 Nicholas J. G. Webster
Nicholas J. G. Webster- 1Medical Research Service, VA San Diego Healthcare System, San Diego, CA, United States
- 2Department of Medicine, School of Medicine, Moores Cancer Center, University of California San Diego, La Jolla, CA, United States
Non-alcoholic fatty liver disease (NAFLD) is becoming increasingly prevalent due to the worldwide obesity epidemic and currently affects one-third of adults or about one billion people worldwide. NAFLD is predicted to affect over 50% of the world’s population by the end of the next decade. It is the most common form of liver disease and is associated with increased risk for progression to a more severe form non-alcoholic steatohepatitis, as well as insulin resistance, type 2 diabetes mellitus, cirrhosis, and eventually hepatocellular carcinoma. This review article will focus on the role of alternative splicing in normal liver physiology and dysregulation in liver disease.
Introduction
Publication of the human genome sequence in 1995, and subsequently other mammalian genomes in the following two decades, has revealed a surprisingly small number of genes that must account for tremendous species diversity. Indeed, recent estimates have suggested that the number of human protein-coding genes may be as low as 19,000 (1). This is surprising given that the Drosophila melanogaster and Caenorhabditis elegans genomes encode 17,000 and 21,733 genes, respectively (2, 3), and even the lowly amoeboflagellate Naegleria gruberi, a free-living unicellular eukaryotic organism, has 15,727 genes (4). These observations posed a diversity paradox for genetics and challenged the one gene-one protein hypothesis. Unlike prokaryotic and lower eukaryotic genes, most mammalian genes are composed of multiple coding exons with intervening non-coding introns of variable length. Very often these exons encode discreet protein modules or substructures. Transcription of these split genes produces a primary transcript that requires further processing to remove the intronic sequences, a process called RNA splicing. Much of our understanding of the mechanism of RNA splicing comes from elegant biochemical and genetic studies in yeast and has been extensively reviewed (5). The presence of exons and introns provides a solution to the diversity paradox by allowing assembly of different proteins by modular construction of RNA transcript isoforms through a process termed alternative splicing (6, 7). The diversity of RNA transcripts is further amplified by the use of alternative transcription start sites and polyadenylation sequences. Transcriptome sequencing has shown that upward of 90% of mammalian genes have multiple transcript isoforms, and an estimated 160,000 alternatively spliced transcripts are protein encoding (8, 9). Although 85% of these genes have a predominant RNA transcript isoform (10), the minor isoforms can have different functions and may play an important role in disease.
Alternative splicing and the generation of protein diversity have broad implications for clinical disease. It is estimated that 50–60% of 31,250 disease-causing mutations in the Human Gene Mutation Database affect splicing (11, 12). Approximately 16% of these mutations are located directly in splice sites (13), and 66% are SNPs, microdeletions, or insertions within exons. While some of these latter mutations have a pathogenic effect by altering protein sequence, a large proportion do not, but rather interfere with splicing by interrupting exonic splicing enhancers or silencers. Cancer has been termed a disease of the genome due to the accumulation of DNA damage and genetic alterations that cumulatively cause transformation and malignancy. Indeed many mutations alter protein function by creating constitutively active oncogenic proteins or disrupting tumor suppressor proteins. However, it is now increasingly recognized that many cancer-associated RNA transcripts do not result from point mutations in the RNA itself, but rather by changes in expression or function of splicing factors that regulate the ordered splicing of primary gene transcripts giving rise to aberrant expression of oncofetal isoforms with greater proliferative capacity.
Mechanism of Alternative RNA Splicing
Much of our understanding of mechanism of RNA splicing comes from genetic studies in yeast and biochemical reconstitution experiments (5, 14). These studies have shown that the ends of an intron are aligned for excision by a complex network of RNA and protein interactions involving both splice sites in a large complex called the spliceosome. Initially the 5′ splice site is recognized by the U1 small nuclear ribonucleoprotein particle (snRNP) by base pairing of the U1 small nuclear RNA to the 5′ splice site. The U2 snRNP is then recruited to the 3′ splice site and branch point sequence by the accessory factor U2AF. The complex containing the pre-RNA, and the U1 and U2 snRNPs is called the pre-spliceosomal complex and defines the intron. This complex then recruits the U4/U5/U6 tri-snRNP, and the spliceosome undergoes a number of rearrangements including replacement of the U4:U6 duplex with a U2:U6 duplex, loss of the U4 snRNP, and displacement of the U1 snRNP on the 5′ splice site by the U6 snRNP to create the catalytically competent splicing complex. The actual splicing reaction then proceeds by two transesterification reactions first by the branch point adenine at the 5′ splice site then by the exonic terminal hydroxyl group at the 3′ splice site resulting in ligation of the two exons and liberation of an intron-lariat structure.
