 Ryan Michael Cassidy
Ryan Michael Cassidy Qingchun Tong
Qingchun Tong- Brown Foundation of the Institute of Molecular Medicine for the Prevention of Human Diseases of McGovern Medical School, Neuroscience Program MD Anderson Cancer Center and UTHealth Graduate School of Biological Sciences, The University of Texas Health Science Center at Houston, Houston, TX, USA
Many of the neurocircuits and hormones known to underlie the sensations of hunger and satiety also substantially alter the activity of the dopaminergic reward system. Much interest lies in the ways that hunger, satiety, and reward tie together, as the epidemic of obesity seems tied to the recent development and mass availability of highly palatable foods. In this review, we will first discuss the basic neurocircuitry of the midbrain and basal forebrain reward system. We will elaborate how several important mediators of hunger—the agouti-related protein neurons of the arcuate nucleus, the lateral hypothalamic nucleus, and ghrelin—enhance the sensitivity of the dopaminergic reward system. Then, we will elaborate how mediators of satiety—the nucleus tractus solitarius, pro-opiomelanocortin neurons of the arcuate nucleus, and its peripheral hormonal influences such as leptin—reduce the reward system sensitivity. We hope to provide a template by which future research may identify the ways in which highly rewarding foods bypass this balanced system to produce excessive food consumption.
Introduction
In evolutionary psychology, a supernormal stimulus or “superstimulus” is some evolutionarily novel concentration of engaging characteristics, which produces a stronger response than the natural one (1). As Pinker described it, strawberry cheesecake is a superstimulus as compared to a Neolithic human diet; it overloads the senses and drives caloric overconsumption, combining the “sweet taste of ripe fruit, the creamy mouth feel of fats and oils from nuts and meat, and the coolness of fresh water” (2). The debate is ongoing as to what exactly differentiates a superstimulus from a regular one or whether it is truly maladaptive to create them or seek them out (1). Nevertheless, an important concept for neuroscience emerges from this discussion; certain systems governing the reaction to rewarding stimulus can be overloaded and their negative feedback component overridden. This hypothesis provides explanation for the panoply of excessive behaviors we cope with as a society, often conceptualized as “behavioral addictions”: drug addiction, internet addiction, porn addiction, food addiction, and so on (3). Even physical activity in some individuals meets the criteria for behavioral addiction, demonstrating the complex nature of this phenomenon (4). It is necessary to provide biological evidence for the existence of the balanced system which these superstimuli overload.
The neurocircuitry and endocrinology underlying hunger and satiety may represent the best studied system in this regard. It clearly produces a physiological balance in certain conditions with regular stimulus, such as a laboratory mouse fed rodent chow its whole life producing a normal weight, and is dysregulated in other conditions that contain superstimuli, such as when that mouse is fed a high-fat, high-carbohydrate diet producing obesity. The signals encoding hunger and satiety alter the brain’s dopaminergic reward system in a multitude of ways. In this review, we will discuss the mechanisms by which these signals, primarily hypothalamic neurocircuits and neuropeptides in combination with peripheral hormones, modulate midbrain dopaminergic activity to alter reward salience and value. By laying out this evidence, we provide a substrate for future research to examine how superstimulus foods, such as cheesecake or high-fat/high-carbohydrate chow, drive so-called hedonic feeding and produce obesity.
The relationship of the midbrain dopaminergic reward system and hypothalamic neurocircuits governing hunger and satiety, hereafter referred to as the reward system and hunger system, is ancient. The patterned expression of genes necessary to produce segmentation of the brain at the mesencephalon (midbrain) and diencephalon (hypothalamus) occurred very early in chordate evolution (5, 6). Both dopamine neuronal receptors and hypothalamic feeding-related peptides and their associated receptors are present in most vertebrates and have similar functions across taxa (7–9). Given this intimate association, it is not surprising that they share a fundamental interdependence. For example, dopamine-deficient mice stop feeding a few weeks after birth; administration of l-DOPA reverses this phenotype and restores normal growth (10). Conversely, knockout of orexin, a hypothalamic neuropeptide associated with hunger, reduces dopamine response to cocaine (11). They also produce cross-sensitization or desensitization; food-restricted, hungry mice have enhanced response and reinforcement to amphetamine or cocaine (drugs which flood the brain with dopamine), and satiety signals such as leptin reduce the drive to seek self-administration of these drugs (12, 13). As will be discussed below, hypothalamic and endocrine components of the hunger system alter the activity of the reward system. To demonstrate this, we will first present a brief overview of the neurocircuitry of the reward system. Then, we will chart the various ways the hunger system interacts with the reward system.
Neurocircuitry of the Reward System
The Ventral Tegmental Area (VTA)
The VTA and substantia nigra pars compacta (SNc) are immediately caudal to the posterior hypothalamus, brace the third ventricle, and contain the major source of dopaminergic outflow to the rest of the brain. The SNc is best known for its role in the nigrostriatal pathway regulating the dorsal striatum in movement, and the VTA for mediating salience, motivation, and reward and aversion-related learning (14, 15). Salience refers to the attention paid to the stimulus; an increase in salience means the stimulus, if identified, will be more likely to draw the organism’s attention. The value of the stimulus, whether it is rewarding or aversive, refers to whether a stimulus induces behavior to acquire it or avoid it, respectively (15). Rewarding stimuli produce a positive valence when acquired and a negative valence when unable to be acquired; the converse is true with aversive stimuli (15).
The VTA dopaminergic neurons are the primary mediators of the behavioral response to a rewarding or aversive stimulus (16). They are not uniform in their activity or projection targets, and thus activation of one neuron may produce substantially different behavioral output than another. This is why studies evaluating the rewarding nature of dopamine often focus on the VTA to nucleus accumbens (NAc) projections specifically; this will be discussed below. However, much effort has been spent elucidating the ways in which local VTA dopaminergic neurons encode reward across brain regions by alteration in firing pattern, increase or decrease in action potential frequency, and projection target. The literature is incomplete on this topic, but discussion of some of these mechanisms sets the stage for further discussion of how the hunger system interfaces with the VTA dopaminergic neurons. For example, in vivo recording of the VTA during a conditioned place preference task suggests that one subset of dopaminergic neurons exhibits phasic activation in response to reward-related cues or reward consumption; another exhibits phasic inhibition in response to aversive stimulus or the absence of reward consumption after a reward cue (17). Tonic activation of dopaminergic neurons can produce the opposite effect of phasic activity on the same target and will decrease reward consumption (18). Thus, a given projection target receives either increased or decreased dopamine input dependent on the valence of the reward. Furthermore, dopaminergic neurons vary in their projection targets; the VTA’s projections are heterogeneous. Dependent on the projection target, an increase in dopamine outflow produces either rewarding or aversive responses (14, 19). As has been well-established, VTA projections to the NAc core (NAcc) and NAc shell (NAcSh) increase dopamine release in response to a rewarding stimulus and induce goal-direct behavior to acquire and consume it (14). Conversely, VTA dopaminergic neurons projecting to the medial prefrontal cortex are activated in response to an aversive stimulus and produce aversive behaviors (19). However, even within the same target, dopaminergic activation can code both types of behaviors; VTA dopaminergic projections to the lateral portion of the NAcSh are activated in response to both rewarding and aversive stimulus (19).
The VTA also possesses neurons releasing the classic neurotransmitters glutamate and GABA. The function of these glutamatergic and GABAergic neurons is less well-known, but recent evidence indicates they also participate in valence-related responses. VTA glutamatergic projections to the lateral habenula (LHb) play a significant role in encoding aversive learning (20). VTA glutamatergic projections to the NAcSh act in concert with the dopaminergic projections to produce reward-mediated behavior (14). Finally, VTA GABAergic neurons projecting to the LHb appear to inhibit this area to enhance positive valence responses (21). Recent analysis has identified that some VTA neurons corelease glutamate and dopamine—it is as yet unknown whether this occurs at the same synapse or at separate synaptic targets (14). Further research is needed to fully evaluate how the classic fast-acting neurotransmitters coordinate with dopaminergic neurons to produce the full suite of valence-related behaviors and alter future learned responses.
The NAc
The NAc is part of the ventral striatum and extended amygdala in the basal forebrain, and it mediates much of the motivated behavior produced in response to VTA dopaminergic outflow after sensation of a rewarding stimulus. Many components of the hunger system act here as well as in the VTA to alter the responsiveness to rewarding stimulus; thus, some description of its components and basic activity follows. The NAc is divided into a medial shell (NAcSh) and lateral core (NAcc). Self-administration of cocaine into the NAcSh is highly rewarding and rapidly produces cue-responsiveness with locomotor sensitization to anticipation of the drug (22–24). Self-administration of cocaine into the NAcc, however, is not reinforcing (22). Phasic activity of VTA dopaminergic projections to the NAcc instead responds to risk and prediction error in response to reward presentation (22, 24, 25). Thus, a basic paradigm can be constructed, where the NAcc responds to the salience, availability, and risk of acquiring the reward to produce motivation to pursue it, and the NAcSh responds to the positive valence of the reward acquisition, learns the cues which associate with the reward, and enhances the future salience of those cues. Interestingly, if dopamine is depleted in the NAc but reward acquisition is low effort, rats will still take the reward; however, if it requires high effort, rats will choose less effort-requiring behaviors (26). Thus, the level of dopamine in the NAc may provide a rough proxy for the amount of motivation an animal has to ignore risk and effort costs of acquiring a reward.
Both the shell and the core are inhibitory on all downstream targets; the vast majority of neurons are the GABAergic medium spiny neurons (MSNs). These are divided by receptor profile. There are D1R-MSNs, possessing excitatory D1R-like dopamine receptors (D1R and D5R), and D2R-MSNs, possessing inhibitory D2R-like dopamine receptors (D2R, D3R, and D4R). A significant minority express both receptor subtypes (27). The projection fields of the NAcSh and NAcc are wide and differ from each other in several important respects for their mediation of behavior. The NAcSh densely projects to the ventromedial ventral pallidum, lateral hypothalamic area (LH), and lateral preoptic area, whereas the NAcc projects to the dorsolateral ventral pallidum, subthalamic nucleus, and substantia nigra pars reticulata (22). The NAcSh shares significant reciprocal connections with feeding-related areas of the hypothalamus, whereas the NAcc primarily interacts with the basal ganglia. Thus, the NAcSh responds more to signals from the hunger system than the NAcc and will feature more prominently in this discussion.
