 Larissa C. Faustino
Larissa C. Faustino Tania M. Ortiga-Carvalho
Tania M. Ortiga-Carvalho- Laboratorio de Endocrinologia Molecular, Instituto de Biofisica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
Cerebellum development is sensitive to thyroid hormone (TH) levels, as THs regulate neuronal migration, differentiation, and myelination. Most effects of THs are mediated by the thyroid hormone receptor (TR) isoforms TRβ1, TRβ2, and TRα1. Studies aimed at identifying TH target genes during cerebellum development have only achieved partial success, as some of these genes do not possess classical TH-responsive elements, and those that do are likely to be temporally and spatially regulated by THs. THs may also affect neurodevelopment by regulating transcription factors that control particular groups of genes. Furthermore, TH action can also be affected by TH transport, which is mediated mainly by monocarboxylate transporter family members. Studies involving transgenic animal models and genome-wide expression analyses have helped to address the unanswered questions regarding the role of TH in cerebellar development. Recently, a growing body of evidence has begun to clarify the molecular, cellular, and functional aspects of THs in the developing cerebellum. This review describes the current findings concerning the effects of THs on cerebellar development and maintenance as well as advances in the genetic animal models used in this field.
Introduction
The thyroid hormones (THs) thyroxine (T4) and 3,5,3′-triiodothyronine (T3) are essential for embryonic development and play critical roles in cellular metabolism, acting primarily through the stimulation of oxygen consumption and basal metabolic rate (1, 2). THs are necessary for proper central nervous system (CNS) development, and they have long been known to regulate neuronal differentiation and migration, synaptogenesis, and myelination (3–6). The cerebellum is located near the rear of the brain stem at the midbrain–hindbrain junction, and this structure is generally thought to coordinate proprioceptive–motor functions, although more recently, it has also been associated with neurocognition (7, 8). The cerebellum was one of the first targets of THs to be identified, and it is a useful model for studying the mechanisms by which THs influence the CNS. In particular, the cerebellum has a relatively homogenous and simple structure with a well-characterized laminar organization and a small number of cell types that develop within spatially defined regions (9–11).
The majority of TH actions are mediated through the binding of T3 to nuclear thyroid hormone receptors (TRs), which act as ligand-modulated transcription factors that modify the expression of target genes (12). Fundamentally, TH nuclear signaling is mediated by interactions between TRs and specific DNA sequences known as thyroid response elements (TREs), which associate with a variety of co-factors within the regulatory regions of target genes (12, 13). TR isoforms are expressed in several brain regions, including the cerebellum (14, 15). However, the target genes of THs and the cells that express genes likely to be involved in cerebellar development and maintenance are still not well-established (6, 16).
In addition to the classical roles of TH in the nucleus, TH can also initiate rapid effects at the cell surface, within mitochondria and via cytoplasmic TRs (17, 18). The fact that brain development in TR knockout (KO) animals is only slightly affected (19) suggests the existence of non-genomic morphogenic roles for TH in the CNS. One of the best characterized non-genomic roles for TH in the brain is illustrated by the induction of actin polymerization in astrocytes by T4 in vitro (20), which is very important for the organization of extracellular neural guidance molecules during neurodevelopmental processes. Finally, TH metabolism and transport, which are mediated mainly by deiodinases (21) and monocarboxylate transporters (22, 23), respectively, have also been shown to be important for cerebellar function.
The aims of this review are to briefly describe the current knowledge concerning the effects of THs on cerebellar development and functional maintenance as well to summarize advances in the genetic animal models used in this field.
The Influence of THs on Cerebellar Ontogenesis
In humans, T3, T4, and TRs are already present within the developing cortex prior to the onset of fetal thyroid gland activity, or gestational week 12, which suggests an important role for maternal TH during this critical window of brain development (24–27). Congenital hypothyroidism leads to structural and intellectual impairment in infants (28). Furthermore, TH administration to human infants with congenital hypothyroidism immediately after birth was shown to promote near-normal intellectual development (29). The majority of studies on the role of THs in neurodevelopment have been carried out in rodent models in which THs, deiodinases, and TRs are present prior to the onset of fetal TH synthesis and secretion (30, 31). Paired box 8 (Pax8) KO mice are a commonly used animal model for studying the effects of postnatal TH on CNS development, as Pax8 is an essential transcription factor for thyroid follicular cell differentiation, and its absence leads to thyroid gland dysgenesis (32). Therefore, the Pax8-KO mouse is a model for congenital hypothyroidism that displays extensive abnormalities in cerebellar development, resulting in an ataxic phenotype (32–34) (Table 1).
