011). Deciphering the role of IS6110 in a highly transmissible Mycobacterium tuberculosis Beijing strain, GC1237. Tuberculosis 91, 117–126. doi: 10.1016/j.tube.2010.12.007
Benjak, A., Sala, C., and Hartkoorn, R. C. (2015). Whole-genome sequencing for comparative genomics and de novo genome assembly. Methods Mol. Biol. 1285, 1–16. doi: 10.1007/978-1-4939-2450-9_1
Caminero, J. A., Pena, M. J., Campos-Herrero, M. I., Rodriguez, J. C., Garcia, I., Cabrera, P., et al. (2001). Epidemiological evidence of the spread of a Mycobacterium tuberculosis strain of the Beijing genotype on Gran Canaria Island. Am. J. Respir. Crit. Care Med. 164, 1165–1170. doi: 10.1164/ajrccm.164.7.2101031
Campos-Neto, A. (2016). Mycobacterium tuberculosis strain 18b, a useful non-virulent streptomycin dependent mutant to study latent tuberculosis as well as for in vivo and in vitro testing of anti-tuberculosis drugs. Tuberculosis 99, 54–55. doi: 10.1016/j.tube.2016.04.006
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Coll, F., McNerney, R., Guerra-Assuncao, J. A., Glynn, J. R., Perdigao, J., Viveiros, M., et al. (2014). A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5:4812. doi: 10.1038/ncomms5812
Cowan, L. S., Mosher, L., Diem, L., Massey, J. P., and Crawford, J. T. (2002). Variable-number tandem repeat typing of Mycobacterium tuberculosis isolates with low copy numbers of IS6110 by using mycobacterial interspersed repetitive units. J. Clin. Microbiol. 40, 1592–1602. doi: 10.1128/JCM.40.5.1592-1602.2002
Gopinath, K., Warner, D. F., and Mizrahi, V. (2015). Targeted gene knockout and essentiality testing by homologous recombination. Methods Mol. Biol. 1285, 131–149. doi: 10.1007/978-1-4939-2450-9_8
Hanekom, M., Mata, D., van Pittius, N. G., van Helden, P., Warren, R., and Hernandez-Pando, R. (2010). Mycobacterium tuberculosis strains with the Beijing genotype demonstrate variability in virulence associated with transmission. Tuberculosis 90, 319–325. doi: 10.1016/j.tube.2010.08.004
Hümpel, A., Gebhard, S., Cook, G. M., and Berney, M. (2010). The SigF regulon in Mycobacterium smegmatis reveals roles in adaptation to stationary phase, heat, and oxidative stress. J. Bacteriol. 192, 2491–2502. doi: 10.1128/JB.00035-10
Jia, X., Yang, L., Dong, M., Chen, S., Lv, L., Cao, D., et al. (2017). The Bioinformatics analysis of comparative genomics of Mycobacterium tuberculosis Complex (MTBC) provides insight into dissimilarities between intraspecific groups differing in host association, virulence, and epitope diversity. Front. Cell. Infect. Microbiol. 7:88. doi: 10.3389/fcimb.2017.00088
Kamerbeek, J., Schouls, L., Kolk, A., Van Agterveld, M., Van Soolingen, D., Kuijper, S., et al. (1997). Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35, 907–914.
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. doi: 10.1038/nprot.2009.86
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lee, J. S., Krause, R., Schreiber, J., Mollenkopf, H. J., Kowall, J., Stein, R., et al. (2008). Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell Host Microbe 3, 97–103. doi: 10.1016/j.chom.2008.01.002
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
López-Calleja, A., Lezcano, M., Vitoria, M., Iglesias, M., Cebollada, A., Lafoz, C., et al. (2007). Genotyping of Mycobacterium tuberculosis over two periods: a changing scenario for tuberculosis transmission. Int. J. Tuberculosis Lung Dis. 11, 1080–1086.