Hunger Neurocircuits Select for Increased Reward System Activity in the Presence of Food
There are numerous and dense interconnections between the hypothalamic nuclei, VTA, and NAc, and a wealth of neuropeptide and neurocircuit data exists to support the powerful influence of the several hypothalamic nuclei and their specific neuronal subtypes on the reward system. The interaction is complex and dynamic, depending upon both the availability of food and the endocrine manipulation of the system, as will be discussed later. A summary of the major hunger and satiety neurocircuits influencing the reward system is shown in Figure 1.
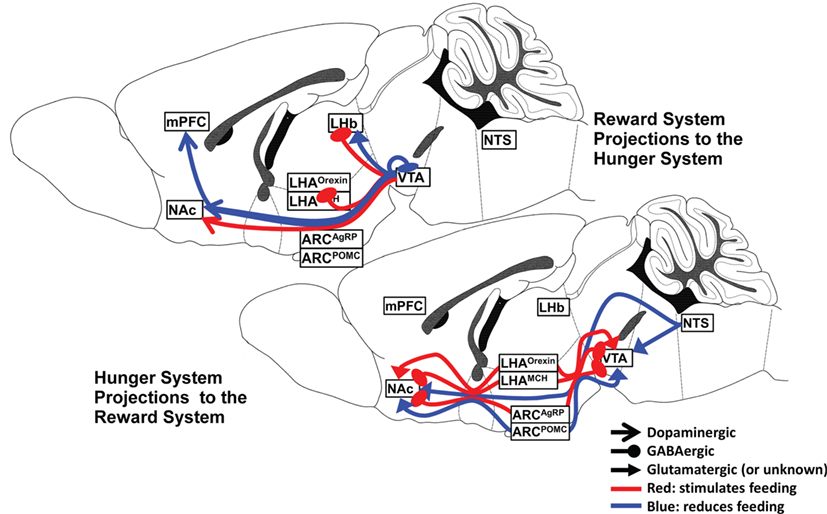
Figure 1. Hunger and satiety neurocircuits interface with the midbrain–basal forebrain reward pathway.
Arcuate Nucleus
The arcuate nucleus of the hypothalamus (Arc) sits adjacent to the third ventricle immediately ventral to the paraventricular hypothalamic nucleus (PVH). These two nuclei share the distinction of integrating central nervous system (CNS) input into the hypothalamic–pituitary axis and are the major source of the “releasing hormones,” which are secreted into the hypophyseal portal veins to alter anterior pituitary production of various hormones. Thus, the Arc possesses a multitude of neuronal populations defined by their neuropeptide content, such as gonadotropin-releasing hormone neurons, growth hormone releasing hormone neurons, kisspeptin neurons, tuberoinfundibular dopamine (TIDA) neurons regulating prolactin release, somatostatin neurons, and so on. Many of these neurons alter feeding behavior, but their interaction with the dopaminergic reward system is poorly understood at this point in time (28). Thus, our discussion will focus on the agouti-related protein (AgRP)/neuropeptide Y (NPY) neurons that govern hunger and TIDA neurons role in feeding behavior. The pro-opiomelanocortin (POMC) neurons that govern satiety will be discussed later.
AgRP/NPY-Expressing Neurons
The AgRP/NPY-expressing neurons are found solely within the Arc (29). NPY acts on NPY receptors (Y1, Y2, Y4, and Y5 which are GiPCRs) (30). AgRP is an inverse agonist of melanocortin receptors (MCRs; MC3R and MC4R, which are GsPCRs and MAPK pathway activators) (31–35). Perhaps because of this, these neurons share with POMC neurons the same set of connections with hypothalamic and extrahypothalamic nuclei (29, 36). Thus, AgRP neurons are strong inhibitors of their downstream targets via GABA release, inverse agonism of MC-Rs, and NPYR-Gi activity. These neurons respond to a wide array of peripheral and central signals of energy balance, such as leptin, ghrelin, low glucose concentration, and gustatory sensation, and are activated during fasting (37–41). Surprisingly, given this role, knockout of AgRP by itself or in combination with NPY does not produce any obvious phenotype either in ad libitum or starvation feeding conditions—only in old age do they demonstrate slightly reduced body weight and adiposity due to increased metabolic rate (42, 43). Furthermore, neonatal destruction of the AgRP/NPY neurons has minimal effect on feeding; only adult ablation of these neurons prevents feeding behavior and leads mice in this condition to starve to death (44–46). Interestingly, several AgRP/NPY neuronal projections are not formed until a week postnatal in mice, such as to the PVH; there are many opportunities for developmental compensation to alter the neonatal AgRP/NPY ablation phenotype, which deserve further study to understand the homeostasis of feeding behavior (47). Much recent effort has been spent on understanding the acute dynamics of AgRP/NPY neuronal activity in hunger and reward.
Optogenetic stimulation produces food seeking and food consuming behaviors, with enhanced risk-taking and reduced anxiety (48–51). Their activity is aversive, as mice avoid the side of a chamber associated with their optogenetic activation (52). AgRP neurons select for food consumption; when activated, they reduce motivation to engage in other behaviors such as social interactions or drinking water when thirsty (51). Sustained AgRP neuronal activity is not necessary to produce feeding, and in vivo recording demonstrates that these neurons stop firing in the presence of food cues (53). Further optogenetic evidence demonstrates that a brief period of activation, prior to presentation with any food stimulus, will produce subsequent feeding, enhanced motivation to work for food, and selection for calorie dense foods (54). As strength of AgRP signal increases, there is a first-order kinetic increase in length of feeding and motivation to work which saturates (54). Stimulating AgRP projections to the PVH, bed nucleus of the stria terminalis, or LH are all individually sufficient to produce this effect (54). However, AgRP neurons also synapse on the VTA and regulate the reward system through this connection. AgRP projections to the VTA inhibit dopaminergic and glutamatergic release in the NAc and reduce the development of long-term potentiation (LTP) (55). It can be argued that AgRP neurons in the Arc reduce activity of the reward system while activating the hunger system, priming it to respond to food and not other stimuli; once food is spotted, cessation of AgRP neuronal activity releases the brakes on the reward system to enhance dopaminergic outflow. The increase in dopamine in the NAc likely increases the willingness to work for food and take risks to acquire it.
TIDA Neurons
The dopaminergic neurons of the Arc regulate the release of prolactin from the anterior pituitary. A subset of TIDA neurons appear to be functionally distinct from governing prolactin; these corelease GABA and dopamine, and deletion of prolactin receptor within this subpopulation has no effect on prolactin secretion regulation (56). A recent study demonstrated that optogenetic stimulation of these neurons produces feeding behavior independent of their stimulation of prolactin release, and inhibition of these neurons reduces body weight (57). Activation of these neurons, which are ghrelin sensitive, inhibited POMC neurons and excited AgRP/NPY neurons. While these are not considered part of the dopaminergic reward system, their role in detecting the intersection of hunger and reward is relevant to this discussion as any manipulation altering whole-brain dopamine systems (such as dopamine reuptake inhibitors such as cocaine) will alter the activity of these neurons and may subtly alter the phenotype.
The Lateral Hypothalamus
One of the major downstream targets of AgRP neurons is the LH. This was classically understood as both a hunger center and reward hot spot from early lesion and electrical stimulation studies. The LH has extensive projections divided by multiple subpopulations of neurons that express various neuropeptides as well as solely fast-acting neurotransmitter neurons (58). Many of these fast-acting projections play a role in mediating hunger and reward. LH glutamatergic projections to the LHb prevent consumption of a conditioned reward of sucrose and has negative valence (59). Inhibition of this same projection has positive valence and induces sucrose consumption. Conversely, LH GABAergic projections appear to mediate consumption; activation of LH GABAergic neurons produces consumption regardless of the target’s food value, such as ethanol, water, saccharin, sucrose, or wood (60). The projection targets of the LH GABAergic neurons may relate to different aspects of this behavior. For example, LH GABAergic projections to the PVH produce directed food consumption (61). However, activation of the LH-VTA GABAergic projections produce non-directed gnawing and licking of immediately available objects, despite the availability of a sucrose reward distant from the mouse (62). These LH-VTA GABAergic neurons inhibit both a subpopulation of dopaminergic and GABAergic neurons within the VTA. Interestingly, the reciprocal VTA-LH projection is observed to be activated in response to reward omission; the disorganized feeding behavior from LH-VTA projection may occur because of the lack of this feedback information (62). Indeed, a recent study evaluating lateral septum inhibitory influence on the LH found that one subset of LH GABAergic neurons are activated during food approach and another during food consumption, indicating that a temporal sequence of GABAergic subpopulation activation occurs to produce successful food consumption (63).
Orexin Neurons
Another reciprocal functional relationship between LH neuronal subtypes can be found in orexin (hypocretin) and melanin-concentrating hormone (MCH) neuropeptide-expressing neurons. Orexin, as the name suggests, is an orexigenic or food consumption-inducing neuropeptide released primarily by glutamatergic neurons in the medial and dorsal portions of the LH. Orexin A and B are co-released from the same neurons and bind to their receptors O1R and O2R, potently inducing arousal via Gs signaling. Orexin release dramatically increases in release in the human amygdala upon waking up or during arousing positive stimuli such as laughing or talking (64). Orexinergic neurons are activated in response to learned reward cues, but not novel objects; for example, activating them can reinstate extinguished drug-seeking behavior (65). Indeed, O2R protein levels in the NAc are elevated for up to 60 days after discontinuation of repeated cocaine administration (66). They are depolarized in response to low glucose, and directly activate VTA dopaminergic neurons which project to both the medial and lateral portions of the NAcSh (67). Orexin receptors also exist within both of these subdivisions; injection of Orexin A into the medial NAcSh induces a sucrose pleasure-associated facial response, whereas injection into the lateral NAcSh induces a sucrose seeking reaction (68). O1R activation increases NMDA glutamate receptor activity, a sign of LTP formation, whereas blockade of O1R in the VTA reduces the rate of self-administration of cocaine or chocolate (11, 69). Thus, orexinergic neurons clearly enhance the seeking and acquisition of a learned reward, partially via activation of the dopaminergic pathway as well as potentially enhancing the reinforcing quality of those rewards.