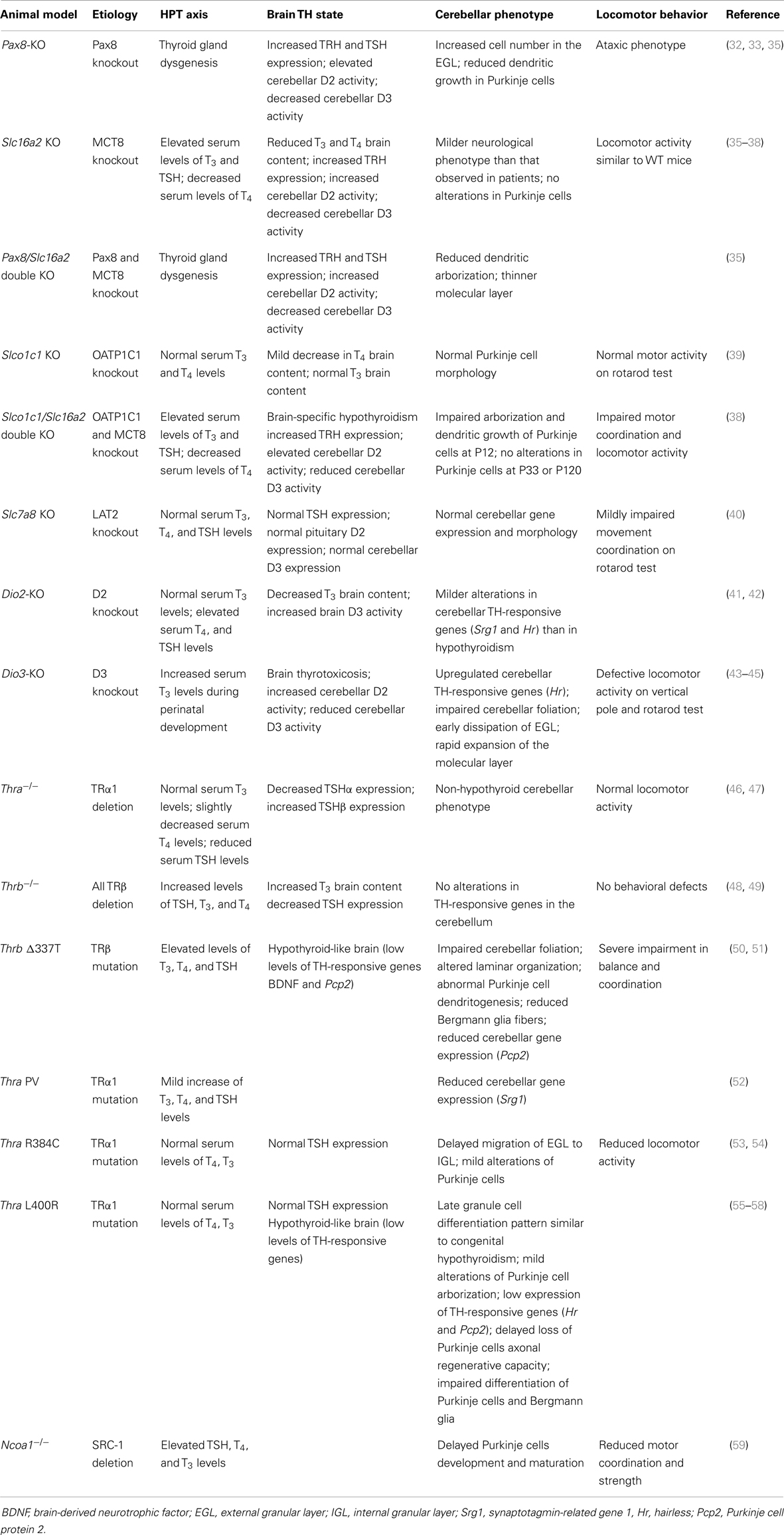
Table 1. Summary of mutant animal models and their cerebellar phenotypes.
Rodent cerebellar development is complete within the first 2–3 weeks after birth, when the cerebellar foliation process, which encompasses the transition from a smooth cerebellar surface to an X lobule cerebellum, is completed (7). It has long been known that cerebellar ontogenesis is closely linked to TH regulation (60–62), although the molecular mechanisms through which THs modulate this process remain unclear. Hypothyroidism results in a number of morphological alterations in the cerebellum, including increased neuronal death within the internal granular layer (IGL), increased perdurance of the external granular layer (EGL), defects in granular cell migration, impaired Purkinje cell dendritogenesis, delayed myelination, defects in the late differentiation pattern of Golgi interneurons and mossy fibers, reduced protrusions of Bergmann glial cells, and increased cell apoptosis (9, 46, 63–65). TH administration prior to the end of postnatal week 2 prevented these structural changes. Moreover, the expression levels of neurotrophins and growth factors, such as BDNF, NT3, and EGF, as well as cell adhesion molecules, such as NCAM and L1, are modified by TH in the developing cerebellum (63, 66–68). For example, TH was shown to promote cerebellar neuronal migration and the differentiation of Bergmann glia by inducing EGF secretion (69).
Perspectives from Transgenic Mouse Models
T3 and T4 enter the cell through plasma membrane transporters, including the monocarboxylate transporter family members MCT8 and MCT10, organic anion transporting peptides (OATP), and carriers of l-amino acids (LATS) (70, 71). Recent studies have indicated that TH transporters such as MCT8, which are found in a subset of neuronal populations (23), may play critical roles in neurodevelopment processes mediated by THs. Patients harboring inactivating mutations in the MCT8 gene (Slc16a2) exhibit Allan–Herndon–Dudley syndrome, which is characterized by psychomotor retardation, lack of speech development, increased serum T3 concentrations, and low T4 levels (72, 73).
Although MCT8-KO mice have been generated, they do not display the same neurological abnormalities observed in human patients (Table 1). This phenomenon is likely due to the presence of other neuronal TH transporters, such as OATP14, LAT1, and LAT2, during earlier stages of mouse brain development that compensate for the absence of MCT8 (36, 74). However, another possible explanation for the difference between the mouse and human phenotypes is that human MCT8 is necessary for the transport of an unknown signaling molecule necessary for CNS development, which is consistent with clinical evidence indicating that the neurological syndromes observed in patients with MCT8 mutations are more severe than those observed in patients with congenital hypothyroidism (36). A recent study performed in MCT8-KO mice demonstrated that 3,5,3′,5′-tetraiodothyroacetic acid (tetrac), a T4 metabolite that is not transported by MCT8 or OATP1C1, is capable of replacing TH during brain development (35). Tetrac can be converted into 3,3′,5-triiodothyroacetic acid (triac) by deiodinase type 2, which can subsequently interact with TRs, thereby replacing T3 activity. Indeed, treatment of MCT8-KO mice with tetrac led to improvements in TH-dependent neuronal differentiation in the striatum, cortex, and cerebellum during the first three postnatal weeks.
