
- Department of Physiology and Biophysics, Medical Center, Stony Brook University, Stony Brook, NY, USA
In neurons, as in a variety of other cell types, the enzyme phosphatidylinositol-3-kinase (PI3K) is a key intermediate that is common to the signaling pathways of a number of peripheral metabolic cues, including insulin and leptin, which are well known to regulate both metabolic and reproductive functions. This review article will explore the possibility that PI3K is a key integrator of metabolic and neural signals regulating gonadotropin releasing hormone (GnRH)/luteinizing hormone (LH) release and explore the hypothesis that this enzyme is pivotal in many disorders where gonadotropin release is at risk. Although the mechanisms mediating the influence of metabolism and nutrition on fertility are currently unclear, the strong association between metabolic disorders and infertility is undeniable. For example, women suffering from anorectic disorders experience amenorrhea as a consequence of malnutrition-induced impairment of LH release, and at the other extreme, obesity is also commonly co-morbid with menstrual dysfunction and infertility. Impaired hypothalamic insulin and leptin receptor signaling is thought to be at the core of reproductive disorders associated with metabolic dysfunction. While low levels of leptin and insulin characterize states of negative energy balance, prolonged nutrient excess is associated with insulin and leptin resistance. Metabolic models known to alter GnRH/LH release such as diabetes, diet-induced obesity, and caloric restriction are also accompanied by impairment of PI3K signaling in insulin and leptin sensitive tissues including the hypothalamus. However, a clear link between this signaling pathway and the control of GnRH release by peripheral metabolic cues has not been established. Investigating the role of the signaling pathways shared by metabolic cues that are critical for a normal reproductive state can help identify possible targets in the treatment of metabolic and reproductive disorders such as polycystic ovarian syndrome.
Introduction
The achievement of reproductive competence as well as the normal sustenance of adult reproductive activity is highly affected by the availability of nutrients. This is particularly evident in female mammals, where physiological processes such as gestation, parturition, lactation, and rearing of the young are energetically demanding. Thus, the hypothalamic pituitary gonadal (HPG) axis limits fertility when nutrient availability is inadequate to support these events. Many studies suggest that negative energy balance inhibits reproduction through the alteration of gonadotropin releasing hormone (GnRH) secretion in the hypothalamus. GnRH neurons are often described as the “master regulators” of the HPG axis meaning that successful reproduction and thus the survival of the species in mammals depends on the ability of these neurons to sense changes in the internal and external environment, including changes in energy status. The intermittent discharge from these specialized neurons in the hypothalamus is in fact responsible for the pulsatile secretion of the gonadotropins luteinizing hormone (LH) and follicle-stimulating hormone (FSH). These in turn act on the gonads to direct gametogenesis and secretion of steroid hormones. Within the brain, the actions of gonadal steroids are then manifested as both feedback regulation of GnRH secretion and the facilitation of sexual behavior.
The mechanisms by which metabolic information is relayed to the hypothalamic GnRH network are not entirely clear. However, various peripheral metabolic signals, such as leptin and insulin, act on specific neuronal populations that in turn affect GnRH neuronal function. This is the case for multiple hypothalamic neuropeptide systems such as neuropeptide Y (NPY), products of pro-opiomelanocortin (POMC), and galanin-like peptide (GALP), among many others (Crown et al., 2007). When these neuronal populations are not able to appropriately respond to metabolic cues, for example in states of malnutrition, obesity, and diabetes, the repercussions are not limited to imbalanced metabolic homeostasis, but can include reproductive dysfunction. Therefore, metabolic hormones have a pivotal role in the central regulation of the HPG axis. Much less is known, however, about the intracellular signaling pathways that integrate the actions of metabolic signals to regulate GnRH/LH release and hence reproductive function. The variety and complexity of signaling networks that affect cell metabolism and neuronal activity make this a daunting task. However, peripheral hormones and metabolic cues such as leptin and insulin share specific signal transduction pathways that in many cases act synergistically or in parallel to affect neuronal function. Therefore, molecules that serve as central integrators of metabolism and reproductive function might be identified by determining which signaling pathways are shared by peripheral metabolic cues known to affect both physiological functions.
The PI3K signaling cascade is well known for its role in mediating insulin and leptin effects on cell and tissue metabolism. This lipid enzyme is activated in classical metabolic tissues such as liver, pancreas, skeletal muscle, and adipose tissue, where it participates in the regulation of carbohydrate and lipid homeostasis (Foukas and Withers, 2010). In addition, central activation of PI3K plays a key role in the effect of leptin and insulin on food intake and glucose homeostasis (Morton et al., 2005; Xu et al., 2005). The critical role that PI3K plays in regulation of metabolism does not exclude the possibility that it also participates in the control of other physiological processes such as reproduction. In fact, the PI3K signaling cascade is a common pathway activated by hormones and growth factors that are also critical for the normal function of the HPG axis, including insulin and leptin. It remains to be determined whether this signaling pathway integrates the effects of these or other hormones and growth factors in the regulation of GnRH release. Furthermore, the brain sites and neuronal populations in which activation of this enzyme is necessary for the regulation of energy homeostasis might not be the same as those involved in the regulation of the HPG axis.
This article reviews the studies supporting the idea that hypothalamic control of GnRH/LH release by peripheral metabolic cues is indispensable for the normal function of the HPG axis. Then the hypothesis that PI3K is a key integrator of metabolic and neural signals regulating GnRH neurosecretion will be explored.
Dysregulation of GnRH/LH Release by Metabolic Imbalance
Negative Energy Balance
Examples of the effects that a dramatic decrease in caloric intake has on the reproductive axis can be found in patients suffering from eating disorders such as anorexia nervosa (AN) and in female athletes undergoing extreme exercise regimens (Poyastro Pinheiro et al., 2007; Attia and Roberto, 2009). In these women, hypothalamic amenorrhea and menstrual irregularities are associated with impairments in gonadotropins, specifically LH release (Attia and Roberto, 2009). These effects are likely due to decreased amplitude and/or frequency of hypothalamic pulses of GnRH.
To better understand the mechanisms by which negative energy balance disrupts reproductive function, a number of in vivo animal models have been developed. These include streptozotocin (STZ)-induced type 1 diabetes (Kovacs et al., 2002, 2003), extreme exercise (Manning and Bronson, 1989), chemical inhibition of metabolic pathways such as glucose oxidation (Nagatani et al., 1996), lipoprivation (Shahab et al., 2006; Sajapitak et al., 2008), as well as chronic and acute food restriction in adult (Dyer et al., 1985; Cameron and Nosbisch, 1991), and peripubertal animals (Bronson, 1986, 1988). The latter model takes advantage of the phenomenon of “catch up” growth in which stopping growth at a particular body weight halts reproductive development and subsequent reinitiation of ad libitum feeding results in rapid growth and reproductive development (Bronson, 1986). These negative metabolic challenges can affect the HPG axis at various levels including decreased gonadotropin secretion, slow follicular development, and inhibited synthesis of gonadal steroids (Schneider, 2004). However, it is the inhibition of the GnRH pulse generator, and hence GnRH release, that is responsible for the dysfunction of the reproductive axis. For instance, infusing GnRH in a pulsatile manner can yield close to normal onset of puberty in food-restricted female rats, even in the total absence of body growth (Bronson, 1986). Likewise, pulsatile treatment with GnRH in food-deprived or food-restricted adult mammals also reinstates HPG normal function (Rojdmark, 1987; Foster et al., 1989; Cameron and Nosbisch, 1991). These studies confirm that positive energy balance is crucial for normal reproductive function, whether the female is an adult or peripubertal, and that proper activity of the GnRH network is essential for this to occur.
Conditions of energy deficit are associated with a drastic decrease in the levels of peripherally produced metabolic cues such as insulin and the adipocyte-derived cytokine leptin. The reduction in the levels of these signals contributes to the decrease in GnRH/LH activity produced by negative energy balance. For example, central infusion of leptin or insulin restores the reproductive deficits observed using in vivo models in which energy balance is severely compromised such as food restriction (Nagatani et al., 1998) or type 1 diabetes (Kovacs et al., 2002), respectively. In addition, leptin treatment improves mean LH pulse frequency and ovulation when administered to AN patients (Chan and Mantzoros, 2005). These studies emphasized the importance of leptin and insulin in the metabolic control of the HPG axis. However, a decrease in the levels of other peripherally produced signals such as insulin-like growth factor-1 (IGF-1), glucose, and lipids may also contribute to the suppression of GnRH activity produced by negative energy balance.
Chronic Excess Nutrition: Obesity
Fewer studies have examined the effects of a state of excess nutrition on GnRH/LH release and its impact on the rest of the HPG axis. Diet-induced obesity (DIO) in rodents has been utilized to mimic the gradual increase in body fat and body weight observed in obese humans. Similar to humans, mice with DIO display fasting hyperglycemia, hyperinsulinemia, and hyperleptinemia accompanied by insulin and/or leptin resistance (Tortoriello et al., 2004; Ghanayem et al., 2010). In female mice, DIO causes infertility, the degree of which depends on the background strain, the duration, and the percentage of fat in the diet. For example, compared to other strains such as C57BL/6, DBA/2J female mice show high susceptibility to high fat diet (HFD)-induced infertility. In these mice, a diet containing 24% fat is sufficient to significantly decrease natural pregnancy rates and follicular activity (Tortoriello et al., 2004). Their fertility defect is likely of central origin, because normal ovulatory responses and pregnancy rates are restored after exogenous gonadotropin stimulation (Tortoriello et al., 2004). Additionally, prolonged hyperleptinemia and central leptin resistance are accompanied by reduced hypothalamic GnRH gene expression (Tortoriello et al., 2004). However, whether reduction in GnRH expression caused by DIO in DBA/2J mice results in diminished hypothalamic GnRH secretion is unknown. In contrast, studies using different strains of mice (i.e., mixed CD1/129vJ/57BL6) have found that elevated LH levels might be the primary mediator of DIO-induced infertility (Brothers et al., 2010). In these mice, 60% fat content in the diet resulted in decreased fertility, lack of regular estrous cyclicity, and reduced follicular activity (Brothers et al., 2010). The elevated LH levels observed in this model of DIO suggest that obesity affects the ability of steroid hormones to exert negative feedback on GnRH secretion. Interestingly, although this diet resulted in peripheral insulin resistance, the pituitary remained responsive to insulin and exhibited a heightened pituitary response to GnRH due to an increase in GnRH receptors (Brothers et al., 2010). In addition, DIO on this genetic background resulted in elevated androgen levels, a characteristic of polycystic ovarian syndrome (PCOS). In this context, it is valid to point out that rodent models of PCOS, such as female rats exposed to prenatal androgens, in addition to developing characteristics of the metabolic syndrome also have elevated androgen and LH levels, suggesting that an acceleration of the GnRH pulse generator mediates infertility (Demissie et al., 2008; Foecking et al., 2008). Unfortunately, studies of DIO effects on male fertility are scarce. In general, male mice are very resistant to the detrimental effects DIO on fertility, even though it also results in similar degrees of obesity, insulin resistance, and hyperleptinemia. In order to observe reduced fertility in males, both a diet with a high fat content (60%) and a prolonged exposure to it are needed (Ghanayem et al., 2010).
In women, obesity is co-morbid with menstrual dysfunction, decreased fertility, and an increased risk of miscarriages (Pasquali et al., 2007). In men, it can result in reduced testosterone (T) levels and erectile dysfunction (Lebinger, 2007). In the latter, increased estradiol-17β (E2) levels from peripheral adipose tissue excess can then lead to secondary hypogonadism through negative feedback on the HPG axis (Pitteloud et al., 2005; Kasturi et al., 2008). Obesity can also exacerbate female reproductive disorders such as PCOS (Lebinger, 2007; Nestler, 2008). PCOS is the most common cause of infertility in women; it is characterized by hyperandrogenemia and elevated LH (Lebinger, 2007; Nestler, 2008). As with states of negative energy balance, alterations in the levels of peripheral metabolic cues, such as hyperinsulinemia and hyperleptinemia, characterize these metabolic disorders and are thought to play a major role in the central dysregulation of GnRH/LH pulsatility that is observed in many cases. Specifically, a decreased sensitivity of the HPG axis to insulin and leptin actions due to insulin and leptin resistance is suspected to be a major contributing factor.
Energy Imbalance and Pubertal Development
The effects of eating and metabolic disorders on the reproductive axis are not limited to adults. Nutritional disturbances during early developmental stages can have adverse consequences on the reproductive axis as well. For instance, malnutrition and sickness in children can cause delayed puberty (Euling et al., 2008). On the other hand, the prevalence of obesity and other metabolic diseases in children and adolescents has increased at alarming rates in recent decades. Several studies suggest that this too represents a metabolic challenge for normal reproductive development. In girls, obesity has been associated with precocious puberty (Buyken et al., 2009), whereas the opposite has been found in boys, with obesity increasing the probability of delayed puberty (Lee et al., 2010). At the neuroendocrine level, puberty starts when GnRH neurons secrete GnRH in a pulsatile manner. Although there are no clear data linking obesity with premature activation of gonadotropin secretion in girls, an increase in adiposity, insulin, and leptin levels during childhood and adolescence can alter the secretion of other steroid hormones such as androgens from ovarian and/or adrenal origin (reviewed in Burt Solorzano and McCartney, 2010). Androgens can in turn alter the feedback mechanisms that normally restrain GnRH/LH secretion. This is one of the reasons childhood obesity has been linked to adult PCOS (Brewer et al., 2010; Burt Solorzano and McCartney, 2010).
