
- 1Department of Internal Medicine, Division of Diabetes, Metabolism, and Endocrinology, Graduate School of Medicine, Kobe University, Kobe, Japan
- 2Department of Medicine, University of Washington, Seattle, WA, USA
- 3Diabetes and Obesity Center of Excellence, University of Washington, Seattle, WA, USA
- 4Department of Internal Medicine, Diabetes Center, Chibune Hospital, Osaka, Japan
It has been demonstrated in rodents and humans that chronic inflammation characterized by macrophage infiltration occurs mainly in adipose tissue or liver during obesity, in which activation of immune cells is closely associated with insulin sensitivity. Macrophages can be classified as classically activated (M1) macrophages that support microbicidal activity or alternatively activated (M2) macrophages that support allergic and antiparasitic responses. In the context of insulin action, M2 macrophages sustain insulin sensitivity by secreting IL-4 and IL-10, while M1 macrophages induce insulin resistance through the secretion of proinflammatory cytokines, such as TNFα. Polarization of M1/M2 is controlled by various dynamic functions of other immune cells. It has been demonstrated that, in a lean state, TH2 cells, Treg cells, natural killer T cells, or eosinophils contribute to the M2 activation of macrophages by secreting IL-4 or IL-10. In contrast, obesity causes alteration of the constituent immune cells, in which TH1 cells, B cells, neutrophils, or mast cells induce M1 activation of macrophages by the elevated secretion of TNFα and IFNγ. Increased secretion of TNFα and free fatty acids from hypertrophied adipocytes also contributes to the M1 activation of macrophages. Since obesity-induced insulin resistance is established by macrophage infiltration and the activation of immune cells inside tissues, identification of the factors that regulate accumulation and the intracellular signaling cascades that define polarization of M1/M2 would be indispensable. Regulation of these factors would lead to the pharmacological inhibition of obesity-induced insulin resistance. In this review, we introduce molecular mechanisms relevant to the pathophysiology and review the most recent studies of clinical applications targeting chronic inflammation.
Introduction
Obesity develops as a consequence of nutritional excess and insufficient exercise; it causes major adverse health outcomes such as type 2 diabetes, cardiovascular diseases, dyslipidemia, chronic kidney diseases, and cancers, which are serious problems worldwide. These pathological states are strongly associated with insulin resistance or hyperinsulinemia. On the basis of efforts over the last two decades, there have been remarkable developments in the investigation of obesity-induced insulin resistance, especially in terms of the mechanisms involved, some of which are expected to lead to treatments of the disease. Among these, low-grade chronic inflammation in obesity is one of the most innovative and newly identified concepts. The metabolic pathway and the immune response pathway, which are strongly evolutionarily conserved among species, have been found to be strongly associated with each other in the development of obesity-induced insulin resistance. In this review, we look back over the initial findings in the research field of inflammation and insulin resistance and discuss recent studies, including those on clinical applications.
Obesity-Induced Chronic Inflammation in Adipose Tissue and Adipokine Secretion
Low-grade chronic inflammation was found to be closely associated with obesity-related metabolic diseases. This association between obesity/type 2 diabetes and inflammation can be traced back to case reports published over a century ago, showing that high-dose sodium salicylate could diminish glycosuria in older diabetic patients (1, 2). Thereafter, several studies also showed that acetylsalicylic acid or sodium salicylate reduced the glucose level and improved glucose tolerance in diabetic patients (3, 4). These reports again drew attention in 1993 with the publication of a report demonstrating in mice that the expression of TNFα in adipose tissue was increased during the development of obesity, while conversely the neutralization of TNFα attenuated insulin resistance (5). Subsequently, the same research group demonstrated that TNFα suppressed insulin signaling by inhibiting insulin receptor tyrosine kinase activity (6) and proposed a model in which inflammation defined as an increased level of TNFα in adipose tissue could be the basis of systemic insulin resistance. Concurrently with these findings, leptin was identified as a secretory bioactive molecule from adipocytes, which regulates food intake and energy expenditure through the hypothalamus (7). This led to the establishment of an innovative concept in which adipose tissue not only simply stores excess energy as triacylglycerol but is also an organ that secretes the biologically active substances referred to as adipokines. Adipokines could directly regulate the insulin sensitivity of remote insulin-sensitive organs including liver and skeletal muscle through the circulation. Deregulated adipokine secretion from the expanded adipose tissue of obese individuals was shown to contribute to the development of systemic insulin resistance and metabolic diseases. Following the discovery of leptin (7), a number of adipokines have been identified; these include IL-6 (8, 9), resistin (10), retinol-binding protein 4 (RBP-4) (11), omentin (12), chemerin (13–15), progranulin (16), and monocyte chemoattractant protein-1 (MCP-1) (17–19). The proinflammatory cytokine TNFα, produced mainly by macrophages that have infiltrated into adipose tissue, can also be considered as an adipokine (5, 20). Given that TNFα activates proinflammatory signal cascades as well as inhibits insulin receptor signaling, this molecule is thought to be a major player linking adipose tissue inflammation and insulin resistance (21, 22). In contrast, in a lean state, a certain level of “healthy” adipokines contributes to insulin sensitivity and adequate glucose homeostasis. For instance, adiponectin is considered a “healthy” adipokine. Adiponectin-deficient mice exhibited insulin resistance (23, 24) along with increased expression of TNFα in adipose tissue (23). Chronic inflammation, especially in adipose tissue, causes impairment of adipokine secretion, leading to systemic insulin resistance. Thus, adipose tissue inflammation and adipokine secretion are strongly associated with each other and coordinately contribute to insulin resistance in obesity.
Macrophage Accumulation in Adipose Tissue
The mechanisms by which TNFα is increased during obesity were unclear until the findings published in 2003 that chronic inflammation observed in rodents and humans was characterized by the accumulation of macrophages into adipose tissue (21, 22). In general, macrophages differentiate in tissue from recruited monocytes and function in innate immunity during host defense. However, these studies demonstrated that macrophages existed even in a lean state, but expanded their populations during the development of obesity in mice and humans (21, 22). It is now considered that macrophages defined as F4/80+ CD11b+ are resident in lean adipose tissue, representing 5% of the stromal vascular fraction (17, 25), but are increased by obesity up to 14–30% (17, 18, 25). In healthy subjects, adipose tissue macrophages show dynamic diversity. Kosteli et al. showed that, although chronic weight loss reduced the macrophage content in adipose tissue, fasting or acute weight loss in turn elicited their accumulation (26). Such conditions seemed to enhance the lipolysis that caused elevation of local free fatty acid (FFA), which induced macrophage accumulation. Infiltrated macrophages incorporate lipids, which act to suppress lipolysis. These findings provide evidence that, not only in a pathological state, but also in physiological circumstances, macrophages in adipose tissue play dynamic roles in the maintenance of homeostasis.
The Role of Chemokines in Adipose Tissue Inflammation and Insulin Resistance
Chemokines are a family of low-molecular-weight proteins with an essential role in leukocyte trafficking during both homeostasis and inflammation. On the basis of their molecular structure, chemokines are divided into two major subgroups: CC chemokine ligand (CCL) and CXC chemokine ligand (CXCL), which bind to CC chemokine receptor (CCR) or CXC chemokine receptor (CXCR), respectively (27). Intriguingly, MCP-1 (also known as CCL2), a representative CC chemokine, was found to be remarkably increased in adipose tissue in obesity (21, 22, 28). We and others sought to investigate whether MCP-1 is a factor that enhances the infiltration of macrophages in adipose tissue. Adipose tissue-specific overexpression of MCP-1 in mice indeed increased macrophage infiltration into adipose tissue and insulin resistance (17, 19), whereas disruption of MCP-1 or its receptor, CCR2, impaired high-fat diet (HFD)-induced migration of macrophages into adipose tissue, thereby reducing adipose tissue inflammation and attenuating insulin resistance (17, 18, 29, 30). These findings suggest that MCP-1 secreted from enlarged adipocytes attracted circulating monocytes to adipose tissue, causing inflammatory characteristics of adipose tissue. Infiltrated monocytes differentiate into macrophages and produce additional inflammatory cytokines, leading to further inflammation. Secreted inflammatory cytokines are supposed to induce insulin resistance in liver and skeletal muscle by functioning as adipokines (Figure 1). In addition, chronic increase in the circulating level of MCP-1 by the administration of recombinant MCP-1 protein induced insulin resistance, macrophage infiltration into adipose tissue, and an increase in hepatic triacylglycerol content without affecting body weight (18). Acute increase in the circulating MCP-1 concentration also induced insulin resistance but not macrophage infiltration into adipose tissue. These findings indicate that an increase in the concentration of MCP-1 in the circulation is sufficient to induce systemic insulin resistance irrespective of adipose tissue inflammation (18). In fact, circulating MCP-1 levels were found to be increased in type 2 diabetic patients compared with normal subjects (31, 32) or to be correlated with HOMA-IR in type 2 diabetic patients (33). On the other hand, studies by others found no difference or even more infiltrated macrophages in adipose tissues in MCP-1-deficient mice, although the reason for the different results is unknown (34, 35). Recent study by Oh et al. provided evidence by employing a new method for quantitative in vivo macrophage tracking, in which monocytes isolated from peripheral blood were labeled ex vivo with fluorescent PKH26 dye and then injected into recipient mice (36). Mice receiving CCR2-deficient monocytes were protected from HFD-induced accumulation of macrophages in adipose tissue and the liver, while transplantation of intact monocytes into MCP-1 knockout mice on an HFD did not cause infiltration of macrophages into the tissues (36). These results all suggest that the MCP-1-CCR2 signaling pathway plays an important role in adipose tissue inflammation (17–19, 29, 30, 36), hepatic steatosis (17, 18, 37, 38), and glucose metabolism (17–19, 29, 30, 36–38) in insulin-resistant model mice. Thus, examination of the factors that induce MCP-1 expression in hypertrophied adipocytes is also important. Ito et al. demonstrated that down-regulation of mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1) increased MCP-1 expression through MAPK activation in cultured adipocytes (39). Furthermore, Kitade et al. demonstrated that the expression of CCR5 in adipose tissue was similarly increased during obesity (40). Genetic deletion of CCR5 in mice resulted in protection against HFD-induced macrophage infiltration, insulin resistance, and hepatic steatosis. Furthermore, alteration of macrophages in adipose tissues was accompanied by polarization to M2. These results were reproduced using a cell-specific approach by employing bone marrow transplantation (40). At present, it is believed that M2 macrophages contribute to maintain insulin sensitivity, while obesity causes a switch to M1 polarization that enhances systemic insulin resistance through the secretion of inflammatory cytokines (41). Subsequently, the contributions of chemokines other than the CCL family, such as CXCL14 (42) or other factors including osteopontin (43), angiopoietin-like protein 2 (Angptl2) (44), serum amyloid A (45), and dietary cholesterol (46), to the accumulation of macrophages in adipose tissue have been demonstrated.
