 Hernan Valdes-Socin1*
Hernan Valdes-Socin1* Matilde Rubio Almanza1
Matilde Rubio Almanza1 Mariana Tomé Fernández-Ladreda1François Guillaume Debray2Vincent Bours2
Mariana Tomé Fernández-Ladreda1François Guillaume Debray2Vincent Bours2 Albert Beckers1
Albert Beckers1
- 1Service of Endocrinology, CHU Liège, University of Liège, Liège, Belgium
- 2Service of Human Genetics, CHU Liège, University of Liège, Liège, Belgium
The neuroendocrine control of reproduction in mammals is governed by a neural hypothalamic network of nearly 1500 gonadotropin-releasing hormone (GnRH) secreting neurons that modulate the activity of the reproductive axis across life. Congenital hypogonadotropic hypogonadism (HH) is a clinical syndrome that is characterized by partial or complete pubertal failure. HH may result from inadequate hypothalamic GnRH axis activation, or a failure of pituitary gonadotropin secretion/effects. In man, several genes that participate in olfactory and GnRH neuronal migration are thought to interact during the embryonic life. A growing number of mutations in different genes are responsible for congenital HH. Based on the presence or absence of olfaction dysfunction, HH is divided in two syndromes: HH with olfactory alterations [Kallmann syndrome (KS)] and idiopathic hypogonadotropic hypogonadism (IHH) with normal smell (normosmic IHH). KS is a heterogeneous disorder affecting 1 in 5000 males, with a three to fivefold of males over females. KS is associated with mutations in KAL1, FGFR1/FGF8, FGF17, IL17RD, PROK2/PROKR2, NELF, CHD7, HS6ST1, FLRT3, SPRY4, DUSP6, SEMA3A, NELF, and WDR11 genes that are related to defects in neuronal migration. These reproductive and olfactory deficits include a variable non-reproductive phenotype, including sensorineural deafness, coloboma, bimanual synkinesis, craniofacial abnormalities, and/or renal agenesis. Interestingly, defects in PROKR2, FGFR1, FGF8, CHD7, DUSP6, and WDR11 genes are also associated with normosmic IHH, whereas mutations in KISS1/KISSR, TAC3/TACR3, GNRH1/GNRHR, LEP/LEPR, HESX1, FSHB, and LHB are only present in patients with normosmic IHH. In this paper, we summarize the reproductive, neurodevelopmental, and genetic aspects of HH in human pathology.
Introduction
Reproductive system development and control in mammals is dependent on specific neurons located in the hypothalamus that secrete gonadotropin-releasing hormone-1 (GnRH-1) and control the pituitary–gonadal axis (Figure 1). During embryogenesis, these neurons originate in the nasal placode and migrate into the forebrain along the olfactory-vomeronasal nerves (1–3). Alterations in this migratory process lead to defective GnRH-1 secretion, resulting in heterogeneous genetic disorders such as idiopathic hypogonadotropic hypogonadism (IHH), and other reproductive diseases characterized by the reduction in or failure of sexual maturation and competence. Another consequence of these migratory neuronal defects can be olfactory dysfunction. Depending of the affected genes, other neurological developmental disorders can also be encountered (1–4).
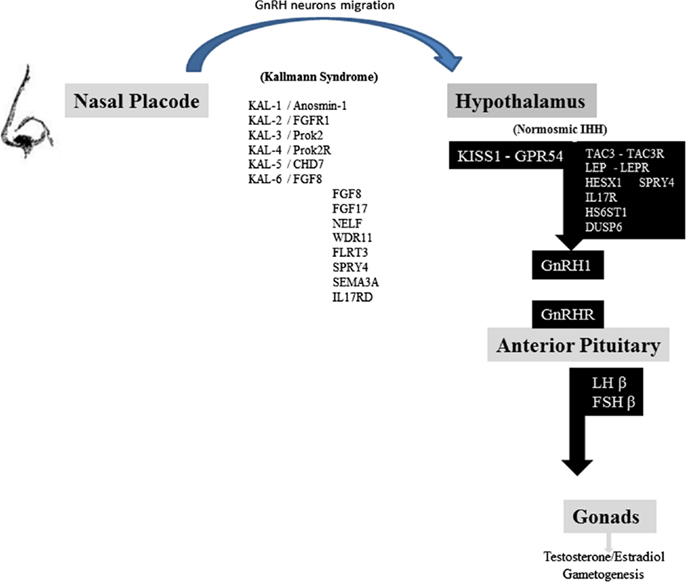
Figure 1. Schematic representation of the reproductive axis and the different genes invalidating mutations participating in Kallman syndrome and nIHH. GnRH neurons migrate from nasal placode to hypothalamus in the first weeks of fetal life. Several genes are represented in the left column: their invalidating mutations are responsible for Kallmann syndrome and olfactory dysfunction. Kiss1 neurons integrate different hormonal, metabolic, circadian inputs from other hypothalamic and brain areas and thus stimulate GnRH neurons firing. Several genes are represented in the right black column, including KISS1-GPR54 system, which invalidating mutations lead to normosmic idiopathic hypogonadotropic hypogonadism. GnRH secretion leads to gonadotropins FSH and LH pituitary release. LH and FSH control sex steroids secretion and gametogenesis [adapted from Valdes-Socin et al. (1), with permission].
Thus, idiopathic hypogonadotropic hypogonadism (IHH) is a genetic disease that can occur with a normal sense of smell (normosmic IHH) or in association with anosmia (Kallmann syndrome; KS). To date, mutations in many genes have been described in relations to KS and/or normosmic IHH (nIHH) (Tables 1 and 2). Hypogonadotropic hypogonadism (HH) can also be found in association with other distinctive clinical syndromic conditions, such as Prader Willi syndrome, that are outside the scope of the current review.
