Activation of Inflammatory and Pro-Thrombotic Pathways in Acute Stress Cardiomyopathy
 Timothy P. Fitzgibbons1*
Timothy P. Fitzgibbons1*
 Yvonne J. K. Edwards2
Yvonne J. K. Edwards2
 Peter Shaw1
Aline Iskandar3
Peter Shaw1
Aline Iskandar3
 Mohamed Ahmed1
Josiah Bote1
Tejen Shah1
Mohamed Ahmed1
Josiah Bote1
Tejen Shah1
 Sumita Sinha4
Robert E. Gerszten4
John F. Keaney Jr.1
Michael R. Zile5
Gerard P. Aurigemma1
Sumita Sinha4
Robert E. Gerszten4
John F. Keaney Jr.1
Michael R. Zile5
Gerard P. Aurigemma1
- 1Department of Medicine, University of Massachusetts Medical School, Worcester, MA, United States
- 2Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA, United States
- 3Division of Cardiovascular Medicine, Maine Medical Center, Portland, ME, United States
- 4Department of Medicine, Beth Israel Deaconess Medical Center, Boston, MA, United States
- 5Ralph H. Johnson Veterans Administration Medical Center, Medical University of South Carolina, Charleston, SC, United States
Stress cardiomyopathy (SCM) is a unique cardiac disorder that more often occurs in women. SCM presents in a similar fashion as acute myocardial infarction (AMI), with chest pain, ECG changes, and congestive heart failure. The primary distinguishing feature is the absence of thrombotic coronary occlusion in SCM. How this reduction in cardiac function occurs in the absence of coronary occlusion remains unknown. Therefore, we tested the hypothesis that a targeted proteomic comparison of patients with acute SCM and AMI might identify relevant mechanistic differences. Blood was drawn in normal controls (n = 6), women with AMI (n = 12), or women with acute SCM (n = 15). Two-week follow-up samples were available in AMI (n = 4) and SCM patients (n = 11). Relative concentrations of 1,310 serum proteins were measured in each of the 48 samples using the SOMAscan assay. Women with AMI had greater myocyte necrosis, as reflected by a higher peak troponin I concentration (AMI 32.03 ± 29.46 vs. SCM 2.68 ± 2.6 ng/ml, p < 0.05). AMI and SCM patients had equivalent reductions in left ventricular ejection fraction [LVEF (%) 39 ± 12 vs. 37 ± 12, p = 0.479]. In follow-up, women with SCM had a greater improvement in cardiac function [LVEF (%) 60 ± 7 vs. 45 ± 13, p < 0.001]. No differentially expressed proteins were detected (absolute log2-fold change >1; q < 0.05) between AMI and SCM in the acute or recovery phase. However, when we compared normal controls to patients with AMI, there was differential expression of 35 proteins. When we compared normal controls to patients with SCM, 45 proteins were differentially expressed. In comparison to normal controls, biological processes such as complement, coagulation, and inflammation were activated in both AMI and SCM. There were four proteins that showed a non-significant trend to be increased in acute SCM vs. AMI (netrin-1, follistatin-like 3, kallikrein 7, kynureninase). Despite a lesser degree of myocardial necrosis than AMI, SCM is characterized by a similar activation of inflammatory, complement, and coagulation pathways. These findings may explain reported thromboembolic complications in the short term and elevated risk of mortality in the long term of SCM.
Introduction
Stress cardiomyopathy (SCM) is a unique form of heart disease that primarily affects women. Described in 1990 by Sato and colleagues, it is now recognized as accounting for 5.9–7.5% of women presenting to the emergency department with acute coronary syndromes (1–4). Although the prognosis is typically good, complications such as heart failure, arrhythmias, and thromboembolic phenomena do occur (5). Recent studies have shown that SCM patients have increased long-term morbidity and mortality (5.6% risk of death per patient year) (6–8). Therefore, this disorder may not be as benign as previously thought.
After almost 30 years of study, the mechanism of SCM is still not understood. Plasma concentrations of norepinephrine, epinephrine, and dopamine are twofold higher than those of AMI (9). Myocardial biopsy in SCM reveals inflammation and myocyte contraction band necrosis, which are features of catecholamine excess (9). In contrast, acute myocardial infarction (AMI) has greater myocyte necrosis (10). Despite these different mechanisms of injury, cardiac function is impaired to an equivalent degree in both conditions (11). The exact mechanism by which LV dysfunction occurs in SCM remains poorly understood.
Due to the presenting signs and symptoms, patients with SCM are referred for urgent coronary angiography, which reveals the absence of atherosclerotic plaque rupture. Coronary angiography exposes the SCM patient to unnecessary risk without therapeutic benefit. A biomarker that could differentiate between AMI and SCM would be useful, allowing for the identification of low risk SCM patients in whom angiography could be avoided. This would be especially helpful in patients with “secondary” SCM (5). “Secondary SCM” frequently occurs in medical intensive care units and is precipitated by critical illnesses such as sepsis (12). Due to the severity of illness in these cases, cardiac interventions such as coronary angiography or the initiation of antiplatelet therapy are often deferred.
There have been no studies examining novel protein biomarkers in SCM. A ratio of serum B-type natriuretic peptide to troponin I (TnI) ≥1,272 is highly specific for SCM (95%) but has limited sensitivity (52%) (13). Soluble suppression of tumorigenicity 2 has been studied to identify SCM in medical intensive care units (12). A circulating profile of plasma microRNAs (miR-16, mir-26a, miR-1, and miR-133a) has shown good sensitivity and specificity for SCM (14). However, the use of plasma microRNAs for diagnostic purposes has not been adopted in clinical practice (15). Furthermore, these studies shed no light into the biologic differences between these two syndromes.