As interest in the effects of obesity on child development has increased, so have studies investigating the effects of diet on puberty. In female rodents, states of negative energy balance delay puberty, whereas a HFD has the opposite effect, advancing pubertal onset. Female rats and mice receiving a HFD immediately after weaning showed advanced timing of vaginal opening (VO) when compared to littermates exposed to a lean diet (Boukouvalas et al., 2008; Brill and Moenter, 2009). In mice, the effect of a HFD on VO is associated with increased insulin levels (Brill and Moenter, 2009). Furthermore, treatment with the insulin sensitizer metformin blocked advancement of VO induced by HFD (Brill and Moenter, 2009). In the same study, treatment with the androgen receptor antagonist flutamide blocked pubertal acceleration while delaying VO in the lean diet fed control group, suggesting that androgens are involved in the pubertal process. It is important to point out that VO is an external marker of puberty onset that reflects exposure to pubertal sex steroid levels, but it does not necessarily imply central activation of gonadotropin secretion. Other parameters, such as day of first estrus and measurement of gonadotropin levels, are needed to further characterize these models. Nevertheless, it seems that an imbalance in insulin levels, which is known to stimulate androgen synthesis, contributes to the “precocious” pubertal onset induced by a HFD. These results also support a model where obesity alters the hypothalamic sensitivity to the restraining effects of steroid negative feedback on GnRH neuronal activity early in development.
These findings indicate that alterations in the activity of the GnRH network underlie many of the detrimental effects that metabolic imbalances have on the HPG axis. These metabolic challenges are also accompanied by profound alterations in the levels and sensitivity to peripheral hormones and metabolic signals, including IGF-1, E2, leptin, and insulin. These hormones have in common the capacity to activate the PI3K pathway in various reproductive tissues including the hypothalamus.
The PI3K Signaling Pathway
Based on their sequence homology, similarities in function, and substrate specificity, PI3Ks are grouped into three principal categories: Classes I, II, and III (Vanhaesebroeck et al., 2005). Class I PI3Ks exist as heterodimers composed of a regulatory/adapter subunit tightly associated with a catalytic subunit. Class I is further divided into Class IA and Class IB. Three isoforms of the Class IA catalytic subunits p110 have been described, namely p110α, p110β, and p110δ, whereas three mammalian genes encode the Class IA adapter subunits p85α, p85β, and p55γ (Vanhaesebroeck et al., 2005). Class IA regulatory subunits bind to tyrosine-phosphorylated proteins through their SH2 (Src homology 2) domains, linking them to tyrosine kinase signaling pathways. In contrast, the Class IB regulatory subunits p84 and p101 lack SH2 domains and instead couple the Class IB catalytic subunit p110γ to G protein-coupled receptors. We will limit our discussion to Class IA PI3Ks, which are the major targets of insulin and leptin actions.
PI3Ks utilize phosphatidylinositol 4,5-bisphosphate (PIP2) as the main substrate to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), which then acts as a second messenger lipid to allosterically modify the activity and/or induce the subcellular relocalization of downstream signaling molecules by binding to their pleckstrin homology (PH) domains (Cantrell, 2001). For example, receptor-mediated phosphorylation of specific tyrosine residues in proteins such as insulin receptor substrates (IRS), produce docking sites that bind to the SH2 domains of p85, hence targeting the catalytic subunit to the membrane (Backer et al., 1992; Carpenter et al., 1993). Activated PI3K can then affect cellular functions through specific signaling pathways such as the serine/threonine protein kinase Akt. Intracellular proteins that are targets of the Akt pathway include glycogen synthase kinase-3 (GSK3β), members of the forkhead family of transcription factors, and the mammalian target of rapamycin (mTOR; Cantrell, 2001; Kozma and Thomas, 2002). These enzymes and transcription factors regulate intracellular metabolic functions such as the generation of glucose and protein synthesis (for a review see Vanhaesebroeck et al., 2005).
Non-receptor tyrosine kinases such as the cytoplasmic Janus tyrosine kinase family (JAKs) as well as other adapter proteins like GRB-2 at the plasma membrane can also initiate PI3K signaling in response to insulin, leptin, and growth factors (Cantrell, 2001). Therefore, the potential for activation by a variety of mechanisms as well as its cross-talk with other signaling pathways such as the Ras/MAPK pathway confers upon this lipid kinase the ability to be involved in a variety of biological functions including reproduction.
PI3K Signaling Molecules and the Control of the HPG Axis
Pharmacological and genetic studies have demonstrated a role of several members of the PI3K signaling pathway in reproductive function (Table 1). Examples include upstream regulators of PI3K signaling such as estrogen receptor alpha (ERα), the IGF-1 receptor (IGF-1R), the insulin receptor (IR), and the leptin receptor (LepR), as well as downstream targets of the Akt branch downstream of PI3K, such as mTOR. Moreover, the steroid hormones E2 and progesterone (P), which are essential for the regulation of puberty, LH release, and sexual behavior, are known to alter the expression and/or activity of members of this signaling pathway such as p85α, IRS-1, and IRS-2. Below is a summary of these findings.
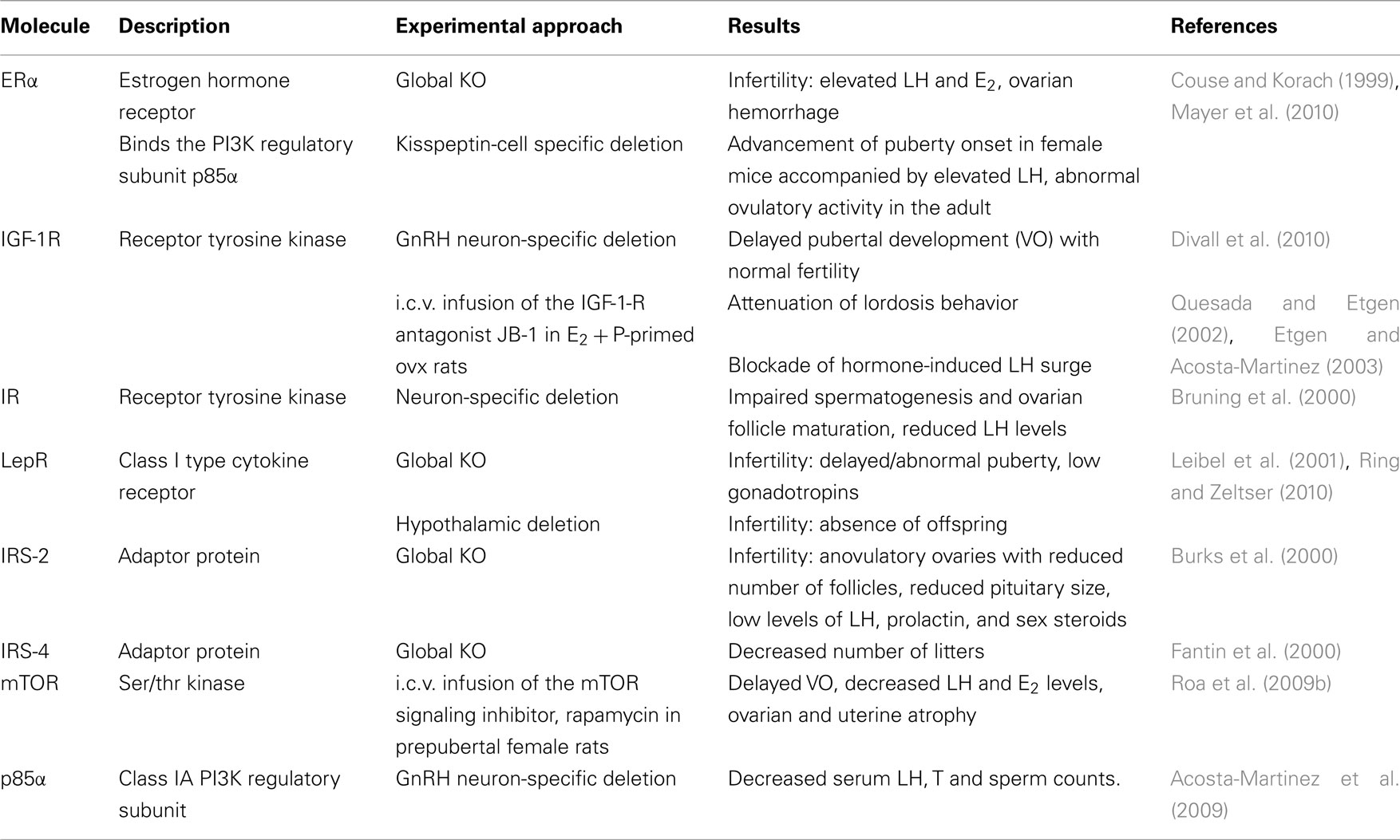
Table 1. Summary of studies on the role of upstream and downstream components of the PI3K signaling pathway in HPG axis function.
Estrogen Receptor α
The steroid hormone E2 is essential for the feedback regulation of gonadotropin release and female reproductive behavior. These E2 effects are dependent on the activation of ERα in specific neuronal populations of the preoptic area–anterior hypothalamus (POA–AH) and the mediobasal hypothalamus–median eminence (MBH–ME, for reviews see (Couse and Korach, 1999; Roepke et al., 2011)). Traditionally, E2 acts through its receptor to regulate the transcription of important genes for reproduction, such as the progesterone receptor. However, E2 is also capable of inducing rapid non-genomic actions. This “non-classical” ERα signaling includes mechanisms in which cytoplasmic or membrane-associated receptor activation is coupled to stimulation of cytoplasmic signaling pathways, including the PI3K signaling cascade (Acosta-Martinez et al., 2009).
Cross-talk between ERα and PI3K signaling occurs at various levels. For example, in vitro PI3K signaling induces the phosphorylation of discrete residues of the endogenous ER, upregulating its transcriptional activity and stability (Campbell et al., 2001). Likewise, E2, presumably acting on membrane ERs, induces PI3K activity, which results in activation of gene transcription in the target cell (Pasapera Limon et al., 2003). In vivo, E2 treatment evokes rapid activation of Akt in the hypothalamus (Burt et al., 2011). ERα is required for the E2 activation of hypothalamic Akt since in ERα KO mice acute E2 treatment is no longer able to stimulate Akt phosphorylation (Burt et al., 2011). There is also biochemical evidence demonstrating that ERα binds to the PI3K regulatory subunit p85α in a ligand-dependent manner (Simoncini et al., 2000; Mendez et al., 2003). The interaction between ERα and p85α results in the activation of Akt and endothelial nitric oxide synthase (eNOS; Simoncini et al., 2000).
The PI3K signaling pathway participates in the E2 facilitation of lordosis behavior, a reflexive posture displayed by female rodents during mating. The PI3K inhibitors wortmannin and LY294002 partially suppressed lordosis when administered during E2 priming (Etgen and Acosta-Martinez, 2003). Interestingly, when both wortmannin and the MAPK inhibitor PD98059 are simultaneously infused, lordosis behavior is completely abolished (Etgen and Acosta-Martinez, 2003). Therefore, the complete inhibition of this E2-dependent behavior requires concurrent suppression of both central MAPK and PI3K signaling. Future studies should examine whether central inhibition of PI3K participates in E2-induced LH release.
Insulin-Like Growth Factor-1 Receptor
The actions of IGF-1 are mediated through activation of its receptor (IGF-1R), a member of the tyrosine kinase receptor family that signals through the IRS/PI3K and the MAPK signaling cascades (Nakae et al., 2001). In addition to its prominent role in the regulation of somatic growth, the IGF-1R signaling pathway participates in the regulation of GnRH release, the progression of puberty, adult reproductive function, and sexual behavior (reviewed in Daftary and Gore, 2005). Although IGF-1 can regulate the HPG axis by acting at the pituitary and gonads, it also acts centrally to stimulate GnRH release and puberty. For example, in peripubertal rats intracerebroventricular (i.c.v.) infusion of IGF-1 increases plasma LH levels, a response that is blocked by central immunoneutralization of GnRH (Hiney et al., 1996).
Recent studies suggest that the IGF-1 effect on advancing pubertal onset is mediated through direct regulation of GnRH neurons, which are known to co-express IGF-1Rs. Male and female mice with the IGF-1R specifically deleted in GnRH neurons (GnRH–IGF-1R KO) experienced delayed pubertal development with normal fertility (Divall et al., 2010). Administration of IGF-1 is not able to advance puberty onset in GnRH–IGF-1R KO females, suggesting that the re-activation of the GnRH pulse generator at puberty is triggered by activation of IGF-1Rs in GnRH neurons. Compared to WT littermates, pre- and peripubertal GnRH–IGF-1R KO mice have a reduced number of GnRH neurons with complex dendritic trees and a significantly lower proportion of GnRH neurons with spines. The authors concluded that IGF-1R regulation of GnRH synaptic structure during the prepubertal period is critical for the induction of GnRH pulsatility at puberty onset (Divall et al., 2010).