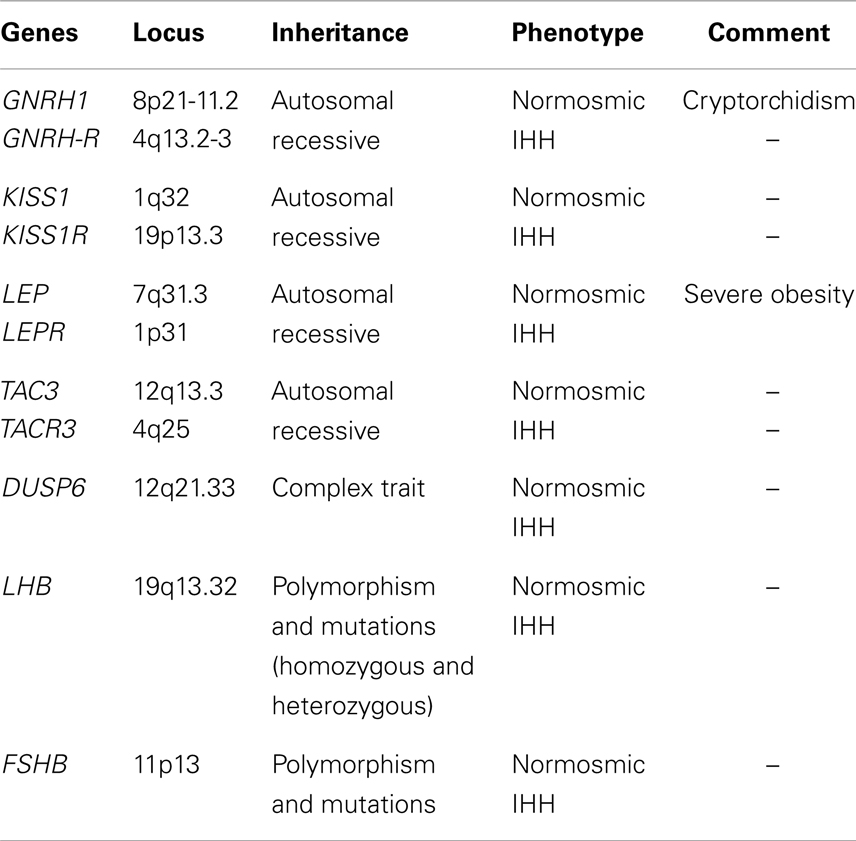
Table 1. Genes and phenotype related only with normosmic IHH.
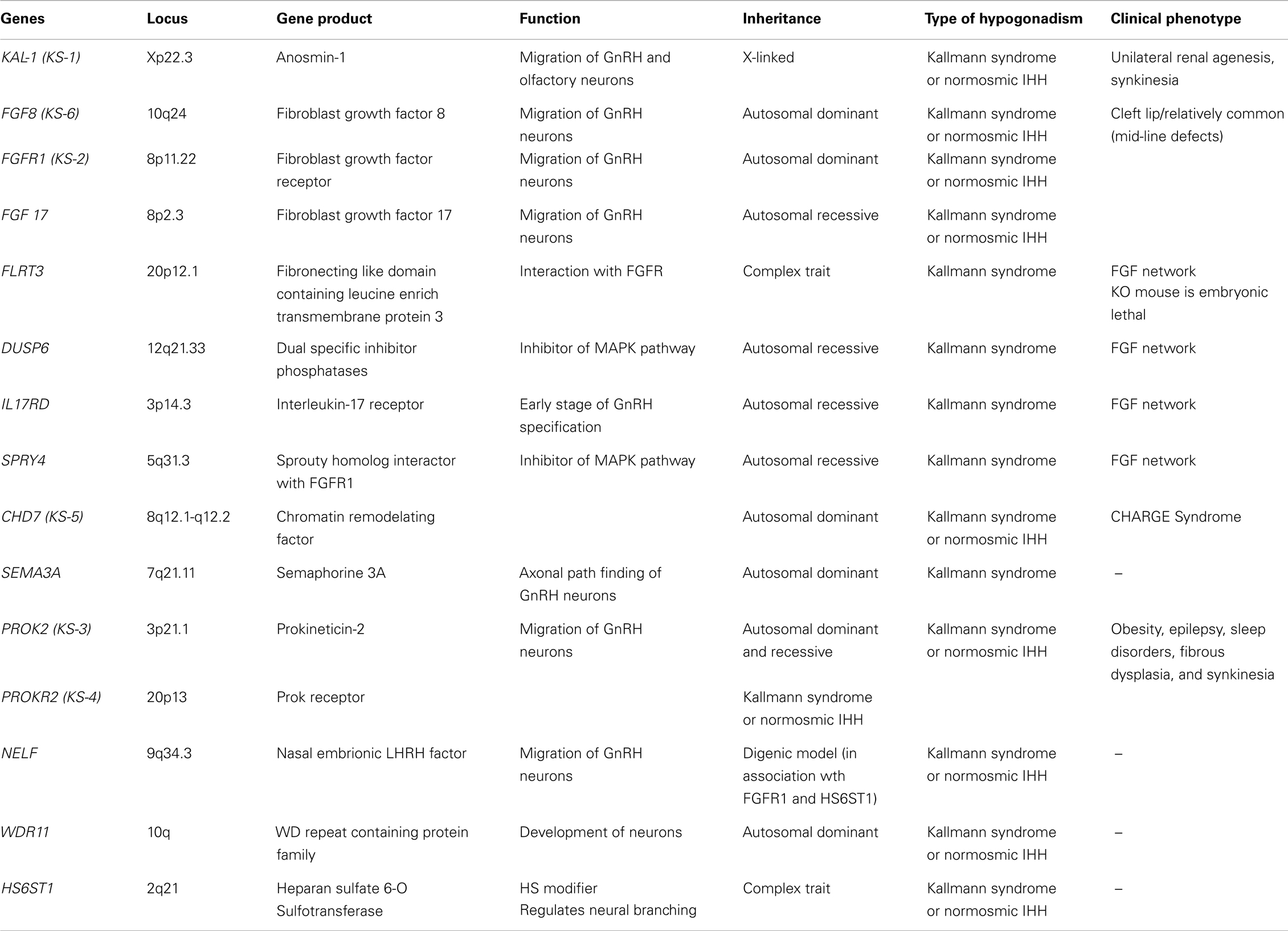
Table 2. Genes, genes product, function, and phenotypes associated to congenital hypogonadism hipogonadotropic with anosmia/hyposmia (KS, Kallmann syndrome).