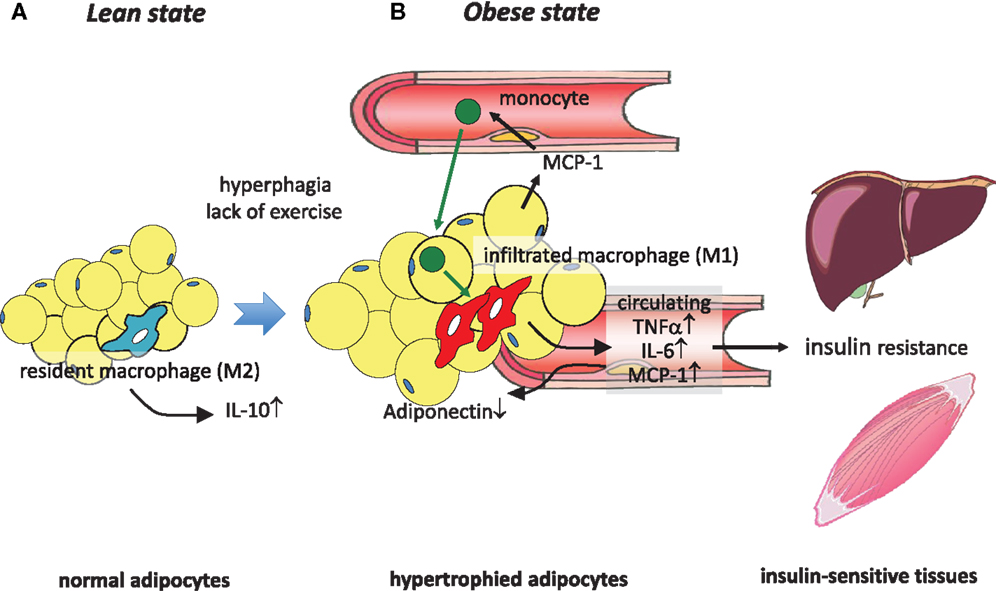
Figure 1. Obesity-induced macrophage infiltration into adipose tissue causes insulin resistance. (A) In adipose tissue in a lean state, most resident macrophages are M2 macrophages that contribute to insulin sensitivity by secreting IL-10. (B) Hyperphagia and lack of exercise cause hypertrophy of adipocytes, which induces MCP-1 secretion to the circulation, leading to the recruitment of circulating monocytes to adipose tissues. These infiltrated monocytes differentiate into activated M1 macrophages, which robustly secrete proinflammatory cytokines such as TNFα, IL-6, and MCP-1, thus contributing to low-grade inflammation in adipose tissue and a decrease of adiponectin. At the same time, these secreted cytokines cause insulin resistance in liver and skeletal muscle by acting as insulin resistance-inducing adipokines.
Inflammatory Activation of Myeloid Cells in the Liver
Following the findings for adipose tissue, the issues of whether obesity can cause hepatic inflammation and whether this inflammation can contribute to hepatic or systemic inflammation became important in this field. Obesity-associated nutrient excess has been linked to inflammation in part via activation of inhibitor of κB kinase β (IKKβ) and subsequent nuclear translocation of nuclear factor κB (NF-κB), one of the key transcriptional mediators of inflammation (47–49). Consumption of an HFD clearly induced proinflammatory activation of Kupffer cells, the resident macrophages of the liver, in mice (50, 51). In addition, inflammatory activation of Kupffer cells was implicated in the pathogenesis of obesity-induced insulin resistance and fatty liver disease (50). Deletion of IKKβ in myeloid cells reduced macrophage-mediated inflammation and improved obesity-associated systemic and hepatic insulin sensitivity (47). Furthermore, chemical deletion of Kupffer cells was demonstrated to cause improved insulin sensitivity during HFD feeding (52). Obesity and insulin resistance are often associated with hepatic steatosis in a large proportion of obese patients. We demonstrated mechanically that overexpression of MCP-1 in adipose tissue caused hepatic steatosis along with adipose tissue inflammation, while systemic deletion of MCP-1 inhibited HFD-induced steatosis (17). In addition, chronic increase of plasma MCP-1 level was also sufficient to induce hepatic steatosis and adipose tissue inflammation (18). These findings suggest that an increase of circulating MCP-1 or adipose tissue inflammation may cause hepatic steatosis. Although HFD feeding caused M1 activation of Kupffer cells in the liver (50, 51), it seemed that the number of Kupffer cells was not increased in obesity (53). Using flow cytometry, it was investigated how a population of myeloid cells (CD11b+) changed during obesity or type 2 diabetes. Kupffer cells, defined as CD45+, F4/80+, were a major subset of myeloid cells in the liver. Obesity rather reduced the number of Kupffer cells, while in turn, the proportion of myeloid cells, defined as CD11b+, CD45+, F4/80low, doubled, from 10.0 to 19.7% (53). Given that these recruited myeloid cells were also characterized by CCR2+, hepatic expression of CCL2/CCR2, which was increased by HFD, seemed to have originated from infiltrated macrophages. By employing bone marrow transplantation from CCR2-deficient mice, it was further demonstrated that the trafficking of the infiltrated cells was dependent on CCR2. In addition, adenoviral overexpression of CCL2 in the liver caused the accumulation of myeloid cells coincident with hepatic steatosis (53). CCR2-dependent recruitment of myeloid cells to the liver (36) and CCL2-dependent development of hepatic steatosis (54) were also demonstrated by other studies. These results also underline the role of the CCL2-CCR2 signaling pathway in the recruitment of myeloid cells to the liver. Taking these findings together, the range of immune cells in the liver is thus complex and heterogeneous, but they are thought to play important roles in both insulin resistance and hepatic steatosis.
Regulation of Kupffer Cell Activation by Endothelial NO Production
Local and systemic insulin resistance has been discussed in relation to the interactions between immune cells and parenchymal cells. We have proposed that endothelial cells could be added to those components with which interactions are shown. We have demonstrated that HFD feeding induced proinflammatory activation of Kupffer cells in wild-type (WT) mice coincident with reduced liver endothelial nitric oxide synthase activity and nitric oxide (NO) content while, conversely, the enhancement of cGMP signaling downstream of endogenous NO by phosphodiesterase-5 inhibition protected Kupffer cells against HFD-induced inflammation (51). Furthermore, proinflammatory activation of Kupffer cells was evident in eNos−/− mice, even on a low-fat diet. Targeted deletion of vasodilator-stimulated phosphoprotein (VASP), a key downstream target of endothelially derived NO, similarly led to a predisposition to hepatic and Kupffer cell inflammation and abrogated the protective effect of NO signaling in both macrophages and hepatocytes studied in a cell culture model (51). These results collectively imply a physiological role for endothelial NO to limit obesity-associated inflammation and insulin resistance in hepatocytes and support a model in which Kupffer cell activation during HFD feeding is dependent on reduced NO signaling (51) (Figure 2). The NO/cGMP/VASP axis was also shown to be relevant in adipose tissue (55).