Finally, central IGF-1Rs regulate adult LH release and sexual receptivity (lordosis) in female rats. Chronic i.c.v. infusion of the highly selective IGF-1R antagonist JB-1 suppresses E2-induced LH release and partially inhibits lordosis behavior (Quesada and Etgen, 2002; Etgen and Acosta-Martinez, 2003). The effect of JB-1 on LH release is not observed in ovariectomized rats given no hormone replacement, suggesting that JB-1 acts primarily to suppress E2-positive feedback (Quesada and Etgen, 2002). Participation of IGF-1R signaling in E2-induced synaptic remodeling in the hypothalamic arcuate nucleus (ARC), which is dependent on IGF-1R activity, may underlie IGF-1 stimulatory effects on adult LH release (Fernandez-Galaz et al., 1999).
Insulin and Leptin Receptors
Transgenic mouse models and in vivo pharmacological studies have demonstrated that leptin and insulin receptor signaling in the brain has a major role in the regulation of the HPG axis. When male and female db/db mice (which have a global LepR mutation) are subjected to a neuron-specific LepR knock in, obesity, diabetes, and infertility are reversed (de Luca et al., 2005). The brain-specific deletion of the IR results in obesity and hypothalamic hypogonadism (Bruning et al., 2000). However, the brain region(s) and the phenotype of the neurons that are responsible for leptin and insulin actions on the reproductive axis are largely unknown. GnRH neurons are unlikely to be direct targets for leptin or insulin in rodents. Recent studies show that the GnRH neuron-specific deletion of either receptor does not affect puberty or adult HPG axis function (Quennell et al., 2009; Divall et al., 2010). Therefore, it is likely that these and other peripheral metabolic cues inform the HPG axis about energetic status via a complex network of neurons and interneurons, many of which are functionally connected to GnRH neurons. In this regard, the combined deletion of IR and LepR in POMC neurons results in high T levels, ovarian abnormalities, and reduced fertility (Hill et al., 2010). Interestingly, a reproductive phenotype is not observed in single deletions of either receptor in POMC neurons (Balthasar et al., 2004; Konner et al., 2007). In view of the fact that deletion of the LepR or the IR in all neurons results in low LH levels, it is likely that the role each receptor has in the control of the HPG axis depends on the phenotype of the neurons in which it is expressed, as well as environmental and genetic circumstances.
Insulin and leptin converge on PI3K signaling to centrally regulate feeding and glucose homeostasis (Niswender et al., 2003). Their receptors activate PI3K through the phosphorylation of tyrosine residues on IRS proteins (Foukas and Withers, 2010). In the case of the LepR this is accomplished through the JAK/signal transducer and activator of transcription (STAT3) signaling pathway, whereas IR autophosphorylation on tyrosine residues provides docking sites for the IRS (Niswender et al., 2003; Foukas and Withers, 2010). Intracerebroventricular infusion of PI3K inhibitors blocks the ability of insulin and leptin to inhibit food intake (Niswender et al., 2003; Morrison et al., 2005). Leptin and insulin reduce appetite by decreasing the production of potent orexigenic neuropeptides such as NPY and agouti related peptide (AgRP), while increasing the expression of anorexigenic neuropeptides such as POMC (Munzberg et al., 2003; Morrison et al., 2005). Leptin regulation of NPY/AgRP transcription depends on PI3K signaling, while that of POMC requires activation of the JAK/STAT3 pathway (Munzberg et al., 2003; Morrison et al., 2005). PI3K is also involved in insulin and leptin effects on the membrane potential and firing rates of these ARC neurons (Plum et al., 2006; Hill et al., 2008; Al-Qassab et al., 2009). Since both leptin and insulin stimulate LH release, it is tempting to speculate that PI3K also serves as an integrator for the effects that these hormones have on the HPG axis.
As discussed previously, leptin and insulin resistance are thought to mediate, in part, the detrimental effects of obesity on the HPG axis. Central leptin resistance is associated with a defective PI3K pathway in the hypothalamus. For example, the ability of leptin to induce hypothalamic PI3K activity is impaired in mice fed a HFD (Metlakunta et al., 2008). These animals are hyperleptinemic, hyperinsulinemic, and have impaired glucose and insulin tolerance. Moreover, leptin fails to induce PI3K activity as early as 4 weeks after a HFD, whereas at this age, phosphorylation of STAT3 by leptin is not impaired (Metlakunta et al., 2008). A recent study using mouse lines with reduced IR expression in the brain and the ovary show that insulin resistant and hyperinsulinemic mice have impaired HPG axis activity (Nandi et al., 2010). This includes altered duration of estrous cycles, aberrant distribution, and morphology of ovarian follicles, and a decrease in pregnancy outcomes. These effects were observed in the absence of body weight gain and hyperglycemia, suggesting that abnormal insulin signaling, independent of adipose tissue mass or hyperglycemia, can affect HPG axis function. Even though these studies demonstrate the importance of IR and LepR signaling for normal HPG axis activity, it is impossible to assess whether defects in IR and LepR signaling in the brain vs. other reproductive tissues, such as the ovary and the pituitary, are responsible for the detrimental effects on the HPG axis. This remains an important question since HFD-induced obesity promotes insulin resistance in the ovary of rats and increases pituitary sensitivity to GnRH in mice (Akamine et al., 2010; Brothers et al., 2010). One way to address the question of whether impaired PI3K signaling in the brain is responsible for the effects of obesity on the HPG axis would be to investigate whether an increase in PI3K activity could rescue or lower LH levels in such models. Possible approaches include the use of brain region-specific adenoviral expression of a constitutively active mutant of PI3K to activate downstream targets such as Akt. This approach was successfully used to demonstrate that hypothalamic PI3K signaling mediates the effects of insulin treatment on glucose levels in rats with STZ-induced diabetes (Gelling et al., 2006).
Insulin Receptor Substrates
The IRS family of proteins consists of four members named IRS-1, -2, -3, and -4 (White, 1998). Upon insulin or IGF-1 receptor activation, IRS proteins undergo rapid tyrosine phosphorylation, allowing them to recruit and activate PI3K (White, 1998, 2002). Global ablation studies have revealed a role for IRS-2 and IRS-4 in the integration of metabolism and reproduction. For example, IRS-2-null female mice are infertile, with deficits at multiple levels of the reproductive axis, including anovulatory ovaries with reduced number of follicles, reduced pituitary size, as well as very low levels of LH, prolactin, and sex steroids (Burks et al., 2000). The infertility caused by the deletion of IRS-2 is not a consequence of abnormal glucose homeostasis as these females were euglycaemic and only mildly insulin resistant. However, IRS-2-null females exhibit increased food intake, mild obesity, and have defective leptin-stimulated hypothalamic STAT3 phosphorylation, suggesting that IRS-2 may integrate the actions of both insulin and leptin on metabolism and reproduction. IRS-4-null mice also exhibit defects in reproduction, although these are not as severe as those for IRS-2. Compared to wild type breeders, the number of litters produced by IRS-4-null breeding pairs is significantly lower (Fantin et al., 2000). Because a reduced frequency of total litters was not observed upon mating the IRS-4-null males with heterozygous females, IRS-4-null females are suspected to be responsible for the decrease in fertility.
Finally, the steroid hormones, E2 and P, regulate the expression and/or phosphorylation levels of specific IRS proteins. For example, in vitro, P induces IRS-2 gene expression in a progesterone receptor-dependent manner (Vassen et al., 1999). E2, through activation of ERα, stimulates tyrosine phosphorylation and nuclear translocation of IRS-1 in vitro and in the uterus (Richards et al., 1996; Panno et al., 2006). The effects that growth factors such as IGF-1 have on gonadotropin release and sexual maturation are modulated and even dependent on sex steroid hormones such as E2 (Quesada and Etgen, 2002; Etgen and Acosta-Martinez, 2003; Daftary and Gore, 2005). Therefore, regulation of IRS proteins may be a mechanism by which steroid hormones modulate tyrosine kinase receptor effects on the HPG axis. Future studies should examine the effect of steroid hormones on IRS proteins in the hypothalamus.
The Mammalian Target of Rapamycin
Mammalian target of rapamycin is activated under nutrient-rich conditions, whereas its kinase activity is attenuated when nutrients (amino acids, glucose, and oxygen) are depleted (Howell and Manning, 2011). mTOR exists in two forms of multiprotein complexes: one that contains raptor protein (mTORC1) and another that contains the protein rictor (mTORC2; Howell and Manning, 2011). The tuberous sclerosis complex of proteins, TSC1 and TSC2, normally blocks mTORC1 activation by a variety of signals (Howell and Manning, 2011). In response to insulin, IGF-1, and other growth factors, PI3K signaling activates mTORC1, at least in part through Akt-mediated phosphorylation of TSC1 and TSC2 (Inoki et al., 2002; Manning et al., 2002). Recent studies have linked central mTORC1 to the control of puberty onset and gonadotropin secretion. In pubertal female rats, acute activation of mTOR by L-leucine stimulates LH secretion and partially rescues the suppression of LH caused by chronic food restriction (Roa et al., 2009b). Conversely, chronic i.c.v. infusion of rapamycin, a potent mTORC1 inhibitor, delayed VO, decreased LH and E2 levels, and produced ovarian and uterine atrophy (Roa et al., 2009b). Central inhibition of mTOR also abolished the positive effects of leptin on puberty onset in food-restricted animals (Roa et al., 2009b). Therefore, mTOR signaling in the brain participates in the control of puberty onset and gonadotropin secretion. Furthermore, its activation may be relevant in the suppression of GnRH/LH release and puberty caused by negative energy balance.
The suppressive effects of mTOR on puberty and gonadotropin secretion in rats were also accompanied by decreased expression of kisspeptin mRNA levels in the ARC, linking this neuropeptide system to the regulation of the HPG axis by nutrients (Roa et al., 2009b). Kisspeptin is a potent stimulator of LH secretion and is critical for the initiation and maintenance of adult reproduction (Castellano et al., 2009). Because a state of positive energy balance is needed for the normal timing and development of the reproductive axis, mTOR’s role in pubertal onset and LH secretion correlates well with its known activation by positive energy status. As described earlier, IGF-1 is a potent stimulator of the PI3K/Akt pathway, and central activation of IGF-1 receptors stimulates LH release and accelerates the onset of puberty in rodents (Daftary and Gore, 2005). IGF-1 also up-regulates hypothalamic kisspeptin gene expression through the activation of Akt (Hiney et al., 2010). Therefore, it is possible that an IGF-1R/PI3K/Akt/mTOR/Kisspeptin pathway plays a role in pubertal onset and GnRH/LH secretion.
Neuropeptide Systems that Convey Energy Balance Information to the GnRH Network
A number of neuropeptide systems have been identified as possible mediators of metabolic signaling to the GnRH network. These include but are not limited to GALP, NPY, and products of POMC (e.g., alpha-melanocyte stimulating hormone, α-MSH) in the ARC (Crown et al., 2007), orexin neurons in the lateral hypothalamic area (LHA; Kohsaka et al., 2001; Small et al., 2003), melanin-concentrating neurons (MCH) in the LHA and zona incerta (Chiocchio et al., 2001; Williamson-Hughes et al., 2005; Murray et al., 2006), nesfatin-1 in the LHA, paraventricular (PVN), and supraoptic (SON) nuclei (Garcia-Galiano et al., 2010), and kisspeptin neurons in the anteroventral periventricular nucleus (AVPV) and the ARC (Castellano et al., 2009). With the exception of kisspeptin, these neuropeptides serve a dual role, being important players for both the central control of feeding and energy homeostasis and of gonadotropin secretion. Importantly, many of the neurons that synthesize these neuropeptides co-express insulin and/or the leptin receptors, and their expression is regulated by metabolic hormones as well as by sex steroids. This is important because conditions of negative energy balance affect the gene and protein expression of these neuropeptides in a brain region-specific fashion. This could explain why the ability of these neuropeptides to either stimulate or suppress LH secretion in the normal female depends on the hormonal milieu (i.e., presence of estrogens), developmental stage (i.e., prepubertal vs. adult), and energetic condition (i.e., fasted vs. normally fed; Table 2).