In this review, we focus on genetic central hypogonadism, which is more frequently encountered in males than in females. Congenital IHH is a clinically and genetically heterogeneous disorder (3, 4). Although sporadic cases predominate, families with congenital IHH have been reported with X-linked, autosomal dominant (AD) or autosomal recessive (AR) inheritance patterns (1–4). In some families, high variability in reproductive and non-reproductive phenotypic features suggests the presence of complex inheritance. In particular, polygenic (digenic or oligogenic) forms and variable forms of transmission can be found in selected cases (6–11). Indeed, further complexity is added by the remarkable observation of reversibility of the phenotype in some cases of genetically determined hypogonadism (12–16).
The Human Reproductive Axis
Normal human reproduction and sexual characteristics rely on an intact hypothalamic–pituitary–gonadal axis (HPG; Figure 1). Hypogonadism is defined as the insufficient production of sex hormones with or without disturbed gametogenesis. HH results from a dysfunction of the hypothalamic–pituitary axis interfering with control of gonadotropin secretion (1–3, 16).
During life, the activity of the HPG axis has a tri-phasic pattern of “on-off-on.” A first phase of activity occurs from the 16th week of intrauterine life as well as in the period between the 4th and 10th weeks of postnatal life (or “mini-puberty”). Mini-puberty is characterized by an increase in gonadotropin and steroid hormone secretion. Gonadotropins and sex hormones levels rise to a lesser extent than in true puberty. After mini-puberty, the HPG axis is repressed (“off”) until puberty, when the system is reactivated (“on”). HPG axis activity is maintained throughout adult life in men whereas in women, menopause intervenes, and low sex steroids and compensatory high gonadotropin levels are characteristic (1–3).
The immediate postnatal period can be a window of opportunity for pediatricians and neonatologists to diagnose certain forms of HH. The congenital gonadotropin deficiency phenotype is variable and depends on the gender, the magnitude of the deficit, and the specific genetic abnormalities (Figure 1). At the time of puberty, the diagnosis of HH may be suspected due to the absence in the onset of puberty and development of secondary sex characteristics in both sexes. In adulthood, gonadotropin deficiency can be suspected in a woman without breast development or who presents with primary amenorrhea. In adult men, gynecomastia, small testes (<14 mL), penile hypoplasia, and/or oligo-azoospermia raise the clinical suspicion of congenital hypogonadism (1–3).
Normosmic Idiopathic Hypogonadotropic Hypogonadism
The genetic abnormalities described below are infrequent or rare (see Table 1). In contrast to KS, patients with nIHH have a normal sense of smell and tend not to have other clinical signs. From a biological point of view, sex steroids secretion and gametogenesis are compromised, but to varying degrees. As it would be expected, reproductive phenotypes are more pronounced in subjects in whom the receptor is inactivated as compared to those harboring hormone inactivating mutations.
GNRH-1 and GNRHR mutations
Gonadotropin-releasing hormone (GnRH) is encoded by the GNRH1 gene, which is located on chromosome 8p21-11.2. GNRH-1 mutations are rare and have been described in only two families (17, 18). A single homozygous mutation (c.18-19insA) affecting the peptide precursor preproGnRH was described in a Romanian family (17). It encoded a truncated and biologically inactive peptide in a male patient and his sister, both of whom had delayed puberty and normal sense of smell. The phenotype was reversed by pulsatile GnRH administration. Another homozygous GNRH1 mutation was identified in a prepubertal boy from Armenia, with cryptorchidism and microphallus (18).
The GNRHR gene (locus on chromosome 4q13.2-3) encodes for the GNRH receptor. There is some variability in clinical expression of GnRHR mutations that is due to a partial loss of function. GNRHR mutations have been described in about 40–50% of familial AR nIHH cases, and in around 17% of sporadic nIHH (1–3).
KISS1 and GFPR54 mutations
The gene KISS1 was described originally as a metastasis suppressor gene but it is a key gene in reproduction. It is localized on chromosome 1q32, encoding a protein called kisspeptin, which is, in turn, processed in four peptides Kp10, Kp13, Kp14, and Kp54. Kisspeptins stimulate GnRH neuronal firing and GnRH secretion, which then triggers an increased release of LH and FSH (Figure 1). The KISS1R gene (locus 19p.13.3), a G-protein-coupled receptor, is also known as the GPR54 gene, and it is the receptor for kisspeptins. GPR54 mutations can be compound heterozygous or homozygous (19–22). Loss of function mutations in KISS1 (20) and GPR54 cause HH in mice and men (19, 21, 22). Moreover, higher serum kisspeptins levels are found in obese hypogonadal men and in central hypogonadism than in controls (23). In patients with GPR54 mutations, GnRH deficiency can be partial or complete, permanent, or reversible, and can have a congenital or adult onset. Six homozygous inactivating mutations have been described in 19 individuals with nIHH: their LH and FSH secretion was blunted but normal secretion was restored after exogenous GnRH stimulation (19, 21, 22). Kisspeptins are highly expressed in placenta during pregnancy; different patterns of spatiotemporal expression of KISS1 and KISSR were described in normal and pathological placentas (24).