A mouse model lacking LAT2 (Slc7a8) was generated to further characterize the role of this transporter in TH physiology. However, LAT2-KO mice exhibited normal cerebral and cerebellar development, with the exception of slight defects in movement coordination on rotarod tests (40) (Table 1).
The iodothyronine deiodinase enzymes D1 (Dio1) and D2 (Dio2) modulate the intracellular availability of the active hormone T3. In particular, D2 catalyzes the conversion of T4 to T3, whereas D3 inactivates T4 and T3 by converting them to T2 and reverse T3 (rT3), respectively (75). Studies have demonstrated that nearly 80% of T3 is generated by local conversion within the brain (3, 5) through the activity of D2, which is primarily found in astrocytes (41). Therefore, the presence of D2 together with increased levels of T3 suggests a role for D2 in supplying the developing brain with T3 derived from maternal T4. However, some unexpected findings in Dio2-KO mice are inconsistent with the hypothesis that D2 is essential for all TH-dependent neurodevelopment processes.
Although Dio2-KO mice display elevated brain T4 levels and reduced T3 content, surprisingly, the observed neurological impairments, which included changes in the cerebellar expression of TH-dependent genes and behavioral defects, were found to be mild compared with those observed in hypothyroidism (42, 76). These data suggest that decreased local T3 production can be largely compensated for by increased T3 uptake from circulation, and indeed, this was later confirmed by experiments carried out in double Dio1/Dio2-KO mice, which demonstrated normal serum T3 concentrations and only mild neurological phenotypes (21). On the other hand, Dio3-KO animals were characterized by high T3 levels during perinatal development, which induced the upregulation of TH-responsive genes in the cerebellum (43, 44). Recently, it was reported that Dio3-KO mice exhibited impaired cerebellar foliation, early premature disappearance of the EGL, rapid expansion of the molecular layer, and abnormal locomotor behavior. Furthermore, the cerebellar phenotypes of these mice could be partially rescued by deletion of the TRα1 isoform (45) (Table 1).
The majority of TH functions are mediated through nuclear TRs, which are members of a superfamily of ligand-modulated transcription factors that can either upregulate or downregulate target gene transcription (2). The consensus for positively regulated genes is that TRs bind to activating TREs both in the presence and absence of T3. In the absence of T3, TR represses target gene transcription by recruiting co-repressors, whereas in the presence of T3, co-repressors are released and co-activators are recruited, leading to transcriptional up regulation (1, 12). In mammals, two different genes encode at least three high-affinity TRs: TR-β1 (Thrb), TR-β2 (Thrb), and TR-α1 (Thra) (77). TR-α1 is the isoform that is predominantly expressed both prenatally and postnatally throughout the brain, including the developing cerebellum, and it is responsible for nearly 80% of total receptor T3 binding (14, 78, 79). In contrast, TR-β expression is confined to a few postnatal neuronal populations, including the paraventricular hypothalamus, cerebellar Purkinje cells, and hippocampal pyramidal and granule cells (80, 81). In rodents, TR-α1 is already present at E11.5 in the neural tube and at E12.5 in the diencephalon and ventral rhombencephalon (14). Both TRα and TRβ are expressed in the cerebellum. TRα is primarily expressed in the early cerebellar neurepithelium, granular cell precursors, and later in the transient EGL, whereas TRβ is predominantly expressed during later stages, notably in the Purkinje cell layer (PCL) and in deep internal layers (14, 81, 82) (Figure 1).
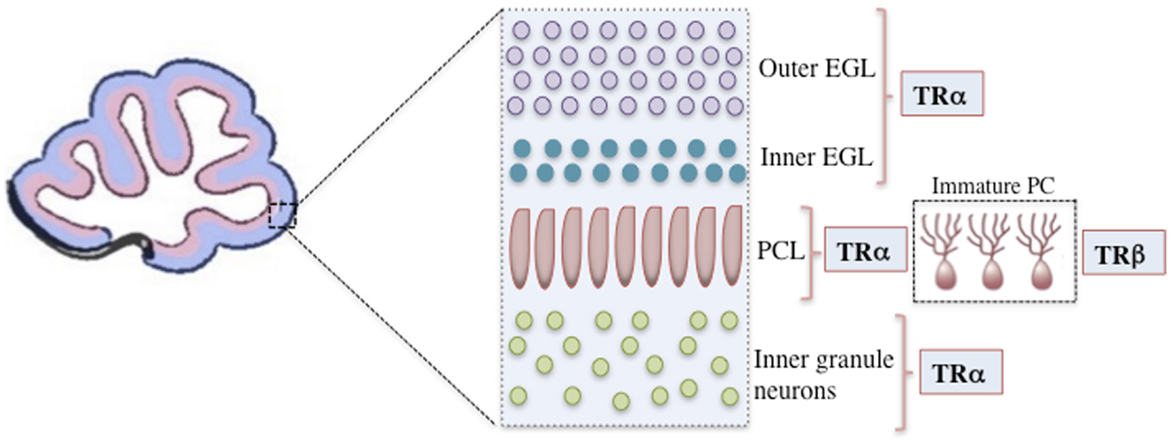
Figure 1. A representation of the mouse cerebellar cortex during the initial postnatal days showing the positions of cells expressing specific TR isoforms. Only the outer EGL, inner EGL, Purkinje cell layer (PCL), and inner granule layer are shown. TRα is primarily expressed in granular cell precursors and subsequently in the transient outer and inner EGL. In the Purkinje cells, TRα is the first isoform to be detected, however, after the second postnatal week TRβ is predominantly expressed. TR, thyroid hormone receptor; EGL, external granular layer; P14, postnatal day 14; PCL, Purkinje cell layer.