MCH Neurons
Melanin-concentrating hormone neurons are released from the anatomically adjacent dorsolateral LH and have a complex reciprocal relationship with orexin neurons. They linked with induction of sleep and directly inhibit orexin neurons via the MCH receptor (MCH1R) Gi protein coupled signaling (70). Their interaction with feeding behavior is complex. Knockdown of MCH1R produces hyperphagia, hyperactivity with increased foraging behaviors; however, these mice have a baseline lower body weight than controls, gain less weight on a high-fat diet, and exhibit an increased metabolic rate (71, 72). Elevation of glucose concentration from fasting levels to fed-state levels inhibits orexin neurons and depolarizes MCH neurons. It appears that MCHergic activity appears to play a role in placing a brake on orexin-induced seeking and consuming behaviors after food is acquired (73). However, injection of MCH into the lateral ventricle increases food consumption, without producing long-term weight gain (74). It appears that MCH neurons integrate olfactory, taste, and gut sensory input about the nutritional value of food and project to the VTA, NAc, and dorsal striatum in order to enhance the rewarding value of nutritionally valuable foods (75, 76). To this end, optogenetic stimulation of MCH neurons increases dopamine levels in the striatum only when paired with active consumption of an artificial sweetener; without stimulation, only caloric foods like sucrose induce this dopamine release (76). Pairing these stimuli produced a future preference for the artificial sweetener over sucrose—opposite of the control mouse preference. As further evidence of this, ablation of MCH neurons prevented the natural spike of dopamine in the striatum after the consumption of sucrose—though, interestingly, it did not ablate preference for sweet flavor over water, indicating that other mechanisms are at play (76). Thus, MCH neuronal activity increases when olfaction, taste, and gut nutrient sensors indicate that the food under active consumption is calorically valuable; it enhances the rewarding value of food by increasing VTA dopaminergic activity in the NAc.
The pattern of MCH1R activation within the NAc is similarly complex and deserves some further discussion. These receptors are coexpressed with D1R and D2R on opioid-producing MSNs. Activation of MCH1R here induces feeding and causes a depressed phenotype on the forced swim test, implying reduced locomotor drive (77, 78). Given that MCH1R is inhibitory, it is unsurprising that MSNs have decreased membrane excitability and reduced AMPA glutamatergic receptor currents; furthermore, antagonism of MCH1R reduces cocaine self-administration or cue-induced reinstatement of cocaine seeking behavior. However, on MSNs which coexpress D1R, D2R, and MCH1R, there is a unique synergy which enhances their firing activity and activates a phosphorylation cascade known to increase NMDA receptor activation (79). Some of this response is opioid dependent, as blockade of any of the three opioid receptors in the NAcSh prevents MCH-induced facial pleasure expression in response to oral gavage of sucrose (80). It appears that MCH1R activity within the NAc may shift the reward system response to enhance immediate food consumption and learning the nutrient value of food and dampen the seeking function of the reward system.
Hunger Enhances Sensitivity to Reward
The above discussion details how several neuronal populations—AgRP/NPY neurons, LH GABAergic neurons, orexin neurons, and MCH neurons—each alter the activity of the reward system in a distinct way as part of their contribution to the sensation of hunger. AgRP neurons reduce reward system sensitivity and inhibit its function until food is detected. The food cue silences their activity, disinhibiting the VTA and NAc to produce a large increase in dopamine release. This increases both the salience and value of the food cue. A subset of LH GABAergic neurons act in concert with orexin neurons to respond to these cues and produce food-seeking behavior, enhancing VTA dopaminergic release into the NAc. Other LH GABAergic neurons act with MCH neurons to also enhance the rewarding value of food and increase learning of nutritionally valuable food-related cues, producing food consumption. Once this process begins, satiety neurocircuits begin to act in order to limit overconsumption, as will be detailed below.
Satiety Neurocircuits Decrease the Activity of the Reward System
The neurocircuitry of satiety is not as well-known as that of hunger. The general paradigm appears to be as follows: peripheral signals of positive energy balance, primarily hormonal, travel to the brain to inhibit the activity of hunger-producing neurocircuits. However, three populations of neurons within the CNS, defined by their neuropeptide content, are activated by these signals and directly influence the reward system. These are the POMC neurons of the Arc, the POMC neurons of the nucleus tractus solitarius (NTS), and the preproglucagon (PPG) neurons of the NTS. The NTS neurons integrate peripheral satiety signals, such as leptin, cholecystokinin (CCK), glucagon-like peptide 1 (GLP1), and gut distention, to induce rapid satiety. The role of POMC neurons is more complex, but appears to reduce the immediate value of the food reward while maintaining future responsiveness to that same reward.
PPG Neurons in NTS
The NTS has multiple functions both related to primary routing of taste information as well as being the routing point for significant amounts of autonomic and peripheral hormonal information entering the brain. It is situated in the dorsal motor vagal nerve complex and receives the vagal sensory afferents. These afferents are activated by GLP1, CCK, and leptin. The first two are released from stomach and intestinal cells and leptin from adipocytes in response to increased nutrient availability. They all synergistically increase the activity of the GLP1 producing neurons (termed PPG, or PPG neurons) in the NTS (81, 82).
The PPG NTS neurons have a wide projection field and synapse directly onto the VTA and NAc (83). Intestinally released GLP1 does not appear to cross the blood–brain barrier (BBB), and GLP1 receptor (GLP1R) signaling in the midbrain and basal forebrain requires NTS-mediated release of GLP1 (84). Activating GLP1R decreases highly palatable food intake and produces long-term weight loss; conversely, inhibiting them increases food consumption (83). Both changes occur due to changes in meal size, not frequency, indicating their role in terminating meal consumption. GLP1R activity in the VTA reduces LTP formation on dopaminergic neuron (85). GLP1 administration significantly increases dopamine transporter expression, increasing dopamine clearance from the synapse (86). GLP1R activity in the reward system may act to place the brakes on dopaminergic response to nutrient contents in the gut and prevent excessive learning of a food reward—or any reward, given the wide applicability of the reward system pathway. Consistent with this prediction, GLP1R knockout mice exhibit enhanced reward learning on the conditioned place preference test and increased cocaine-induced locomotion—receptor agonism had the inverse effect (87, 88). Thus, the PPG NTS neurons clearly mediate a dopamine-dampening function in the reward system and use this as a mechanism to reduce the reward salience and value of food or other objects.
POMC Neurons in the NTS and the Arcuate Nucleus
The role of POMC neurons is more complex, and much effort has been expended to explore their role in satiety. POMC is a precursor polypeptide which is cleaved by prohormone convertase 1 or 2 into α-, β-, or γ-melanocyte-stimulating hormone (MSH), corticotropin, and β-endorphin (89). These neuropeptides act via the MCR family, the best studied of which in regard to satiety are MC3R and MC4R. Deletion of MC4R produces obesity and hyperphagia; furthermore, the most common syndromic cause of obesity, Prader–Willi, occurs in part due to reduced cleavage of POMC (90, 91). The NTS and Arc POMC neurons both drive satiety via their actions on their receptors, but at different time scales. Optogenetic stimulation of the POMC NTS immediately halts feeding, but long-term activation does not produce weight loss (92). Stimulation of POMC Arc neurons has no immediate effect on feeding, but over many hours reduces feeding and produces weight loss (37). Both populations of POMC neurons project to the NAc and VTA—however, evidence is lacking on what effect direct stimulation of these projections produces (36). Both GABA-expressing and glutamate-expressing neurons have been found within the POMC neurons of the arcuate nucleus; but, the effects of fast neurotransmission from these neurons are not well explored (93). Instead, this discussion will focus on the pharmacological and receptor-related data that exist on the role of MC3R and MC4R in these regions.
MCR Signaling
The pharmacological effects of MCR activation are complex and their signaling mechanisms require some discussion before elaborating on their influence on the reward system. Multiple endogenous ligands for melanocortins exist. AgRP is an inverse agonist of MC3R and MC4R. α-, β-, and γ-MSHs and adrenocorticotropin hormone are produced from POMC by alternative cleavage and have varying affinities for each MCR type. Their downstream signaling activity is significantly altered dependent on their heterodimerization with other receptors such as dopamine receptors or ghrelin receptors. MC4R can couple with Gs, Gq, or Gi/o protein complexes dependent upon allosteric binding-related conformational changes and thus produce increases in intracellular calcium, increased cAMP signaling, or inhibit these same pathways (33). The two neuron-activating pathways, Gs and Gq, mediate distinct physiologic effects; knockout of PVH Gq, but not Gs, prevented the efficacy of MC4R agonism to prevent food intake (94). Conversely, the ghrelin receptor, when coupled with either MC3R or MC4R, selects for Gs signaling in both proteins in the PVH, for example, emphasizing that in conditions of increased hunger, MSH activity may select for activation (95). One of the most commonly used non-selective synthetic MCR agonists, melanotan II (MTII), also selects for Gs signaling (33, 95). AgRP, discussed above, is an inverse agonist of MC4R and reduces cAMP levels independent of the presence of any agonist (33–35). Hypothetically, AgRP activity may select for the Gi signaling conformation of MC4R, as it both antagonizes the ligand-binding site and binds to the allosteric site to cause a conformational change; it also induces receptor internalization and alters MC4R-mediated inhibition of L-type calcium channel activity (31–33, 96). While it has been demonstrated conclusively that intracerebroventricular infusion of the MC3R/MC4R agonist α-MSH suppresses feeding and infusion of AgRP suppresses feeding, the multiple signaling pathways invoked by each ligand as compared to MTII are important considerations while evaluating the pharmacological data for melanocortin influence on the reward system (97).