TAC3R and TAC3 mutations
The TACR3 gene (chromosome 4q25) encodes the neurokinin 3 receptor (NK3R) and the TAC3 gene (chromosome 12q13.3) encodes neurokinin B (NKB), its endogenous ligand. nIHH caused by mutations in TAC3 and TAC3R have an AR heritance (25). As well as the GPR54/Kisspeptin system, TACR3/TAC3 pathway stimulates GnRH neurons. In initial studies, defects in either TAC3 or TACR3 were found in 11 patients from 5 of 10 families studied, but in none of 50 sporadic cases (25–27). Francou et al. studied the gonadotropin axis dysfunction associated with nCHH due to TAC3/TACR3 mutations: it was related to a low GnRH pulsatile frequency leading to a low frequency of alpha-subunit pulses and to an elevated FSH/LH ratio (27). They suggested that this ratio might be useful for pre-screening nCHH patients for TAC3/TACR3 mutations. In another broad cohort of normosmic IHH patients, 7 of the 16 males and 5 of the 7 females with TACR3/TAC3 mutations were assessed after discontinuation of therapy: 6 of the 7 males and 4 of the 5 females demonstrated evidence for reversibility of their hypogonadotropism (14).
Leptin (Ob) and leptin receptor mutations
Leptin is an adipocyte secreted protein that ensures a link between body fat and the reproductive axis. HH and severe obesity are seen in humans and ob/ob mice with genetic leptin deficiency. There are at least 12 patients with leptin deficiency and homozygous mutations. In such cases, recombinant leptin administration restores gonadotropin secretion and dramatically reduces body mass index. Defects in the leptin receptor are more common, being identified in 3% of severe early onset obesity patients. Interestingly, the leptin receptor is expressed on kisspeptin neurons whereas leptin administration induces the expression of Kiss-1 in ob/ob mice (3, 28).
LHB mutations
The LHB subunit gene is located at chromosome 19q13.32. Five mutations have been published up to now; clinical and molecular data are summarized in Table 3. The syndrome of preserved spermatogenesis with androgenic failure (now known to be due to LH deficiency) was described for the first time by Pasqualini and Bur in 1950 (29). The term “fertile eunuch” was then coined to describe these men.
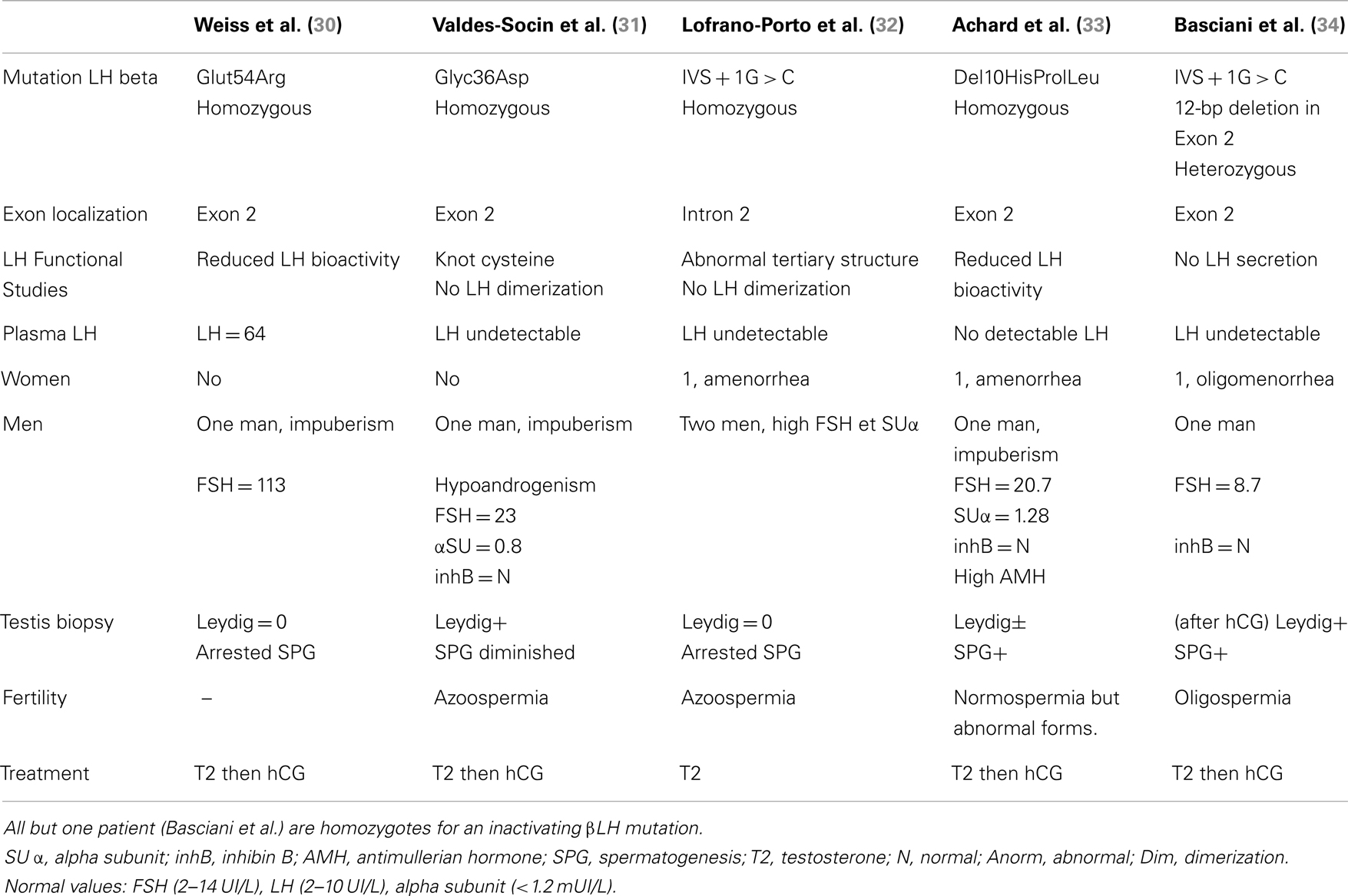
Table 3. Clinical, biological, pathological, and genetic studies in patients with LH deficiency.