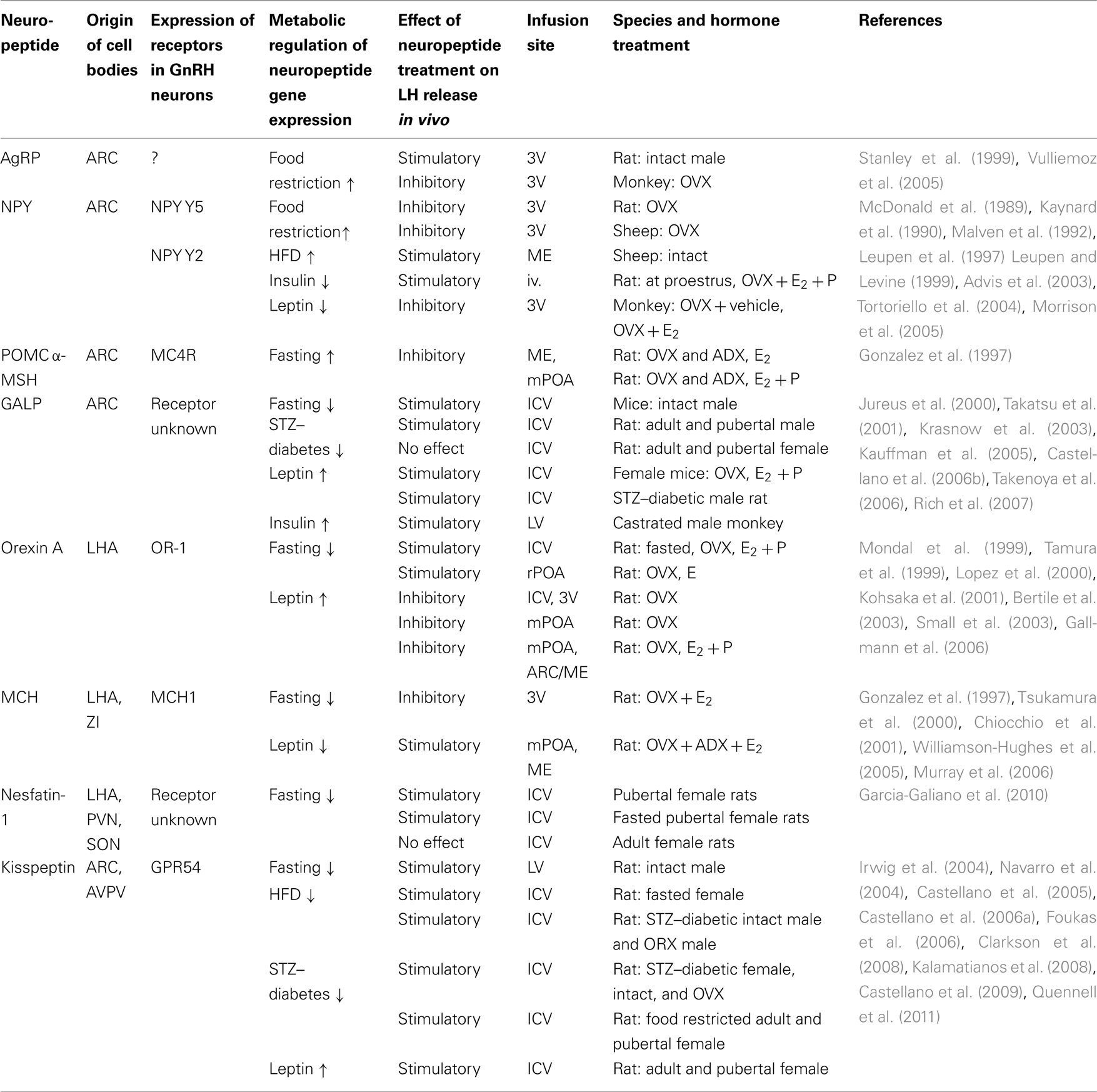
Table 2. Examples of hypothalamic neuropeptide regulation of LH release.
Each of these neuropeptide systems represents a functional neuroanatomical pathway that can send projections to areas containing GnRH neurons, in many cases directly influencing their activity. A review of the effects that each of these neuropeptides has on the reproductive axis is beyond the scope of this article. Instead, the kisspeptin system will be used to illustrate mechanisms by which the indirect regulation of GnRH neuronal activity by metabolic signals occurs (Figure 1).
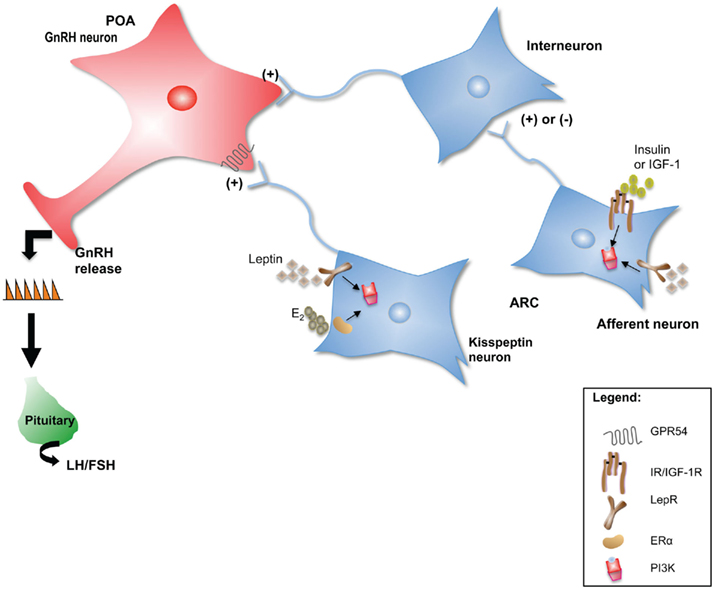
Figure 1. Neuropeptide neurons such as kisspeptin could serve as intermediaries for peripheral metabolic cues to communicate changes in energy status to the GnRH network. In addition, metabolic signals could be “sensed” by interneurons that in turn communicate with GnRH neurons to regulate their activity. Several upstream regulators of PI3K signaling such as the LepR, IR, IGF-1R, and ERα, are critical for the normal initiation and maintenance of reproductive function in mammals. In this model, PI3K serves as an integrator of E2, leptin, insulin, and IGF-1 indirect effects on GnRH neuronal function and hence on the HPG axis.
Kisspeptin: Linking Metabolic Signals to the Control of GnRH Neuronal Function
As introduced earlier, kisspeptin and its receptor, the G protein-coupled receptor GPR-54, have a pivotal role in the regulation of the HPG axis in mammals (for review see Roa et al., 2009a; Roseweir and Millar, 2009). Kisspeptin neurons have been proposed to represent a link between systemic metabolic signals such as leptin and central maintenance of reproductive function. For example, in rodents, the detrimental effects that negative energy balance has on the maintenance or the development of the female reproductive axis is completely rescued by central kisspeptin therapy. The models studied to date include chronic subnutrition of adult rats (Clarkson et al., 2008), peripubertal female rats subjected to food restriction (Castellano et al., 2005; Clarkson et al., 2008), and STZ-induced diabetes in male and female rats (Castellano et al., 2006a, 2009). In addition to their suppressive effects on LH release and gonadal function, these states of negative energy balance are accompanied by suppression of hypothalamic kisspeptin mRNA levels (Castellano et al., 2006a, 2009; Kalamatianos et al., 2008). In some cases, i.c.v. infusion of leptin can rescue both kisspeptin expression and gonadotropin levels (Castellano et al., 2006a).
Models of obesity accompanied by leptin resistance also result in suppression of hypothalamic kisspeptin mRNA (Foukas et al., 2006; Quennell et al., 2011). Compared to WT mice, ob/ob mice have a significantly reduced kisspeptin mRNA level, and leptin administration increases the levels of kisspeptin mRNA in these mice (Foukas et al., 2006). Almost half of cells expressing kisspeptin mRNA in the ARC nucleus express the LepR, implying that kisspeptin neurons are direct targets of leptin (Foukas et al., 2006). However, the kisspeptin cell-specific deletion of the LepR does not affect puberty or fertility in female mice, suggesting that a leptin-kisspeptin signaling pathway is not essential for normal HPG activity (Donato et al., 2011). On the other hand, previous studies using metabolic challenges, such as undernutrition and DIO, suggest that this pathway is more relevant under such metabolic challenges.
Electrophysiological studies have recently shown that the ability of kisspeptin to stimulate GnRH neuronal activity is modulated by neuropeptide systems involved in the regulation of feeding and energy balance. This is the case for the orexigenic neuropeptide MCH. MCH fiber projections are in close apposition with a large number of GnRH cell bodies throughout the POA–AH of the rat (Williamson-Hughes et al., 2005). In addition, more than 50% of GnRH neurons co-expressed the MCH receptor, MCHR1 (Williamson-Hughes et al., 2005). Using whole-cell patch clamp recordings in brain slices from established lines of GFP mice, MCH was shown to strongly inhibit kisspeptin-activated GnRH neurons but not kisspeptin-insensitive GnRH neurons (Wu et al., 2009). These effects are mediated via a direct postsynaptic mechanism. Therefore, the MCH neuropeptide system may have a role in conveying information about energy balance to the HPG axis by modulating the activity of kisspeptin-sensitive GnRH neurons.
Kisspeptin can also interact with other appetite-regulating neuropeptide systems, such as POMC and NPY neurons in the ARC. Many kisspeptin-immunoreactive axonal boutons terminate on or near POMC neurons (Fu and van den Pol, 2010). Moreover, kisspeptin was found to potently and directly excite POMC neurons (Fu and van den Pol, 2010). In contrast, kisspeptin inhibits NPY neurons through a presynaptic mechanism based on enhancing GABA-mediated inhibitory synaptic tone (Fu and van den Pol, 2010). Since both POMC and NPY are known regulators of GnRH release, the ability of kisspeptin to regulate their neuronal function provides an additional mechanism by which metabolic information is conveyed to the HPG axis, i.e., neuropeptides serving as a relay station downstream of kisspeptin.
In summary, the ability of kisspeptin to stimulate GnRH release can be directly regulated by peripheral metabolic signals such as leptin, or by other neuropeptide systems known to regulate GnRH release (Figure 1). Leptin and insulin directly regulate the gene expression and/or neuronal activity of many of these appetite-regulating neuropeptides, such as POMC and NPY. PI3K signaling plays a key role in the actions of both metabolic cues on these neurons. However, additional challenges include how to differentiate between PI3K metabolic and reproductive effects and how to identify in which neuronal population PI3K signaling acts to affect HPG axis activity. The cre/loxP system has offered a wealth of information about the phenotype of the neurons in which insulin and leptin receptors act to regulate metabolism and in some cases reproduction. In the case of PI3K, this has entailed the cell-specific ablation of its catalytic (p110) or regulatory (p85) subunits.
Targeting Class IA PI3K Regulatory and Catalytic Subunits in Hypothalamic Neurons
The cell-specific deletion of members of the class I PI3K family has confirmed the role of this enzyme as a key integrator of the central effects of insulin and leptin on feeding and metabolic control. These studies have focused on neuronal populations in the ARC, such as POMC and AgRP neurons, well known for their involvement in the central control of feeding and body weight. The phenotype observed depends on the type of neuron being targeted (i.e., NPY vs. POMC), as well as the PI3K component (regulatory vs. catalytic subunit). For example, while mice with a POMC-specific deletion of p110α have increased body weight and adipose tissue, mice with a POMC-specific deletion of p85α showed resistance to DIO (Hill et al., 2009). These alterations in energy homeostasis are often accompanied by the prevention of insulin- and leptin-stimulated electrophysiological responses (Hill et al., 2008; Al-Qassab et al., 2009).
No reproductive phenotype has been reported in mice with POMC- or AgRP-specific deletion of class IA PI3K catalytic or regulatory subunits. The same is true in studies of mice with specific deletion of the leptin or insulin receptor in POMC- or NPY/AgRP-expressing neurons. Unfortunately, many of these studies were carried out in males, whose reproductive axis is more resistant to the effects of metabolic disturbance. Moreover, the expression of a reproductive phenotype in these models might depend on the age of the animal or might be observed only in response to a metabolic challenge. This is the case for female mice with POMC- and NPY/AgRP-specific deletion of the LepR, which show reduced fertility with age (Israel and Chua, 2010). Finally, while POMC and NPY/AgRP neurons are important for the normal regulation of the HPG axis, and alterations in their gene expression could contribute to the detrimental effects that metabolic disorders have on fertility, gene manipulation studies in other neuronal populations that are essential for pubertal and adult GnRH/LH release are necessary in order to provide information about PI3K’s role in the control of the HPG axis. Candidate neurons include GnRH and kisspeptin.
While central regulation of the HPG axis by leptin and insulin is likely due to actions on interneurons that ultimately control GnRH neurosecretion, other signals such as steroid hormones and growth factors might exert direct regulation of GnRH neurons either during development or in the adult. As indicated earlier, many of the factors that regulate GnRH release also have the capacity to activate cell signaling pathways mediated by PI3K. Examples include E2 through ERα and IGF-1 through IGF-1Rs. To investigate whether PI3K signaling mediates the direct regulation of GnRH neurons by these and other factors, we used conditional gene targeting to ablate the expression of p85α in GnRH neurons.
Surprisingly, the GnRH neuron-specific deletion of p85α did not affect the female reproductive axis. Instead, we observed a male-specific phenotype consisting of decreased serum LH, T, and sperm counts (Acosta-Martinez et al., 2009). Furthermore, the same phenotype was observed when we generated the GnRH neuron-specific deletion of p85α on a global p85β KO background. Hence, p85β does not substitute for p85α activity toward PI3K function in GnRH neurons. These findings suggest that p85α in GnRH neurons participates in normal GnRH neurosecretory activity in the male mouse. It remains unknown whether p85α and PI3K activity in GnRH neurons are required in this regard during development, in adulthood, or throughout the lifespan.