MC4R is found on D1R MSNs in the NAc. Application of α-MSH to ex vivo brain slices produces a decreased post-synaptic ratio of AMPA/NMDA glutamatergic receptor signaling and fewer excitatory post-synaptic currents—a sign of long-term depression (LTD) (98). These MC4R-linked D1R MSNs synapse onto LH GABAergic neurons; optogenetic activation of these prevents feeding (99). This reduction in feeding also occurs by MCR activity in the VTA; injection of MTII decreases sucrose consumption (100). Thus, MC4R activity may increase activity of these neurons to inhibit feeding, but lose their synaptic strength to prevent excessive inhibition of feeding behavior and simultaneously reduce their rewarding value. This coincides with evidence that administration of MC4R shRNA (knockdown) prevents the natural decrease in reward seeking induced by a chronic stress paradigm (98). Furthermore, blockade of NAc MC4R prevents chronic stress-elicited anhedonia, a known low-dopamine phenomenon (98).
However, in other conditions MCRs may play a role in reward-mediated learning and sensitivity to reward. After 2 h, intraventricular injection of MTII reduced the threshold for brain self-stimulation (101). Furthermore, mice with knockdown or inhibition of MC4R display reduced cocaine-induced reinforcement and reduced locomotor sensitization (102, 103). Injection of alpha-MSH into the posterior VTA increases the activity of MC4R-expressing dopaminergic neurons and induces ethanol self-administration (104). These neurons coexpress MC3R, and agonism of these neurons with γ-MSH increases motivation to work for sucrose reward in a dopamine-dependent fashion (105). Indeed, it appears that the level of glucose increase in response to food intake induces excitatory synaptic plasticity in a subpopulation of POMC neurons, which may enhance some of these downstream dopaminergic responses (106).
Thus, the multiple combinations of POMC cleavage products, the heterodimerization and allosteric-binding configurations of MCRs, and the differences in activity dependent on brain region all indicate that further evaluation of intracellular signaling pathways is needed. However, a possible synthesis may be as follows. MC4R signaling reduces ongoing feeding via its action on D1R MSNs in the NAc, with a naturally decaying signal because of the development of LTD. Simultaneously, in other regions and neuronal subpopulations, MC3R and MC4R enhance synaptic plasticity to encode future responses to that food reward—hence, knockdown of these receptors reduces reward consumption. The balance of melanocortin-mediated LTP and LTD in striatal neurons is important to halt ongoing food consumption, but appropriately encourage future responsiveness to rewarding food cues. One demonstration of this phenomena arises when hyperphagic MC4R knockout mice have induced second deletion of synapse-associated protein 90/post-synaptic density protein 95-associated protein 3 (SAPAP3) in the striatum (90). This double deletion both cures the compulsive grooming behavior of these mice and normalizes their weight. Deletion of SAPAP3 induces excessive LTD formation, and one may postulate that this helps reduce the excessive excitatory synapses formed in the VTA by unchecked MC3R signaling that in turn promote feeding behavior (107).
Food-Related Hormones Influence Dopaminergic Activity and Sensitivity to Reward
The above discussion has focused on the interaction between neurons encoding hunger and satiety and the reward system. However, these are all interoceptive and are responsible for the integration of internal and external sensory information to produce unified behaviors. As is to be expected, the sensory input into this system is both neuronal (gut distention, pain, olfaction, taste, etc.) and endocrine. While the vast majority of endocrine pathways are altered in some way in response to variations in energy stores and the presence or absence of food in the gut, the best studied of these which appear to act directly on the reward system are the hunger hormone ghrelin and the satiety hormone leptin.
Ghrelin
Ghrelin is a peptide hormone first discovered as an endogenous ligand of the growth hormone secretagogue receptor (GHS1R), produced in the oxyntic glands of the stomach (108). Copies of ghrelin mRNA and ghrelin immunoreactivity are found in abundance in the antrum and fundus of the stomach and to a lesser degree in the duodenum, jejunum, ileum, and pancreas (109–111). Both peripheral and intracerebroventricular injections of ghrelin produce feeding in mice, and knockout of neuronal GHS1R prevents the development of diet-induced obesity; this places the CNS activity of ghrelin as a primary mediator of ghrelin’s orexigenic effect (112, 113).
The mechanics governing ghrelin release and the neuronal response to ghrelin are complex and deserve some consideration. Ghrelin secretion occurs in ultradian pulses, peaking immediately before onset of meals and declining soon after, with the greatest rise occurring overnight preceding breakfast (114). Several neurotransmitters, hormones, and metabolic signals also affect ghrelin release; acetylcholine, CCK, gastrointestinal peptide, and low glucose concentration enhance it, whereas insulin, gastrin, somatostatin, GLP-1, and increasing glucose concentration inhibit its release (115–117). Once ghrelin is produced, it can undergo post-translational modification with fatty acid linkage (octanoylation). This appears to affect ghrelin transport across the BBB; human octanoylated ghrelin and mouse des-octanoyl ghrelin are preferentially transported into the brain, whereas mouse octanoylated ghrelin (with two amino acid differences) is preferentially transported into the blood (118). The rate of BBB transport is enhanced by elevated serum triglycerides and fasting and blunted in aging, indicating physiological state modulation of this mechanism (119). There is also some evidence suggesting that a subpopulation of ghrelin-producing neurons may exist within the hypothalamus itself, which would not be affected by BBB transport and would have entirely unique, as yet undescribed, regulation (38). Finally, GHS1R-Gq-induced calcium flux (i.e., the signal strength) is attenuated by its heterodimerization with serotonin (specifically 5-HT2C), dopamine (D1R), and melanocortin (MC3R) receptors (120). Given that GHS1R possesses one of the highest levels of basal Gq activity of any GPCR, this dimerization may be important for reducing basal signaling except in the presence of the appropriate ligands for each receptor, potentiating the signal strength of the ghrelin-GHS1R active conformation or increasing GHS1R Gq-signaling via dedimerization after dopamine-D1R or serotonin-5-HT2C interactions (121).
After integrating the influences of pulsivity, BBB transport, and heterodimerization considered, ghrelin induces feeding behavior by depolarizing neurons expressing GHS1R. Chief among these, AgRP/NPY neurons express GHS1R-Gq-coupled signaling; this produces feeding behavior and reduced thermogenesis (112, 122). Thus, in the absence of any food cues, ghrelin actually reduces sensitivity to reward, acting via AgRP/NPY neurons and inhibiting the VTA and NAc as described earlier. However, GHS1R is also found in the VTA and laterodorsal VTA, a source of midbrain acetylcholine; injecting ghrelin into either of these places increases locomotor activity, food consumption, and NAc dopamine levels (123). Indeed, ghrelin enhances the rewarding value of high fat diet in ad libitum-fed mice (124). If no food is consumed after VTA ghrelin injection, VTA GABAergic neurons increase activity and reduce the release of dopamine into the NAc (123). These effects occur both due to ghrelin action on the VTA cell body and by alteration of pre-synaptic activity, especially the LH as orexin-deficient mice are resistant to the effect of ghrelin (40, 124–126).
These effects may be cross-sensitive for non-food rewards such as alcohol and amphetamines. In recovering alcoholics, ghrelin injection increases craving for alcohol; coincident with this, ghrelin receptor blockade attenuates both alcohol and amphetamine-induced locomotion sensitization (127–130). However, once sensitization has occurred, blockade of ghrelin transport into the brain does not appear to alter alcohol-induced locomotor activity or expression of conditioned place preference in rats (131). Thus, ghrelin may act to enhance learning of non-food rewards, but not be necessary to express this preference (132). Notably, GHS1R is found in the hippocampus; here it may enhance dopamine-induced synaptic plasticity within the hippocampus, promoting retention of the food-related reward via GHS1R-D1R heterodimer interactions (133).
Ghrelin action in gaging reward sensitivity can be summarized in three ways. First, by acting on AgRP/NPY neurons, it selects for food-seeking and food-consuming behavior and ignoring of other rewards. Second, by acting on the VTA, it increases the amplitude of dopamine release once cue-mediated silencing of AgRP/NPY neurons disinhibits these regions. Third, by acting on the hippocampus, it promotes dopamine-induced synaptic plasticity and future salience of the reward cue. Given the complexity and numerous levels of signal modification in the ghrelin system, future research will undoubtedly expand upon this story.
Leptin
Leptin is a signal of positive energy balance and appears to exert a dampening effect on the reward system. Leptin is a protein hormone released by white adipose tissue in pulses, highest around midnight in humans; the amplitude of each pulse directly correlates with the total amount of adipose tissue (114, 134). Glucose, insulin, glucocorticoids, TNF-alpha, and IL-6 all increase leptin release (134). Leptin acts on its receptors (leptin receptor, LepR), which are located in the CNS, particularly the hypothalamus. It can influence neuronal activity from the blood via action on the circumventricular organs and vagal afferents and is also translocated across the BBB via the action of tanycytes (135). The Arc, ventromedial and dorsomedial hypothalamus, LH, and NTS most densely express LepRs, and leptin action can be either inhibitory or excitatory on these neurons to inhibit food consumption and induce satiety (136). LepR also exists within the VTA on the dopaminergic projections to the extended central amygdala; exciting these neurons reduces food intake (137–139). This is consistent with the central amygdala’s role in mediating the stress response.
Other populations of LepR expressing dopaminergic neurons have their activity inhibited by leptin and produce a general reward-dampening effect. Direct administration of leptin into the VTA and into the Arc increases the threshold for brain self-stimulation reward and decreases food intake (140). Conversely, knockdown or inhibition of LepRs within the VTA increases dopamine release onto the NAc and enhances cocaine-conditioned place preference (13). LepR depletion within the NAc core mediates the same effect (13). Interestingly, it also prevented D2R agonism from reducing cocaine reinforcement, implying that there is some synergy between LepR inhibitory action and D2R’s reduction of synaptic plasticity mechanisms (13). This may occur via LepR activation of the signal transducer and activator of transcription 3 signaling pathway in the VTA, which reduces both feeding behavior and motivation to exercise (141). Finally, the hyperleptinemia of diet-induced obesity is also sufficient to produce the generalized reward-dampening effects described above. Mice with diet-induced obesity have reduced drive to self-administer cocaine, express less amphetamine conditioned place preference, and are less likely to express operant response to sucrose (142, 143). These mice are demonstrated to have reduced dopamine turnover in the NAc (142, 143). Interestingly, alcohol consumption does not appear to follow this paradigm. Leptin administration significantly increased the consumption of ethanol in mice exposed to an ethanol deprivation-then-reinstatement procedure (144). High leptin levels at onset of alcohol withdrawal in humans were predictive of cravings and alcohol relapse; anti-opioid receptor drugs used to prevent relapse reduced leptin levels (145). Future research is necessary to understand how alcohol bypasses the dopaminergic reward system pathway to induce opioid release in a leptin-potentiated manner. Nevertheless, leptin clearly induces satiety by directly reducing activity of both the hunger system and the reward system, with generalizable reduction of the salience and value of both food and non-food rewards.