In affected men, sexual differentiation was normal, but the absence of or significantly reduced LH secretion restrained the induction of puberty and altered Leydig cell proliferation and maturation (30–34). These males have impaired spermatogenesis, ranging from azoospermia to oligospermia, which has been linked to the lack of LH stimulation and low intratesticular testosterone action (5, 30–35). In 2004, we described a man with a homozygous missense mutation (G36D) in the LHB subunit gene that abrogated subunit dimerization and rendered LH biologically and immunologically inactive (31). Treatment with human chorionic gonadotropin (hCG) induced near normalization of testicular structure (5). The patient and his wife conceived a child by intracytoplasmic sperm injection from ejaculated sperm. The male heterozygous child had normal LH, FSH, and testosterone levels, at the age of 4 weeks (5, 35).
In women, LHB mutations lead to a normal pubertal development but they can have primary amenorrhea and micropolycystic ovaries (32–34).
FSHB mutations
The β subunit of FSH (FSHB) is located at chromosome 11p13. Three men and four women with inactivating FSH mutations have been reported. Men have normal pubertal development although they have azoospermia, whereas women have abnormal pubertal maturation; in these patients high level of LH are found whereas FSH is low/undetectable. Estrogen and progesterone concentrations are low (1, 2).
Gonadotropins receptor (LHR and FSHR) mutations
Inactivating mutations affecting the gonadotropin receptors contrast with those affecting their ligands in that they are invariably associated with hypergonadotropic hypogonadism; hence, they are not discussed here.
Kallmann Syndrome
Kallmann syndrome, involving the characteristic features of HH and anosmia was noted in the historical literature long before being properly characterized as a genetic disorder. A man with delayed puberty and the lack of olfactory bulbs was reported over 150 years ago by the Spanish doctor Aureliano Maestre de San Juan (1828–1890). The German Franz Kallmann (1897–1965) completed in the 1940s a description of hypogonadism and anosmia in two families, establishing the genetic basis of transmission. The Swiss scientist, Georges de Morsier (1894–1982) provided the neuropathological description of the syndrome. KS has a prevalence of 1/5000, with a clear male predominance (1–3, 36, 37). Several mechanisms of inheritance and molecular mutations are described here after and summarized in Table 2.
Anosmin-1 (KAL-1) mutations
The KAL-1 gene is located on the X chromosome at Xp22.3. KAL-1 encodes anosmin-1, a glycoprotein playing an in important role in kidney, respiratory tract, digestive system, and brain embryogenesis (1–3, 37, 38). Anosmin-1 is an adhesion molecule located on cell surface, consistent with the underlying defect of embryonic neuronal migration in KS. Anosmin-1 is mainly involved in growth and migration of GnRH, mitral olfactory cells, and Purkinje cerebellum neurons. Mutations in KAL-1 gene cause 14% of familial cases of KS and 11% of cases of sporadic cases (1–3, 38). KAL-1 mutations lead to HH with or without anosmia and may include synkinesis (mirror movements), unilateral renal aplasia, and mid-line abnormalities such as cleft lip/palate (37, 38).
FGF8 (KAL-6), FGF17, and FGFR1 (KAL-2) mutations
The FGF8 gene (also known as KAL-6) is located on chromosome 10q24. Fibroblast growth factors (FGF) interact with FGF tyrosine kinase receptors to mediate growth and development. FGF8 participates in gastrulation, regionalization of the brain, and organogenesis of the limb and face as an embryonic epithelial factor. FGF8 and its receptor FGFR1 are involved in GnRH neuron migration. FGF8 inactivating mutations can lead to both KS and nIHH with an AD inheritance. Triallelic inheritance has also been described. In addition, cleft lip or palate and other mid-line defects have been described in patients with FGF8 and FGFR1 mutations. Other features such as corpus callosum hypoplasia-agenesis or nose, ear, and finger abnormalities are more specific of FGFR1 defects (37, 39, 40).
FGF17 is located at chromosome 8p2.3 and FGF17 has a strong sequence identity with FGF8. FGF17 might be implicated in GnRH neuron biology as an alternative to ligand FGF8b. Miraoui et al. have identified FGF17 heterozygous mutations in three patients with congenital HH and anosmia and in another individual. In a sporadic male patient with congenital, HH without anosmia (41).
FGFR1 is located at 8p11.22-p11.23 and FGFR1 mutations have been identified in 10% of KS. FGFR1 related KS has an AD inheritance, associated with incomplete penetrance and interfamilial variability. FGFR1 encodes for type 1 FGF receptor, which is expressed in several embryonic tissues. The activation of the FGF–FGFR complex requires two FGF ligands. FLRT3 (Fibronecting like domain containing leucine enrich transmembrane protein 3) also interacts with FGFR (see Table 2). In addition, the binding of heparin or HS: heparan sulfate proteoglycan (see HS6ST1 gene later) have been shown to be essential for FGF receptor dimerization and function (38). Mice with Fgfr1−/− mutations and patients with loss-of-function mutations in FGFR1 have defective GnRH neuron migration (31). Thus, loss of function mutations in FGFR1 which is involved in the development of the face, lead to the abnormal morphogenesis of the olfactory bulb, while specific gain-of function mutations in FGFR1 cause craniosynostosis (37, 39–41).