Thra- and Thrb-KO mouse models, which exhibit abrogated nuclear signaling, have been created to address the roles of different TR isoforms in proper brain development and function (47, 48, 83). However, it was reported that these mice exhibit only a mild neurological phenotype compared with hypothyroid animals, indicating that the absence of T3 binding (unliganded TR) is more harmful to the CNS than the absence of TR isoforms (46, 84) (Table 1). Later, Thra- and Thrb-knock-in mutant mice expressing dominant-negative TRs were generated, and it was reported that these mice were phenotypically distinct from TR-KO mice (50, 53–55). Specifically, in mice harboring the Thrb Δ337T mutation – a point mutation in the ligand-binding domain that prevents T3 binding but not binding to DNA or co-factors (85) – cerebellar morphogenesis was similar to that observed in congenital hypothyroidism, presumably because TR remained constitutively bound to its co-repressors, thereby mimicking a hypothyroid state (50). Hashimoto et al. (50) demonstrated that Thrb Δ337T mice displayed impairments in balance and coordination, reductions in the molecular and PCLs, and decreases in the number and branching of Purkinje cells, which may account for the decreased cerebellar size observed in these mutant animals.
Therefore, functional TR-β is required for TH-dependent cerebellar development, which was further demonstrated by the phenotypes observed in Thrb Δ337T mutant mice, including defects in cerebellar foliation, altered laminar organization, abnormal Purkinje cell dendritogenesis, and reduced Bergmann glia fibers (51). Cerebellar foliation is characterized by the presence of 10 well-formed lobules and sub-lobules (7). In Thrb Δ337T homozygotes at postnatal day (PND) 21, researchers observed decreases in the molecular and granular layers as well as a failure in the subdivision of lobule VI, which is subdivided into sub-lobules VIa and VIb in wild-type and heterozygous animals. During PND 9, which is the initial period of cerebellar development, Thrb Δ337T mice fail to form fissures between lobules VI–VII, and lobule IX is also severely affected. During both the initial and final stages of cerebellar foliation, the Thrb Δ337T mutation leads to extreme defects in fissure and lobule formation (51). Unfortunately, the identification of direct target genes that are regulated by TH in the developing brain using RNA-based techniques has been problematic. However, recent studies using chromatin immunoprecipitation combined with DNA microarray analysis (ChIP on chip) identified a large number of TR-β binding sites and target genes in the developing mouse cerebellum, reinforcing the role of TR-β in mediating gene transcription through TH in this brain structure (86, 87). Chatonnet et al. introduced TR-α1 and TR-β1 into a neural cell line lacking endogenous TRs and demonstrated that the majority of the T3 target genes analyzed were regulated by both TR-α1 and TR-β1. Nevertheless, a significant number of the analyzed genes showed strong preferences for one receptor isoform over the other (88).
In the cerebellum of mice carrying a cell-specific L400R mutation in the ligand-binding domain of TR-α1 Thra L400R), which prevents histone acetyltransferase recruitment and facilitates the permanent recruitment of co-repressors, there is a delay in the pattern of granule cell differentiation similar to what is observed in congenital hypothyroid animals; however, Purkinje cell arborization is not strongly affected in these mutants (55). Another study involving Thra L400R mice highlighted the importance of TRα-dependent signaling in postnatal brain development by showing that it promotes the secretion of neurotrophins from astrocytes and Purkinje cells and that it maintains adult brain function by limiting the proliferation of oligodendrocyte precursor cells (56). Late in their development, these mutant mice displayed a loss of axonal regenerative capacity in Purkinje cells, which is thought to play a role in the brain maturation process. These data indicate an important role for TR-α1 in mediating T3-induced inhibition of axonal regeneration in Purkinje cells (57). In addition, it was very recently reported that the L400R mutation primarily affects the differentiation of two specific cerebellar cell populations, Purkinje cells, and Bergmann glia, which indicates that the autonomous effects of TH on these cells indirectly impact global cerebellar cortex development (58). In Purkinje cells, T3 acts through TR-α1 to promote dendritic tree development and the secretion of neurotrophic factors, whereas in Bergmann glia, T3 promotes the development and organization of radial fibers and the alignment of cell bodies within the PCL (58) (Table 1). In humans, a role for TR-α1 in brain development is supported by descriptions of patients with cognitive impairment phenotypes similar to those observed in congenital hypothyroidism who harbor primary mutations in the THRA gene (89, 90).
Taken together, these data suggest that TR-α and TR-β function together to mediate the processes of cerebellar ontogenesis controlled by THs. Compared with Thrb mutants, Thra-knock-in mice show more severe cerebellar defects, indicating that TR-α may play a key role in regulating the expression of target genes involved in cerebellar ontogeny (52). Other relevant mutant animal models with impaired neurological phenotypes also exist, such as Ncoa1-KO animals. Steroid receptor co-activator 1, which is encoded by the Ncoa1 gene, has been shown to modulate TH activity via specific TR isoforms (91, 92). This co-activator is highly expressed in the cerebellum; thus, Ncoa1-KO mice exhibit cerebellar abnormalities that are similar to those observed in congenital hypothyroid mice (59).