Limitations in the Study of the Interface Between the Hunger and Reward Systems
The above reviewed evidence is not comprehensive, as the relationship between the hunger system and reward system is of great interest and under active research with exciting new evidence unveiled daily. Given the ancient association of these two systems and the necessity of proper balance of food consumption to sustain life, it is understandable that nearly every neuronal and endocrine system appears to alter food consumption and manipulate the reward system in some way. Furthermore, the developmental activity of these systems may differ from their adult activity, and disruptions to these systems may result in significant compensation over the developmental period. The techniques utilized to study feeding and reward behavior are also limited by the necessity of overactivation, for an extended period, a single node within the feeding system. Given the interconnected nature of these neuronal circuits within the hunger system, continuous activation of any part likely engages the other portions and produces the unified behavioral response. However, it is likely that each neuronal pathway described above does not independently induce or inhibit food or reward consumption, but instead activates a certain portion of a “reward consumption sequence.” A clear example of this is how LH-VTA GABAergic activation induces gnawing and licking behaviors, but not sucrose seeking (62). The nature of this reward consumption sequence remains to be uncovered, and will likely require further molecular characterization of each of these components of the reward sequence, in vivo recording of their activity, and more advanced optogenetic and chemogenetic stimulation techniques to better approximate physiological activity.
Conclusion
The hunger system manipulates the reward system to increase the salience and value of food and enhance future response to rewarding food cues. Ghrelin and AgRP neurons prime the reward system to activate only in the presence of food cues. LH fast neurotransmitters and orexin neurons produce food-seeking paradigm in response to a learned cue. Other LH GABAergic neurons and MCH neurons increase food consumption, reducing reward seeking behavior and producing reward consuming behavior by their action on the VTA and NAc. Early satiety signals like GLP1 and CCK put the brakes on food consumption by dampening reward system activity, reducing its value. POMC neuronal activity produces a balanced synaptic plasticity to respond effectively to future food stimulus, while gradually inducing satiety. Late satiety signals like leptin dampen reward system activity generally to reduce effort for acquiring food or other rewards while in a positive energy state. A summary of these effects is presented in Table 1.
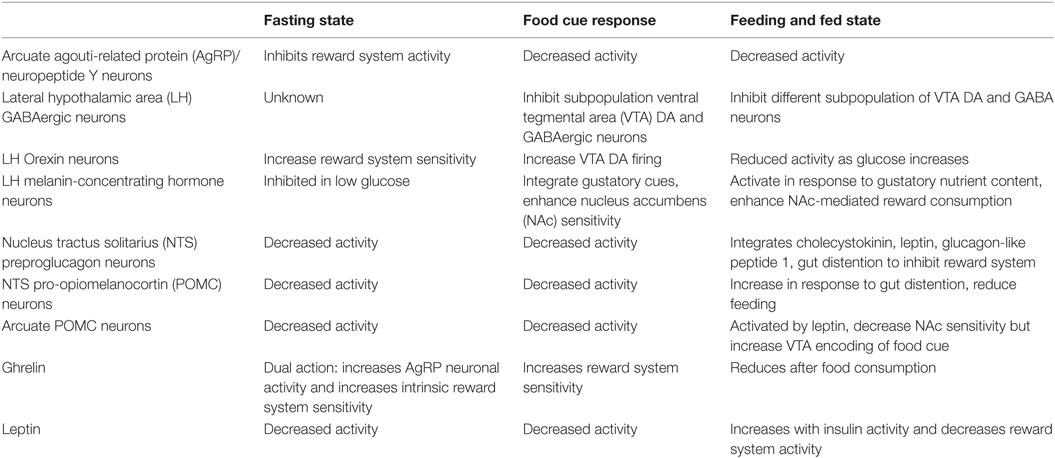
Table 1. Hunger system activity and influence on the reward system.
With this paradigm in mind, we can return to the superstimulus concept and observe how extreme rewards may bypass this balanced relationship of hunger systems enhancing food rewards to increase food consumption, and satiety systems reducing food rewards to reduce consumption. Highly palatable foods can induce food consumption in fed mice, even with adult ablation of AgRP neurons—a disruption which typically produces starvation (146). This relies on dopaminergic tone to produce the feeding response. A natural extension of this finding is this: if the reward’s learned value is high enough to stimulate a large dopamine spike upon sighting, overcoming the reward-dampening influence of satiety circuits and hormones, this triggers the feeding sequence. It can be speculated that normally, AgRP neurons via their aversive, inhibitory action sensitize the reward system so that when it is finally released, even an intrinsically low-value food cue produces a large spike in dopaminergic activity. This is in keeping with the old saying that “hunger is the best sauce.” Thus, it may not be that there are separate hedonic and homeostatic feeding mechanisms, but instead that highly palatable foods bypass the natural inhibition of the reward system by the hunger system.
Author Contributions
RC prepared and wrote the manuscript. QT revised the manuscript and provided critical input for new ideas and content to be added to the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Work in the Tong lab was supported by NIH R01DK092605 and R01DK109934, UT BRAIN Initiative and CTSA UL1 TR000371, Welch Foundation (L-AU0002), Grand-in-aid from American Heart Association (15GRNT22370024), and Basic Research Award (1-15-BS-184) from American Diabetes Association; QT is the holder of Cullen Chair in Molecular Medicine at McGovern Medical School. RC is supported by the Center for Clinical and Translational Sciences at UTHealth (4TL1TR000369-10).
Abbreviations
AgRP, agouti-related protein; Arc, arcuate nucleus of the hypothalamus; CCK, cholecystokinin; D1R, D2R, dopamine receptors; GHS1R, growth hormone secretagogue 1 receptor (ghrelin receptor); GLP1/GLP1R, glucagon-like peptide 1/receptor; LepR, leptin receptor; LH, lateral hypothalamic area; LTD, long-term depression; LTP, long-term potentiation; MC3R, MC4R, melanocortin receptors; MCH/MCH1R, melanin concentrating hormone/receptor; MSH, melanocyte-stimulating hormone (natural MCR agonist); MSN, medium spiny neuron; MTII, melanotan II (non-selective MCR agonist); NAc, nucleus accumbens; NAcc, nucleus accumbens core; NAcSh, nucleus accumbens shell; NTS, nucleus tractus solitarius; O1R, O2R, orexin receptors; POMC, pro-opiomelanocortin; PPG, preproglucagon; PVH, paraventricular hypothalamic nucleus; VTA, ventral tegmental area.
References
1. De Block A, Du Laing B. Amusing ourselves to death? Superstimuli and the evolutionary social sciences. Philos Psychol (2010) 23(6):821–43. doi:10.1080/09515089.2010.529048
3. Longo DL, Volkow ND, Koob GF, McLellan AT. Neurobiologic advances from the brain disease model of addiction. N Engl J Med (2016) 374(4):363–71. doi:10.1056/NEJMra1511480
4. Freimuth M, Moniz S, Kim SR. Clarifying exercise addiction: differential diagnosis, co-occurring disorders, and phases of addiction. Int J Environ Res Public Health (2011) 8(10):4069–81. doi:10.3390/ijerph8104069
5. Murakami Y, Kuratani S. Brain segmentation and trigeminal projections in the lamprey; with reference to vertebrate brain evolution. Brain Res Bull (2008) 75(2):218–24. doi:10.1016/j.brainresbull.2007.10.057
6. Pani AM, Mullarkey EE, Aronowicz J, Assimacopoulos S, Grove EA, Lowe CJ. Ancient deuterostome origins of vertebrate brain signalling centres. Nature (2012) 483(7389):289–94. doi:10.1038/nature10838
7. Jackson PJ, Douglas NR, Chai B, Binkley J, Sidow A, Barsh GS, et al. Structural and molecular evolutionary analysis of agouti and agouti-related proteins. Chem Biol (2006) 13(12):1297–305. doi:10.1016/j.chembiol.2006.10.006
8. Västermark Å, Krishnan A, Houle ME, Fredriksson R, Cerdá-Reverter JM, Schiöth HB. Identification of distant agouti-like sequences and re-evaluation of the evolutionary history of the agouti-related peptide (AgRP). PLoS One (2012) 7(7):e40982. doi:10.1371/journal.pone.0040982
9. Callier S, Snapyan M, Crom S, Prou D, Vincent J-D, Vernier P. Evolution and cell biology of dopamine receptors in vertebrates. Biol Cell (2003) 95(7):489–502. doi:10.1016/S0248-4900(03)00089-3
10. Zhou Q-Y, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell (1995) 83(7):1197–209. doi:10.1016/0092-8674(95)90145-0
11. España RA, Oleson EB, Locke JL, Brookshire BR, Roberts DC, Jones SR. The hypocretin-orexin system regulates cocaine self-administration via actions on the mesolimbic dopamine system. Eur J Neurosci (2010) 31(2):336–48. doi:10.1111/j.1460-9568.2009.07065.x
12. Zheng D, De Vaca SC, Carr KD. Food restriction increases acquisition, persistence and drug prime-induced expression of a cocaine-conditioned place preference in rats. Pharmacol Biochem Behav (2012) 100(3):538–44. doi:10.1016/j.pbb.2011.10.021
13. Shen M, Jiang C, Liu P, Wang F, Ma L. Mesolimbic leptin signaling negatively regulates cocaine-conditioned reward. Transl Psychiatry (2016) 6(12):e972. doi:10.1038/tp.2016.223
14. Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci (2017) 18(2):73–85. doi:10.1038/nrn.2016.165
15. Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron (2010) 68(5):815–34. doi:10.1016/j.neuron.2010.11.022
16. Bariselli S, Glangetas C, Tzanoulinou S, Bellone C. Ventral tegmental area subcircuits process rewarding and aversive experiences. J Neurochem (2016) 139(6):1071–80. doi:10.1111/jnc.13779