PROK2 and PROKR2 mutations
PROK2 (locus 3p21.1) and PROKR2 (locus 20p13) genes encode for prokineticin-2 and its receptor (42). Prok2 and prokr2 gene knockout mice both have agenesis or hypoplasia of the olfactory bulbs, in association with HH. In this model, there is also abnormal GnRH neuron migration (43). Its heritance can be AD or AR. Mutations in these genes are described in up to 6% of KS and 3% of nIHH (1–3, 42). In pituitary deficits associated with septo-optical dysplasia, McCabe et al. (44), described a patient with a heterozygous L173R mutation in PROKR2 gene, while its healthy mother is homozygous for this mutation. As some controversy exists on the pathogenic role of some PROKR2 mutations, the prevalence given should be interpreted cautiously. Indeed, digenic mutations are encountered (i.e., PROKR2 and KAL1), while heterozygous patients (i.e., AD transmission) are present in families with healthy relatives presenting the same genotype. There are no reliable accessory pathognomonic features of PROK2/PROKR2 function loss: patients have been described with obesity, sleep disorder, fibrous dysplasia, epilepsy, and synkinesia (2, 3, 43, 44).
NELF mutations
The NELF gene is located at chromosome 9q34.3. This gene encodes the nasal embryonic LHRH factor. The NELF gene is detected in olfactory sensory cells and GnRH cells during embryonic development. It constitutes a guidance molecule for the olfactory axon and GnRH neurons across the nasal region (45). NELF mutations have been described in patients with KS, in association with mutations in FGFR1 or HS6ST1, indicating digenic inheritance. More studies are necessary to confirm a relationship between NELF and any reproductive and olfactory disorders (2).
WDR11 mutations
The WDR11 locus is at chromosome 10q26.12 and its heritance is AD. It encodes murine Wdr11 that is expressed in the developing olfactory and GnRH migratory pathway and in the adult hypothalamus. WDR11 biological function is not well understood: however, Kim et al. identified five different heterozygous mutations in nIHH and KS patients. WDR11 probably also plays an important role in puberty (46).
CHD7 mutations
The CHD7 gene that encodes a chromatin-remodeling factor is located on chromosome 8q12.1. Mutations (AD inheritance) of this gene can cause CHARGE syndrome (Colobomata, Heart Anomalies, Choanal Atresia, Retardation, Genital, and Ear anomalies). CHD7 was screened in nearly 200 patients: 7 KS and nIHH patients were found, 3 of them with olfactory abnormalities. CHD7 mutations were identified in 6% of KS and 6% of nIHH, respectively (1, 2, 47, 48). Laittinen et al. described in 2012, a KS patient with a truncating CHD7 mutation that underwent a reversal of central hypogonadism after therapy discontinuation (15).
HS6ST1 mutations
The HS6ST1 gene (locus 2q.21) encodes a 6-O-sulfation enzyme, which is a member of the heparan sulfate enzyme family. The protein is involved in normal neuronal development and may play a role in limb development. In nematodes, HS 6-O-sulfate interacts with anosmin-1 and it is involved in function of FGFR1 and FGF8.
HS6ST1 shows complex inheritance patterns, not following autosomal or recessive transmission. HS6ST1 mutations were found in KS patients in combination with mutations affecting the FGFR1 gene. HS6ST1 mutations were found in patients who had nIHH or variable degrees of olfactory dysfunction (KS) as well as with either normal or abnormal olfactory structures (49).
IL17RD, DUSP6, and SPRY4 mutations
The IL17RD gene (locus 3p14.3) encodes a membrane protein belonging to the interleukin-17 receptor (IL-17R) protein family. In a study with eight patients with congenital hypogonadism all had KS, 7/8 had absent puberty, 6/8 showed congenital hearing loss. One IL17RD allelic defect is likely to be insufficient, meaning that additional affected alleles in the same and/or other genes must be present to create the phenotype of KS with hearing loss (41).
DUSP6 (locus 12q22-q23) encodes a member of the dual specificity protein phosphatase subfamily. They negatively regulate members of the mitogen-activated protein (MAP) kinase superfamily (25) Three patients were described with DUSP6 and FGFR1 heterozygous mutation; they were hypogonadic, while one had hearing loss and the two others had abnormal speech. DUSP6−/− mice are however viable and fertile (41).
SPRY4 (locus 5q.31.3) gene encodes a protein (sprouty homolog 4), which is an inhibitor of the receptor-transduced mitogen-activated protein kinase (MAPK) signaling pathway. It is positioned upstream of RAS gene activation and impairs the formation of active GTP-RAS. Diseases associated with SPRY4 include germ cell cancer, and testicular cancer. Miraoui et al. identified four anosmic patients with congenital HH (three females and one male) with heterozygosity for a c.530A-G transition in exon 3 of the SPRY4 gene. Another female patient had a heterozygosity for a c.910G-A transition in exon 3 of the SPRY4 gene. These mutations were not found in 155 controls. One of the patients also had hearing loss and another one had abnormal dentition (41).
HESX1 mutations
The HESX1 gene (locus 3p14.3) encodes a protein that is a transcriptional repressor in the developing forebrain and pituitary gland (27). HESX1 plays an important role in the temporal and sequential development of the forebrain, hypothalamus, optic nerve, and posterior pituitary (28). Mutations in HESX1 have also been described in isolated growth hormone deficiency and combined pituitary deficiency (50, 51). HESX1 mutations have been described in 1.4% of IHH/KS patients (50, 51), but as in PROKR2 mutations this prevalence should be interpreted cautiously.
SEMA3A mutations
The SEMA3A gene (7q21.11) encodes the semaphorin 3A protein, which regulates axonal path finding and participates in GnRH migration. Deletions and mutations of the SEMA3A gene validate a role for SEMA3A in KS. Moreover, SEMA3A knockout mice exhibit GnRH dependent hypogonadism and abnormal olfactory bulb innervation (52).