Although we did not identify the cause of this male-biased effect, we speculate that PI3K signaling in GnRH neurons participates in the organizational effects of E2 during development. E2, which is aromatized from testicular androgens in the male, establishes sex differences in synaptic connections and neuronal morphology that are ultimately responsible for the differentiation of the male brain from the female brain in mammals (Schwarz et al., 2008). In the developing hypothalamus, activation of ERs by E2 promotes dendritic spine formation via rapid, non-genomic activation of PI3K (Schwarz et al., 2008). Because fetal GnRH neurons may express ERs (Sharifi et al., 2002), it is possible that this mechanism may be activated in GnRH neurons during the prenatal/neonatal surges of T production that occur in males. The subsequent changes in synaptic connectivity might support higher levels of GnRH neurosecretory activity or increased responsiveness to synaptic inputs in adulthood. Such a mechanism might explain the observation that the GnRH pulse generator operates at a higher frequency in males than in females of many species, and that prenatal androgen exposure leads to an acceleration of GnRH pulsatility in female rodents, sheep, and monkeys (reviewed in Foecking et al., 2008).
Besides steroid hormones, PI3K signaling is activated by and regulates the function of other molecules that affect GnRH neuronal activity through direct mechanisms. This is the case of IGF-1Rs and ATP-sensitive potassium (KATP) channels, both of which are expressed in GnRH neurons (Zhang et al., 2007a; Brothers et al., 2010). While GnRH neuronal expression of IGF-1Rs is important for normal pubertal timing, KATP channels confer responsiveness to glucose in GnRH neurons and regulate GnRH/LH release in an E2-dependent manner (Zhang et al., 2007a; Huang et al., 2008). One can speculate that a reproductive phenotype in GnRH-specific p85α KO females might be observed after metabolic challenges such as fasting or DIO. On the other hand, it is unknown whether p85α’s effects on male GnRH neurons are mediated through p110-independent or -dependent mechanisms. Hence, it is possible that a GnRH-specific deletion of p110 catalytic subunits might alter GnRH neuronal activity resulting in a reproductive phenotype in females.
Neuronal populations that express ERα and are important for GnRH/LH release or sexual behavior are also good candidates to investigate the role of PI3K in the control of reproduction. A good candidate for this is kisspeptin neurons. The majority of kisspeptin neurons co-express ERα, with more than 60% in the AVPV and more than 90% in the ARC (Mayer et al., 2010). In addition, the kisspeptin cell-specific deletion of ERα results in precocious puberty (advanced VO and LH hypersecretion) and the arrest of subsequent pubertal maturation (Mayer et al., 2010). Therefore, in addition to relaying metabolic information from the periphery, PI3K might participate in the direct actions of E2 on kisspeptin neuronal activity and hence GnRH/LH secretion (Figure 1). Through the use of transgenic mouse models, experiments in our lab are underway to investigate this possibility.
Inflammation and PI3K Signaling
Obesity is considered to be a chronic inflammatory state. With time, adipose tissue becomes infiltrated by macrophages, resulting in increased circulating pro-inflammatory cytokines such as transforming growth factor-alpha (TNF-α) and interleukin (IL)-6 (Thaler et al., 2010). The chronic exposure to circulating cytokines as well as to excess glucose or fatty acids can activate intracellular inflammatory pathways in other cell types and organs including the liver, muscle, endothelial cells, and neurons (Thaler and Schwartz, 2010; Thaler et al., 2010). It has been hypothesized that hypothalamic inflammation resulting from the prolonged consumption of a HFD leads to obesity through the development of central insulin and leptin resistance (Thaler and Schwartz, 2010). These are states associated with infertility in both humans and animal models of DIO. Therefore, chronic immune signals might also reduce the sensitivity of signaling pathways important for the central control of the HPG axis.
In humans, a reproductive disorder associated with a state of chronic low-grade inflammation (LGI) is PCOS (Repaci et al., 2011). Markers of LGI in PCOS patients include elevated TNF-α (Gonzalez et al., 1999), IL-18 (Escobar-Morreale et al., 2004), IL-6 (Repaci et al., 2011), and C-reactive protein (CRP; Kelly et al., 2001; Benson et al., 2008). Many of these inflammatory signals are associated with insulin resistance and high circulating insulin levels. Insulin resistance, although not part of the diagnostic criteria, is present in a large percentage of women diagnosed with PCOS, regardless of obesity (Repaci et al., 2011). It has been speculated that LGI might be the link between hyperandrogenism, insulin resistance, and the long-term consequences of PCOS. For example, TNF-α is a known inducer of insulin resistance, presumably through its ability to interfere with insulin receptor signaling (Feinstein et al., 1993; Uysal et al., 1997). While the focus of these studies has been on peripheral insulin-responsive organs such as muscles and adipose tissue, the effects that long-term TNF-α levels might have on insulin receptor signaling in reproductive organs has not yet been investigated. This is of importance since both TNF-α and insulin are known to modulate ovarian steroid production as well as induce cell proliferation in interstitial theca cells in vitro (Roby and Terranova, 1990; Spaczynski et al., 1999). Interestingly, the ability of insulin to stimulate androgen synthesis by the ovary is preserved in PCOS patients (Nestler et al., 1998). In fact, the higher concentration of insulin in women with PCOS is thought to amplify the effects of LH on androgen production by theca cells. In in vitro studies using human ovarian theca cells, insulin augmentation of 17 α-hydroxylase activity, a key enzyme for androgen biosynthesis, is mediated by PI3K (Munir et al., 2004). Therefore, ovarian PI3K signaling could potentially contribute to the hyperandrogenemia observed in PCOS. However, whether or not inflammatory signals such as TNF-α modulate PI3K-mediated androgen production by the ovary is not known.
A role of inflammation and insulin resistance in PCOS is also supported by the marked improvement PCOS patients show after treatment with insulin sensitizers such as metformin. Long-term treatment with metformin increases ovulation, improves menstrual cyclicity, and reduces serum androgen levels (Lebinger, 2007; Nestler, 2008). Metformin treatment also significantly reduces circulating levels of CRP and white blood cells (Morin-Papunen et al., 2003; Diamanti-Kandarakis et al., 2006; Orio et al., 2007). The anti-inflammatory actions of metformin include the inhibition of IL-1β-induced release of IL-6 and IL-8 in endothelial cells, human vascular smooth cells, and macrophages (Morin-Papunen et al., 2003; Diamanti-Kandarakis et al., 2006; Orio et al., 2007).
The studies cited above focus on the role of inflammation and insulin resistance in peripheral tissues, like the ovaries and endothelial cells. However, the role that central inflammation and the resultant insulin resistance might play in the pathogenesis of PCOS, especially on GnRH/LH hypersecretion, is unknown. It is possible that central actions of metformin contribute to its beneficial effects on HPG axis function, as this drug can cross the blood–brain barrier. Here, rodent models of HFD-induced obesity and infertility could be used to determine if centrally administered metformin normalizes LH release.
The mechanisms by which dietary lipids and other nutrients become more immunogenic by overnutrition are not completely understood. It is clear, however, that many of the players involved in the immune response toward disease, by as yet unidentified mechanisms, also promote insulin and leptin resistance. In this context, it is worth mentioning that insulin has anti-inflammatory effects that are independent of its function in regulating glucose homeostasis. In in vivo models of endotoxemia, such as that induced by acute injection of bacterially derived lipopolysaccharide (LPS), insulin is able to reduce inflammation (decreased plasma levels of IL-6, TNF-α, monocyte chemotactic protein) and mortality (Kidd et al., 2008). In this model, pharmacological or genetic inhibition of the PI3K–Akt pathway suppresses insulin’s anti-inflammatory actions (Zhang et al., 2007b; Kidd et al., 2008; Luyendyk et al., 2008). Recent studies support a role for hypothalamic inflammation and the resultant insulin and leptin resistance as triggering factors of hyperphagia and obesity (Thaler and Schwartz, 2010). Future studies should investigate if chronic levels of pro-inflammatory cytokines in the brain affect PI3K signaling in hypothalamic centers involved in the control of both reproduction and metabolism such as the ARC. This could reveal a possible mechanism by which insulin sensitizers such as metformin restore normal gonadotropin levels in PCOS patients.
Summary and Conclusion
Central dysregulation of GnRH neurosecretion underlies the reproductive dysfunction observed during states of metabolic imbalance, such as malnutrition and chronic nutritional excess. The inability of peripheral hormones and metabolic cues to regulate key hypothalamic centers plays a major role in the negative effects that these states have on the HPG axis. Hormones and growth factors key for the central control of GnRH release, puberty, and sexual behavior utilize the PI3K signaling pathway to regulate a variety of physiological functions including metabolism. Examples include insulin, leptin, IGF-1, and E2, whose levels and capacity to act in the brain is severely compromised during conditions of disturbed energy homeostasis (Figure 2). In rodents, PI3K participates in the central control of female sexual behavior (Etgen and Acosta-Martinez, 2003) and gonadotropin release (Acosta-Martinez et al., 2009). A direct link between impaired PI3K signaling and the detrimental effects that a disturbed energy homeostasis has on the HPG axis has not been established. However, PI3K plays a central role mediating the effects of insulin and leptin on feeding and metabolism. Moreover, these metabolic signals also utilized PI3K to regulate the neuronal activity and expression of neuropeptide systems that are important regulators of GnRH release, such as NPY and POMC. To test the hypothesis that PI3K signaling represents a link between metabolism and reproduction, experimental approaches similar to those used to investigate its role in the central control of metabolism should be used. For example, the use of the Cre/LoxP system to alter PI3K gene expression in neuronal populations critical for the control of gonadotropin release, such as kisspeptin, will continue to shed light into the role of this enzyme in the control of reproduction. Similarly, adenoviral gene therapy could be used to investigate if the detrimental effects of negative energy balance and obesity on LH release is prevented or restored by activation of PI3K signaling in specific brain regions, like the POA and the ARC.
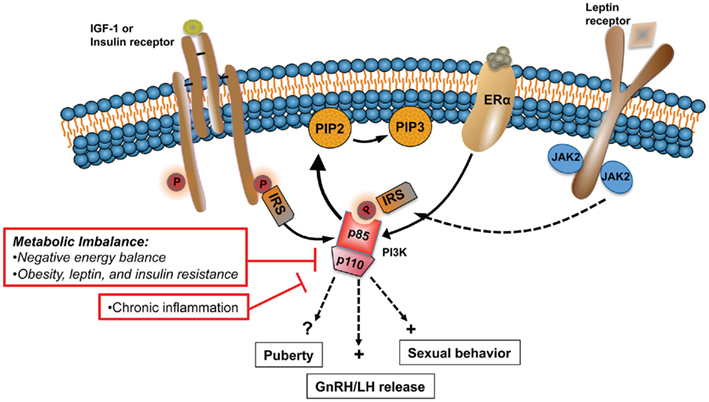
Figure 2. States of metabolic imbalance such as malnutrition and obesity have detrimental effects on the HPG axis, principally altering normal GnRH pulsatile release. Alterations in the levels and sensitivity to peripheral hormones and metabolic signals, including IGF-1, E2, leptin, and insulin, play a major role in the dysfunction of the HPG axis during chronic metabolic disturbances. PI3K is a major signaling pathway activated by the same upstream regulators that are affected during such states. In rodents, PI3K participates in the central control of sexual behavior and LH release. In this model altered PI3K signaling is linked to the negative effects that metabolic imbalance has on the HPG axis.
Finally, chronic inflammation has been identified recently as an important component of the pathophysiology of obesity and the metabolic syndrome. These and other metabolic conditions have a major impact on fertility. Therefore, it is important to identify the intracellular mechanisms that are at play during chronic inflammation within the CNS. Because a direct consequence of inflammation is central insulin and leptin resistance, signaling pathways shared by these hormones, such as PI3K, should be investigated.
The use of pharmacological agents that target different classes of PI3K is already being considered as a therapeutic strategy to treat cancer and metabolic diseases (Holmes, 2011). The insights described within this review highlight the need to also address PI3K and its role in the central control of reproduction. Disorders that span both metabolic and reproductive function, such as PCOS, may have some potential for PI3K pharmacological targeting.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Dr. Anne M. Etgen, Dr. Wanda V. Carriles, Lisa Ballou, and Matt Beymer for their invaluable comments and editorial support. This work was supported by the Eunice Kennedy Shriver National Institute of Child and Human Development Grant, 5R00HD055446-04 and the Office of the Vice President for Research at Stony Brook University.
References
Acosta-Martinez, M., Luo, J., Elias, C., Wolfe, A., and Levine, J. E. (2009). Male-biased effects of gonadotropin-releasing hormone neuron-specific deletion of the phosphoinositide 3-kinase regulatory subunit p85alpha on the reproductive axis. Endocrinology 150, 4203–4212.
Advis, J. P., Klein, J., Kuljis, R. O., Sarkar, D. K., McDonald, J. M., and Conover, C. A. (2003). Regulation of gonadotropin releasing hormone release by neuropeptide Y at the median eminence during the preovulatory period in ewes. Neuroendocrinology 77, 246–257.