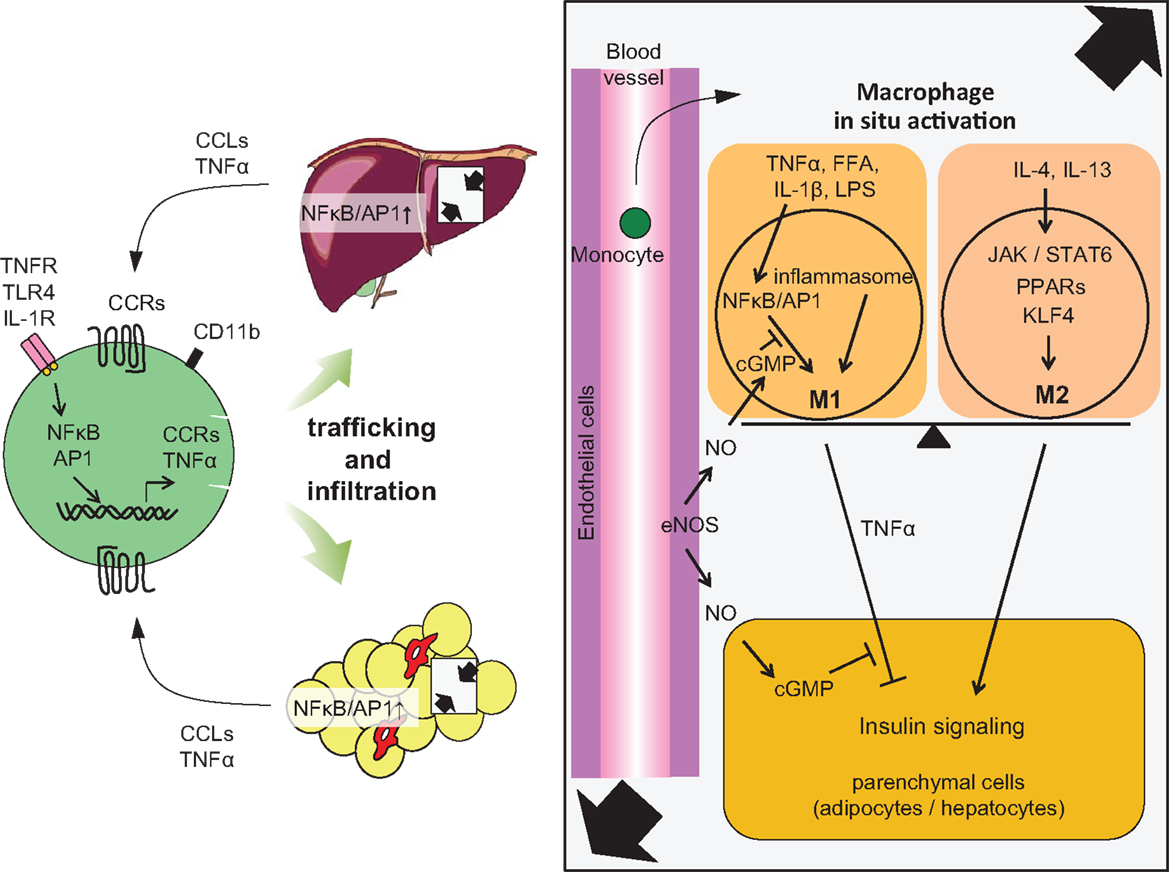
Figure 2. Accumulation of monocytes/macrophages in adipose tissue and liver, and activation in the tissues. Trafficking: During obesity, adipocytes exhibit hypertrophy, while liver incorporates substantial FFAs, both of which cause tissue inflammation, activation of NF-κB, and AP1 signaling, leading to increased secretion of inflammatory chemokines and cytokines, including CCLs and TNFα. Elevated secretion of CCLs (e.g., MCP-1) elicits the accumulation of CCR-positive monocytes to the site of inflammation, particularly CCR2+ for adipose and liver, but CCR5+ for adipose tissue. In situ activation: In a lean state, resident tissue macrophages display the M2 phenotype, which is achieved and sustained through the JAK/STAT6 pathway in response to IL-4 or IL-13 stimuli. These stimuli are derived from resident TH2 cells, Treg cells, eosinophils, and mast cells. PPARs and KLF4 also induce M2 activation. In turn, obesity and subsequent elevation of tissue FFA or inflammatory cytokines stimulate NF-κB and AP1 signaling, which causes switching of the phenotype to M1, leading to further secretion of TNFα. Signal from inflammasome also activates M1 activation. M1 activation of macrophages can be suppressed by endothelial NO/cGMP signaling. M2 macrophages contribute to insulin sensitivity in neighboring parenchymal cells, while M1 induces insulin resistance, with the M1/M2 balance determining tissue and/or systemic insulin sensitivity.
Constituent Cells Other than Macrophages in Obesity-Induced Inflammation: Interactions Among Immune Cells During Inflammation in Adipose Tissue
The role of macrophages in adipose tissue inflammation has been clearly demonstrated. Besides these cells, additional leukocyte subpopulations have recently been demonstrated to be involved in obesity and insulin resistance, such as T cells, B cells, eosinophils, neutrophils, mast cells, and natural killer cells. The involvement of multiple leukocyte subpopulations underlines the complexity of obesity-associated adipose tissue inflammation (Figure 3).
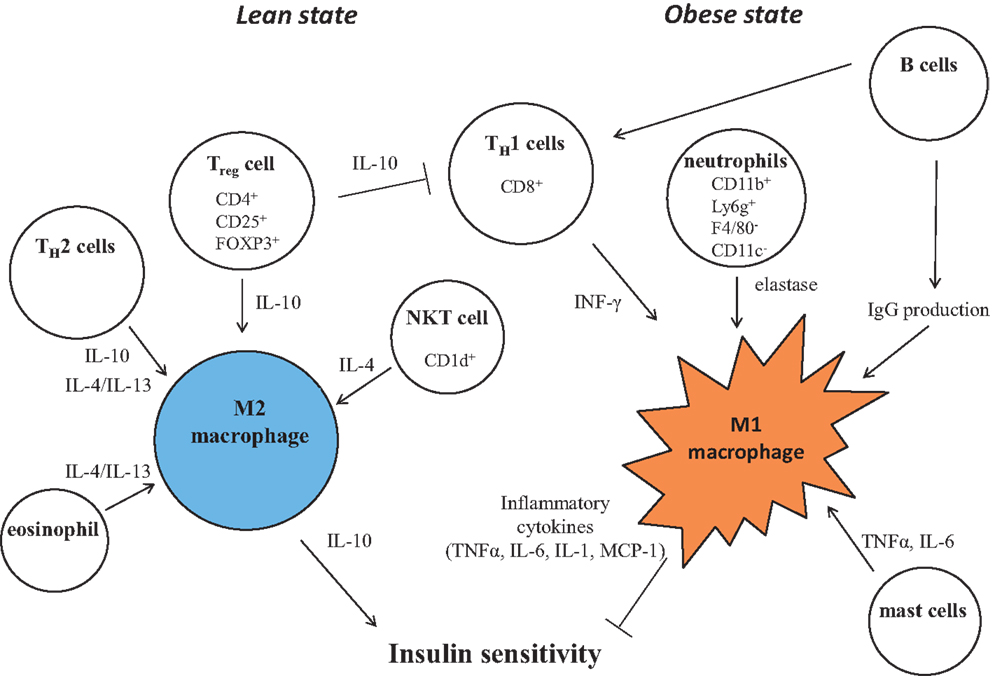
Figure 3. Control of M1/M2 polarization by neighboring immune cells. In a lean state, resident T cells consist of TH2 cells, Treg cells, and NKT cells. Combined with resident eosinophils, these cells sustain the M2 activation of macrophages through secreting IL-4, IL-10, and IL-13. As obesity progresses, alteration of constituent immune cells occurs, in which the numbers of TH2 cells and Treg cells decline, while in turn, TH1 cells and B cells increase. In addition to these more prevalent cells, neutrophils and mast cells induce M1 activation of macrophages by increased secretion of elastase, TNF, IFNγ, IL-6, and pathogenic IgG. B cells also activate T cells.
T Cells
Although macrophage infiltration in adipose tissue has been demonstrated in both mice and humans (56), little is known about the sequence of events that lead to the macrophage accumulation in adipose tissue. Research attempting to investigate which surface antigens of immune cells are associated with inflammation and insulin resistance revealed the involvement of CD11c-positive cells (57). Myeloid-specific deletion of CD11c in mice protected against HFD-induced accumulation of macrophages in adipose tissue and exhibited insulin sensitivity compared with the controls (57). Next, T cells (CD4+, CD8+) were found to be increased in adipose tissue during obesity (58–60). In a lean state, CD4+ helper T cells and regulatory T (Treg) cells (CD4+, CD25+, Foxp3+) were predominant; however, prior to the accumulation of macrophages (F4/80+, CD11b−), CD8+ T cells infiltrated coincidentally with a reduction of the number of Treg (25). The administration of CD8 antibody to WT mice fed an HFD attenuated macrophage infiltration and insulin resistance. Although CD8 knockout mice were protected against HFD-induced accumulation of macrophages, restoration of CD8+ T-cells increased macrophage infiltration. Similar results were obtained by other groups (61, 62). It is now considered that, in a lean state, CD4+ CD25+ Foxp3+ Treg cells induce alternative activation of monocyte/macrophages (63), which is characterized by the expression of macrophage mannose receptor (MMR) or intracellular activity of arginase (64). T helper type 2 (TH2) cells expressing IL-4 and IL-13 also induce M2 activation of macrophages that secrete IL-10, whereas macrophages are M1-activated through IFNγ by T helper type 1 (TH1) cells and through IL-17 by TH17 cells. Recently, peroxisome proliferator-activated receptor γ (PPARγ) activity in Treg cells has been shown to be important to reduce chronic inflammation in adipose tissue (65).