Conclusion and Perspectives
Kallmann syndrome and nIHH have the potential to unravel the processes behind normal embryonic development and reproductive neuroendocrine maturation (2). The complex biological path from childhood to the onset of human puberty is still incompletely understood (1–3). The molecular mechanism behind IHH remains unknown in a large number of cases. Over the past decade, the same genetic mutations have been described in associated with both KS and nIHH. Moreover, a significant clinical heterogeneity is seen in isolated GnRH deficiency, which might be explained to some extent by oligogenicity (6, 7, 9). In addition, over 60% of central hypogonadic patients have no identifiable mutations, suggesting that yet more disease loci remain to be discovered (1–3). These unidentified genes warrant an integrated research including clinicians, geneticists, and biological investigators to pursue further understanding of these fascinating cases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the National Fund of Scientific Research (FNRS, Brussels, Belgium), for their financial support and Dr AF Daly for kindly reviewing the manuscript.
References
1. Valdes-Socin S, Debray FG, Parent AS, Lebrethon MC, Bourguignon JP, Bours V, et al. How to explore…congenital isolated hypogonadotropic hypogonadism? Rev Med Liege (2010) 65(11):634–41.
2. Semple R, Topaloglu KA. The recent genetics of hypogonadotrophic hypogonadism – novel insights and new questions. Clin Endocrinol (Oxf) (2010) 72(4):427–35. doi: 10.1111/j.1365-2265.2009.03687.x
3. Nachtigall LB, Boepple PA, Pralong FP, Crowley WF Jr. Adult-onset idiopathic hypogonadotropic hypogonadism – a treatable form of male infertility. N Engl J Med (1997) 336:410–5. doi:10.1056/NEJM199702063360604
4. Lippincott MF, True C, Seminara SB. Use of genetic models of idiopathic hypogonadotrophic hypogonadism in mice and men to understand the mechanisms of disease. Exp Physiol (2013) 98:1522–7. doi:10.1113/expphysiol.2013.071910
5. Valdes-Socin H, Salvi R, Thiry A, Daly AF, Pralong FP, Gaillard R, et al. Testicular effects of isolated luteinizing hormone deficiency and reversal by long-term human chorionic gonadotropin treatment. J Clin Endocrinol Metab (2009) 94:3–4. doi:10.1210/jc.2008-1584
6. Trarbach EB, Costa EM, Versiani B, de Castro M, Matias Baptista MT, Mendes Garmes H, et al. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab (2006) 91:4006–12. doi:10.1210/jc.2005-2793
7. Pitteloud N, Acierno JS Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A (2006) 103:6281–6. doi:10.1073/pnas.0600962103
8. Pitteloud N, Hayes FJ, Boepple PA, DeCruz S, Seminara SB, MacLaughlin DT, et al. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab (2002) 87:152–60. doi:10.1210/jcem.87.1.8131
9. Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A (2010) 107:15140–4. doi:10.1073/pnas.1009622107
10. Fukami M, Maruyama T, Dateki S, Sato N, Yoshimura Y, Ogata T. Hypothalamic dysfunction in a female with isolated hypogonadotropic hypogonadism and compound heterozygous TACR3 mutations and clinical manifestation in her heterozygous mother. Horm Res Paediatr (2010) 73:477–81. doi:10.1159/000313373
11. Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, et al. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab (2012) 97:136–44. doi:10.1210/jc.2011-2041
12. Raivio J, Falardeau A, Dwyer R, Quinton FJ, Hayes VA, Hughes LW, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med (2007) 357:863–73. doi:10.1056/NEJMoa066494
13. Pitteloud N, Acierno JS Jr, Meysing AU, Dwyer AA, Hayes FJ, Crowley WF Jr. Reversible Kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab (2005) 90:1317–22. doi:10.1210/jc.2004-1361
14. Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab (2010) 95:2857–67. doi:10.1210/jc.2009-2320
15. Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One (2012) 7:e39450. doi:10.1371/journal.pone.0039450
16. Cariboni A, Maggi R. Kallmann’s syndrome, a neuronal migration defect. Cell Mol Life Sci (2006) 63(21):2512–26. doi:10.1007/s00018-005-5604-3
17. Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med (2009) 360:2742–8. doi:10.1056/NEJMoa0900136
18. Chan YM, de Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, Tsiaras S, et al. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A (2009) 106:11703–8. doi:10.1073/pnas.0903449106
19. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A (2003) 100:10972–6. doi:10.1073/pnas.1834399100
20. Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med (2012) 366:629–35. doi:10.1056/NEJMoa1111184
21. Brioude F, Bouligand J, Francou B, Fagart J, Roussel R, Viengchareun S, et al. Two families with normosmic congenital hypogonadotropic hypogonadism and biallelic mutations in KISS1R (KISS1 receptor): clinical evaluation and molecular characterization of a novel mutation. PLoS One (2013) 8(1):e53896. doi:10.1371/journal.pone.0053896
22. Tenenbaum-Rakover Y, Commenges-Ducos M, Iovane A, Aumas C, Admoni O, de Roux N. Neuroendocrine phenotype analysis in five patients with isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of GPR54. J Clin Endocrinol Metab (2007) 92:1137–44. doi:10.1210/jc.2006-2147
23. Valdes-Socin H, Cavalier E, Beckaert A, Carlisi A, Chavez M, Beckers A. High plasma kisspeptins in obese men with acquired hypogonadism and in men with congenital hypogonadism: pilot study of plasma kisspeptins variations before and after hCG and testosterone administration. Ann Endocrinol (Paris) (2011).