Concluding Remarks
It has been known for decades that cerebellar development is regulated by THs. Although the molecular mechanisms through which THs impact CNS development are becoming better understood, primarily due to studies in genetic animal models, many issues remain to be addressed. Only a few T3 targets in neural cells have been described to date, it is important to identify additional direct target genes of THs and to determine how these genes are temporally and spatially regulated during specific neurodevelopment. Finally, the rapid non-genomic actions of THs and the role of the recently described thyronine derivatives require further analysis. Therefore, additional studies will be necessary before our model of TH activity within the developing cerebellum is complete.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Grants and fellowships: FAPERJ to Larissa C. Faustino; CNPq and FAPERJ to Tania M. Ortiga-Carvalho.
References
1. Lazar M. Thyroid hormone action: a binding contact. J Clin Invest (2003) 122:497–9. doi: 10.1172/JCI19479
2. Brent GA. Mechanisms of thyroid hormone action. J Clin Invest (2012) 122(9):3035–43. doi:10.1172/JCI60047
3. Anderson GW, Schoonover CM, Jones SA. Control of thyroid hormone action in the developing rat brain. Thyroid (2003) 13(11):1039–56. doi:10.1089/105072503770867219
4. de Escobar GM, Obregón MJ, del Rey FE. Maternal thyroid hormones early in pregnancy and fetal brain development. Best Pract Res Clin Endocrinol Metab (2004) 18(2):225–48. doi:10.1016/j.beem.2004.03.012
5. Bernal J. Thyroid hormones and brain development. Vitam Horm (2005) 71:95–122. doi:10.1016/S0083-6729(05)71004-9
6. Diez D, Grijota-Martinez C, Agretti P, Marco GD, Tonacchera M, Pinchera A, et al. Thyroid hormone action in the adult brain: gene expression profiling of the effects of single and multiple doses of triiodo-l-thyronine in the rat striatum. Endocrinology (2008) 149(8):3989–4000. doi:10.1210/en.2008-0350
7. Altman J, Bayer SA. Development of the Cerebellar System in Relation to its Evolution, Structure, and Functions. Boca Raton: CRC Press (1997).
8. Tavano A, Grasso R, Gagliardi C, Triulzi F, Bresolin N, Fabbro F, et al. Disorders of cognitive and affective development in cerebellar malformations. Brain (2007) 130(10):2646–60. doi:10.1093/brain/awm201
9. Legrand J. Effects of thyroid hormones on central nervous system development. In: Yanai J, editor. Neurobehavioral Teratology. Amsterdam: Elsevier (1984). p. 31–363.
10. Sillitoe RV, Joyner AI. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol (2007) 23:549–77. doi:10.1146/annurev.cellbio.23.090506.123237
11. Koibuchi N, Ikeda Y. Hormones and cerebellar development. In: Manto M, Gruol D, Schmahmann JD, Koibuchi N, editors. Handbook of the Cerebellum and Cerebellar Disorders. New York, NY: Springer (2013). p. 319–39.
12. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31(2):139–70. doi:10.1210/er.2009-0007
13. Liu Y, Xia X, Fondell JD, Yen PM. Thyroid hormone-regulated target genes have distinct patterns of coactivator recruitment and histone acetylation. Mol Endocrinol (2006) 20(3):483–90. doi:10.1210/me.2005-0101
14. Bradley DJ, Towle HC, Young WS. Spatial and temporal expression of alpha- and beta-thyroid hormone receptor mRNAs, including the beta-2 subtype in the mammalian nervous system. J Neurosci (1992) 12:2288–302.
15. Chan S, Kachilele S, McCabe CJ, Tannahill LA, Boelaert K, Gittoes NJ, et al. Early expression of thyroid hormone deiodinases and receptors in human fetal cerebral cortex. Brain Res Dev Brain Res (2002) 138:109–16. doi:10.1016/S0165-3806(02)00459-5
16. Quignodon L, Grijota-Martinez C, Compe E, Guyot R, Allioli N, Laperriere D, et al. A combined approach identifies a limited number of new thyroid hormone target genes in post-natal mouse cerebellum. J Mol Endocrinol (2007) 39:17–28. doi:10.1677/JME-06-0054
17. Davis PJ, Davis FB, Cody V. Membrane receptors mediating thyroid hormone action. Trends Endocrinol Metab (2005) 16(9):429–35. doi:10.1016/j.tem.2005.09.0072
18. Farwell AP, Dubord-Tomasetti S, Pietrzykowski AZ, Leonard JL. Dynamic nongenomic actions of thyroid hormone in the developing rat brain. Endocrinology (2006) 147(5):2567–74. doi:10.1210/en.2005-1272
19. Gothe S, Wang Z, Ng L, Kindblom JM, Barros AC, Ohlsson C, et al. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev (1999) 13:1329–41. doi:10.1101/gad.13.10.1329
20. Farwell AP, Dubord-Tomasetti SA, Pietrzykowski AZ, Stachelek SJ, Leonard JL. Regulation of cerebellar neuronal migration and neurite outgrowth by thyroxine and 3,3′,5′-triiodothyronine. Brain Res Dev Brain Res (2005) 154(11):121–35. doi:10.1016/j.devbrainres.2004.07.016
21. Galton VA, Schneider MJ, Clark AS, St Germain DL. Life without thyroxin to 3,5,3′-triiodothyronine conversion: studies in mice devoid of 5′-deiodinases. Endocrinology (2009) 150:2958–63. doi:10.1210/en.2008-1572