Akamine, E. H., Marcal, A. C., Camporez, J. P., Hoshida, M. S., Caperuto, L. C., Bevilacqua, E., and Carvalho, C. R. (2010). Obesity induced by high-fat diet promotes insulin resistance in the ovary. J. Endocrinol. 206, 65–74.
Al-Qassab, H., Smith, M. A., Irvine, E. E., Guillermet-Guibert, J., Claret, M., Choudhury, A. I., Selman, C., Piipari, K., Clements, M., Lingard, S., Chandarana, K., Bell, J. D., Barsh, G. S., Smith, A. J., Batterham, R. L., Ashford, M. L., Vanhaesebroeck, B., and Withers, D. J. (2009). Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell Metab. 10, 343–354.
Attia, E., and Roberto, C. A. (2009). Should amenorrhea be a diagnostic criterion for anorexia nervosa? Int. J. Eat. Disord. 42, 581–589.
Backer, J. M., Myers, M. G. Jr., Shoelson, S. E., Chin, D. J., Sun, X. J., Miralpeix, M., Hu, P., Margolis, B., Skolnik, E. Y., Schlessinger, J., and White, M. F. (1992). Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 11, 3469–3479.
Balthasar, N., Coppari, R., McMinn, J., Liu, S. M., Lee, C. E., Tang, V., Kenny, C. D., McGovern, R. A., Chua, S. C. Jr., Elmquist, J. K., and Lowell, B. B. (2004). Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42, 983–991.
Benson, S., Janssen, O. E., Hahn, S., Tan, S., Dietz, T., Mann, K., Pleger, K., Schedlowski, M., Arck, P. C., and Elsenbruch, S. (2008). Obesity, depression, and chronic low-grade inflammation in women with polycystic ovary syndrome. Brain Behav. Immun. 22, 177–184.
Bertile, F., Oudart, H., Criscuolo, F., Maho, Y. L., and Raclot, T. (2003). Hypothalamic gene expression in long-term fasted rats: relationship with body fat. Biochem. Biophys. Res. Commun. 303, 1106–1113.
Boukouvalas, G., Antoniou, K., Papalexi, E., and Kitraki, E. (2008). Post weaning high fat feeding affects rats’ behavior and hypothalamic pituitary adrenal axis at the onset of puberty in a sexually dimorphic manner. Neuroscience 153, 373–382.
Brewer, M., Pawelczak, M., Kessler, M., and Shah, B. (2010). A review of polycystic ovarian syndrome in adolescents. Minerva Pediatr. 62, 459–473.
Brill, D. S., and Moenter, S. M. (2009). Androgen receptor antagonism and an insulin sensitizer block the advancement of vaginal opening by high-fat diet in mice. Biol. Reprod. 81, 1093–1098.
Bronson, F. H. (1986). Food-restricted, prepubertal, female rats: rapid recovery of luteinizing hormone pulsing with excess food, and full recovery of pubertal development with gonadotropin-releasing hormone. Endocrinology 118, 2483–2487.
Bronson, F. H. (1988). Effect of food manipulation on the GnRH-LH-estradiol axis of young female rats. Am. J. Physiol. 254(Pt 2), R616–R621.
Brothers, K. J., Wu, S., DiVall, S. A., Messmer, M. R., Kahn, C. R., Miller, R. S., Radovick, S., Wondisford, F. E., and Wolfe, A. (2010). Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metab. 12, 295–305.
Bruning, J. C., Gautam, D., Burks, D. J., Gillette, J., Schubert, M., Orban, P. C., Klein, R., Krone, W., Müller-Wieland, D., and Kahn, C. R. (2000). Role of brain insulin receptor in control of body weight and reproduction. Science 289, 2122–2125.
Burks, D. J., Font de Mora, J., Schubert, M., Withers, D. J., Myers, M. G., Towery, H. H., Altamuro, S. L., Flint, C. L., and White, M. F. (2000). IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 407, 377–382.
Burt, J., Alberto, C. O., Parsons, M. P., and Hirasawa, M. (2011). Local network regulation of orexin neurons in the lateral hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R572–R580.
Burt Solorzano, C. M., and McCartney, C. R. (2010). Obesity and the pubertal transition in girls and boys. Reproduction 140, 399–410.
Buyken, A. E., Karaolis-Danckert, N., and Remer, T. (2009). Association of prepubertal body composition in healthy girls and boys with the timing of early and late pubertal markers. Am. J. Clin. Nutr. 89, 221–230.
Cameron, J. L., and Nosbisch, C. (1991). Suppression of pulsatile luteinizing hormone and testosterone secretion during short term food restriction in the adult male rhesus monkey (Macaca mulatta). Endocrinology 128, 1532–1540.
Campbell, R. A., Bhat-Nakshatri, P., Patel, N. M., Constantinidou, D., Ali, S., and Nakshatri, H. (2001). Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J. Biol. Chem. 276, 9817–9824.
Cantrell, D. A. (2001). Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 114(Pt 8), 1439–1445.
Carpenter, C. L., Auger, K. R., Chanudhuri, M., Yoakim, M., Schaffhausen, B., Shoelson, S., and Cantley, L. C. (1993). Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem. 13, 9478–9483.
Castellano, J. M., Navarro, V. M., Fernández-Fernández, R., Nogueiras, R., Tovar, S., Roa, J., Vazquez, M. J., Vigo, E., Casanueva, F. F., Aguilar, E., Pinilla, L., Dieguez, C., and Tena-Sempere, M. (2005). Changes in hypothalamic KiSS-1 system and restoration of pubertal activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology 146, 3917–3925.
Castellano, J. M., Navarro, V. M., Fernández-Fernández, R., Roa, J., Vigo, E., Pineda, R., Dieguez, C., Aguilar, E., Pinilla, L., and Tena-Sempere, M. (2006a). Expression of hypothalamic KiSS-1 system and rescue of defective gonadotropic responses by kisspeptin in streptozotocin-induced diabetic male rats. Diabetes 55, 2602–2610.
Castellano, J. M., Navarro, V. M., Fernández-Fernández, R., Roa, J., Vigo, E., Pineda, R., Steiner, R. A., Aguilar, E., Pinilla, L., and Tena-Sempere, M. (2006b). Effects of galanin-like peptide on luteinizing hormone secretion in the rat: sexually dimorphic responses and enhanced sensitivity at male puberty. Am. J. Physiol. Endocrinol. Metab. 291, E1281–E1289.
Castellano, J. M., Navarro, V. M., Roa, J., Pineda, R., Sánchez-Garrido, M. A., García-Galiano, D., Vigo, E., Dieguez, C., Aguilar, E., Pinilla, L., and Tena-Sempere, M. (2009). Alterations in hypothalamic KiSS-1 system in experimental diabetes: early changes and functional consequences. Endocrinology 150, 784–794.
Chan, J. L., and Mantzoros, C. S. (2005). Role of leptin in energy-deprivation states: normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet 366, 74–85.
Chiocchio, S. R., Gallardo, M. G., Louzan, P., Gutnisky, V., and Tramezzani, J. H. (2001). Melanin-concentrating hormone stimulates the release of luteinizing hormone-releasing hormone and gonadotropins in the female rat acting at both median eminence and pituitary levels. Biol. Reprod. 64, 1466–1472.
Clarkson, J., d’Anglemont de Tassigny, X., Moreno, A. S., Colledge, W. H., and Herbison, A. E. (2008). Kisspeptin-GPR54 signaling is essential for preovulatory gonadotropin-releasing hormone neuron activation and the luteinizing hormone surge. J. Neurosci. 28, 8691–8697.
Couse, J. F., and Korach, K. S. (1999). Reproductive phenotypes in the estrogen receptor-alpha knockout mouse. Ann. Endocrinol. (Paris) 60, 143–148.
Crown, A., Clifton, D. K., and Steiner, R. A. (2007). Neuropeptide signaling in the integration of metabolism and reproduction. Neuroendocrinology 86, 175–182.
Daftary, S. S., and Gore, A. C. (2005). IGF-1 in the brain as a regulator of reproductive neuroendocrine function. Exp. Biol. Med. (Maywood) 230, 292–306.
de Luca, C., Kowalski, T. J., Zhang, Y., Elmquist, J. K., Lee, C., Kilimann, M. W., Ludwig, T., Liu, S. M., and Chua, S. C. Jr. (2005). Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J. Clin. Invest. 115, 3484–3493.
Demissie, M., Lazic, M., Foecking, E. M., Aird, F., Dunaif, A., and Levine, J. E. (2008). Transient prenatal androgen exposure produces metabolic syndrome in adult female rats. Am. J. Physiol. Endocrinol. Metab. 295, E262–E268.
Diamanti-Kandarakis, E., Paterakis, T., Alexandraki, K., Piperi, C., Aessopos, A., Katsikis, I., Katsilambros, N., Kreatsas, G., and Panidis, D. (2006). Indices of low-grade chronic inflammation in polycystic ovary syndrome and the beneficial effect of metformin. Hum. Reprod. 21, 1426–1431.
Divall, S. A., Williams, T. R., Carver, S. E., Koch, L., Brüning, J. C., Kahn, C. R., Wondisford, F., Radovick, S., and Wolfe, A. (2010). Divergent roles of growth factors in the GnRH regulation of puberty in mice. J. Clin. Invest. 120, 2900–2909.
Donato, J. Jr., Cravo, R. M., Frazão, R., Gautron, L., Scott, M. M., Lachey, J., Castro, I. A., Margatho, L. O., Lee, S., Lee, C., Richardson, J. A., Friedman, J., Chua, S. Jr., Coppari, R., Zigman, J. M., Elmquist, J. K., and Elias, C. F. (2011). Leptin’s effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J. Clin. Invest. 121, 355–368.
Dyer, R. G., Mansfield, S., Corbet, H., and Dean, A. D. (1985). Fasting impairs LH secretion in female rats by activating an inhibitory opioid pathway. J. Endocrinol. 105, 91–97.
Escobar-Morreale, H. F., Botella-Carretero, J. I., Villuendas, G., Sancho, J., and San Millán, J. L. (2004). Serum interleukin-18 concentrations are increased in the polycystic ovary syndrome: relationship to insulin resistance and to obesity. J. Clin. Endocrinol. Metab. 89, 806–811.
Etgen, A. M., and Acosta-Martinez, M. (2003). Participation of growth factor signal transduction pathways in estradiol facilitation of female reproductive behavior. Endocrinology 144, 3828–3835.
Euling, S. Y., Herman-Giddens, M. E., Lee, P. A., Selevan, S. G., Juul, A., Sørensen, T. I., Dunkel, L., Himes, J. H., Teilmann, G., and Swan, S. H. (2008). Examination of US puberty-timing data from 1940 to 1994 for secular trends: panel findings. Pediatrics 121(Suppl. 3), S172–S191.
Fantin, V. R., Wang, Q., Lienhard, G. E., and Keller, S. R. (2000). Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 278, E127–E133.
Feinstein, R., Kanety, H., Papa, M. Z., Lunenfeld, B., and Karasik, A. (1993). Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J. Biol. Chem. 268, 26055–26058.
Fernandez-Galaz, M. C., Naftolin, F., and Garcia-Segura, L. M. (1999). Phasic synaptic remodeling of the rat arcuate nucleus during the estrous cycle depends on insulin-like growth factor-I receptor activation. J. Neurosci. Res. 55, 286–292.
Foecking, E. M., McDevitt, M. A., Acosta-Martínez, M., Horton, T. H., and Levine, J. E. (2008). Neuroendocrine consequences of androgen excess in female rodents. Horm. Behav. 53, 673–692.
Foster, D. L., Ebling, F. J., Micka, A. F., Vannerson, L. A., Bucholtz, D. C., Wood, R. I., Suttie, J. M., and Fenner, D. E. (1989). Metabolic interfaces between growth and reproduction. I. Nutritional modulation of gonadotropin, prolactin, and growth hormone secretion in the growth-limited female lamb. Endocrinology 125, 342–350.
Foukas, L. C., Claret, M., Pearce, W., Okkenhaug, K., Meek, S., Peskett, E., Sancho, S., Smith, A. J., Withers, D. J., and Vanhaesebroeck, B. (2006). Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441, 366–370.
Foukas, L. C., and Withers, D. J. (2010). Phosphoinositide signalling pathways in metabolic regulation. Curr. Top. Microbiol. Immunol. 346, 115–141.
Fu, L. Y., and van den Pol, A. N. (2010). Kisspeptin directly excites anorexigenic proopiomelanocortin neurons but inhibits orexigenic neuropeptide Y cells by an indirect synaptic mechanism. J. Neurosci. 30, 10205–10219.
Gallmann, E., Arsenijevic, D., Williams, G., Langhans, W., and Spengler, M. (2006). Effect of intraperitoneal CCK-8 on food intake and brain orexin-A after 48 h of fasting in the rat. Regul. Pept. 133, 139–146.