17. Cohen JY, Haesler S, Vong L, Lowell BB, Uchida N. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature (2012) 482(7383):85–8. doi:10.1038/nature10754
18. Mikhailova MA, Bass CE, Grinevich VP, Chappell AM, Deal AL, Bonin KD, et al. Optogenetically-induced tonic dopamine release from VTA-nucleus accumbens projections inhibits reward consummatory behaviors. Neuroscience (2016) 333:54–64. doi:10.1016/j.neuroscience.2016.07.006
19. Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron (2011) 70:855–62. doi:10.1016/j.neuron.2011.03.025
20. Root DH, Mejias-Aponte CA, Qi J, Morales M. Role of glutamatergic projections from ventral tegmental area to lateral habenula in aversive conditioning. J Neurosci (2014) 34(42):13906–10. doi:10.1523/JNEUROSCI.2029-14.2014
21. Stamatakis AM, Jennings JH, Ung RL, Blair GA, Weinberg RJ, Neve RL, et al. A unique population of ventral tegmental area neurons inhibits the lateral habenula to promote reward. Neuron (2013) 80(4):1039–53. doi:10.1016/j.neuron.2013.08.023
22. Zahm DS. Functional-anatomical implications of the nucleus accumbens core and shell subterritories. Ann N Y Acad Sci (1999) 877:113–28. doi:10.1111/j.1749-6632.1999.tb09264.x
23. Wheeler RA, Aragona BJ, Fuhrmann KA, Jones JL, Day JJ, Cacciapaglia F, et al. Cocaine cues drive opposing context-dependent shifts in reward processing and emotional state. Biol Psychiatry (2011) 69(11):1067–74. doi:10.1016/j.biopsych.2011.02.014
24. Saddoris MP, Cacciapaglia F, Wightman RM, Carelli RM. Differential dopamine release dynamics in the nucleus accumbens core and shell reveal complementary signals for error prediction and incentive motivation. J Neurosci (2015) 35(33):11572–82. doi:10.1523/JNEUROSCI.2344-15.2015
25. Sugam JA, Day JJ, Wightman RM, Carelli RM. Phasic nucleus accumbens dopamine encodes risk-based decision-making behavior. Biol Psychiatry (2012) 71(3):199–205. doi:10.1016/j.biopsych.2011.09.029
26. Salamone JD, Correa M, Farrar A, Mingote SM. Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology (Berl) (2007) 191(3):461–82. doi:10.1007/s00213-006-0668-9
27. Yager LM, Garcia AF, Wunsch AM, Ferguson SM. The ins and outs of the striatum: role in drug addiction. Neuroscience (2015) 301:529–41. doi:10.1016/j.neuroscience.2015.06.033
28. Waterson MJ, Horvath TL. Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab (2015) 22(6):962–70. doi:10.1016/j.cmet.2015.09.026
29. Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, et al. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat 1. Endocrinology (1999) 140(3):1408–15. doi:10.1210/endo.140.3.6544
30. Colmers WF, Bleakman D. Effects of neuropeptide Y on the electrical properties of neurons. Trends Neurosci (1994) 17(9):373–9. doi:10.1016/0166-2236(94)90046-9
31. Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev (2010) 31(4):506–43. doi:10.1210/er.2009-0037
32. Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science (1997) 278(5335):135–8. doi:10.1126/science.278.5335.135
33. Yang Y. Structure, function and regulation of the melanocortin receptors. Eur J Pharmacol (2011) 660(1):125–30. doi:10.1016/j.ejphar.2010.12.020
34. Tolle V, Low MJ. In vivo evidence for inverse agonism of agouti-related peptide in the central nervous system of proopiomelanocortin-deficient mice. Diabetes (2007) 57(1):86–94. doi:10.2337/db07-0733
35. Nijenhuis WA, Oosterom J, Adan RA. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol (2001) 15:164–71. doi:10.1210/me.15.1.164
36. Wang D, He X, Zhao Z, Feng Q, Lin R, Sun Y, et al. Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat (2015) 9:40. doi:10.3389/fnana.2015.00040
37. Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci (2011) 14(3):351–5. doi:10.1038/nn.2739
38. Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron (2003) 37(4):649–61. doi:10.1016/S0896-6273(03)00063-1
39. Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep (2012) 13(12):1079–86. doi:10.1038/embor.2012.174
40. Schaeffer M, Langlet F, Lafont C, Molino F, Hodson DJ, Roux T, et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci U S A (2013) 110(4):1512–7. doi:10.1073/pnas.1212137110
41. Takahashi KA, Cone RD. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology (2005) 146(3):1043–7. doi:10.1210/en.2004-1397
42. Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, et al. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol (2002) 22(14):5027–35. doi:10.1128/MCB.22.14.5027-5035.2002
43. Wortley KE, Anderson KD, Yasenchak J, Murphy A, Valenzuela D, Diano S, et al. Agouti-related protein-deficient mice display an age-related lean phenotype. Cell Metab (2005) 2(6):421–7. doi:10.1016/j.cmet.2005.11.004
44. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science (2005) 310(5748):683–5. doi:10.1126/science.1115524
45. Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci (2005) 8(10):1289–91. doi:10.1038/nn1548
46. Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, et al. Postembryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J (2005) 21(3):1–22. doi:10.1096/fj.04-3434fje
47. Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci (2004) 24(11):2797–805. doi:10.1523/JNEUROSCI.5369-03.2004
48. Tong Q, Ye C-P, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci (2008) 11(9):998–1000. doi:10.1038/nn.2167
49. Dietrich MO, Zimmer MR, Bober J, Horvath TL. Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell (2015) 160(6):1222–32. doi:10.1016/j.cell.2015.02.024
50. Nakajima K, Cui Z, Li C, Meister J, Cui Y, Fu O, et al. Gs-coupled GPCR signalling in AgRP neurons triggers sustained increase in food intake. Nat Commun (2016) 7:10268. doi:10.1038/ncomms10268
51. Burnett CJ, Li C, Webber E, Tsaousidou E, Xue SY, Brüning JC, et al. Hunger-driven motivational state competition. Neuron (2016) 92(1):187–201. doi:10.1016/j.neuron.2016.08.032
52. Betley JN, Xu S, Cao ZF, Gong R, Magnus CJ, Yu Y, et al. Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature (2015) 521(7551):180–5. doi:10.1038/nature14416
53. Chen Y, Lin YC, Kuo TW, Knight ZA. Sensory detection of food rapidly modulates arcuate feeding circuits. Cell (2015) 160(5):829–41. doi:10.1016/j.cell.2015.01.033
54. Chen Y, Lin YC, Zimmerman CA, Essner RA, Knight ZA. Hunger neurons drive feeding through a sustained, positive reinforcement signal. Elife (2016) 5:e18640. doi:10.7554/eLife.18640
55. Dietrich MO, Bober J, Ferreira JG, Tellez LA, Mineur YS, Souza DO, et al. AgRP neurons regulate development of dopamine neuronal plasticity and nonfood-associated behaviors. Nat Neurosci (2012) 15(8):1108–10. doi:10.1038/nn.3147
56. Brown RSE, Kokay IC, Phillipps HR, Yip SH, Gustafson P, Wyatt A, et al. Conditional deletion of the prolactin receptor reveals functional subpopulations of dopamine neurons in the arcuate nucleus of the hypothalamus. J Neurosci (2016) 36(35):9173–85. doi:10.1523/JNEUROSCI.1471-16.2016
57. Zhang X, van den Pol AN. Hypothalamic arcuate nucleus tyrosine hydroxylase neurons play orexigenic role in energy homeostasis. Nat Neurosci (2016) 19(10):1341–7. doi:10.1038/nn.4372
58. Stuber GD, Wise RA. Lateral hypothalamic circuits for feeding and reward. Nat Neurosci (2016) 19(2):198–205. doi:10.1038/nn.4220
59. Stamatakis AM, Van Swieten M, Basiri ML, Blair GA, Kantak P, Stuber GD. Lateral hypothalamic area glutamatergic neurons and their projections to the lateral habenula regulate feeding and reward. J Neurosci (2016) 36(2):302–11. doi:10.1523/JNEUROSCI.1202-15.2016
60. Navarro M, Olney JJ, Burnham NW, Mazzone CM, Lowery-Gionta EG, Pleil KE, et al. Lateral hypothalamus GABAergic neurons modulate consummatory behaviors regardless of the caloric content or biological relevance of the consumed stimuli. Neuropsychopharmacology (2016) 41(6):1505–12. doi:10.1038/npp.2015.304
61. Wu Z, Kim ER, Sun H, Xu Y, Mangieri LR, Li DP, et al. GABAergic projections from lateral hypothalamus to paraventricular hypothalamic nucleus promote feeding. J Neurosci (2015) 35(8):3312–8. doi:10.1523/JNEUROSCI.3720-14.2015
62. Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, et al. Decoding neural circuits that control compulsive sucrose seeking. Cell (2015) 160(3):528–41. doi:10.1016/j.cell.2015.01.003
63. Carus-Cadavieco M, Gorbati M, Ye L, Bender F, van der Veldt S, Kosse C, et al. Gamma oscillations organize top-down signalling to hypothalamus and enable food seeking. Nature (2017) 542:232–6. doi:10.1038/nature21066
64. Blouin AM, Fried I, Wilson CL, Staba RJ, Behnke EJ, Lam HA, et al. Human hypocretin and melanin-concentrating hormone levels are linked to emotion and social interaction. Nat Commun (2013) 4:1547. doi:10.1038/ncomms2461
65. Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature (2005) 437(7058):556–9. doi:10.1038/nature04071