22. Friesema EC, Gueters A, Biebermann H, Krude H, von Moers A, Reeser M, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet (2004) 364:1435–7. doi:10.1016/S0140-6736(04)17226-7
23. Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology (2005) 146:1701–6. doi:10.1210/en.2004-1179
24. Obregón MJ, Mallol J, Pastor R, de Escobar GM, del Rey GE. l-Thyroxine and 3,5,3′-triiodo-l-thyronine in rat embryos before onset of fetal thyroid function. Endocrinology (1984) 144:305–7. doi:10.1210/endo-114-1-305
25. Piosik PA, van Groenigen M, van Doorn J, Baas F, de Vijlder JJ. Effects of maternal thyroid status on thyroid hormones and growth in congenitally hypothyroid goat fetuses during the second half of gestation. Endocrinology (1997) 138:5–11. doi:10.1210/endo.138.1.4843
26. Contempré B, Jauniaux E, Calvo R, Jurkovic D, Campbell S, de Escobar GM. Detection of thyroid hormones in human embryonic cavities during the first trimester of pregnancy. J Clin Endocrinol Metab (1993) 77:1719–22. doi:10.1210/jcem.77.6.8263162
27. Kester MH, Martinez de Mena R, Obregon MJ, Marinkovic D, Howatson A, Visser TJ, et al. Iodothyronine levels in the human developing brain: major regulatory roles of iodothyronine deiodinases in different areas. J Clin Endocrinol Metab (2004) 89:3117–28. doi:10.1210/jc.2003-031832
28. Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid (1993) 3:59–69. doi:10.1089/thy.1993.3.59
29. LaFranchi S. Congenital hypothyroidism: etiologies, diagnosis, and management. Thyroid (1999) 9:735–40. doi:10.1089/thy.1999.9.735
30. Porterfield SP, Hendrich CE. The role of thyroid hormones in prenatal and neonatal neurological development – current perspectives. Endocr Rev (1993) 14:94–106. doi:10.1210/edrv-14-1-94
31. Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab (2007) 3:249–59. doi:10.1038/ncpendmet0424
32. Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet (1998) 19:87–90. doi:10.1038/ng0598-87
33. Poguet AL, Legrand C, Feng X, Yen PM, Meltzer P, Samarut J, et al. Microarray analysis of knockout mice identifies cyclin D2 as a possible mediator for the action of thyroid hormone during the postnatal development of the cerebellum. Dev Biol (2003) 254:188–99. doi:10.1016/S0012-1606(02)00039-8
34. Friedrichsen S, Christ S, Heuer H, Schäfer MK, Mansouri A, Bauer K, et al. Regulation of iodothyronine deiodinases in the Pax8−/− mouse model of congenital hypothyroidism. Endocrinology (2003) 144(3):777–84. doi:10.1210/en.2002-220715
35. Horn S, Kersseboom S, Mayer S, Muller J, Groba C, Trajkovic-Arsic M, et al. Tetrac can replace thyroid hormone during brain development in mouse mutants deficient in the thyroid hormone transporter Mct8. Endocrinology (2013) 154(2):968–79. doi:10.1210/en.2012-1628
36. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology (2006) 147:4036–43. doi:10.1210/en.2006-0390
37. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest (2007) 117(3):627–35. doi:10.1172/JCI28253
38. Mayerl S, Müller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest (2014) 124(5):1987–99. doi:10.1172/JCI70324
39. Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H. Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology (2012) 153(3):1528–37. doi:10.1210/en.2011-1633
40. Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Gruters A, et al. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochem J (2011) 439(2):249–55. doi:10.1042/BJ20110759
41. Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A (1997) 94:10391–6. doi:10.1073/pnas.94.19.10391
42. Galton VA, Wood ET, Germain EAS, Withrow CA, Aldrich G, Germain GMS, et al. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology (2007) 148:3080–8. doi:10.1210/en.2006-1727
43. Hernandez A, Martinez ME, Fiering S, Galton VA, Germain DS. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest (2006) 116(2):476–84. doi:10.1172/JCI26240
44. Hernandez A, Martinez E, Fiering S, Galton VA, St Germain DL. Type 3 deiodinase deficiency results in functional abnormalities at multiple levels of the thyroid axis. Endocrinology (2007) 148:5680–7. doi:10.1210/en.2007-0652
45. Peeters RP, Hernandez A, Lily N, Michelle M, David SS, Mei P, et al. Cerebellar abnormalities in mice lacking type 3 deiodinase and partial reversal of phenotype by deletion of thyroid hormone receptor alpha1. Endocrinology (2013) 154(1):550–61. doi:10.1210/en.2012-1738
46. Morte B, Manzano J, Scanlan T, Vennström B, Bernal J. Deletion of the thyroid hormone receptor α1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Natl Acad Sci U S A (2002) 99(6):3985–9. doi:10.1073/pnas.062413299
47. Wilkstrom L, Johansson C, Salto C, Barlow C, Campo-Barros A, Baas F, et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J (1998) 17:455–61. doi:10.1093/emboj/17.2.455
48. Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: evidence for tissue-specific modulation of receptor function. EMBO J (1996) 15(12):3006–15.