Garcia-Galiano, D., Navarro, V. M., Roa, J., Ruiz-Pino, F., Sánchez-Garrido, M. A., Pineda, R., Castellano, J. M., Romero, M., Aguilar, E., Gaytán, F., Diéguez, C., Pinilla, L., and Tena-Sempere, M. (2010). The anorexigenic neuropeptide, nesfatin-1, is indispensable for normal puberty onset in the female rat. J. Neurosci. 30, 7783–7792.
Gelling, R. W., Morton, G. J., Morrison, C. D., Niswender, K. D., Myers, M. G. Jr., Rhodes, C. J., and Schwartz, M. W. (2006). Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 3, 67–73.
Ghanayem, B. I., Bai, R., Kissling, G. E., Travlos, G., and Hoffler, U. (2010). Diet-induced obesity in male mice is associated with reduced fertility and potentiation of acrylamide-induced reproductive toxicity. Biol. Reprod. 82, 96–104.
Gonzalez, F., Thusu, K., Abdel-Rahman, E., Prabhala, A., Tomani, M., and Dandona, P. (1999). Elevated serum levels of tumor necrosis factor alpha in normal-weight women with polycystic ovary syndrome. Metab. Clin. Exp. 48, 437–441.
Gonzalez, M. I., Baker, B. I., and Wilson, C. A. (1997). Stimulatory effect of melanin-concentrating hormone on luteinising hormone release. Neuroendocrinology 66, 254–262.
Hill, J. W., Elias, C. F., Fukuda, M., Williams, K. W., Berglund, E. D., Holland, W. L., Cho, Y. R., Chuang, J. C., Xu, Y., Choi, M., Lauzon, D., Lee, C. E., Coppari, R., Richardson, J. A., Zigman, J. M., Chua, S., Scherer, P. E., Lowell, B. B., Brüning, J. C., and Elmquist, J. K. (2010). Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 11, 286–297.
Hill, J. W., Williams, K. W., Ye, C., Luo, J., Balthasar, N., Coppari, R., Cowley, M. A., Cantley, L. C., Lowell, B. B., and Elmquist, J. K. (2008). Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Invest. 118, 1796–1805.
Hill, J. W., Xu, Y., Preitner, F., Fukuda, M., Cho, Y. R., Luo, J., Balthasar, N., Coppari, R., Cantley, L. C., Kahn, B. B., Zhao, J. J., and Elmquist, J. K. (2009). Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology 150, 4874–4882.
Hiney, J. K., Srivastava, V., Nyberg, C. L., Ojeda, S. R., and Dees, W. L. (1996). Insulin-like growth factor I of peripheral origin acts centrally to accelerate the initiation of female puberty. Endocrinology 137, 3717–3728.
Hiney, J. K., Srivastava, V. K., and Les Dees, W. (2010). Insulin-like growth factor-1 stimulation of hypothalamic KiSS-1 gene expression is mediated by Akt: effect of alcohol. Neuroscience 166, 625–632.
Howell, J. J., and Manning, B. D. (2011). mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 22, 94–102.
Huang, W., Acosta-Martinez, M., and Levine, J. E. (2008). Ovarian steroids stimulate adenosine triphosphate-sensitive potassium (KATP) channel subunit gene expression and confer responsiveness of the gonadotropin-releasing hormone pulse generator to KATP channel modulation. Endocrinology 149, 2423–2432.
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657.
Irwig, M. S., Fraley, G. S., Smith, J. T., Acohido, B. V., Popa, S. M., Cunningham, M. J., Gottsch, M. L., Clifton, D. K., and Steiner, R. A. (2004). Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology 80, 264–272.
Israel, D., and Chua, S. Jr. (2010). Leptin receptor modulation of adiposity and fertility. Trends Endocrinol. Metab. 21, 10–16.
Jureus, A., Cunningham, M. J., McClain, M. E., Clifton, D. K., and Steiner, R. A. (2000). Galanin-like peptide (GALP) is a target for regulation by leptin in the hypothalamus of the rat. Endocrinology 141, 2703–2706.
Kalamatianos, T., Grimshaw, S. E., Poorun, R., Hahn, J. D., and Coen, C. W. (2008). Fasting reduces KiSS-1 expression in the anteroventral periventricular nucleus (AVPV): effects of fasting on the expression of KiSS-1 and neuropeptide Y in the AVPV or arcuate nucleus of female rats. J. Neuroendocrinol. 20, 1089–1097.
Kasturi, S. S., Tannir, J., and Brannigan, R. E. (2008). The metabolic syndrome and male infertility. J. Androl. 29, 251–259.
Kauffman, A. S., Buenzle, J., Fraley, G. S., and Rissman, E. F. (2005). Effects of galanin-like peptide (GALP) on locomotion, reproduction, and body weight in female and male mice. Horm. Behav. 48, 141–151.
Kaynard, A. H., Pau, K. Y., Hess, D. L., and Spies, H. G. (1990). Third-ventricular infusion of neuropeptide Y suppresses luteinizing hormone secretion in ovariectomized rhesus macaques. Endocrinology 127, 2437–2444.
Kelly, C. C., Lyall, H., Petrie, J. R., Gould, G. W., Connell, J. M., and Sattar, N. (2001). Low grade chronic inflammation in women with polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 86, 2453–2455.
Kidd, L. B., Schabbauer, G. A., Luyendyk, J. P., Holscher, T. D., Tilley, R. E., Tencati, M., and Mackman, N. (2008). Insulin activation of the phosphatidylinositol 3-kinase/protein kinase B (Akt) pathway reduces lipopolysaccharide-induced inflammation in mice. J. Pharmacol. Exp. Ther. 326, 348–353.
Kohsaka, A., Watanobe, H., Kakizaki, Y., Suda, T., and Schiöth, H. B. (2001). A significant participation of orexin-A, a potent orexigenic peptide, in the preovulatory luteinizing hormone and prolactin surges in the rat. Brain Res. 898, 166–170.
Konner, A. C., Janoschek, R., Plum, L., Jordan, S. D., Rother, E., Ma, X., Xu, C., Enriori, P., Hampel, B., Barsh, G. S., Kahn, C. R., Cowley, M. A., Ashcroft, F. M., and Brüning, J. C. (2007). Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 5, 438–449.
Kovacs, P., Morales, J. C., and Karkanias, G. B. (2003). Central insulin administration maintains reproductive behavior in diabetic female rats. Neuroendocrinology 78, 90–95.
Kovacs, P., Parlow, A. F., and Karkanias, G. B. (2002). Effect of centrally administered insulin on gonadotropin-releasing hormone neuron activity and luteinizing hormone surge in the diabetic female rat. Neuroendocrinology 76, 357–365.
Kozma, S. C., and Thomas, G. (2002). Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. Bioessays 24, 65–71.
Krasnow, S. M., Fraley, G. S., Schuh, S. M., Baumgartner, J. W., Clifton, D. K., and Steiner, R. A. (2003). A role for galanin-like peptide in the integration of feeding, body weight regulation, and reproduction in the mouse. Endocrinology 144, 813–822.
Lebinger, T. G. (2007). Metformin and polycystic ovary syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 14, 132–140.
Lee, J. M., Kaciroti, N., Appugliese, D., Corwyn, R. F., Bradley, R. H., and Lumeng, J. C. (2010). Body mass index and timing of pubertal initiation in boys. Arch. Pediatr. Adolesc. Med. 164, 139–144.
Leibel, R. L., Chua, S. C., and Rosenbaum, M. (2001). “Obesity,” in The Metabolic and Molecular Bases of Inherited Disease, Vol. III, eds. C. Scriver, A. Beaudet, W. Sly, and D. Valle (New York: McGraw-Hill), 3965–4028.
Leupen, S. M., Besecke, L. M., and Levine, J. E. (1997). Neuropeptide Y Y1-receptor stimulation is required for physiological amplification of preovulatory luteinizing hormone surges. Endocrinology 138, 2735–2739.
Leupen, S. M., and Levine, J. E. (1999). Role of protein kinase C in facilitation of luteinizing hormone (LH)-releasing hormone-induced LH surges by neuropeptide Y. Endocrinology 140, 3682–3687.
Lopez, M., Seoane, L., García, M. C., Lago, F., Casanueva, F. F., Señarís, R., and Diéguez, C. (2000). Leptin regulation of prepro-orexin and orexin receptor mRNA levels in the hypothalamus. Biochem. Biophys. Res. Commun. 269, 41–45.
Luyendyk, J. P., Schabbauer, G. A., Tencati, M., Holscher, T., Pawlinski, R., and Mackman, N. (2008). Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J. Immunol. 180, 4218–4226.
Malven, P. V., Haglof, S. A., and Degroot, H. (1992). Effects of intracerebral administration of neuropeptide-Y on secretion of luteinizing hormone in ovariectomized sheep. Brain Res. Bull. 28, 871–875.
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162.
Manning, J. M., and Bronson, F. H. (1989). Effects of prolonged exercise on puberty and luteinizing hormone secretion in female rats. Am. J. Physiol. 257(Pt 2), R1359–R1364.
Mayer, C., Acosta-Martinez, M., Dubois, S. L., Wolfe, A., Radovick, S., Boehm, U., and Levine, J. E. (2010). Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proc. Natl. Acad. Sci. U.S.A. 107, 22693–22698.
McDonald, J. K., Lumpkin, M. D., and DePaolo, L. V. (1989). Neuropeptide-Y suppresses pulsatile secretion of luteinizing hormone in ovariectomized rats: possible site of action. Endocrinology 125, 186–191.
Mendez, P., Azcoitia, I., and Garcia-Segura, L. M. (2003). Estrogen receptor alpha forms estrogen-dependent multimolecular complexes with insulin-like growth factor receptor and phosphatidylinositol 3-kinase in the adult rat brain. Brain Res. Mol. Brain Res. 112, 170–176.
Metlakunta, A. S., Sahu, M., and Sahu, A. (2008). Hypothalamic phosphatidylinositol 3-kinase pathway of leptin signaling is impaired during the development of diet-induced obesity in FVB/N mice. Endocrinology 149, 1121–1128.
Mondal, M. S., Nakazato, M., Date, Y., Murakami, N., Yanagisawa, M., and Matsukura, S. (1999). Widespread distribution of orexin in rat brain and its regulation upon fasting. Biochem. Biophys. Res. Commun. 256, 495–499.
Morin-Papunen, L., Rautio, K., Ruokonen, A., Hedberg, P., Puukka, M., and Tapanainen, J. S. (2003). Metformin reduces serum C-reactive protein levels in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 88, 4649–4654.
Morrison, C. D., Morton, G. J., Niswender, K. D., Gelling, R. W., and Schwartz, M. W. (2005). Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. Am. J. Physiol. Endocrinol. Metab. 289, E1051–E1057.
Morton, G. J., Gelling, R. W., Niswender, K. D., Morrison, C. D., Rhodes, C. J., and Schwartz, M. W. (2005). Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2, 411–420.
Munir, I., Yen, H. W., Geller, D. H., Torbati, D., Bierden, R. M., Weitsman, S. R., Agarwal, S. K., and Magoffin, D. A. (2004). Insulin augmentation of 17alpha-hydroxylase activity is mediated by phosphatidyl inositol 3-kinase but not extracellular signal-regulated kinase-1/2 in human ovarian theca cells. Endocrinology 145, 175–183.
Munzberg, H., Huo, L., Nillni, E. A., Hollenberg, A. N., and Bjørbaek, C. (2003). Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology 144, 2121–2131.
Murray, J. F., Hahn, J. D., Kennedy, A. R., Small, C. J., Bloom, S. R., Haskell-Luevano, C., Coen, C. W., and Wilson, C. A. (2006). Evidence for a stimulatory action of melanin-concentrating hormone on luteinising hormone release involving MCH1 and melanocortin-5 receptors. J. Neuroendocrinol. 18, 157–167.
Nagatani, S., Bucholtz, D. C., Murahashi, K., Estacio, M. A., Tsukamura, H., Foster, D. L., and Maeda, K. I. (1996). Reduction of glucose availability suppresses pulsatile luteinizing hormone release in female and male rats. Endocrinology 137, 1166–1170.
Nagatani, S., Guthikonda, P., Thompson, R. C., Tsukamura, H., Maeda, K. I., and Foster, D. L. (1998). Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology 67, 370–376.
Nakae, J., Kido, Y., and Accili, D. (2001). Distinct and overlapping functions of insulin and IGF-I receptors. Endocr. Rev. 22, 818–835.
Nandi, A., Wang, X., Accili, D., and Wolgemuth, D. J. (2010). The effect of insulin signaling on female reproductive function independent of adiposity and hyperglycemia. Endocrinology 151, 1863–1871.
Navarro, V. M., Fernandez-Fernandez, R., Castellano, J. M., Roa, J., Mayen, A., Barreiro, M. L., Gaytan, F., Aguilar, E., Pinilla, L., Dieguez, C., and Tena-Sempere, M. (2004). Advanced vaginal opening and precocious activation of the reproductive axis by KiSS-1 peptide, the endogenous ligand of GPR54. J. Physiol. (Lond.) 561(Pt 2), 379–386.
Nestler, J. E. (2008). Metformin for the treatment of the polycystic ovary syndrome. N. Engl. J. Med. 358, 47–54.