B Cells
The accumulation of B cells was observed in adipose tissue of mice fed an HFD before macrophage and T-cell accumulation (66). In addition, diet-induced obese mice lacking B cells were protected from metabolic abnormalities despite weight gain (67). B-cell effects on glucose metabolism were associated with the activation of proinflammatory macrophages and T cells and the production of pathogenic IgG antibodies. In fact, treatment of mice fed an HFD with a B-cell-depleting CD20 antibody ameliorated abnormality in glucose metabolism and adipose tissue inflammation, whereas the transfer of IgG from mice with diet-induced obesity rapidly induced insulin resistance and glucose intolerance (67). Recently, obese B-cell-null mice were reported to exhibit decreased systemic inflammation, inflammatory B- and T-cell cytokines, adipose tissue inflammation, and insulin resistance compared with obese WT mice (68). This was associated with an increased percentage of anti-inflammatory regulatory T cells. B cells from type 2 diabetes subjects promote proinflammatory T-cell function through contact-dependent mechanisms, suggesting that B cells regulate inflammation in type 2 diabetes by modulating T-cell functions (68).
Eosinophils
In addition to TH2 or Treg cells, eosinophils have been shown to exist in lean adipose tissue and participate in the maintenance of M2 activation through secreting IL-4 (69). By using eosinophil-deficient and hypereosinophilic mice, Wu et al. showed that eosinophil-derived IL-4 and IL-13 determined the M2 activation of macrophages in adipose tissue and contributed to insulin sensitivity. Hypereosinophilic mice displayed improved insulin sensitivity, while eosinophil-deficient mice exhibited increased fat together with impaired glucose tolerance and insulin resistance (69).
Neutrophils
Neutrophils are rare in a lean state; however, an HFD elicits the accumulation of neutrophils (CD11b+ Ly6g+ F4/80− CD11c−), which seem to induce local insulin resistance by secreting elastase (70). The deletion of neutrophil elastase in HFD-induced obese mice led to reduced macrophage content and inflammation. These changes were coincident with improvement of glucose tolerance and increased insulin sensitivity. Intriguingly, neutrophil elastase can degrade IRS-1 protein and cause insulin resistance in adipocytes (70). Similar results were obtained in a very recent study by another group (71). In humans, an increased blood level of myeloperoxidase, a marker of neutrophils, in obese women (72), and increased activity of neutrophils in obese subjects have also been noted (71, 73).
Mast Cells
Mast cell invasion was also detected in adipose tissue in obese mice (74). Mast cell-deficient mice (KitW-sh/W-sh mice) were protected from HFD-induced body weight gain and the increase of proinflammatory cytokines and chemokines along with the improvement of glucose metabolism and energy expenditure due to the up-regulation of UCP-1 expression in BAT (74). Similar effects were observed in the treatment of mice with a mast cell-stabilizing agent. Mast cells were supposed to promote diet-induced obesity and glucose intolerance by the production of IL-6 and IFNγ. Mast cells are also involved in obesity-induced adipose tissue inflammation and insulin resistance. Weight gain of mast-cell-deficient mice during HFD was decreased compared with that of control mice (75). Mechanistically, prostaglandin J2 (PGJ2) produced by mast cells in response to high-glucose enhanced adipocyte differentiation by PPARγ activation, leading to obesity (75).
Natural Killer T Cells
Natural killer T (NKT) cells are innate-like T lymphocytes that recognize glycolipid antigens and have been implicated in autoimmunity, microbial infection, and cancer and hence represent an important immunotherapeutic target (76). Similar to eosinophils, NKT cells have been shown to reside in lean adipose tissue, in which they contribute to sustain the M2 activation of macrophages by stimulating IL-4/STAT6 signaling (77, 78). Schipper et al. demonstrated that CD1d-null mice whose NKT cells were not activated displayed a distinctive insulin resistance phenotype even on a low-fat diet without overt adipose tissue inflammation (79). Activation of NKT cells has thus been demonstrated to modulate polarization toward M2, resulting in improved glucose metabolism (78–80). Unlike in mouse studies, the role of NKT cells during obesity and adipose tissue inflammation in humans remains unclear. An unaltered number of circulating NKT cells in obesity (80, 81) and significantly lower numbers of circulating NKT cells in obese patients have been documented (82).
Cell Signaling in Macrophages that Defines M1 and M2 Activation
Macrophages are terminally differentiated cells of the mononuclear phagocyte system that include dendritic cells, circulating blood monocytes, and committed myeloid progenitor cells in the bone marrow. Local environmental factors are known to affect the properties, functions, and activation state of macrophages. In general, macrophage activation is defined across two separate polarization states, M1 (proinflammatory) and M2 (anti-inflammatory) states. M1 or “classically activated” macrophages are induced by proinflammatory mediators such as lipopolysaccharide (LPS), TNFα, and IFN-γ. M1 macrophages are also associated with enhanced proinflammatory cytokine production (TNFα, IL-6, IL-1). On the other hand, M2 or “alternatively activated” macrophages have low proinflammatory characteristics and instead generate high levels of anti-inflammatory cytokines, for example, IL-10. Since the attenuation of macrophage M1 activation and the maintenance of M2 activity are believed to be important for intact glucose metabolism, there has been research focusing on intracellular signaling that determines proinflammatory or alternative activation in macrophages (Figures 2 and 3).
M1 Macrophages
M1 activation of macrophages is established mainly through the IKKβ/NF-κB and Jun N-terminal kinase (JNK) 1/activator protein 1(AP1) system. Obesity induces adipose tissue inflammation, which results in high levels of proinflammatory cytokines and chemokines. In particular, TNFα is a representative inflammatory cytokine that causes lipolysis in adipose tissue. Thereby, plasma FFA levels are usually elevated in obesity. FFAs released from adipocytes through lipolysis have been shown to be capable of serving as ligands for the toll-like receptor 4 (TLR4) complex (83). TLRs are initially indispensable for innate immune cells to recognize intruding pathogens and trigger an appropriate immune response. Among them, TLR4 is a high-affinity receptor for LPS, which is a component of the cell walls of gram-negative bacteria (84). TLR4 signaling activated by FFA induces the expression of a large number of proinflammatory target genes and drives M1 activation by regulating the transcriptional factors including NF-κB, AP1, and interferon-regulatory factor (IRF) family members. TNFα also drives M1 activation by inducing proinflammatory genes through activating NF-κB and AP1 transcriptional factors. For instance, lipid infusion caused the accumulation of macrophages in adipose tissue accompanied by insulin resistance in WT control mice, but this was not the case in TLR4-deficient mice (83). Hematopoietic cell-specific deletion of TLR4 in mice attenuated HFD-induced insulin resistance in adipose and the liver (85). Activated TLR4 signaling induced a classical inflammatory response, which led to the recruitment of macrophages. In this way, macrophages activated to M1 by FFA through TLR4-mediated signaling secrete TNFα, which in turn enhances lipolysis in neighboring adipocytes, leading to further production of FFA. This vicious cycle or paracrine loop mediated by TNFα and FFA between adipocytes and macrophages in obese adipose tissue induces further adipose tissue inflammation (86). In addition, TNFα and FFA inhibit insulin receptor signaling via the increase of serine phosphorylation of IRS-1. Recently, a liver secretory glycoprotein, fetuin-A, was demonstrated to play a crucial role as an endogenous ligand for TLR4 in FFA-induced inflammation and insulin resistance in adipocytes (87). The serum concentration of fetuin-A was significantly increased in obese diabetic patients compared with that in non-obese non-diabetic human subjects. Next, myeloid differentiation primary response protein 88 (MYD88), the primary mediator of TLR and IL1 receptor signaling, has been investigated to clarify whether this is also involved in the FFA-induced insulin resistance. However, MyD88 deficiency in mice exacerbated diet-induced glucose intolerance and hyperlipidemia (88). There is therefore a conflict regarding the activity of the TLR4/MyD88 axis in diet-induced obesity and insulin resistance, which remains to be elucidated in future studies.
M2 Macrophages
The activation of M2 macrophages is basically maintained by the signaling of the IL-4/JAK/STAT6 pathway. The administration of IL-4 to mice induces M2 activation of macrophages, thereby attenuating HFD-induced insulin resistance (89). IL-10 secreted by M2 macrophages enhances insulin signaling, including that in the liver, thereby having a protective role against obesity-induced insulin resistance (90). Taking these findings together, the activation of IL-4 signaling is considered to be a promising target to suppress insulin resistance and thus studies to identify molecular mediators are underway. We describe here several factors involved in M2 activation.