49. Sandhofer C, Schwartz HL, Mariash CN, Forrest D, Oppenheimer JH. Beta receptor isoforms are not essential for thyroid hormone-dependent acceleration of PCP-2 and myelin basic protein gene expression in the developing brains of neonatal mice. Mol Cell Endocrinol (1998) 137:109–15. doi:10.1016/S0303-7207(98)00005-7
50. Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci U S A (2001) 98:3998–4003. doi:10.1073/pnas.051454698
51. Portella A, Carvalho F, Faustino LC, Ortiga-Carvalho TM, Gomes FC. Thyroid hormone receptor beta mutation causes severe impairment of cerebellar development. Mol Cell Neurosci (2010) 44(1):68–77. doi:10.1016/j.mcn.2010.02.004
52. Itoh Y, Esaki T, Kaneshige M, Suzuki H, Cook M, Sokoloff L, et al. Brain glucose utilization in mice with a targeted mutation in the thyroid hormone alpha or beta receptor gene. Proc Natl Acad Sci U S A (2001) 98(17):9913–8. doi:10.1073/pnas.171319498
53. Tinnikov A, Nordström K, Thorén P, Kindblom JM, Malin S, Rozell B, et al. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor alpha1. EMBO J (2002) 21:5079–87. doi:10.1093/emboj/cdf523
54. Venero C, Guadaño-Ferraz A, Herrero AI, Nordström K, Manzano J, del Escobar GM, et al. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev (2005) 19(18):2152–63. doi:10.1101/gad.346105
55. Quignodon L, Vincent S, Winter H, Samarut J, Flamant F. A point mutation in the activation function 2 domain of thyroid hormone receptor alpha1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol Endocrinol (2007) 21:2350–60. doi:10.1210/me.2007-0176
56. Picou F, Fauquier T, Chatonnet F, Flamant F. A biomodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol Endocrinol (2012) 26:608–18. doi:10.1210/me.2011-1316
57. Avci HX, Lebrun C, Wehrle R, Doulazmi M, Chatonnet F, Morel MP, et al. Thyroid hormone triggers the developmental loss of axonal regenerative capacity via thyroid hormone receptor α1 and Krüppel-like factor 9 in Purkinje cells. Proc Natl Acad Sci U S A (2012) 109(35):14206–11. doi:10.1073/pnas.1119853109
58. Fauquier T, Chatonnet F, Picou F, Lamonerie T, Flamant F. Purkinje cells and Bergmann glia are primary targets of the TRα1 thyroid hormone receptor during mouse cerebellum postnatal development. Development (2014) 141:166–75. doi:10.1242/dev.103226
59. Nishihara E, Yoshida-Komiya H, Chan CS, Liao L, Davis RL, O’Malley BW, et al. SRC-1 null mice exhibit moderate motor dysfunction and delayed development of cerebellar Purkinje cells. J Neurosci (2003) 23(1):213–22.
60. Nicholson JL, Altman J. The effects of early hypo- and hyperthyroidism on the development of rat cerebellar cortex. I. Cell proliferation and differentiation. Brain Res (1972) 44(1):13–23. doi:10.1016/0006-8993(72)90362-9
61. Lauder JM. The effects of early hypo- and hyperthyroidism on the development of rat cerebellar cortex. III. Kinetics of cell proliferation in the external granular layer. Brain Res (1977) 126:31–51. doi:10.1016/0006-8993(77)90213-X
62. Lauder JM. Effects of early hypo- and hyperthyroidism on development of rat cerebellar cortex. IV. The parallel fibers. Brain Res (1978) 142(1):25–39. doi:10.1016/0006-8993(78)90174-9
63. Heuer H, Mason CA. Thyroid hormone induces cerebellar Purkinje cell dendritic development via the thyroid hormone receptor α1. J Neurosci (2003) 23(33):10604–12.
64. Martinez R, Gomes FC. Proliferation of cerebellar neurons induced by astrocytes treated with thyroid hormone is mediated by a cooperation between cell contact and soluble factors and involves the epidermal growth factor-protein kinase a pathway. J Neurosci (2005) 80:341–9. doi:10.1523/jneurosci.3358-03.2004
65. Wang Y, Zhong J, Xu H, Wei W, Dong J, Yu F, et al. Perinatal iodine deficiency and hypothyroidism increase cell apoptosis and alter doublecortin and reelin protein expressions in rat cerebellum. Arch Med Res (2012) 43(4):255–64. doi:10.1016/j.arcmed.2012.05.002
66. Alvarez-Dolado M, Cuadrado A, Navarro-Yubero C, Sonderegger P, Furley AJ, Bernal J, et al. Regulation of the L1 cell adhesion molecule by thyroid hormone in the developing brain. Mol Cell Neurosci (2000) 16:499–514. doi:10.1006/mcne.2000.0879
67. Martinez R, Gomes FCA. Neuritogenesis induced by thyroid hormone-treated astrocytes is mediated by epidermal growth factor/mitogen-activated protein kinase-phosphatidylinositol 3-kinase pathways and involves modulation of extracellular matrix proteins. J Biol Chem (2002) 277:49311–8. doi:10.1074/jbc.M209284200
68. Chakraborty G, Magagna-Poveda A, Parrat C, Umans JG, MacLusky NJ, Scharfman HE. Reduced hippocampal brain-derived neurotrophic factor (BDNF) in neonatal rats after prenatal exposure to propylthiouracil (PTU). Endocrinology (2012) 153(3):1311–6. doi:10.1210/en.2011-1437
69. Martinez R, Eller C, Viana NB, Gomes FC. Thyroid hormone induces cerebellar neuronal migration and Bergmann glia differentiation through epidermal growth factor/mitogen-activated protein kinase pathway. Eur J Neurosci (2011) 33(1):26–35. doi:10.1111/j.1460-9568.2010.07490
70. van der Deure WM, Peeters RP, Visser TJ. Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OATPs, and the effects of genetic variation in these transporters. J Mol Endocrinol (2010) 44(1):1–11. doi:10.1677/JME-09-0042