We hypothesized that there would be differences between circulating proteins in SCM and AMI that might elucidate the underlying mechanisms. Therefore, the purpose of our study was twofold. The first aim was to perform an unbiased comparison of circulating proteins in SCM and AMI in order to provide insight into the pathophysiology of SCM. Our second aim was to identify putative protein biomarkers of SCM.
In order to achieve these aims, we utilized recently described DNA aptamer technology to perform our screen (SOMAscan V1.3K, SOMAlogics) (16–18). This assay uses single stranded DNA aptamers to specifically bind 1,310 predetermined proteins in a single sample. The advantages of this technology include higher throughput and precision than traditional proteomic methods such as mass spectrometry or antibody based methods (16, 17).
Using this assay, we found that women presenting with either SCM or AMI have an increase in proteins related to complement, coagulation, and inflammation. The unexpected and profound activation of inflammatory and pro-thrombotic pathways in SCM may contribute to the increased risk of mortality attributed to this disorder (7, 8).
Materials and Methods
Patient Enrollment
Women who presented with non-ST or ST elevation myocardial infarction and had coronary angiography demonstrating either culprit LAD disease or apical variant SCM were prospectively enrolled. Detailed presenting features of the AMI group are shown in Table 1. We specifically limited our study to patients with culprit LAD disease and apical SCM in order to control for the differences between echocardiograms and clinical features that have been demonstrated with variants of SCM (19). All of the SCM patients met the Mayo Clinic Criteria for SCM (20). The normal control group for proteomic analyses consisted of 6 subjects (3 females, 3 males) with a mean age of 51.33 ± 10.11 years. They were healthy ambulatory persons without diabetes, hyperlipidemia, hypertension, or known coronary disease and taking no prescription medications. Written informed consent was obtained according to protocol #00000941 as approved by the University of Massachusetts Medical School IRB.
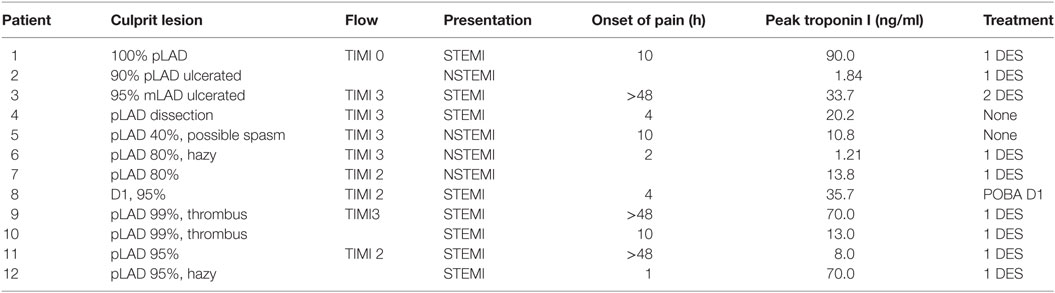
Table 1. Angiographic characteristics of acute myocardial infarction group.
Serum Isolation and Proteomic Analysis
Non-fasting venous blood was drawn (after cardiac catheterization but within 24 h of presentation) into SST tubes, spun, and frozen at −80°C. Acute samples were analyzed in 12 AMI patients and 15 SCM patients. Follow-up samples were drawn in clinic at 2 weeks follow-up (AMI n = 4, SCM n = 11). We chose to sample blood at 2 weeks follow-up because this is the median time to recovery of LV function in SCM (9). Samples were shipped to an outside laboratory on dry ice where assays were performed using SOMAscan reagents according to the manufacturer’s protocol (16). The menu of the 1,310 protein aptamers included in the assay is available online.1 The median intra-assay and inter-assay coefficient of variation was 2.1%.
Echocardiography
Complete 2D echocardiograms were performed within 24 h of presentation and in follow-up (2 weeks) in patients with AMI and SCM. Ejection fraction, 2D volumes, and linear dimensions were measured according to ASE guidelines (21).
Statistical Methods
Somalogics SOMAScan technology was used to measure levels of 1,310 predetermined proteins included in the assay (see text footnote 1) in 48 patient samples using 1,310. Protein expression levels were expressed as normalized counts [relative fluorescent units (RFUs)] for the 48 patient samples. Data were analyzed using Somalogics’ SomaSuite software (version 1.0.3a). The non-parametric two-sided Mann–Whitney U-test was used to analyze the normalized expression levels, to determine statistically significant differentially expressed proteins for each pairwise group comparison. The p-value, q-value, and other statistics were calculated for each protein. The q-values are calculated using the false discovery rate (FDR) method (22). The fold change values for each pairwise comparison are computed on the average values in the log2 scale. Statistically differentially expressed proteins were defined if the following criteria are met (absolute log2-fold change >1; q < 0.05). The raw data for this study were deposited in the Gene Expression Omnibus and can be downloaded at https://www.ncbi.nlm.nih.gov/geo/info/linking.html (Accession GSE95368).
In order to determine signaling pathways altered in each condition, functional enrichment analysis was carried out using the WEB-based GEne SeT AnaLysis Toolkit (Webgestalt) (23). The Kyoto Encyclopedia of Genes and Genomes database was selected for enrichment analysis, with the proteome selected as the reference set. The hypergeometric test was used for enrichment evaluation for the lists of statistically differentially expressed proteins. The Benjamini and Hochberg method was used to calculate the adjusted p-values (q) and the significance cutoff filter was set to q < 0.05 (24).
The complex heatmap package (version 1.12.0) was used to generate a heatmap (Figure 1) and the following parameters were used (clustering distance rows = “maximum,” clustering method rows = “ward.D”) (25). By determining if a protein was differentially expressed in one or more of the 10 possible comparisons (Figure 1), a list of proteins of interest was curated. This list comprising 64 proteins, together with the protein expression levels averaged for each protein in each of the medical conditions, was used as input to generate a heatmap. Five clusters are generated; for each cluster, the members were identified (Figure 1).