66. Zhang G-C, Mao L-M, Liu X-Y, Wang JQ. Long-lasting up-regulation of orexin receptor type 2 protein levels in the rat nucleus accumbens after chronic cocaine administration. J Neurochem (2007) 103(1):400–7. doi:10.1111/j.1471-4159.2007.04748.x
67. Sheng Z, Santiago AM, Thomas MP, Routh VH. Metabolic regulation of lateral hypothalamic glucose-inhibited orexin neurons may influence midbrain reward neurocircuitry. Mol Cell Neurosci (2014) 62:30–41. doi:10.1016/j.mcn.2014.08.001
68. Castro DC, Terry RA, Berridge KC. Orexin in rostral hotspot of nucleus accumbens enhances sucrose “liking” and intake but scopolamine in caudal shell shifts “liking” toward “disgust” and “fear”. Neuropsychopharmacology (2016) 41(8):2101–11. doi:10.1038/npp.2016.10
69. Borgland SL, Chang SJ, Bowers MS, Thompson JL, Vittoz N, Floresco SB, et al. Orexin A/hypocretin-1 selectively promotes motivation for positive reinforcers. J Neurosci (2009) 29(36):11215–25. doi:10.1523/JNEUROSCI.6096-08.2009
70. Gao XB, Van Den Pol AN. Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol (2001) 533(1):237–52. doi:10.1111/j.1469-7793.2001.0237b.x
71. Eiler WJA, Chen Y, Slieker LJ, Ardayfio PA, Statnick MA, Witkin JM. Consequences of constitutive deletion of melanin-concentrating hormone-1 receptors for feeding and foraging behaviors of mice. Behav Brain Res (2017) 316:271–8. doi:10.1016/j.bbr.2016.09.028
72. Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A (2002) 99(5):3240–5. doi:10.1073/pnas.052706899
73. Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci (2005) 25(9):2429–33. doi:10.1523/JNEUROSCI.4925-04.2005
74. Rossi M, Choi SJ, O’Shea D, Miyoshi T, Ghatei MA, Bloom SR. Melanin-concentrating hormone acutely stimulates feeding, but chronic administration has no effect on body weight. Endocrinology (1997) 138(1):351–5. doi:10.1210/endo.138.1.4887
75. Pérez CA, Stanley SA, Wysocki RW, Havranova J, Ahrens-Nicklas R, Onyimba F, et al. Molecular annotation of integrative feeding neural circuits. Cell Metab (2011) 13(2):222–32. doi:10.1016/j.cmet.2010.12.013
76. Domingos AI, Sordillo A, Dietrich MO, Liu ZW, Tellez LA, Vaynshteyn J, et al. Hypothalamic melanin concentrating hormone neurons communicate the nutrient value of sugar. Elife (2013) 2:e01462. doi:10.7554/eLife.01462
77. Chung S, Hopf FW, Nagasaki H, Li CY, Belluzzi JD, Bonci A, et al. The melanin-concentrating hormone system modulates cocaine reward. Proc Natl Acad Sci U S A (2009) 106(16):6772–7. doi:10.1073/pnas.0811331106
78. Georgescu D, Sears RM, Hommel JD, Barrot M, Bolaños CA, Marsh DJ, et al. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci (2005) 25(11):2933–40. doi:10.1523/JNEUROSCI.1714-04.2005
79. Sears RM, Liu RJ, Narayanan NS, Sharf R, Yeckel MF, Laubach M, et al. Regulation of nucleus accumbens activity by the hypothalamic neuropeptide melanin-concentrating hormone. J Neurosci (2010) 30(24):8263–73. doi:10.1523/JNEUROSCI.5858-09.2010
80. Lopez CA, Guesdon B, Baraboi E-D, Roffarello BM, Hétu M, Richard D. Involvement of the opioid system in the orexigenic and hedonic effects of melanin-concentrating hormone. Am J Physiol Regul Integr Comp Physiol (2011) 301(4):R1105–11. doi:10.1152/ajpregu.00076.2011
81. Cheung GW, Kokorovic A, Lam CK, Chari M, Lam TK. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab (2009) 10(2):99–109. doi:10.1016/j.cmet.2009.07.005
82. Baggio LL, Drucker DJ. Biology of Incretins: GLP-1 and GIP. Gastroenterology (2007) 132(6):2131–57. doi:10.1053/j.gastro.2007.03.054
83. Alhadeff AL, Rupprecht LE, Hayes MR. GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. Endocrinology (2012) 153(2):647–58. doi:10.1210/en.2011-1443
84. Alhadeff AL, Mergler BD, Zimmer DJ, Turner CA, Reiner DJ, Schmidt HD, et al. Endogenous glucagon-like peptide-1 receptor signaling in the nucleus tractus solitarius is required for food intake control. Neuropsychopharmacology (2016). doi:10.1038/npp.2016.246
85. Mietlicki-Baase EG, Ortinski PI, Rupprecht LE, Olivos DR, Alhadeff AL, Pierce RC, et al. The food intake-suppressive effects of glucagon-like peptide-1 receptor signaling in the ventral tegmental area are mediated by AMPA/kainate receptors. Am J Physiol Endocrinol Metab (2013) 305(11):E1367–74. doi:10.1152/ajpendo.00413.2013
86. Reddy IA, Pino JA, Weikop P, Osses N, Sørensen G, Bering T, et al. Glucagon-like peptide 1 receptor activation regulates cocaine actions and dopamine homeostasis in the lateral septum by decreasing arachidonic acid levels. Transl Psychiatry (2016) 6(5):e809. doi:10.1038/tp.2016.86
87. Harasta AE, Power JM, von Jonquieres G, Karl T, Drucker DJ, Housley GD, et al. Septal glucagon-like peptide 1 receptor expression determines suppression of cocaine-induced behavior. Neuropsychopharmacology (2015) 40(8):1969–78. doi:10.1038/npp.2015.47
88. Schmidt HD, Mietlicki-Baase EG, Ige KY, Maurer JJ, Reiner DJ, Zimmer DJ, et al. Glucagon-like peptide-1 receptor activation in the ventral tegmental area decreases the reinforcing efficacy of cocaine. Neuropsychopharmacology (2016) 41(7):1917–28. doi:10.1038/npp.2015.362
89. Benjannet S, Rondeau N, Day R, Chrétien M, Seidah NG. PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proc Natl Acad Sci U S A (1991) 88(9):3564–8. doi:10.1073/PNAS.88.9.3564
90. Xu P, Grueter BA, Britt JK, McDaniel L, Huntington PJ, Hodge R, et al. Double deletion of melanocortin 4 receptors and SAPAP3 corrects compulsive behavior and obesity in mice. Proc Natl Acad Sci U S A (2013) 110(26):10759–64. doi:10.1073/pnas.1308195110
91. Burnett LC, LeDuc CA, Sulsona CR, Paull D, Rausch R, Eddiry S, et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J Clin Invest (2017) 127(1):293–305. doi:10.1172/JCI91307
92. Zhan C, Zhou J, Feng Q, Zhang JE, Lin S, Bao J, et al. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J Neurosci (2013) 33(8):3624–32. doi:10.1523/JNEUROSCI.2742-12.2013
93. Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ. Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci (2009) 29(43):13684–90. doi:10.1523/JNEUROSCI.3770-09.2009
94. Li YQ, Shrestha Y, Pandey M, et al. Gq/11α and Gsα mediate distinct physiological responses to central melanocortins. J Clin Invest (2016) 126(1):40–9. doi:10.1172/JCI76348
95. Rediger A, Piechowski CL, Habegger K, Grüters A, Krude H, Tschöp MH, et al. MC4R dimerization in the paraventricular nucleus and GHSR/MC3R heterodimerization in the arcuate nucleus: is there relevance for body weight regulation? Neuroendocrinology (2012) 95(4):277–88. doi:10.1159/000334903
96. Agosti F, Cordisco Gonzalez S, Martinez Damonte V, Tolosa MJ, Di Siervi N, Schioth HB, et al. Melanocortin 4 receptor constitutive activity inhibits L-type voltage-gated calcium channels in neurons. Neuroscience (2017) 346:102–12. doi:10.1016/j.neuroscience.2017.01.007
97. Pandit R, van der Zwaal EM, Luijendijk MC, Brans MA, van Rozen AJ, Oude Ophuis RJ, et al. Central melanocortins regulate the motivation for sucrose reward. PLoS One (2015) 10(3):e0121768. doi:10.1371/journal.pone.0121768
98. Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature (2012) 487(7406):183–9. doi:10.1038/nature11160
99. O’Connor EC, Kremer Y, Lefort S, Harada M, Pascoli V, Rohner C, et al. Accumbal D1R neurons projecting to lateral hypothalamus authorize feeding. Neuron (2015) 88(3):553–64. doi:10.1016/j.neuron.2015.09.038
100. Yen HH, Roseberry AG. Decreased consumption of rewarding sucrose solutions after injection of melanocortins into the ventral tegmental area of rats. Psychopharmacology (Berl) (2015) 232(1):285–94. doi:10.1007/s00213-014-3663-6
101. Cabeza de Vaca S, Kim G-YY, Carr KD. The melanocortin receptor agonist MTII augments the rewarding effect of amphetamine in ad-libitum-fed and food-restricted rats. Psychopharmacology (Berl) (2002) 161(1):77–85. doi:10.1007/s00213-002-0998-1
102. Cui H, Lutter M. The expression of MC4Rs in D1R neurons regulates food intake and locomotor sensitization to cocaine. Genes Brain Behav (2013) 12(6):658–65. doi:10.1111/gbb.12057
103. Hsu R, Taylor JR, Newton SS, Alvaro JD, Haile C, Han G, et al. Blockade of melanocortin transmission inhibits cocaine reward. Eur J Neurosci (2005) 21(8):2233–42. doi:10.1111/j.1460-9568.2005.04038.x
104. Shelkar GP, Kale AD, Singh U, Singru PS, Subhedar NK, Kokare DM. Alpha-melanocyte stimulating hormone modulates ethanol self-administration in posterior ventral tegmental area through melanocortin-4 receptors. Addict Biol (2015) 20(2):302–15. doi:10.1111/adb.12126
105. Pandit R, Omrani A, Luijendijk MC, de Vrind VA, Van Rozen AJ, Ophuis RJ, et al. Melanocortin 3 receptor signaling in midbrain dopamine neurons increases the motivation for food reward. Neuropsychopharmacology (2016) 41(9):2241–51. doi:10.1038/npp.2016.19