Peroxisome proliferator-activated receptor γ
Macrophage-specific deletion of PPARγ in mice impaired M2 activation despite the mice being on a chow diet (91). In these mice, adiponectin expression was decreased. This change was accompanied by reduced oxidative phosphorylation in liver and skeletal muscle, which might have contributed to the insulin resistance in these tissues. Another study demonstrated that macrophage-specific PPARγ-deficient mice showed glucose intolerance and insulin resistance in a lean state. These mice had increased inflammatory markers in adipose tissue, liver, and skeletal muscle and showed decreased effects of thiazolidinediones, indicating a requirement for PPARγ in macrophages for intact insulin sensitivity in muscle/liver and a full antidiabetic effect of thiazolidinediones (92). Odegaard et al. demonstrated that peroxisome proliferator-activated receptor δ (PPARδ) mediated the effects of a Th2 cytokine, IL-4, to direct the expression of the alternative phenotype in Kupffer cells and adipose tissue macrophages of lean mice (50). Adoptive transfer of PPARδ−/− bone marrow into WT mice conversely diminished the alternative activation of hepatic macrophages, causing hepatic dysfunction and systemic insulin resistance (50). Collectively, PPARs are thought to be required for the maturation of M2 activation and the resulting insulin sensitivity.
Krüppel-like factor 4
In addition to PPARs, another nuclear receptor, Krüppel-like factor 4 (KLF4), has been implicated in M2 activation in macrophages (93). In macrophages, KLF4 is suppressed by LPS stimulation, while it is increased by IL-4. Macrophage-specific KLF4 knockout mice display M1 activation and M2 disactivation. Owing to reduced fatty acid oxidation, the mice are susceptible to becoming obese and exhibit glucose intolerance and insulin resistance. In contrast, forced expression of KLF4 in RAW cultured macrophages resulted in M2 activation and resistance to M1 polarization by stimulation of LPS. Importantly, mRNA expression of KLF4 in adipose tissue is reduced in human obesity. Moreover, mRNA expression of KLF4 is not only positively associated with adiponectin expression in adipose tissue but also with well-defined M2 markers, such as CD206 and CCL18 in the stromal vascular fraction of adipose tissue. It has also been documented that IL-4 activates STAT6, leading to transcriptional activation of KLF4 to induce M2 genes (93).
AMP-activated protein kinase
AMP-activated protein kinase (AMPK) is an evolutionarily conserved sensor of cellular energy status that is activated by low energy status (increased cellular AMP/ADP:ATP ratio) and consists of an α catalytic subunit and βγ regulatory subunits. This molecule has also been shown to be crucial for the maintenance of M2 activation (94). Galic et al. tested the effect of AMPK β1 loss in macrophages in vivo by transplantation of bone marrow from WT or β1(−/−) mice into WT recipients. When challenged with an HFD, mice that received β1(−/−) bone marrow displayed enhanced adipose tissue macrophage inflammation and liver insulin resistance compared with animals that received WT bone marrow (94). Taking these findings together, the activation of AMPK and increased fatty acid oxidation in macrophages might provide an avenue for the treatment of type 2 diabetes.
Sirtuin 1
Sirtuin 1 (SIRT1), the mammalian homolog of yeast silent information-regulator 2 (Sir2), is an NAD+-dependent histone deacetylase that has been implicated in the regulation of lifespan under calorie restriction (95) or energy metabolism during fasting (96); thus, it is believed to be a promising target for type 2 diabetes (95, 97). Besides these findings, anti-inflammatory effects have also been demonstrated, showing that SIRT1 deacetylates NFκB and suppresses its transcriptional activity by inhibiting nuclear translocation (98). SIRT1 levels are markedly reduced in adipose tissue of obese humans and mice (99, 100). HFD was also found to result in cleavage of SIRT1 protein (101). In fact, upon the reduction of SIRT1 in fat by antisense oligonucleotides to levels similar to those seen during overnutrition, macrophage recruitment to adipose tissue was significantly increased. Similar results were obtained in fat-specific SIRT1 knockout mice. In contrast, overexpression of SIRT1 in mice prevented HFD-induced accumulation of macrophages (102). Furthermore, it was found that the SIRT1 expression level in human subcutaneous fat was inversely related to the number of adipose tissue macrophages. Mechanistically, others demonstrated that SIRT1 regulated intracellular inflammatory signaling at the levels of JNK and IKK (103). In addition, AMPK was also reported to regulate lipid-induced inflammation negatively through SIRT1 (104). Taken together, these findings indicate that SIRT1 might exert an insulin-sensitizing effect partially through the suppression of inflammation.
Involvement of Inflammasome in Obesity-Induced Inflammation
The mechanisms by which obesity induces macrophage activation despite the absence of any infection or autoimmune processes remained unclear. Although some mechanisms including hypoxia (105, 106) and autophagy (107–109) have been proposed for the induction of inflammation, in this review, we would like to focus on a new concept, the involvement of inflammasome in adipose tissue inflammation and insulin resistance. External or internal stimuli are recognized by pattern recognition receptors (PRRs). External stimuli, particularly pathogen-associated molecular patterns (PAMPs), are detected not only by TLRs but also by inflammasome, which is a protein complex consisting of caspase-1, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and nucleotide-binding oligomerization (NOD)-like receptors (NLRs) (110). Among these components, different pathogens are recognized by distinct constituents of NLRs. For instance, bacterial infection is recognized by nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-1 (NLRP1), NLRP3, NLRP4, and absent in melanoma 2 (AIM2). Viral infection is recognized by NLRP3 and AIM3. Fungal or parasitic infection is recognized by NLRP3 (110). But all of these infections cause the activation of caspase-1, which eventually leads to the processing and secretion of proinflammatory cytokines, including IL-1β and IL-18 (110). A unique feature of inflammasome is its additive ability to recognize internal stimuli as danger signals. For instance, uric acid, silica, fatty acids, and ATP in cytoplasm are detected as non-microbial-originated damage-associated molecular pattern molecules (DAMPs) by NLRs (110). Since mRNA expression of NLRP3 in adipose tissue correlates with IL-1β, body weight, and blood glucose level in rodents and humans (111), Vandanmagsar et al. tested whether NLRP3 played important roles during the development of chronic inflammation in obesity. Using NLRP3 knockout mice, they showed that NLRP3 sensed ceramide as a danger signal that activated caspase-1, which enhanced IL-1β secretion, thereby inducing T-cell activation (111). Target deletion of NLRP3 in mice displayed improved glucose tolerance and increased insulin sensitivity. These results were accompanied by the appearance of small adipocytes, reduced M1 activation, and enhanced insulin signaling in liver, adipose tissue, and skeletal muscle. Elevated ceramide, saturated fatty acid, reactive oxygen species (ROS), and mitochondrial dysfunction caused activation of inflammasome in macrophages (108, 112). The resulting activation of caspase-1 and subsequent secretion of IL-1β then interfere with insulin signaling, whereas inhibition of caspase-1 has been demonstrated to attenuate insulin resistance coincident with improved function of adipocytes (108, 112, 113). In humans, elevated levels of circulating IL-18 in patients with type 2 diabetes have been demonstrated (114), along with a suppressive effect of calorie restriction and resulting weight loss on the reduced expression of adipose NLRP3 in type 2 diabetes (111), and marked reduction of both adipose and liver expression of IL-1β in morbidly obese subjects by laparoscopic adjustable gastric banding surgery (114).
Therapeutic Interventions
The basis of therapeutic interventions in inflammation and insulin resistance is to prevent or to ameliorate obesity by physical exercise and diet control. They can also present the beneficial effects to the improvement of inflammation irrespective of body weight loss. In addition, the significance of chronic inflammation and its molecular mechanisms during the development of type 2 diabetes has been demonstrated and, in mice, suppression of inflammation-related molecules has successfully improved glucose intolerance. On the basis of this evidence, clinical trials targeting inflammation-related molecules have started. Thus, at first we would like to introduce the contribution of exercise and diet to the amelioration of inflammation. Next, we describe the current circumstances concerning several clinical applications of anti-inflammatory drugs.
Exercise and Diet
Although exercise is generally admitted to be effective to attenuate obesity and sustain health, single session of exercise has been reported to trigger an increase in proinflammatory cytokine release together with leukocytosis and increased plasma concentration of CRP (115). Regular and chronic exercise, however, has been reported to be associated with reduction of inflammatory markers such as CRP, IL-6, and TNFα (115–118). Physical (aerobic + resistance) exercise was also associated with increase in anti-inflammatory substances, such as IL-4 and IL-10 in type 2 diabetic patients with metabolic syndrome (118). Among many types of exercise, Oliveira et al. compared the effect of 12 weeks training with three different types of exercise (aerobic training, strength training, and combined training) on subjects with type 2 diabetes, demonstrating that the aerobic training program caused significant up-regulation in antioxidant enzymes (119). Accordingly, exercise-dependent improvement of glucose tolerance seems to be related with suppression of inflammation and oxidative stress (116).
Dietary calorie restriction is well recognized to be beneficial to ameliorate obesity-induced inflammation through weight loss. In addition to this, dietary composition has also been demonstrated to be important for the improvement of inflammation. Dietary bioactive compounds, such as polyphenols and certain fatty acids suppress systemic and adipose tissue inflammation. Polyphenols such as resveratrol exhibited anti-inflammatory effects via suppression of NF-κB (120) and extracellular signaling regulated kinase pathway (121) as well as via activating SIRT1 (122). Resveratrol has also been shown to activate AMPK independent of SIRT1 (123). Therefore, resveratrol may be a promising candidate in anti-inflammatory therapy (124). For instance, resveratrol supplementation for 30 days decreased blood glucose levels and inflammation markers along with improvement of HOMA index in healthy obese men irrespective of body weight (125). In addition, dietary polyunsaturated fatty acids, such as eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA) possess anti-inflammatory effects. Mechanistically, these include activation of AMPK and PPARγ (126). These polyunsaturated fatty acids also inhibits NF-κB pathway by activation of G-protein coupled receptor (GPR) 120 (127). In fact, n-3 polyunsaturated fatty acids (EPA and DHA) decreased adipose tissue and systemic inflammation in severe obese non-diabetic patients and improved lipid metabolism (128). EPA was demonstrated to reduce body weight at least by suppressing lipogenesis in the liver (129).