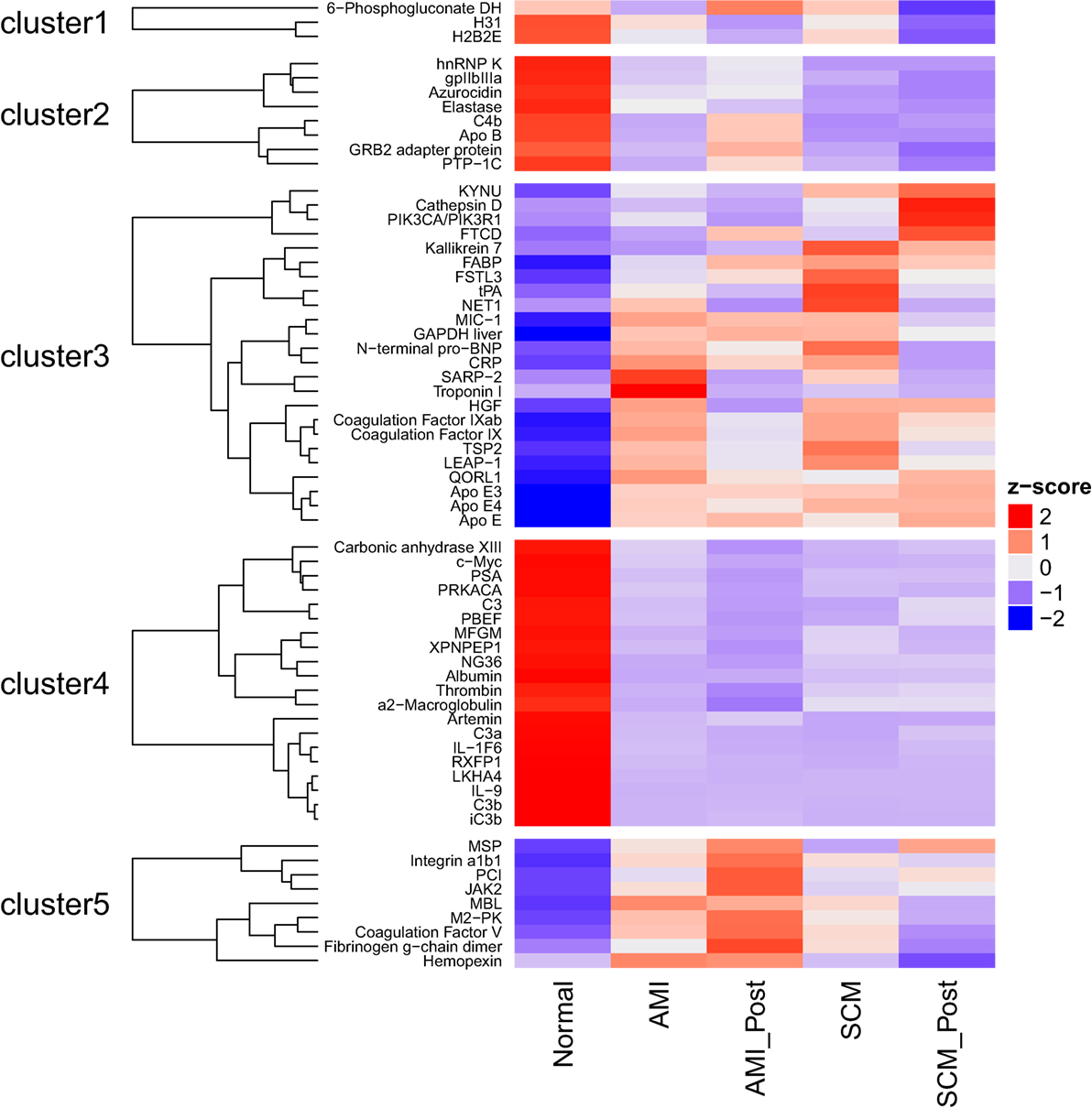
Figure 1. The heatmap shows the serum protein expression in the five groups [normal, the acute myocardial infarction conditions (AMI, AMI_Post) and the stress cardiomyopathy conditions (SCM, SCM_Post)]. By determining if a protein was differentially expressed in one or more of the ten possible comparisons, a list of 64 proteins was generated. Differentially regulated proteins were defined using the following filters (absolute log2 fold change >1; q < 0.05). The average level of each protein, in each of these conditions, was used to generate the heat-map. Bright red is increased expression, and deep purple is decreased expression (Z-score). For example, concentrations of N-terminal pro-BNP (cluster 3) are increased in AMI and SCM compared to normal.
Results
Clinical Characteristics
The baseline clinical and echocardiographic characteristics are shown in Tables 1–3. Compared to AMI, women with SCM had lower systolic and diastolic blood pressure on admission (112.2 ± 16.5 vs. 126.3 ± 21.9 and 66.4 ± 12.1 vs. 74.8 ± 11.6 mm Hg, p < 0.05 for both). Left ventricular end diastolic pressure was elevated in both groups (18.7 ± 9.4 vs. 14.9 ± 14.3 mm Hg, p = 0.36). Women with AMI had higher peak values of TnI (32.03 ± 29.46 vs. 2.68 ± 2.6 ng/ml, p < 0.05) and CPK (1,206 ± 1,348 vs. 104 ± 63.5 ng/ml, p < 0.05) during their hospitalization.