What defines whether an exon is recognized in a primary RNA transcript? While U1 and U2 snRNPs can interact across short introns to define the intron in typical in vitro splicing reactions, this interaction is much less efficient when the size of the intron increases above 250 nucleotides (15). As most introns are kilobases in length, yet the average size of an exon is ~200 nt, definition of the splice sites in vivo is generally thought to occur across exons rather than introns, a process termed exon definition (16). Thus, exons are defined by binding of U1 and U2 snRNP across the exon in the primary transcript followed by the long-range splice site pairing across the intron to assemble functional spliceosomes. Support for this exon-definition model comes from the finding that mutation of the downstream 5′ splice site on an exon can alter splicing of the upstream intron, so the sequential splicing of introns is coordinated and does not occur independent of each other.
How does this process allow for the use of different exons or splice sites during alternative splicing? In general, alternative exons contain weak splice sites that are not recognized efficiently (14). For genes that are co-transcriptionally spliced, this can be explained by a kinetic competition for assembly of the U1–U2 complex across alternative exons, or for non-co-transcriptionally spliced genes, this could be explained by the differences in the stability of the resulting complexes. Whether these weak exons are recognized is determined to a large extent by the presence of cis-acting binding sites for RNA-binding proteins within the exon or adjacent introns in the primary RNA transcript (17–19). Two of the most well-studied families of RNA-binding splicing regulators are the SR proteins (16 members) and the hnRNPs (20 members) (20–24), but there are also a number of less-studied families of RNA-binding proteins that regulate splicing, including the CELF/BRUNOL family, the Zinc-finger proteins, and the RBM family (25–27). All proteins contain RNA-binding domains allowing sequence specific-binding to RNA. Proteins of the SR family have an RNA recognition motif (RRM) at the amino-terminus, and a C-terminal domain that is enriched in arginine/serine dipeptides (RS domain) and heavily phosphorylated. hnRNP proteins show greater structural diversity than SR proteins, with RRM, RGG (arginine/glycine rich box), or KH (K homology box) RNA-binding domains. Additionally, the hnRNPs have auxiliary functional domains, which mediate protein–protein interactions and/or localization, and are divergent in protein sequence and structure (28). Both SR proteins and hnRNPs can promote or inhibit exon recognition depending on sequence context, thus modulating the usage of alternative exons or splice sites (14, 29, 30).
Alternative Splicing in Liver Development
While liver-specific transcriptional regulation is well established and has been studied for decades, liver-specific alternative splicing is less well understood. Alternative splicing has traditionally been studied on a gene-by-gene basis, which required prior knowledge of the gene transcripts, but the development of high-throughput array and RNA sequencing (RNA-seq) technologies has allowed an unbiased assessment of alternative splicing events (Table 1) (31). In a recent study, Nellore et al. aligned 21,504 human RNA-seq samples from the Sequence Read Archive to the human genome and compared exon–exon junctions to the known gene annotation databases (32). Approximately 19% of splice junctions (56,861) that were found in at least 1,000 samples were not previously annotated, indicating that a great deal of transcript diversity is still to be discovered. Brain, liver, and testis show the greatest diversity in transcripts with ~35–40% of genes showing alternative exon or splice site usage (33).