71. Schweizer U, Kohrle J. Function of thyroid hormone transporters in the central nervous system. Biochim Biophys Acta (2013) 1830(7):3965–73. doi:10.1016/j.bbagen.2012.07.015
72. Friesema ECH, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab (2006) 2(9):512–23. doi:10.1038/ncpendmet0262
73. Schwartz C, Stevenson RE. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrinol Metab (2007) 21(2):307–21. doi:10.1016/j.beem.2007.03.009
74. Friesema ECH, Visser WE, Visser TJ. Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol (2010) 322:107–13. doi:10.1016/j.mce.2010.01.016
75. Bianco AC, Kim BW. Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest (2006) 116(10):2571–9. doi:10.1172/JCI29812
76. Schneider MJ, Fiering SN, Pallud SE, Parlow AF, Germain DLS, Galton VA. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol (2001) 15:2137–48. doi:10.1210/me.15.12.2137
77. Tata JR. The road to nuclear receptors of thyroid hormone. Biochim Biophys Acta (2013) 1830(7):3860–6. doi:10.1016/j.bbagen.2012.02.017
78. Ercan-Fang S, Schwartz HL, Oppenheimer JH. Isoform-specific 3,5,3′-triiodothyronine receptor binding capacity and messenger ribonucleic acid content in rat adenohypophysis: effect of thyroidal state and comparison with extrapituitary tissues. Endocrinology (1996) 137:3228–33. doi:10.1210/endo.137.8.8754744
79. Gil-Ibanez P, Morte B, Bernal J. Role of thyroid hormone receptor subtypes alpha and beta on gene expression in the cerebral cortex and striatum of postnatal mice. Endocrinology (2013) 154:1940–7. doi:10.1210/en.2012-2189
80. Bradley DJ, Young WS, Weinberger C. Differential expression of alpha and beta thyroid hormone receptor genes in rat brain and pituitary. Proc Natl Acad Sci U S A (1989) 86(18):7250–4. doi:10.1073/pnas.86.18.7250
81. Strait KA, Schwartz HL, Seybokd VS, Ling NC, Oppenheimer JH. Immunofluorescence localization of thyroid hormone receptor protein beta 1 and variant alpha 2 in selected tissues: cerebellar Purkinje cells as a model for beta 1 receptor-mediated developmental effects of thyroid hormone in brain. Proc Natl Acad Sci U S A (1991) 88:3887–91. doi:10.1073/pnas.88.9.3887
82. Mellström B, Naranjo JR, Santos A, Gonzalez AM, Bernal J. Independent expression of the alpha and beta c-erbA genes in developing rat brain. Mol Endocrinol (1991) 5:1339–50. doi:10.1210/mend-5-9-1339
83. Gauthier K, Chassande O, Plateroti M, Roux JP, Legrand C, Pain B, et al. Different functions for the thyroid hormone receptors TRalpha and TRbeta in the control of thyroid hormone production and post-natal development. EMBO J (1999) 18:623–31. doi:10.1093/emboj/18.3.623
84. Flamant F, Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab (2003) 14:85–90. doi:10.1016/S1043-2760(02)00043-7
85. Usala SJ, Menke JB, Watson TL, Wondisford FE, Weintraub BD, Berard J, et al. A homozygous deletion in the c-erbA beta thyroid hormone receptor gene in a patient with generalized thyroid hormone resistance: isolation and characterization of the mutant receptor. Mol Endocrinol (1991) 5:327–35. doi:10.1210/mend-5-3-327
86. Dong H, Yauk CL, Rowan-Carroll A, You SH, Zoeller RT, Lambert I, et al. Identification of thyroid hormone receptor binding sites and target genes using ChIP-on-chip in developing mouse cerebellum. PLoS One (2009) 4(2):e4610. doi:10.1371/journal.pone.0004610
87. Gagne R, Green JR, Dong H, Wade MG, Yauk CL. Identification of thyroid hormone receptor binding sites in developing mouse cerebellum. BMC Genomics (2013) 14:341. doi:10.1186/1471-2164-14-341
88. Chatonnet F, Guyot R, Benoit G, Flamant F. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc Natl Acad Sci U S A (2013) 110(8):E766–75. doi:10.1073/pnas.1210626110
89. Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, et al. A mutation in the thyroid hormone receptor alpha gene. N Engl J Med (2012) 366:243–9. doi:10.1056/NEJMoa1110296
90. van Mullem A, van Heerebeek R, Chrysis D, Visser E, Medici M, Andrikoula M, et al. Clinical phenotype and mutant TRα1. N Engl J Med (2012) 366:1451–3. doi:10.1056/NEJMc1113940
91. Weiss RE, Xu J, Ning GJ, O’Malley B, Refetoff SV. Mice deficient in the steroid receptor coactivator 1 (SRC-1) are resistant to thyroid hormone. EMBO J (1999) 18:1900–4. doi:10.1093/emboj/18.7.1900
Keywords: thyroid hormones, genes, cerebellum, brain development, animal models
Citation: Faustino LC and Ortiga-Carvalho TM (2014) Thyroid hormone role on cerebellar development and maintenance: a perspective based on transgenic mouse models. Front. Endocrinol. 5:75. doi: 10.3389/fendo.2014.00075
Received: 26 February 2014; Accepted: 02 May 2014;
Published online: 20 May 2014.
Edited by:
Frédéric Flamant, Ecole Normale Supérieure de Lyon, FranceReviewed by:
Heike Heuer, Leibniz Institute for Age Research – Fritz Lipmann Institute, GermanyIsabelle Dusart, Centre National de la Recherche Scientifique, France
Copyright: © 2014 Faustino and Ortiga-Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tania M. Ortiga-Carvalho, Laboratorio de Endocrinologia Molecular, Instituto de Biofisica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Av. Carlos Chagas Filho, s/n Cidade Universitária, Rio de Janeiro 21941-902, Brazil e-mail: taniaort@biof.ufrj.br