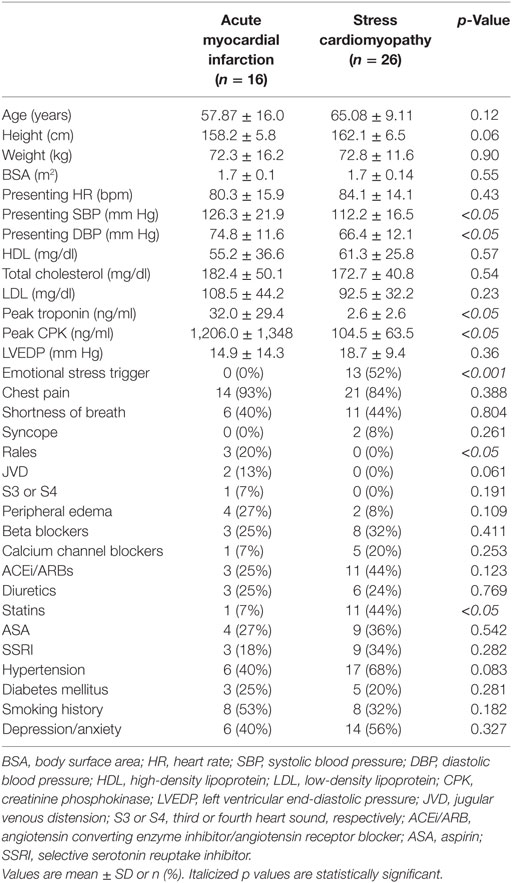
Table 2. Clinical characteristics of study participants.
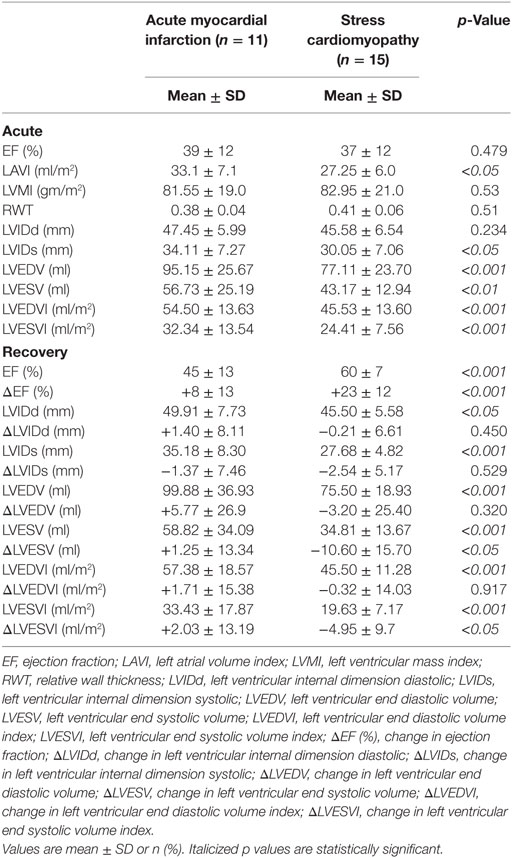
Table 3. Acute and recovery echocardiographic data.
Echocardiographic Characteristics
Stress cardiomyopathy and AMI patients had equivalent reductions in systolic function [LVEF (%)] in the acute phase (37 ± 12 vs. 39 ± 12, p = 0.47) (Table 3). However, SCM patients had smaller indexed left ventricular systolic (LVESVI 24.41 ± 7.56 vs. 32.34 ± 13.54 ml/m2, p < 0.001) and diastolic (LVEDVI 45.53 ± 13.60 vs. 54.50 ± 13.63 ml/m2, p < 0.001) volumes than AMI. SCM patients also had a greater indexed left atrial volume than AMI (LAVI 33.1 ± 7.7 vs. 27.25 ± 6.0, p < 0.05).
Stress cardiomyopathy patients had a significantly greater improvement in LV systolic function in follow-up, as shown by the mean LVEF (%) (60 ± 7 vs. 45 ± 13, p < 0.001), interval change in ΔLVEF (%) (+23 ± 12 vs. +8 ± 13, p < 0.001), and interval reduction in LVESVI (ΔLVESVI −4.95 ± 9.7 vs. +2.0 ± 13.19 ml/m2, p < 0.05).
Proteomic Analyses
There were no proteins differentially expressed (absolute log2-fold change >1; q < 0.05) between AMI and SCM in the acute phase (Table 4). This was true even when we lowered the threshold for differential regulation to q < 0.05 with any fold change (Table 4). When we compared normal controls vs. AMI, there were 35 proteins with differential expression (absolute log2-fold change >1; q < 0.05) (Figure 1; Table 4). In the normal vs. SCM comparison, 45 proteins were differentially regulated (absolute log2-fold change >1; q < 0.05) (Figure 1; Table 4).
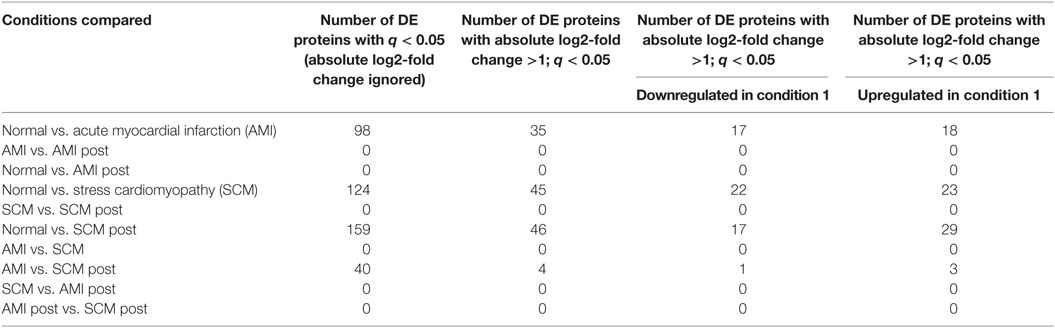
Table 4. Number of differentially expressed (DE) proteins between pairwise comparisons.