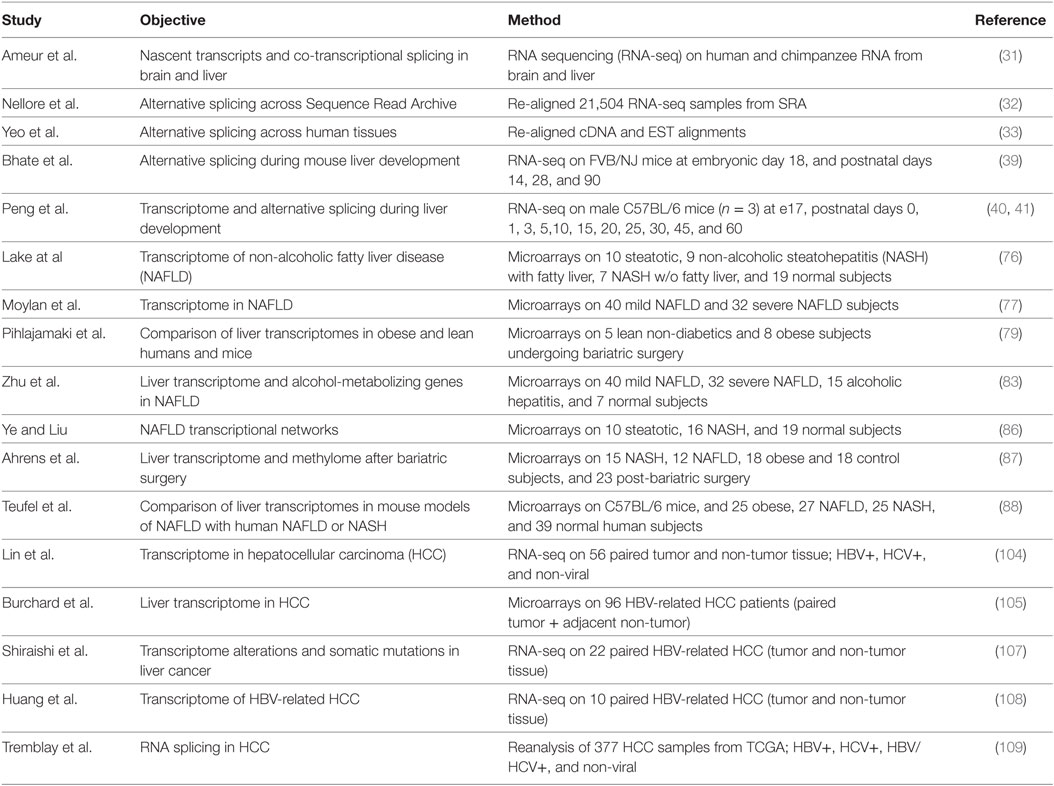
Table 1. Studies reporting alterations of RNA splicing factor expression or alternative splicing in liver.
Changes in alternative RNA splicing have been detected during the development of many tissues including the brain, heart, and skeletal muscle, and more recently in liver (31). Fetal liver does not perform a metabolic function as nutrients are provided from the mother via the placenta. Instead the fetal liver supports hematopoiesis in the embryo (34). Hepatocytes in the embryo are proliferative but they growth arrest and differentiate after birth as the liver takes on a metabolic function (35–37). Hematopoiesis also switches from the liver to the bone marrow during late gestation. The transcription factors regulating this transition in hepatocytes and cholangiocytes have been well documented (37, 38), but many genes also show a switch in fetal-to-adult RNA isoform expression reflecting changes in alternative splicing. Bhate et al. profiled the mouse liver transcriptomes at embryonic day 18 and postnatal days 14 and 28 and at 3 months by RNA-seq (39). In addition to 4,882 changes in gene expression between e18 and adult, the authors found 529 genes that underwent a change in RNA splicing and 214 genes that underwent a change in polyadenylation. The majority of these changes in alternative splicing were conserved between mouse liver and in human fetal (22 weeks) and adult (51 years) liver tissue.
A more extensive study by Peng et al. profiled mouse liver transcriptomes at embryonic day 18, and postnatal days 0, 1, 3, 5, 10, 15, 20, 25, 30, 45, and 60 of mouse liver development (40, 41). They found 7,289 genes that were differentially expressed at some point during development, and 829 of these had multiple annotated splicing variants with 90 being differentially expressed. In addition, they found evidence for 2,383 novel splice isoforms, of which 1,455 were detected at multiple times suggesting that there is a great deal of liver transcript information yet to be annotated. As might be expected, both studies indicated that genes associated with amino acid, fatty acid, cholesterol, bile, glucose, steroid, urea, and drug metabolism were upregulated in adult liver, whereas those associated with hematopoiesis, DNA repair and metabolism, cell cycle, and chromosome reorganization were downregulated. The changes in alternative splicing were not the result of altered cell populations in the liver as the majority (88%) were still observed in purified hepatocytes (39). A number of splicing factors decreased in expression, including Celf1, Celf2, Mbnl1, Ptbp1, Srsf1, 2, 3, 4, 6, 7, and 10, Hnrnpa1, and Hnrnph, but Esrp2 was increased in both studies.