106. Hu J, Jiang L, Low MJ, Rui L. Glucose rapidly induces different forms of excitatory synaptic plasticity in hypothalamic POMC neurons. PLoS One (2014) 9(8):e105080. doi:10.1371/journal.pone.0105080
107. Chen M, Wan Y, Ade K, Ting J, Feng G, Calakos N. Sapap3 deletion anomalously activates short-term endocannabinoid-mediated synaptic plasticity. J Neurosci (2011) 31(26):9563–73. doi:10.1523/JNEUROSCI.1701-11.2011
108. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature (1999) 402(6762):656–60. doi:10.1038/45230
109. Ghelardoni S, Carnicelli V, Frascarelli S, Ronca-Testoni S, Zucchi R. Ghrelin tissue distribution: comparison between gene and protein expression. J Endocrinol Invest (2006) 29(2):115–21. doi:10.1007/BF03344083
110. Gnanapavan S. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab (2002) 87(6):2988–2988. doi:10.1210/jc.87.6.2988
111. Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet (2005) 20(5):368–78. doi:10.2133/dmpk.20.368
112. Shintani M, Ogawa Y, Ebihara K, Aizawa-Abe M, Miyanaga F, Takaya K, et al. Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes (2001) 50(2):227–32. doi:10.2337/diabetes.50.2.227
113. Lee JH, Lin L, Xu P, Saito K, Wei Q, Meadows HE, et al. Neuronal deletion of ghrelin receptor almost completely prevents diet-induced obesity. Diabetes (2016) 65(8):2169–78. doi:10.2337/db15-1587
114. Yildiz BO, Suchard MA, Wong M-L, Mccann SM, Licinio J. Alterations in the Dynamics of Circulating Ghrelin, Adiponectin, and Leptin in Human Obesity. (2004). Available from: http://www.pnas.org.ezproxyhost.library.tmc.edu/content/101/28/10434.full.pdf
115. Shrestha YB, Wickwire K, Giraudo SQ. Direct effects of nutrients, acetylcholine, CCK, and insulin on ghrelin release from the isolated stomachs of rats. Peptides (2009) 30(6):1187–91. doi:10.1016/j.peptides.2009.02.001
116. Sakata I, Park WM, Walker AK, Piper PK, Chuang JC, Osborne-Lawrence S, et al. Glucose-mediated control of ghrelin release from primary cultures of gastric mucosal cells Glucose-mediated control of ghrelin release from primary cultures of gastric mucosal cells. Am J Physiol Endocrinol Metab (2012) 302(10):E1300–10. doi:10.1152/ajpendo.00041.2012
117. Lippl F, Kircher F, Erdmann J, Allescher H-D, Schusdziarra V. Effect of GIP, GLP-1, insulin and gastrin on ghrelin release in the isolated rat stomach. Regul Pept (2004) 119(1):93–8. doi:10.1016/j.regpep.2004.01.003
118. Banks WA, Tschop M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther (2002) 302(2):822–7. doi:10.1124/jpet.102.034827
119. Banks WA, Burney BO, Robinson SM. Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood-brain barrier. Peptides (2008) 29(11):2061–5. doi:10.1016/j.peptides.2008.07.001
120. Schellekens H, van Oeffelen WE, Dinan TG, Cryan JF. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. J Biol Chem (2013) 288(1):181–91. doi:10.1074/jbc.M112.382473
121. Damian M, Mary S, Maingot M, M’Kadmi C, Gagne D, Leyris JP, et al. Ghrelin receptor conformational dynamics regulate the transition from a preassembled to an active receptor: Gq complex. Proc Natl Acad Sci U S A (2015) 112(5):1601–6. doi:10.1073/pnas.1414618112
122. Wu CS, Bongmba OYN, Yue J, Lee JH, Lin L, Saito K, et al. Suppression of GHS-R in AgRP neurons mitigates diet-induced obesity by activating thermogenesis. Int J Mol Sci (2017) 18(4):832. doi:10.3390/ijms18040832
123. Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol (2007) 12(1):6–16. doi:10.1111/j.1369-1600.2006.00041.x
124. Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, et al. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry (2010) 67(9):880–6. doi:10.1016/j.biopsych.2009.10.030
125. Olszewski PK, Li D, Grace MK, Billington CJ, Kotz CM, Levine AS. Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides (2003) 24(4):597–602. doi:10.1016/S0196-9781(03)00105-0
126. Seoane LM, López M, Tovar S, Casanueva FF, Señarís R, Diéguez C. Agouti-related peptide, neuropeptide Y, and somatostatin-producing neurons are targets for ghrelin actions in the rat hypothalamus. Endocrinology (2003) 144(2):544–51. doi:10.1210/en.2002-220795
127. Jerlhag E, Egecioglu E, Landgren S, Salomé N, Heilig M, Moechars D, et al. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A (2009) 106(27):11318–23. doi:10.1073/pnas.0812809106
128. Suchankova P, Engel JA, Jerlhag E. Sub-chronic ghrelin receptor blockade attenuates alcohol- and amphetamine-induced locomotor stimulation in mice. Alcohol Alcohol (2016) 51(2):121–7. doi:10.1093/alcalc/agv100
129. Haass-Koffler CL, Aoun EG, Swift RM, de la Monte SM, Kenna GA, Leggio L. Leptin levels are reduced by intravenous ghrelin administration and correlated with cue-induced alcohol craving. Transl Psychiatry (2015) 5(9):e646. doi:10.1038/tp.2015.140
130. Leggio L, Zywiak WH, Fricchione SR, Edwards SM, de la Monte SM, Swift RM, et al. Intravenous ghrelin administration increases alcohol craving in alcohol-depe1ndent heavy drinkers: a preliminary investigation. Biol Psychiatry (2014) 76(9):734–41. doi:10.1016/j.biopsych.2014.03.019
131. Jerlhag E, Ivanoff L, Vater A, Engel JA. Peripherally circulating ghrelin does not mediate alcohol-induced reward and alcohol intake in rodents. Alcohol Clin Exp Res (2014) 38(4):959–68. doi:10.1111/acer.12337
132. Gomez JL, Cunningham CL, Finn DA, Young EA, Helpenstell LK, Schuette LM, et al. Differential effects of ghrelin antagonists on alcohol drinking and reinforcement in mouse and rat models of alcohol dependence. Neuropharmacology (2015) 97:182–93. doi:10.1016/j.neuropharm.2015.05.026
133. Kern A, Mavrikaki M, Ullrich C, Albarran-Zeckler R, Brantley AF, Smith RG. Hippocampal dopamine/DRD1 signaling dependent on the ghrelin receptor. Cell (2015) 163(5):1176–90. doi:10.1016/j.cell.2015.10.062
134. Park H-KK, Ahima RS. Physiology of leptin: energy homeostasis, neuroendocrine function and metabolism. Metabolism (2015) 64:24–34. doi:10.1016/j.metabol.2014.08.004
135. Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an erk-gated conduit for leptin into the brain. Cell Metab (2014) 19:293–301. doi:10.1016/j.cmet.2013.12.015
136. Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest (1996) 98(5):1101–6. doi:10.1172/JCI118891
137. Domingos AI, Vaynshteyn J, Voss HU, Ren X, Gradinaru V, Zang F, et al. Leptin regulates the reward value of nutrient. Nat Neurosci (2011) 14(12):1562–8. doi:10.1038/nn.2977
138. Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron (2006) 51(6):801–10. doi:10.1016/j.neuron.2006.08.023
139. Leshan RL, Opland DM, Louis GW, Leinninger GM, Patterson CM, Rhodes CJ, et al. Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci (2010) 30(16):5713–23. doi:10.1523/JNEUROSCI.1001-10.2010
140. Bruijnzeel AW, Corrie LW, Rogers JA, Yamada H. Effects of insulin and leptin in the ventral tegmental area and arcuate hypothalamic nucleus on food intake and brain reward function in female rats. Behav Brain Res (2011) 219(2):254–64. doi:10.1016/j.bbr.2011.01.020
141. Fernandes MF, Matthys D, Hryhorczuk C, Sharma S, Mogra S, Alquier T, et al. Leptin suppresses the rewarding effects of running via STAT3 signaling in dopamine neurons. Cell Metab (2015) 22(4):741–9. doi:10.1016/j.cmet.2015.08.003
142. Wellman PJ, Nation JR, Davis KW. Impairment of acquisition of cocaine self-administration in rats maintained on a high-fat diet. Pharmacol Biochem Behav (2007) 88(1):89–93. doi:10.1016/j.pbb.2007.07.008
143. Davis JF, Tracy AL, Schurdak JD, Tschöp MH, Lipton JW, Clegg DJ, et al. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav Neurosci (2008) 122(6):1257–63. doi:10.1037/a0013111
144. Kiefer F, Jahn H, Wolf K, Kampf P, Knaudt K, Wiedemann K. Free-choice alcohol consumption in mice after application of the appetite regulating peptide leptin. Alcohol Clin Exp Res (2001) 25(5):787–9. doi:10.1111/j.1530-0277.2001.tb02280.x
145. Kiefer F, Jahn H, Otte C, Demiralay C, Wolf K, Wiedemann K. Increasing leptin precedes craving and relapse during pharmacological abstinence maintenance treatment of alcoholism. J Psychiatr Res (2005) 39(5):545–51. doi:10.1016/j.jpsychires.2004.11.005
Keywords: hunger, satiety, obesity, hypothalamus, reward, superstimulus, dopamine
Citation: Cassidy RM and Tong Q (2017) Hunger and Satiety Gauge Reward Sensitivity. Front. Endocrinol. 8:104. doi: 10.3389/fendo.2017.00104
Received: 03 March 2017; Accepted: 02 May 2017;
Published: 18 May 2017
Edited by:
Serge H. Luquet, Paris Diderot University, FranceReviewed by:
Virginie Tolle, Institut national de la santé et de la recherche médicale (INSERM), FranceMiguel López, Universidade de Santiago de Compostela, Spain
Copyright: © 2017 Cassidy and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan Michael Cassidy, ryan.m.cassidy@uth.tmc.edu