Most of the proteins were commonly differentially regulated in both SCM and AMI compared to controls [Figure 1 (cluster 4), Figure 2; Table 5]. However, there were four candidate proteins that tended to be increased in acute SCM compared to AMI and controls. These proteins were kynureninase (KYNU), K7, follistatin-like 3, and netrin-1 (NET1) [Figure 1 (cluster 3); Table 5].
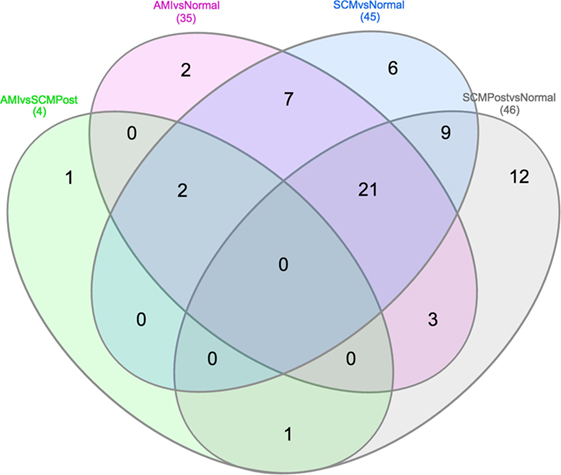
Figure 2. Venn diagram showing differential and overlapping protein expression between the conditions compared (Table 4). As shown in the middle of the diagram, 30 (7+21+2) proteins are commonly differentially regulated in AMI and SCM compared to normal controls. Specific proteins from Venn diagram subsections are listed in Table 5.

Table 5. Specific proteins from Venn diagram subsection.
The heat map (Figure 1) and bar plots of significantly altered proteins (Figures S1 and S2 in Supplementary Material) demonstrated the expected rise and fall of traditional markers of necrosis (TNNI3, CK-MB), inflammation (CRP), and wall stress (NPPB) in the acute and follow-up phase. There was a strong positive correlation between the hospital laboratory measured peak TnI (nanogram per milliliter) and the SOMAscan measured TnI (RFU) (ρ = 0.79, p < 0.001) (Figure 3).
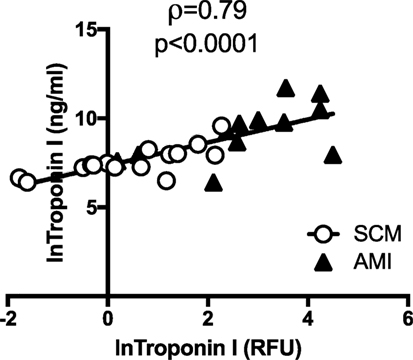
Figure 3. SOMAscan measured troponin correlates well with hospital-based measurement. There was a strong and significant correlation between peak troponin I (TnI) (ng/ml) measured in the hospital laboratory and the TnI [relative fluorescent units (RFU)] as measured by SOMAscan. Open circles represent peak TnI in stress cardiomyopathy (SCM) patients (n = 15) and black triangles represent peak TnI in acute myocardial infarction (AMI) patients (n = 12).
Pathway analysis of the differentially regulated proteins in each group revealed a large degree of similarity (Tables 6 and 7). The complement and coagulation pathway displayed the greatest enrichment ratio in both groups (Tables 6 and 7). There was significant overlap among other inflammatory pathways, nine of which were commonly increased in SCM and AMI. Several pathways were unique to SCM: focal adhesion, extracellular matrix–receptor interaction, malaria, regulation of actin cytoskeleton, arrythmogenic right ventricular cardiomyopathy, axon guidance, and Wnt signaling (Table 7).
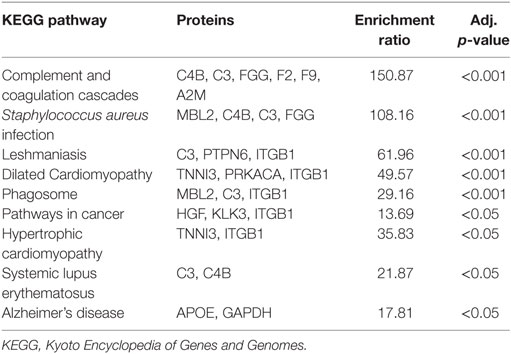
Table 6. KEGG pathway enrichment analysis: normal vs. acute myocardial infarction.
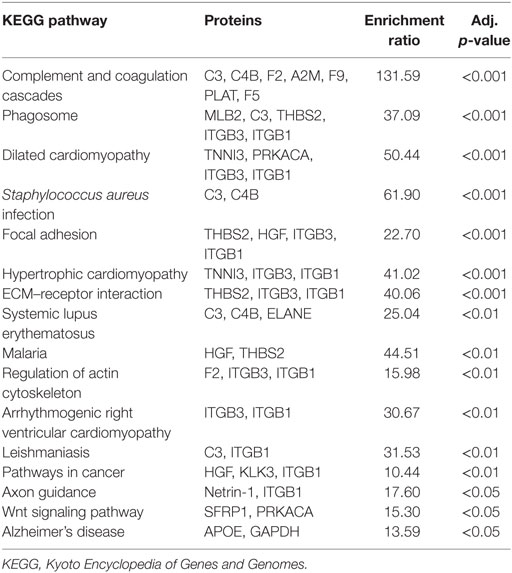
Table 7. KEGG pathway enrichment analysis: normal vs. stress cardiomyopathy.