Luo, T., Comas, I., Luo, D., Lu, B., Wu, J., Wei, L., et al. (2015). Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. Proc. Natl. Acad. Sci. U.S.A. 112, 8136–8141. doi: 10.1073/pnas.1424063112
Merker, M., Blin, C., Mona, S., Duforet-Frebourg, N., Lecher, S., Willery, E., et al. (2015). Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 47, 242–249. doi: 10.1038/ng.3195
Nattestad, M., and Schatz, M. C. (2016). Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023. doi: 10.1093/bioinformatics/btw369
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ribeiro, S. C., Gomes, L. L., Amaral, E. P., Andrade, M. R., Almeida, F. M., Rezende, A. L., et al. (2014). Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J. Clin. Microbiol. 52, 2615–2624. doi: 10.1128/JCM.00498-14
Rodrigue, S., Brodeur, J., Jacques, P.-É., Gervais, A. L., Brzezinski, R., and Gaudreau, L. (2007). Identification of mycobacterial σ factor binding sites by chromatin immunoprecipitation assays. J. Bacteriol. 189, 1505–1513. doi: 10.1128/JB.01371-06
Rodríguez-Castillo, J. G., Pino, C., Niño, L. F., Rozo, J. C., Llerena-Polo, C., Parra-López, C. A., et al. (2017). Comparative genomic analysis of Mycobacterium tuberculosis Beijing-like strains revealed specific genetic variations associated with virulence and drug resistance. Infect. Genet. Evol. 54, 314–323. doi: 10.1016/j.meegid.2017.07.022
Shabbeer, A., Cowan, L. S., Ozcaglar, C., Rastogi, N., Vandenberg, S. L., Yener, B., et al. (2012). TB-Lineage: an online tool for classification and analysis of strains of Mycobacterium tuberculosis complex. Infect. Genet. Evol. 12, 789–797. doi: 10.1016/j.meegid.2012.02.010
Shimono, N. (2003). Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc. Natl. Acad. Sci. U.S.A. 100, 15918–15923. doi: 10.1073/pnas.2433882100
Siu, G. K. H., Yam, W. C., Zhang, Y., and Kao, R. Y. (2014). An upstream truncation of the furA-katG operon confers high-level isoniazid resistance in a Mycobacterium tuberculosis clinical isolate with no known resistance-associated mutations. Antimicrob. Agents Chemother. 58, 6093–6100. doi: 10.1128/AAC.03277-14
Siu, G. K., Tam, Y. H., Ho, P. L., Lee, A. S., Que, T. L., Tse, C. W., et al. (2011). Direct detection of isoniazid-resistant Mycobacterium tuberculosis in respiratory specimens by multiplex allele-specific polymerase chain reaction. Diagn. Microbiol. Infect. Dis. 69, 51–58. doi: 10.1016/j.diagmicrobio.2010.08.021
Smith, I. (2003). Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 16, 463–496. doi: 10.1128/CMR.16.3.463-496.2003
Soto, C. Y., Menéndez, M. C., Pérez, E., Samper, S., Gómez, A. B., García, M. J., et al. (2004). IS6110 mediates increased transcription of the phoP virulence gene in a multidrug-resistant clinical isolate responsible for tuberculosis outbreaks. J. Clin. Microbiol. 42, 212–219. doi: 10.1128/JCM.42.1.212-219.2004
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Weniger, T., Krawczyk, J., Supply, P., Niemann, S., and Harmsen, D. (2010). MIRU-VNTRplus: a web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 38(Web Server issue), W326–W331. doi: 10.1093/nar/gkq351
Wong, K. C., Leong, W. M., Law, H. K., Ip, K. F., Lam, J. T., Yuen, K. Y., et al. (2007). Molecular characterization of clinical isolates of Mycobacterium tuberculosis and their association with phenotypic virulence in human macrophages. Clin. Vaccine Immunol. 14, 1279–1284. doi: 10.1128/CVI.00190-07
Keywords: Mycobacterium tuberculosis, hypervirulent, macrophage, Pacbio, lineage2, comparative genomics, phylogenetic, virulence
Citation: Rajwani R, Yam WC, Zhang Y, Kang Y, Wong BKC, Leung KSS, Tam KKG, Tulu KT, Zhu L and Siu GKH (2018) Comparative Whole-Genomic Analysis of an Ancient L2 Lineage Mycobacterium tuberculosis Reveals a Novel Phylogenetic Clade and Common Genetic Determinants of Hypervirulent Strains. Front. Cell. Infect. Microbiol. 7:539. doi: 10.3389/fcimb.2017.00539
Received: 04 October 2017; Accepted: 26 December 2017;
Published: 12 January 2018.
Edited by:
Jianjun Sun, University of Texas at El Paso, United StatesReviewed by:
Srinand Sreevatsan, Michigan State University, United StatesXiao-Yang Zhi, Yunnan University, China
Copyright © 2018 Rajwani, Yam, Zhang, Kang, Wong, Leung, Tam, Tulu, Zhu and Siu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilman Kit Hang Siu, gilman.siu@polyu.edu.hk