Clinical Applications of Anti-Inflammatory Drugs
Aspirin/salsalate
It has been reported that high-dose sodium salicylate or acetylsalicylic acid could diminish glycosuria or improve the blood glucose level in diabetic patients (1–4). Given that IKKβ is a key downstream mediator of insulin resistance and its blockade by salicylates attenuated hyperglycemia, hyperinsulinemia, and dyslipidemia in obese rodents (130, 131), Hundal et al. asked whether high-dose aspirin (∼7 g/day) could ameliorate insulin resistance and improve glucose tolerance in patients with type 2 diabetes (132). They demonstrated that this treatment for 2 weeks resulted in marked reduction of metabolic parameters including fasting glucose, basal rate of hepatic glucose production, and insulin-stimulated peripheral glucose uptake, despite no change in body weight. A large randomized trial, the National Institute of Diabetes and Digestive and Kidney Diseases-sponsored Targeting Inflammation with Salsalate, Non-acetylated Prodrug of Salicylate, in Type 2 Diabetes (TINSAL-T2D) trial, recently concluded that salsalate lowered hemoglobin A1c (HbA1c) levels and improved glycemic control in patients with type 2 diabetes (133). In a single-masked run-in period, patients were randomly assigned to receive placebo or salsalate at a dosage of 3.0, 3.5, or 4.0 g/day for 14 weeks (27 patients each) in addition to their current therapy. Mean HbA1c changes were −0.36% (P = 0.02) at 3.0 g/day, −0.34% (P = 0.02) at 3.5 g/day, and −0.49% (P = 0.001) at 4.0 g/day compared with placebo (133). The number of patients studied and the trial duration were insufficient to warrant recommending the use of salsalate for type 2 diabetes; however, it appears warranted to target this molecule in further investigations.
IL-1β
Reducing the activity of inflammasome and suppressing IL-1β secretion might be targets to attenuate insulin resistance in diabetes. Randomized clinical trials have shown that the blockade of IL-1β signaling by anakinra, a recombinant human IL-1 receptor antagonist, reduced systemic inflammation and improved glycemia of type 2 diabetes (134–136).
TNFα
Etanercept is a dimeric recombinant form of the extracellular domain of the human p75 TNFα receptor 2 fused to the Fc fragment of human immunoglobulin G1 (IgG1) and acts as a TNFα antagonist by interfering with the binding of TNFα to its cellular receptors and thus blocks the inflammatory response (137). Several studies have been conducted to test whether this biopharmaceutical improves glucose tolerance in patients with type 2 diabetes; however, despite a suppressive effect on systemic inflammation, the attenuation of glucose tolerance or insulin resistance has not yet been achieved (137–139). These results might be attributable to the distinct role of TNFα between rodents (5) and humans (137–139). Alternatively, antagonism of TNFα by other drugs remains hopeful in future studies.
Concluding Remarks
Following the discovery of chronic inflammation characterized by macrophage accumulation in adipose tissue, an explosion of studies in the past decade have begun to reveal the contributions of inflammation to the development of insulin resistance and subsequent metabolic abnormalities in other tissues, such as liver (47–52) and most recently brain (140). Adipose tissue, liver, and the hematopoietic system are evolutionarily derived from the same tissue. This developmental heritage can underlie the link between obesity-induced adipose tissue and hepatic inflammation (56) (Figure 2). Studies using flow cytometry subsequently identified the relative importance of other immune cells, including T cells, B cells, eosinophils, neutrophils, mast cells, and NKT cells, during the development of chronic inflammation. At present, besides the identification of constituent immune cells, an avenue intended to reveal how these neighboring immune cells modulate the inflammatory signals in macrophages has being created. In order to reveal the significance of inflammation during the development of type 2 diabetes, the identification of both factors that regulate trafficking of macrophages and intracellular molecules that control inflammatory activation in macrophages would be indispensable. Since there might be substantial differences in the nature of inflammation between rodents and humans and since clinical applications have not yet achieved excellent results, the question remains of how much the inhibition of inflammation contributes to improving glucose homeostasis. In future, there is a need for translational research that applies evidence from mice to human subjects. Because chronic inflammation is also involved in the development of atherosclerosis, rheumatoid arthritis, cancers, and neurodegenerative diseases, the suppression of inflammation can be a desirable therapy for type 2 diabetes. However, simple reduction of inflammation cannot be a beneficial approach as innate immunity is a radical form of homeostasis to deal with pathogenic infections. In addition, since the pathophysiology does not develop via a single molecule, multilayered targeting of various molecules without affecting physiological immune function has to be achieved. The location, timing, and degree of suppression all have to be controlled. Although recent studies have shed light on the pathophysiological roles of inflammation in diabetes, substantial efforts are required to achieve clinical application in human subjects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work was supported by a grant for the Intellectual Cluster Formation Project and the Twentieth Century COE Program “Center of Excellence for Signal Transduction Disease: Diabetes Mellitus as a Model” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to Masato Kasuga), by the Manpei Suzuki Diabetes Foundation (to Sanshiro Tateya), by the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to Sanshiro Tateya), by The Naito Foundation (to Sanshiro Tateya), by NIH Grant DK-073878 (to Francis Kim), and by a grant from the John L. Locke Jr. Charitable Trust and from the Kenneth H. Cooper Endowed Professorship in Preventive Cardiology (to Francis Kim). Sanshiro Tateya wrote the manuscript, contributed to discussions, and reviewed and edited the manuscript. Francis Kim and Yoshikazu Tamori interpreted the data and wrote, reviewed, and edited the manuscript.
References
1. Ebstein W. Zur therapie des diabetes mellitus, insbesondere uber die anwendeng der salicylauren natron bei demselben. Klin Wochenschr (1876) 13:337–40.
2. Williamson RT. On the treatment of glycosuria and diabetes mellitus with sodium salicylate. Br Med J (1901) 1:760–2. doi:10.1136/bmj.1.2100.760
3. Reid J, Macdougall AI, Andrews MM. Aspirin and diabetes mellitus. Br Med J (1957) 2:1071–4. doi:10.1136/bmj.2.5053.1071
4. Reid J, Lightbody TD. The insulin equivalence of salicylate. Br Med J (1959) 1:897–900. doi:10.1136/bmj.1.5126.897
5. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259:87–91. doi:10.1126/science.7678183
6. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science (1996) 271:665–8. doi:10.1126/science.271.5249.665
7. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi:10.1038/372425a0
8. Hoene M, Weigert C. The role of interleukin-6 in insulin resistance, body fat distribution and energy balance. Obes Rev (2008) 9:20–9.
9. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science (2008) 322:1539–43. doi:10.1126/science.1160794
10. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature (2001) 409:307–12. doi:10.1038/35053000
11. Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature (2005) 436:356–62. doi:10.1038/nature03711
12. Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab (2006) 290:E1253–61. doi:10.1152/ajpendo.00572.2004
13. Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD, et al. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem (2007) 282:28175–88. doi:10.1074/jbc.M700793200
14. Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, et al. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology (2007) 148:4687–94. doi:10.1210/en.2007-0175
15. Takahashi M, Takahashi Y, Takahashi K, Zolotaryov FN, Hong KS, Kitazawa R, et al. Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett (2008) 582:573–8. doi:10.1016/j.febslet.2008.01.023
16. Matsubara T, Mita A, Minami K, Hosooka T, Kitazawa S, Takahashi K, et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab (2012) 15:38–50. doi:10.1016/j.cmet.2011.12.002
17. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116:1494–505. doi:10.1172/JCI26498
18. Tateya S, Tamori Y, Kawaguchi T, Kanda H, Kasuga M. An increase in the circulating concentration of monocyte chemoattractant protein-1 elicits systemic insulin resistance irrespective of adipose tissue inflammation in mice. Endocrinology (2010) 151:971–9. doi:10.1210/en.2009-0926
19. Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem (2006) 281:26602–14. doi:10.1074/jbc.M601284200
20. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature (1997) 389:610–4. doi:10.1038/39335
21. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi:10.1172/JCI19246
22. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112:1821–30. doi:10.1172/JCI19451
23. Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med (2002) 8:731–7. doi:10.1038/nm724
24. Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem (2002) 277:25863–6. doi:10.1074/jbc.C200251200
25. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15:914–20. doi:10.1038/nm.1964
26. Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest (2010) 120:3466–79. doi:10.1172/JCI42845
27. Bruserud Ø, Kittang AO. The chemokine system in experimental and clinical hematology. Curr Top Microbiol Immunol (2010) 341:3–12. doi:10.1007/82_2010_18
28. Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A (2003) 100:7265–72. doi:10.1073/pnas.1133870100
29. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest (2006) 116:115–24. doi:10.1172/JCI24335C1
30. Ito A, Suganami T, Yamauchi A, Degawa-Yamauchi M, Tanaka M, Kouyama R, et al. Role of CC chemokine receptor 2 in bone marrow cells in the recruitment of macrophages into obese adipose tissue. J Biol Chem (2008) 283:35715–23. doi:10.1074/jbc.M804220200
31. Piemonti L, Calori G, Mercalli A, Lattuada G, Monti P, Garancini MP, et al. Fasting plasma leptin, tumor necrosis factor-alpha receptor 2, and monocyte chemoattracting protein 1 concentration in a population of glucose-tolerant and glucose-intolerant women: impact on cardiovascular mortality. Diabetes Care (2003) 26:2883–9. doi:10.2337/diacare.26.10.2883
32. Mine S, Okada Y, Tanikawa T, Kawahara C, Tabata T, Tanaka Y. Increased expression levels of monocyte CCR2 and monocyte chemoattractant protein-1 in patients with diabetes mellitus. Biochem Biophys Res Commun (2006) 344:780–5. doi:10.1016/j.bbrc.2006.03.197
33. Kouyama K, Miyake K, Zenibayashi M, Hirota Y, Teranishi T, Tamori Y, et al. Association of serum MCP-1 concentration and MCP-1 polymorphism with insulin resistance in Japanese individuals with obese type 2 diabetes. Kobe J Med Sci (2008) 53:345–54.