Discussion
Although the pathophysiology, natural history, and outcomes of AMI and SCM are different, they do share common features. First, both are characterized by acutely decreased cardiac function (11). Second, each condition can be complicated by heart failure, arrhythmia, or thromboembolic events. Due to these similarities, women presenting with SCM are often referred for coronary angiography (5). However, in low risk cases, and in cases of “secondary SCM,” a biomarker would be useful. One of the aims of our study was to discover putative biomarkers of SCM; we identified four candidates (FSTL3, Kallikrein7, KYNU, and NET1). A second aim was to learn about the pathophysiology of SCM by comparing serum proteins to normal patients and those with AMI. We were surprised to find no differences between SCM and AMI in the acute phase. Both conditions were characterized by a robust increase in proteins related to inflammation, coagulation, and complement.
The characteristics of both study groups were consistent with prior studies. Patients with AMI had a greater peak TnI and a lower LVEF (%) on follow-up (7, 9, 11). SCM patients had smaller indexed LV diastolic and systolic volumes in both the acute and recovery phase. SCM patients had normalization of their systolic function in recovery, as demonstrated by a greater increase in LVEF (%) (Table 2).
In regards to our proteomic analysis, we wish to emphasize that the SOMAscan 1.3 K measures a predetermined panel of 1,310 proteins, which is only 6.5% of the approximately 20,000 protein encoding genes in the genome. Therefore, there are many changes that were undetected by our assay. For example, tissue inhibitor of metalloproteinase 4 has previously been shown to be increased in SCM compared to AMI (26). However, tissue inhibitor of matrix metalloproteinase (TIMP)-4 specific aptamers were not included in the version of the SOMAscan that we used; therefore, this difference was not detected. Of the proteins that were included, many were commonly elevated in both AMI and SCM, particularly those related to complement, coagulation, and innate immunity. This was unexpected, as the degree of myocardial necrosis in SCM is less than in AMI (7, 9). The trigger for this acute inflammatory response remains unclear.
Complement is an arm of the innate immune system that aids in the detection and clearance of self and bacterial antigens (27). It was demonstrated in 1990 that complement is activated in AMI (27). This is the first report of complement activation in SCM. Local hypoxia may play a role in complement activation and neutrophil chemoattraction to sites of ischemia (27). Complement activation can trigger endothelial adhesion molecule expression, and subsequent leukocyte rolling, arrest, and diapedesis. It is notable that there are several small studies indicating that patients with SCM have greater endothelial dysfunction than those with AMI even in the subacute phase (28–30). This dysfunction could be due to low-level complement activation, increased leukocyte adhesion, and release of reactive oxygen species.
Complement is known to interact with the coagulation cascade (27). The increased levels of coagulation factor V (F5) and tissue plasminogen activator (PLAT, tPA) in SCM indicate that a pro-thrombotic state may be present. Patients with SCM have inducible hyperviscosity and there are multiple reports of increased thromboembolic events in the acute phase of SCM (31–33).
Several candidate proteins demonstrated a non-significant trend to be increased in acute SCM compared to AMI and controls [NET1, FSTL3, kallikrein 7 (KLK7), KYNU] (Figure 1, cluster 3). Netrin-1 is a neuronal growth factor that was originally reported in 1991 (34). NET1 promotes endothelial cell nitric oxide production and cell growth and migration in vitro via the receptor deleted in colorectal cancer (DCC) (35). NET1 has also been shown to reduce infarct size in mice (36). There is interest in using NET1 for the treatment of postischemic conditioning in AMI.
Follistatin-like 3 (FSTL3) was increased in SCM compared to controls. FSTL3 inhibits transforming growth factor beta (TGFβ) signaling (37). FSTL3 is upregulated in the myocardium and endothelium of failing human hearts (38). Cardiac specific ablation of FSTL3 reduces cardiac hypertrophy and fibrosis in response to transverse aortic constriction (37). FSTL3 may contribute to myocyte hypertrophy in response to stress stimuli, and the increased levels of FSTL3 in SCM may promote cardiac hypertrophy and fibrosis. This is consistent with prior data demonstrating that SCM has a matrix metalloproteinase (MMP)/tissue inhibitor of matrix metalloproteinase (TIMP) profile similar to hypertensive heart disease, which is distinct from AMI (26). We observed the same MMP/TIMP profile in our study (data not shown); however, the differences did not reach statistical significance. This is likely because the prior study used ELISA to measure MMP/TIMPs (26).
Kynureninase is an enzyme involved in tryptophan metabolism. It degrades 3-hydroxy kynurenine to 3-hydroxyanthranillic acid (39). Kynurenine is an endothelium-derived relaxation factor induced in sepsis (40). Whether or not KYNU is truly elevated in SCM will be the topic of future studies. Finally, KLK7 is a serine proteinase belonging to the human kallikrein gene family, of which there are 15 members (41). The kallikrein system is broadly expressed and has also been demonstrated to play a role in cardiac remodeling (42).
Limitations
We acknowledge that our study has a small sample size and this may lead to false negative results. The AMI group included patients with both infarction and ischemia; this led to a large SD in the peak TnI value for the AMI group (Table 1). Differences in medications given during the hospital stay may have influenced our results, in particular, the administration of heparin (17). The majority of our study participants were Caucasian women; therefore, the applicability of other races is limited. Many of our patients are referred from outside hospitals and were not available for the follow-up blood draw and echocardiogram. Unfortunately, we could not run the SOMAscan on all our samples due to cost limitations. Despite these limitations, we feel that our observations are original and make an important contribution to the literature.
Conclusion
In summary, despite different pathophysiologic mechanisms, the circulating proteome of AMI and SCM is similar. Activation of complement and coagulation pathways in SCM has not been previously reported and highlights the pro-thrombotic and inflammatory state of acute SCM. The stimulus for inflammation in SCM is unclear and will be the subject of future investigation. NET1, FSTL3, and KYNU, and KLK7 are putative markers of SCM that require prospective validation.