Nestler, J. E., Jakubowicz, D. J., de Vargas, A. F., Brik, C., Quintero, N., and Medina, F. (1998). Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J. Clin. Endocrinol. Metab. 83, 2001–2005.
Niswender, K. D., Morrison, C. D., Clegg, D. J., Olson, R., Baskin, D. G., Myers, M. G. Jr., Seeley, R. J., and Schwartz, M. W. (2003). Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 52, 227–231.
Orio, F., Manguso, F., Di Biase, S., Falbo, A., Giallauria, F., Labella, D., Tolino, A., Lombardi, G., Colao, A., and Palomba, S. (2007). Metformin administration improves leukocyte count in women with polycystic ovary syndrome: a 6-month prospective study. Eur. J. Endocrinol. 157, 69–73.
Panno, M. L., Mauro, L., Marsico, S., Bellizzi, D., Rizza, P., Morelli, C., Salerno, M., Giordano, F., and Andò, S. (2006). Evidence that the mouse insulin receptor substrate-1 belongs to the gene family on which the promoter is activated by estrogen receptor alpha through its interaction with Sp1. J. Mol. Endocrinol. 36, 91–105.
Pasapera Limon, A. M., Herrera-Munoz, J., Gutiérrez-Sagal, R., and Ulloa-Aguirre, A. (2003). The phosphatidylinositol 3-kinase inhibitor LY294002 binds the estrogen receptor and inhibits 17beta-estradiol-induced transcriptional activity of an estrogen sensitive reporter gene. Mol. Cell. Endocrinol. 200, 199–202.
Pasquali, R., Patton, L., and Gambineri, A. (2007). Obesity and infertility. Curr. Opin. Endocrinol. Diabetes Obes. 14, 482–487.
Pitteloud, N., Hardin, M., Dwyer, A. A., Valassi, E., Yialamas, M., Elahi, D., and Hayes, F. J. (2005). Increasing insulin resistance is associated with a decrease in Leydig cell testosterone secretion in men. J. Clin. Endocrinol. Metab. 90, 2636–2641.
Plum, L., Ma, X., Hampel, B., Balthasar, N., Coppari, R., Münzberg, H., Shanabrough, M., Burdakov, D., Rother, E., Janoschek, R., Alber, J., Belgardt, B. F., Koch, L., Seibler, J., Schwenk, F., Fekete, C., Suzuki, A., Mak, T. W., Krone, W., Horvath, T. L., Ashcroft, F. M., and Brüning, J. C. (2006). Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Invest. 116, 1886–1901.
Poyastro Pinheiro, A., Thornton, L. M., Plotonicov, K. H., Tozzi, F., Klump, K. L., Berrettini, W. H., Brandt, H., Crawford, S., Crow, S., Fichter, M. M., Goldman, D., Halmi, K. A., Johnson, C., Kaplan, A. S., Keel, P., LaVia, M., Mitchell, J., Rotondo, A., Strober, M., Treasure, J., Woodside, D. B., Von Holle, A., Hamer, R., Kaye, W. H., and Bulik, C. M. (2007). Patterns of menstrual disturbance in eating disorders. Int. J. Eat. Disord. 40, 424–434.
Quennell, J. H., Howell, C. S., Roa, J., Augustine, R. A., Grattan, D. R., and Anderson, G. M. (2011). Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology 152, 1541–1550.
Quennell, J. H., Mulligan, A. C., Tups, A., Liu, X., Phipps, S. J., Kemp, C. J., Herbison, A. E., Grattan, D. R., and Anderson, G. M. (2009). Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology 150, 2805–2812.
Quesada, A., and Etgen, A. M. (2002). Functional interactions between estrogen and insulin-like growth factor-I in the regulation of alpha 1B-adrenoceptors and female reproductive function. J. Neurosci. 22, 2401–2408.
Repaci, A., Gambineri, A., and Pasquali, R. (2011). The role of low-grade inflammation in the polycystic ovary syndrome. Mol. Cell. Endocrinol. 335, 30–41.
Rich, N., Reyes, P., Reap, L., Goswami, R., and Fraley, G. S. (2007). Sex differences in the effect of prepubertal GALP infusion on growth, metabolism and LH secretion. Physiol. Behav. 92, 814–823.
Richards, R. G., DiAugustine, R. P., Petrusz, P., Clark, G. C., and Sebastian, J. (1996). Estradiol stimulates tyrosine phosphorylation of the insulin-like growth factor-1 receptor and insulin receptor substrate-1 in the uterus. Proc. Natl. Acad. Sci. U.S.A. 93, 12002–12007.
Ring, L. E., and Zeltser, L. M. (2010). Disruption of hypothalamic leptin signaling in mice leads to early-onset obesity, but physiological adaptations in mature animals stabilize adiposity levels. J. Clin. Invest. 120, 2931–2941.
Roa, J., Castellano, J. M., Navarro, V. M., Handelsman, D. J., Pinilla, L., and Tena-Sempere, M. (2009a). Kisspeptins and the control of gonadotropin secretion in male and female rodents. Peptides 30, 57–66.
Roa, J., Garcia-Galiano, D., Varela, L., Sánchez-Garrido, M. A., Pineda, R., Castellano, J. M., Ruiz-Pino, F., Romero, M., Aguilar, E., López, M., Gaytan, F., Diéguez, C., Pinilla, L., and Tena-Sempere, M. (2009b). The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology 150, 5016–5026.
Roby, K. F., and Terranova, P. F. (1990). Effects of tumor necrosis factor-alpha in vitro on steroidogenesis of healthy and atretic follicles of the rat: theca as a target. Endocrinology 126, 2711–2718.
Roepke, T. A., Ronnekleiv, O. K., and Kelly, M. J. (2011). Physiological consequences of membrane-initiated estrogen signaling in the brain. Front. Biosci. 16, 1560–1573.
Rojdmark, S. (1987). Increased gonadotropin responsiveness to gonadotropin-releasing hormone during fasting in normal subjects. Metab. Clin. Exp. 36, 21–26.
Roseweir, A. K., and Millar, R. P. (2009). The role of kisspeptin in the control of gonadotrophin secretion. Hum. Reprod. Update 15, 203–212.
Sajapitak, S., Iwata, K., Shahab, M., Uenoyama, Y., Yamada, S., Kinoshita, M., Bari, F. Y., I’Anson, H., Tsukamura, H., and Maeda, K. (2008). Central lipoprivation-induced suppression of luteinizing hormone pulses is mediated by paraventricular catecholaminergic inputs in female rats. Endocrinology 149, 3016–3024.
Schwarz, J. M., Liang, S. L., Thompson, S. M., and McCarthy, M. M. (2008). Estradiol induces hypothalamic dendritic spines by enhancing glutamate release: a mechanism for organizational sex differences. Neuron 58, 584–598.
Shahab, M., Sajapitak, S., Tsukamura, H., Kinoshita, M., Matsuyama, S., Ohkura, S., Yamada, S., Uenoyama, Y., I’Anson, H., and Maeda, K. (2006). Acute lipoprivation suppresses pulsatile luteinizing hormone secretion without affecting food intake in female rats. J. Reprod. Dev. 52, 763–772.
Sharifi, N., Reuss, A. E., and Wray, S. (2002). Prenatal LHRH neurons in nasal explant cultures express estrogen receptor beta transcript. Endocrinology 143, 2503–2507.
Simoncini, T., Hafezi-Moghadam, A., Brazil, D. P., Ley, K., Chin, W. W., and Liao, J. K. (2000). Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541.
Small, C. J., Goubillon, M. L., Murray, J. F., Siddiqui, A., Grimshaw, S. E., Young, H., Sivanesan, V., Kalamatianos, T., Kennedy, A. R., Coen, C. W., Bloom, S. R., and Wilson, C. A. (2003). Central orexin A has site-specific effects on luteinizing hormone release in female rats. Endocrinology 144, 3225–3236.
Spaczynski, R. Z., Arici, A., and Duleba, A. J. (1999). Tumor necrosis factor-alpha stimulates proliferation of rat ovarian theca-interstitial cells. Biol. Reprod. 61, 993–998.
Stanley, S. A., Small, C. J., Kim, M. S., Heath, M. M., Seal, L. J., Russell, S. H., Ghatei, M. A., and Bloom, S. R. (1999). Agouti related peptide (Agrp) stimulates the hypothalamo pituitary gonadal axis in vivo and in vitro in male rats. Endocrinology 140, 5459–5462.
Takatsu, Y., Matsumoto, H., Ohtaki, T., Kumano, S., Kitada, C., Onda, H., Nishimura, O., and Fujino, M. (2001). Distribution of galanin-like peptide in the rat brain. Endocrinology 142, 1626–1634.
Takenoya, F., Guan, J.-L., Kato, M., Sakuma, Y., Kintaka, Y., Kitamura, Y., Kitamura, S., Okuda, H., Takeuchi, M., Kageyama, H., and Shioda, S. (2006). Neural interaction between galanin-like peptide (GALP)-and luteinizing hormone-releasing hormone (LHRH)-containing neurons. Peptides 27, 2885–2893.
Tamura, T., Irahara, M., Tezuka, M., Kiyokawa, M., and Aono, T. (1999). Orexins, orexigenic hypothalamic neuropeptides, suppress the pulsatile secretion of luteinizing hormone in ovariectomized female rats. Biochem. Biophys. Res. Commun. 264, 759–762.
Thaler, J. P., Choi, S. J., Schwartz, M. W., and Wisse, B. E. (2010). Hypothalamic inflammation and energy homeostasis: resolving the paradox. Front. Neuroendocrinol. 31, 79–84.
Thaler, J. P., and Schwartz, M. W. (2010). Minireview: inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology 151, 4109–4115.
Tortoriello, D. V., McMinn, J., and Chua, S. C. (2004). Dietary-induced obesity and hypothalamic infertility in female DBA/2J mice. Endocrinology 145, 1238–1247.
Tsukamura, H., Thompson, R. C., Tsukahara, S., Ohkura, S., Maekawa, F., Moriyama, R., Niwa, Y., Foster, D. L., and Maeda, K. (2000). Intracerebroventricular administration of melanin-concentrating hormone suppresses pulsatile luteinizing hormone release in the female rat. J. Neuroendocrinol. 12, 529–534.
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., and Hotamisligil, G. S. (1997). Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614.
Vanhaesebroeck, B., Ali, K., Bilancio, A., Geering, B., and Foukas, L. C. (2005). Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 30, 194–204.
Vassen, L., Wegrzyn, W., and Klein-Hitpass, L. (1999). Human insulin receptor substrate-2 (IRS-2) is a primary progesterone response gene. Mol. Endocrinol. 13, 485–494.
Vulliemoz, N. R., Xiao, E., Xia-Zhang, L., Wardlaw, S. L., and Ferin, M. (2005). Central infusion of agouti-related peptide suppresses pulsatile luteinizing hormone release in the ovariectomized rhesus monkey. Endocrinology 146, 784–789.
White, M. F. (1998). The IRS-signaling system: a network of docking proteins that mediate insulin and cytokine action. Recent Prog. Horm. Res. 53, 119–138.
White, M. F. (2002). IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283, E413–E422.
Williamson-Hughes, P. S., Grove, K. L., and Smith, M. S. (2005). Melanin concentrating hormone (MCH): a novel neural pathway for regulation of GnRH neurons. Brain Res. 1041, 117–124.
Wu, M., Dumalska, I., Morozova, E., van den Pol, A., and Alreja, M. (2009). Melanin-concentrating hormone directly inhibits GnRH neurons and blocks kisspeptin activation, linking energy balance to reproduction. Proc. Natl. Acad. Sci. U.S.A. 106, 17217–17222.
Xu, A. W., Kaelin, C. B., Takeda, K., Akira, S., Schwartz, M. W., and Barsh, G. S. (2005). PI3K integrates the action of insulin and leptin on hypothalamic neurons. J. Clin. Invest. 115, 951–958.
Zhang, C., Bosch, M. A., Levine, J. E., Rønnekleiv, O. K., and Kelly, M. J. (2007a). Gonadotropin-releasing hormone neurons express K(ATP) channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J. Neurosci. 27, 10153–10164.
Keywords: PI3K, LH, GnRH, obesity, insulin, leptin, puberty, metabolism
Citation: Acosta-Martínez M (2012) PI3K: an attractive candidate for the central integration of metabolism and reproduction. Front. Endocrin. 2:110. doi: 10.3389/fendo.2011.00110
Received: 16 September 2011;
Accepted: 13 December 2011;
Published online: 24 January 2012.
Edited by:
Jennifer Wootton Hill, University of Toledo College of Medicine, USAReviewed by:
Raul Miguel Luque Huertas, University of Cordoba, SpainCsaba Fekete, Institute of Experimental Medicine, Hungary
Ji Luo, National Institute of Health, USA
Copyright: © 2012 Acosta-Martínez. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Maricedes Acosta-Martínez, Department of Physiology and Biophysics, Medical Center, Stony Brook University, Stony Brook, NY 11794-8661, USA. e-mail: maricedes.acosta@stonybrook.edu