34. Kirk EA, Sagawa ZK, McDonald TO, O’Brien KD, Heinecke JW. Monocyte chemoattractant protein deficiency fails to restrain macrophage infiltration into adipose tissue [corrected]. Diabetes (2008) 57:1254–61. doi:10.2337/db07-1061
35. Inouye KE, Shi H, Howard JK, Daly CH, Lord GM, Rollins BJ, et al. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes (2007) 56:2242–50. doi:10.2337/db07-0425
36. Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased macrophage migration into adipose tissue in obese mice. Diabetes (2012) 61:346–54. doi:10.2337/db11-0860
37. Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, et al. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol (2008) 28:2195–201. doi:10.1161/ATVBAHA.108.168633
38. Yang SJ, IglayReger HB, Kadouh HC, Bodary PF. Inhibition of the chemokine (C-C motif) ligand 2/chemokine (C-C motif) receptor 2 pathway attenuates hyperglycaemia and inflammation in a mouse model of hepatic steatosis and lipoatrophy. Diabetologia (2009) 52:972–81. doi:10.1007/s00125-009-1309-8
39. Ito A, Suganami T, Miyamoto Y, Yoshimasa Y, Takeya M, Kamei Y, et al. Role of MAPK phosphatase-1 in the induction of monocyte chemoattractant protein-1 during the course of adipocyte hypertrophy. J Biol Chem (2007) 282:25445–52. doi:10.1074/jbc.M701549200
40. Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes (2012) 61:1680–90. doi:10.2337/db11-1506
41. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117:175–84. doi:10.1172/JCI29881
42. Nara N, Nakayama Y, Okamoto S, Tamura H, Kiyono M, Muraoka M, et al. Disruption of CXC motif chemokine ligand-14 in mice ameliorates obesity-induced insulin resistance. J Biol Chem (2007) 282:30794–803. doi:10.1074/jbc.M700412200
43. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest (2007) 117:2877–88. doi:10.1172/JCI31986
44. Tabata M, Kadomatsu T, Fukuhara S, Miyata K, Ito Y, Endo M, et al. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metab (2009) 10:178–88. doi:10.1016/j.cmet.2009.08.003
45. Han CY, Subramanian S, Chan CK, Omer M, Chiba T, Wight TN, et al. Adipocyte-derived serum amyloid A3 and hyaluronan play a role in monocyte recruitment and adhesion. Diabetes (2007) 56:2260–73. doi:10.2337/db07-0218
46. Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A, et al. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol (2008) 28:685–91. doi:10.1161/ATVBAHA.107.157685
47. Arkan M, Hevener A, Greten F, Maeda S, Li Z, Long J, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med (2005) 11:191–8. doi:10.1038/nm1185
48. Cai D, Yuan M, Frantz D, Melendez P, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med (2005) 11:183–90. doi:10.1038/nm1166
49. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest (2006) 116:1793–801. doi:10.1172/JCI29069
50. Odegaard J, Ricardo-Gonzalez R, Red Eagle A, Vats D, Morel C, Goforth M, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab (2008) 7:496–507. doi:10.1016/j.cmet.2008.04.003
51. Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan-Stevenson V, Daum G, et al. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes (2011) 60:2792–801. doi:10.2337/db11-0255
52. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube J, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes (2010) 59:347–57. doi:10.2337/db09-0016
53. Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes (2010) 59:916–25. doi:10.2337/db09-1403
54. Nio Y, Yamauchi T, Iwabu M, Okada-Iwabu M, Funata M, Yamaguchi M, et al. Monocyte chemoattractant protein-1 (MCP-1) deficiency enhances alternatively activated M2 macrophages and ameliorates insulin resistance and fatty liver in lipoatrophic diabetic A-ZIP transgenic mice. Diabetologia (2012) 55:3350–8. doi:10.1007/s00125-012-2710-2
55. Handa P, Tateya S, Rizzo NO, Cheng AM, Morgan-Stevenson V, Han CY, et al. Reduced vascular nitric oxide-cGMP signaling contributes to adipose tissue inflammation during high-fat feeding. Arterioscler Thromb Vasc Biol (2011) 31:2827–35. doi:10.1161/ATVBAHA.111.236554
56. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
57. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab (2008) 8:301–9. doi:10.1016/j.cmet.2008.08.015
58. Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation (2007) 115:1029–38. doi:10.1161/CIRCULATIONAHA.106.638379
59. Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes (Lond) (2008) 32:451–63. doi:10.1038/sj.ijo.0803744
60. Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol (2008) 28:1304–10. doi:10.1161/ATVBAHA.108.165100
61. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med (2009) 15:921–9. doi:10.1038/nm.2001
62. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15:930–9. doi:10.1038/nm.2002
63. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A (2007) 104:19446–51. doi:10.1073/pnas.0706832104
64. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi:10.2741/2692
65. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature (2012) 486:549–53. doi:10.1038/nature11132
66. Duffaut C, Galitzky J, Lafontan M, Bouloumié A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun (2009) 384:482–5. doi:10.1016/j.bbrc.2009.05.002
67. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med (2011) 17:610–7. doi:10.1038/nm.2353
68. Defuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A (2013) 110:5133–8. doi:10.1073/pnas.1215840110
69. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332:243–7. doi:10.1126/science.1201475
70. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012): doi:10.1038/nm.2885
71. Mansuy-Aubert V, Zhou QL, Xie X, Gong Z, Huang JY, Khan AR, et al. Imbalance between neutrophil elastase and its inhibitor α1-antitrypsin in obesity alters insulin sensitivity. inflammation, and energy expenditure. Cell Metab (2013) 17:534–48. doi:10.1016/j.cmet.2013.03.005
72. Andrade VL, Petruceli E, Belo VA, Andrade-Fernandes CM, Caetano Russi CV, Bosco AA, et al. Evaluation of plasmatic MMP-8, MMP-9, TIMP-1 and MPO levels in obese and lean women. Clin Biochem (2012) 45:412–5. doi:10.1016/j.clinbiochem.2012.01.008
73. Trellakis S, Rydleuskaya A, Fischer C, Canbay A, Tagay S, Scherag A, et al. Low adiponectin, high levels of apoptosis and increased peripheral blood neutrophil activity in healthy obese subjects. Obes Facts (2012) 5:305–18. doi:10.1159/000339452
74. Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med (2009) 15:940–5. doi:10.1038/nm.1994
75. Tanaka A, Nomura Y, Matsuda A, Ohmori K, Matsuda H. Mast cells function as an alternative modulator of adipogenesis through 15-deoxy-delta-12, 14-prostaglandin J2. Am J Physiol Cell Physiol (2011) 301:C1360–7. doi:10.1152/ajpcell.00514.2010
76. Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol (2009) 9:28–38. doi:10.1038/nri2451
77. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, et al. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem (2012) 287:13561–71. doi:10.1074/jbc.M112.350066
78. Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem (2012) 287:24378–86. doi:10.1074/jbc.M112.371807
79. Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest (2012) 122:3343–54. doi:10.1172/JCI62739
80. Scanga CB, Verde TJ, Paolone AM, Andersen RE, Wadden TA. Effects of weight loss and exercise training on natural killer cell activity in obese women. Med Sci Sports Exerc (1998) 30:1666–71. doi:10.1097/00005768-199812000-00002
81. Nieman DC, Henson DA, Nehlsen-Cannarella SL, Ekkens M, Utter AC, Butterworth DE, et al. Influence of obesity on immune function. J Am Diet Assoc (1999) 99:294–9. doi:10.1016/S0002-8223(99)00077-2
82. Lynch LA, O’Connell JM, Kwasnik AK, Cawood TJ, O’Farrelly C, O’Shea DB. Are natural killer cells protecting the metabolically healthy obese patient? Obesity (Silver Spring) (2009) 17:601–5. doi:10.1038/oby.2008.565
83. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116:3015–25. doi:10.1172/JCI28898
84. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282:2085–8. doi:10.1126/science.282.5396.2085
85. Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab (2009) 10:419–29. doi:10.1016/j.cmet.2009.09.006
86. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol (2005) 25:2062–8. doi:10.1161/01.ATV.0000183883.72263.13
87. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med (2012). doi:10.1038/nm.2851 [Epub ahead of print].