Ethics Statement
This study was carried out in accordance with the recommendations of “Committee for the Protection of Human Subjects in Research” with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Institutional Review Board of UMASS Medical Center, IRB docket #00000941.
Author Contributions
TF designed the study, interpreted the data, drafted and revised the manuscript, approved the final version, and is accountable for all aspects of the work. YE, PS, AI, RG, SS, JB, TS, and MA acquired the data, revised the manuscript, approved the final version, and are accountable for all aspects. JK, MZ, and GA made substantial contributions to the study design, revised the manuscript, approved the final version, and are accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank the UMASS Cardiovascular Clinical Research Team for their assistance in obtaining samples and Evan Mills from SOMAlogics for helping to coordinate sample analysis.
Funding
TF is supported by AHA Grant 12FTF11260010 and a UMASS Faculty Scholar Award. MZ is supported by NIH grants R56HL123478 and 1R01HL123478-01A1. RG is supported by NIH grants U24DK112340 and HL132320. JK is supported by NIH grants HL092122 and HL098407.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/article/10.3389/fcvm.2017.00049/full#supplementary-material.
Footnote
References
1. Sato HTH, Uchida T. Takotsubo-type cardiomyopathy due to multivessel spasm. In: Kodama KHK, Hon M, editors. Clinical Aspect of Myocardial Injury: From Ischemia to Heart Failure. Tokoyo: Kagakuhyouronsya Company (1990). p. 56–64.
2. Ghadri JR, Ruschitzka F, Luscher TF, Templin C. Takotsubo cardiomyopathy: still much more to learn. Heart (2014) 100(22):1804–12. doi:10.1136/heartjnl-2013-304691
3. Sy F, Basraon J, Zheng H, Singh M, Richina J, Ambrose JA. Frequency of takotsubo cardiomyopathy in postmenopausal women presenting with an acute coronary syndrome. Am J Cardiol (2013) 112(4):479–82. doi:10.1016/j.amjcard.2013.04.010
4. Wedekind H, Moller K, Scholz KH. [Tako-tsubo cardiomyopathy. Incidence in patients with acute coronary syndrome]. Herz (2006) 31(4):339–46. doi:10.1007/s00059-006-2822-x
5. Lyon AR, Bossone E, Schneider B, Sechtem U, Citro R, Underwood SR, et al. Current state of knowledge on takotsubo syndrome: a Position Statement from the Taskforce on Takotsubo Syndrome of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail (2016) 18(1):8–27. doi:10.1002/ejhf.424
6. Ghadri JR, Cammann VL, Templin C. The International Takotsubo Registry: rationale, design, objectives, and first results. Heart Fail Clin (2016) 12(4):597–603. doi:10.1016/j.hfc.2016.06.010
7. Templin C, Ghadri JR, Diekmann J, Napp LC, Bataiosu DR, Jaguszewski M, et al. Clinical features and outcomes of takotsubo (stress) cardiomyopathy. N Engl J Med (2015) 373(10):929–38. doi:10.1056/NEJMoa1406761
8. Tornvall P, Collste O, Ehrenborg E, Jarnbert-Petterson H. A case-control study of risk markers and mortality in takotsubo stress cardiomyopathy. J Am Coll Cardiol (2016) 67(16):1931–6. doi:10.1016/j.jacc.2016.02.029
9. Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med (2005) 352(6):539–48. doi:10.1056/NEJMoa043046
10. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol (2014) 11(5):255–65. doi:10.1038/nrcardio.2014.28
11. Medeiros K, O’Connor MJ, Baicu CF, Fitzgibbons TP, Shaw P, Tighe DA, et al. Systolic and diastolic mechanics in stress cardiomyopathy. Circulation (2014) 129(16):1659–67. doi:10.1161/CIRCULATIONAHA.113.002781
12. Yang HS, Kim HJ, Shim HJ, Kim SJ, Hur M, Di Somma S, et al. Soluble ST2 and troponin I combination: useful biomarker for predicting development of stress cardiomyopathy in patients admitted to the medical intensive care unit. Heart Lung (2015) 44(4):282–8. doi:10.1016/j.hrtlng.2015.04.010
13. Randhawa MS, Dhillon AS, Taylor HC, Sun Z, Desai MY. Diagnostic utility of cardiac biomarkers in discriminating takotsubo cardiomyopathy from acute myocardial infarction. J Card Fail (2014) 20(5):377.e25–31. doi:10.1016/j.cardfail.2014.01.015
14. Jaguszewski M, Osipova J, Ghadri JR, Napp LC, Widera C, Franke J, et al. A signature of circulating microRNAs differentiates takotsubo cardiomyopathy from acute myocardial infarction. Eur Heart J (2014) 35(15):999–1006. doi:10.1093/eurheartj/eht392
15. Navickas R, Gal D, Laucevicius A, Taparauskaite A, Zdanyte M, Holvoet P. Identifying circulating microRNAs as biomarkers of cardiovascular disease: a systematic review. Cardiovasc Res (2016) 111(4):322–37. doi:10.1093/cvr/cvw174
16. Hathout Y, Brody E, Clemens PR, Cripe L, DeLisle RK, Furlong P, et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A (2015) 112(23):7153–8. doi:10.1073/pnas.1507719112
17. Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, et al. Aptamer-based proteomic profiling reveals novel candidate biomarkers and pathways in cardiovascular disease. Circulation (2016) 134(4):270–85. doi:10.1161/CIRCULATIONAHA.116.021803
18. Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One (2010) 5(12):e15004. doi:10.1371/journal.pone.0015004
19. Ghadri JR, Cammann VL, Napp LC, Jurisic S, Diekmann J, Bataiosu DR, et al. Differences in the clinical profile and outcomes of typical and atypical takotsubo syndrome: data from the International Takotsubo Registry. JAMA Cardiol (2016) 1(3):335–40. doi:10.1001/jamacardio.2016.0225
20. Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (tako-tsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J (2008) 155(3):408–17. doi:10.1016/j.ahj.2007.11.008
21. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr (2015) 28(1):1–39.e14. doi:10.1016/j.echo.2014.10.003
22. Storey JD. A direct approach to false discovery rates. J R Stat Soc B Stat Methodol (2002) 64(3):479–98. doi:10.1111/1467-9868.00346
23. Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res (2013) 41(Web Server issue):W77–83. doi:10.1093/nar/gkt439
24. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B Methodol (1995) 57(1):289–300.
25. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics (2016) 32(18):2847–9. doi:10.1093/bioinformatics/btw313
26. Essa EM, Zile MR, Stroud RE, Rice A, Gumina RJ, Leier CV, et al. Changes in plasma profiles of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in stress-induced cardiomyopathy. J Card Fail (2012) 18(6):487–92. doi:10.1016/j.cardfail.2012.04.002
27. Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol (2005) 23:821–52. doi:10.1146/annurev.immunol.23.021704.115835
28. Naegele M, Flammer AJ, Enseleit F, Roas S, Frank M, Hirt A, et al. Endothelial function and sympathetic nervous system activity in patients with takotsubo syndrome. Int J Cardiol (2016) 224:226–30. doi:10.1016/j.ijcard.2016.09.008
29. Martin EA, Prasad A, Rihal CS, Lerman LO, Lerman A. Endothelial function and vascular response to mental stress are impaired in patients with apical ballooning syndrome. J Am Coll Cardiol (2010) 56(22):1840–6. doi:10.1016/j.jacc.2010.03.107
30. Vasilieva E, Vorobyeva I, Lebedeva A, Urazovskaya I, Kalinskaya A, Skrypnik D, et al. Brachial artery flow-mediated dilation in patients with tako-tsubo cardiomyopathy. Am J Med (2011) 124(12):1176–9. doi:10.1016/j.amjmed.2011.05.033
31. Kurisu S, Inoue I, Kawagoe T, Ishihara M, Shimatani Y, Nakama Y, et al. Incidence and treatment of left ventricular apical thrombosis in tako-tsubo cardiomyopathy. Int J Cardiol (2011) 146(3):e58–60. doi:10.1016/j.ijcard.2008.12.208
32. El-Battrawy I, Behnes M, Hillenbrand D, Haghi D, Hoffmann U, Papavassiliu T, et al. Prevalence, clinical characteristics, and predictors of patients with thromboembolic events in takotsubo cardiomyopathy. Clin Med Insights Cardiol (2016) 10:117–22. doi:10.4137/CMC.S38151
33. Cecchi E, Parodi G, Giglioli C, Passantino S, Bandinelli B, Liotta AA, et al. Stress-induced hyperviscosity in the pathophysiology of takotsubo cardiomyopathy. Am J Cardiol (2013) 111(10):1523–9. doi:10.1016/j.amjcard.2013.01.304
34. Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell (1994) 78(3):409–24. doi:10.1016/0092-8674(94)90420-0
35. Nguyen A, Cai H. Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc Natl Acad Sci U S A (2006) 103(17):6530–5. doi:10.1073/pnas.0511011103
36. Zhang J, Cai H. Netrin-1 prevents ischemia/reperfusion-induced myocardial infarction via a DCC/ERK1/2/eNOS s1177/NO/DCC feed-forward mechanism. J Mol Cell Cardiol (2010) 48(6):1060–70. doi:10.1016/j.yjmcc.2009.11.020
37. Shimano M, Ouchi N, Nakamura K, Oshima Y, Higuchi A, Pimentel DR, et al. Cardiac myocyte-specific ablation of follistatin-like 3 attenuates stress-induced myocardial hypertrophy. J Biol Chem (2011) 286(11):9840–8. doi:10.1074/jbc.M110.197079
38. Lara-Pezzi E, Felkin LE, Birks EJ, Sarathchandra P, Panse KD, George R, et al. Expression of follistatin-related genes is altered in heart failure. Endocrinology (2008) 149(11):5822–7. doi:10.1210/en.2008-0151
39. Chen Y, Guillemin GJ. Kynurenine pathway metabolites in humans: disease and healthy states. Int J Tryptophan Res (2009) 2:1–19.
40. Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med (2010) 16(3):279–85. doi:10.1038/nm.2092
41. Diamandis EP, Yousef GM, Luo LY, Magklara A, Obiezu CV. The new human kallikrein gene family: implications in carcinogenesis. Trends Endocrinol Metab (2000) 11(2):54–60. doi:10.1016/S1043-2760(99)00225-8
Keywords: acute myocardial infarction, coagulation, inflammation, stress cardiomyopathy, women
Citation: Fitzgibbons TP, Edwards YJK, Shaw P, Iskandar A, Ahmed M, Bote J, Shah T, Sinha S, Gerszten RE, Keaney JF Jr., Zile MR and Aurigemma GP (2017) Activation of Inflammatory and Pro-Thrombotic Pathways in Acute Stress Cardiomyopathy. Front. Cardiovasc. Med. 4:49. doi: 10.3389/fcvm.2017.00049
Received: 09 May 2017; Accepted: 13 July 2017;
Published: 03 August 2017
Edited by:
Junjie Xiao, Shanghai University, ChinaReviewed by:
Kumaravelu Jagavelu, Central Drug Research Institute, Indiazhuyuan Liu, Nanjing Medical University, China
Copyright: © 2017 Fitzgibbons, Edwards, Shaw, Iskandar, Ahmed, Bote, Shah, Sinha, Gerszten, Keaney, Zile and Aurigemma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy P. Fitzgibbons, timothy.fitzgibbons@umassmed.edu