24. Valdes-Socin H, Munaut C, Chavez M, Chantrain F, Delbecque K, Delvenne P, et al. Expression and spatio-temporal distribution of KISS1 and its receptor KISSR in human normal and pathological placentae. Ann Endocrinol (Paris) (2012) 4:374. doi:10.1016/j.ando.2012.07.494
25. Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan D, Porter KM, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet (2009) 41:354–8. doi:10.1038/ng.306
26. Guran T, Tolhurst G, Bereket A, Rocha N, Porter K, Turan S, et al. A novel missense mutation in the first extracellular loop of the neurokinin B receptor causes hypogonadotropic hypogonadism. J Clin Endocrinol Metab (2009) 94:3633–9. doi:10.1210/jc.2009-0551
27. Francou B, Bouligand J, Voican A, Amazit L, Trabado S, Fagart J, et al. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One (2011) 6:e25614. doi:10.1371/journal.pone.0025614
28. Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med (2007) 356:237–47. doi:10.1056/NEJMoa063988
29. Pasqualini RQ, Bur GE. Síndrome hipoandrogénico con gametogénesis conservada: clasificación de la insuficiencia testicular. Rev Asoc Med Argent (1950) 64:15–30.
30. Weiss J, Axelrod L, Whitcomb RW, Harris PE, Crowley WF, Jameson JL. Hypogonadism caused by a single amino acid substitution in the subunit of luteinizing hormone. N Engl J Med (1992) 326:179–83. doi:10.1056/NEJM199201163260306
31. Valdes-Socin H, Salvi R, Daly AF, Gaillard RC, Quatresooz P, Tebeu P-M, et al. Hypogonadism in a patient with a mutation in the luteinizing hormone-subunit gene. N Engl J Med (2004) 351:2619–25. doi:10.1056/NEJMoa040326
32. Lofrano-Porto A, Barra GB, Giacomini LA, Nascimento PP, Latronico AC, Casulari LA, et al. Luteinizing hormone mutation and hypogonadism in men and women. N Engl J Med (2007) 357:897–904. doi:10.1056/NEJMoa071999
33. Achard C, Courtillot C, Lahuna O, Me’duri G, Soufir JC, Lie’re P, et al. Normal spermatogenesis in a man with mutant luteinizing hormone. N Engl J Med (2009) 361:1856–63. doi:10.1056/NEJMoa0805792
34. Basciani S, Watanabe M, Mariani S, Passeri M, Persichetti A, Fiore D, et al. Hypogonadism in a patient with two novel mutations of the luteinizing hormone –subunit gene expressed in a compound heterozygous form. J Clin Endocrinol Metab (2012) 97:3031–8. doi:10.1210/jc.2012-1986
35. Daly A, Valdes-Socin H, Beckers A. A tall man with hypogonadism. In: Wartofsky L, editor. Diagnostic Dilemmas: Images in Endocrinology. Washington DC: The Endocrine Society Press (2011). p. 204–7.
36. Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, Wunderle V, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell (1991) 67:423–35. doi:10.1016/0092-8674(91)90193-3
37. Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab (2004) 89:1079–88. doi:10.1210/jc.2003-030476
38. Tsai PS, Gill JC. Mechanisms of disease: insights into X-linked and autosomal-dominant Kallmann syndrome. Nat Clin Pract Endocrinol Metab (2006) 2:160–71. doi:10.1038/ncpendmet0119
39. Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet (2003) 33:463–5. doi:10.1038/ng1122
40. Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet (2006) 2(10):e175. doi:10.1371/journal.pgen.0020175
41. Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, et al. Mutations in FGF17, IL17RD, DUPS6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet (2013) 92:725–43. doi:10.1016/j.ajhg.2013.04.008
42. Cole LW, Sidis Y, Zhang CK, Quinton R, Plummer L, Pignatelli D, et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab (2008) 93:3551–9. doi:10.1210/jc.2007-2654
43. Matsumoto S, Yamazaki C, Masumoto K, Nagano M, Naito M, Soga T, et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci U S A (2006) 103:4140–5. doi:10.1073/pnas.0508881103
44. McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O, et al. Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab (2013) 98:547–57. doi:10.1210/jc.2012-3067
45. Kramer PR, Wray S. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev (2000) 14:1824–34.
46. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet (2010) 87(4):465–79. doi:10.1016/j.ajhg.2010.08.018
47. Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet (2008) 83:511–9. doi:10.1016/j.ajhg.2008.09.005
48. Bergman JE, de Ronde W, Jongmans MC, Wolffenbuttel BH, Drop SL, Hermus A, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab (2012) 97(5):E858–62. doi:10.1210/jc.2011-2652
49. Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci U S A (2011) 108:11524–9. doi:10.1073/pnas.1102284108
50. McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou A, et al. HESX1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol Metab (2007) 92:691–7. doi:10.1210/jc.2006-1609
51. Newbern K, Natrajan N, Kim H-G, Chorich LP, Halvorson LM, Cameron RS, et al. Identification of HESX1 mutations in Kallmann syndrome. Fertil Steril (2013) 99:1831–7. doi:10.1016/j.fertnstert.2013.01.149
Keywords: reproduction, male, Kallman syndrome, hypogonadotropic hypogonadism, olfaction, kisspeptin, genetics
Citation: Valdes-Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M, Debray FG, Bours V and Beckers A (2014) Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front. Endocrinol. 5:109. doi: 10.3389/fendo.2014.00109
Received: 31 March 2014; Accepted: 24 June 2014;
Published online: 09 July 2014.
Edited by:
Gianluca Tamagno, St Columcille’s Hospital, IrelandReviewed by:
Alexandru Saveanu, Aix Marseille University, FranceThomas King, Mater Misericordiae University Hospital, Ireland
Copyright: © 2014 Valdes-Socin, Rubio Almanza, Tomé Fernández-Ladreda, Debray, Bours and Beckers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hernan Valdes-Socin, Service of Endocrinology, Centre Hospitalier Universitaire, Rue de l’Hôpital 1, Liège 4000, Belgium e-mail: hg.valdessocin@chu.ulg.ac.be