88. Hosoi T, Yokoyama S, Matsuo S, Akira S, Ozawa K. Myeloid differentiation factor 88 (MyD88)-deficiency increases risk of diabetes in mice. PLoS ONE (2010) 5(9):e12537. doi:10.1371/journal.pone.0012537
89. Ricardo-Gonzalez RR, Red Eagle A, Odegaard JI, Jouihan H, Morel CR, Heredia JE, et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci U S A (2010) 107:22617–22. doi:10.1073/pnas.1009152108
90. Cintra DE, Pauli JR, Araújo EP, Moraes JC, de Souza CT, Milanski M, et al. Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J Hepatol (2008) 48:628–37. doi:10.1016/j.jhep.2007.12.017
91. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi:10.1038/nature05894
92. Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest (2007) 117:1658–69. doi:10.1172/JCI31561
93. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest (2011) 121:2736–49. doi:10.1172/JCI45444
94. Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest (2011) 121:4903–15. doi:10.1172/JCI58577
95. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol (2012) 13:225–38. doi:10.1038/nrm3293
96. Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nat Rev Endocrinol (2009) 5:367–73. doi:10.1038/nrendo.2009.101
97. Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature (2007) 450:712–6. doi:10.1038/nature06261
98. Yeung F, Hoberg J, Ramsey C, Keller M, Jones D, Frye R, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J (2004) 23:2369–80. doi:10.1038/sj.emboj.7600244
99. Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J Biol Chem (2006) 281:39915–24. doi:10.1074/jbc.M607215200
100. Costa Cdos S, Hammes TO, Rohden F, Margis R, Bortolotto JW, Padoin AV, et al. SIRT1 transcription is decreased in visceral adipose tissue of morbidly obese patients with severe hepatic steatosis. Obes Surg (2010) 20:633–9. doi:10.1007/s11695-009-0052-z
101. Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab (2012) 16:180–8. doi:10.1016/j.cmet.2012.07.003
102. Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, et al. SirT1 regulates adipose tissue inflammation. Diabetes (2011) 60:3235–45. doi:10.2337/db11-0616
103. Yoshizaki T, Schenk S, Imamura T, Babendure J, Sonoda N, Bae E, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab (2010) 298:E419–28. doi:10.1152/ajpendo.00417.2009
104. Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem (2010) 285:19051–9. doi:10.1074/jbc.M110.123620
105. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes (2007) 56:901–11. doi:10.2337/db06-0911
106. Sung HK, Doh KO, Son JE, Park JG, Bae Y, Choi S, et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab (2013) 17:61–72. doi:10.1016/j.cmet.2012.12.010
107. Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A (2009) 106:19860–5.
108. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi:10.1038/ni.1831
109. Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, et al. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology (2012) 153:5866–74. doi:10.1210/en.2012-1625
110. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13:325–32. doi:10.1038/ni.2231
111. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17:179–88. doi:10.1038/nm.2279
112. Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab (2010) 12:593–605. doi:10.1016/j.cmet.2010.11.011
113. Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulangé A, Capeau J, et al. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia (2006) 49:2162–73. doi:10.1007/s00125-006-0335-z
114. Moschen AR, Molnar C, Enrich B, Geiger S, Ebenbichler CF, Tilg H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol Med (2011) 17:840–5. doi:10.2119/molmed.2010.00108
115. Kasapis C, Thompson PD. The effects of physical activity on serum C-reactive protein and inflammatory markers: a systematic review. J Am Coll Cardiol (2005) 45:1563–9. doi:10.1016/j.jacc.2004.12.077
116. de Lemos ET, Oliveira J, Pinheiro JP, Reis F. Regular physical exercise as a strategy to improve antioxidant and anti-inflammatory status: benefits in type 2 diabetes mellitus. Oxid Med Cell Longev (2012) 2012:741545. doi:10.1155/2012/741545
117. Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol (2005) 98:1154–62. doi:10.1152/japplphysiol.00164.2004
118. Balducci S, Zanuso S, Nicolucci A, Fernando F, Cavallo S, Cardelli P, et al. Anti-inflammatory effect of exercise training in subjects with type 2 diabetes and the metabolic syndrome is dependent on exercise modalities and independent of weight loss. Nutr Metab Cardiovasc Dis (2010) 20:608–17. doi:10.1016/j.numecd.2009.04.015
119. de Oliveira VN, Bessa A, Jorge ML, Oliveira RJ, de Mello MT, De Agostini GG, et al. The effect of different training programs on antioxidant status, oxidative stress, and metabolic control in type 2 diabetes. Appl Physiol Nutr Metab (2012) 37:334–44. doi:10.1139/h2012-004
120. Manna SK, Mukhopadhyay A, Aggarwal BB. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J Immunol (2000) 164:6509–19.
121. El-Mowafy AM, White RE. Resveratrol inhibits MAPK activity and nuclear translocation in coronary artery smooth muscle: reversal of endothelin-1 stimulatory effects. FEBS Lett (1999) 451:63–7. doi:10.1016/S0014-5793(99)00541-4
122. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature (2003) 425:191–6. doi:10.1038/nature01960
123. Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A (2007) 104:7217–22. doi:10.1073/pnas.0610068104
124. Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab (2010) 298:E751–60. doi:10.1152/ajpendo.00745.2009
125. Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab (2011) 14:612–22. doi:10.1016/j.cmet.2011.10.002
126. Figueras M, Olivan M, Busquets S, López-Soriano FJ, Argilés JM. Effects of eicosapentaenoic acid (EPA) treatment on insulin sensitivity in an animal model of diabetes: improvement of the inflammatory status. Obesity (Silver Spring) (2011) 19:362–9. doi:10.1038/oby.2010.194
127. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142:687–98. doi:10.1016/j.cell.2010.07.041
128. Itariu BK, Zeyda M, Hochbrugger EE, Neuhofer A, Prager G, Schindler K, et al. Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: a randomized controlled trial. Am J Clin Nutr (2012) 96:1137–49. doi:10.3945/ajcn.112.037432
129. Sato A, Kawano H, Notsu T, Ohta M, Nakakuki M, Mizuguchi K, et al. Antiobesity effect of eicosapentaenoic acid in high-fat/high-sucrose diet-induced obesity: importance of hepatic lipogenesis. Diabetes (2010) 59:2495–504. doi:10.2337/db09-1554
130. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature (1998) 396:77–80. doi:10.1038/23948
131. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science (2001) 293:1673–7. doi:10.1126/science.1061620
132. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest (2002) 109:1321–6. doi:10.1172/JCI14955
133. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, et al. TDTIUSiTDS: The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med (2010) 152:346–57. doi:10.7326/0003-4819-152-6-201003160-00004
134. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356:1517–26. doi:10.1056/NEJMoa065213
135. Osborn O, Brownell SE, Sanchez-Alavez M, Salomon D, Gram H, Bartfai T. Treatment with an Interleukin 1 beta antibody improves glycemic control in diet-induced obesity. Cytokine (2008) 44:141–8. doi:10.1016/j.cyto.2008.07.004
136. Donath MY, Mandrup-Poulsen T. The use of interleukin-1-receptor antagonists in the treatment of diabetes mellitus. Nat Clin Pract Endocrinol Metab (2008) 4:240–1. doi:10.1038/ncpendmet0783
137. Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK. Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med (2006) 166:902–8. doi:10.1001/archinte.166.8.902
138. Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, et al. Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res (2005) 42:517–25. doi:10.1159/000088261
139. Lo J, Bernstein LE, Canavan B, Torriani M, Jackson MB, Ahima RS, et al. Effects of TNF-alpha neutralization on adipocytokines and skeletal muscle adiposity in the metabolic syndrome. Am J Physiol Endocrinol Metab (2007) 293:E102–9. doi:10.1152/ajpendo.00089.2007
Keywords: obesity, chronic inflammation, insulin resistance, adipose tissue, TNFα, macrophages
Citation: Tateya S, Kim F and Tamori Y (2013) Recent advances in obesity-induced inflammation and insulin resistance. Front. Endocrinol. 4:93. doi: 10.3389/fendo.2013.00093
Received: 10 May 2013; Paper pending published: 28 May 2013;
Accepted: 11 July 2013; Published online: 08 August 2013.
Edited by:
Tsuguhito Ota, Kanazawa University, JapanReviewed by:
Undurti Narasimha Das, UND Life Sciences, USAVenu Lagishetty, University of California Los Angeles, USA
Copyright: © 2013 Tateya, Kim and Tamori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshikazu Tamori, Department of Internal Medicine, Division of Diabetes, Metabolism, and Endocrinology, Graduate School of Medicine, Kobe University, 7-5-1 Kusunoki-cho, Chuo-ku, Kobe 650-0017, Japan e-mail: tamori@med.kobe-u.ac.jp