A number of these genes have been studied in vitro, knocked out in the whole animal or deleted in hepatocytes (Table 2). Surprisingly, the Mbnl and Celf family proteins were identified in fetal liver. These proteins are expressed highly in muscle and have been studied extensively for their involvement in myotonic dystrophy (MD) (42–44). The whole-body knockout of Mbnl1 causes muscle and eye abnormalities reminiscent of MD (45). Subsequently, two studies reported that loss of Mbnl2 had no muscle phenotype but a third reported myotonia (46–48). Interestingly, one Mbnl2 knockout mouse had a brain phenotype with impaired hippocampal plasticity and synaptic transmission consistent with high-level expression of Mbnl2 in the brain (48). Loss of neither family member is associated with liver defects, however, but MBNL1 has been reported to regulate hematopoiesis in the fetal liver (49) potentially by regulating splicing of the Ndel1 gene. In contrast, CUGBP1 (Celf1) is highly expressed in the liver but its effects appear unrelated to its role as a splicing factor, but rather are due to its role as a translational regulator as it forms a complex with eIF2 to support translation of proteins involved in liver function and regulates hepatic stellate cell activation (50–55).
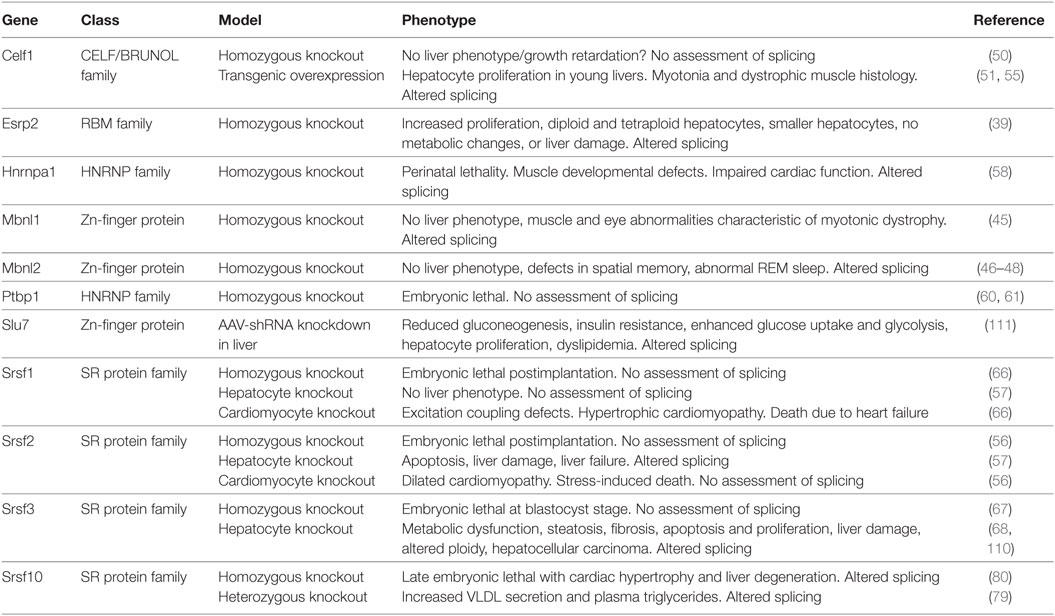
Table 2. Genetic manipulation of RNA-binding proteins in vivo.
Mice with complete deletion of Srsf2 die just after embryo implantation but mice with hepatocyte-specific deletion of Srsf2 are viable and have normal size at birth (56, 57). The mice fail to thrive, however, and die by 2–3 weeks of age. The cause of death is liver failure. In contrast, hepatocyte-specific deletion of Srsf1 did not have a phenotype and the mice were healthy. Livers in the newborn Srsf2 KO mice appear normal in size and color but by day 11 the livers are pale and firmer. Histologically, the livers show hepatocyte ballooning with periportal fibrosis and inflammation. The liver failure is likely due to the lack of proliferation of hepatocytes in the neonatal liver, and increased apoptosis possibly due to endoplasmic reticulum and oxidative stress. Metabolically, the knockout livers show steatosis and lack glycogen. RNA-seq analysis indicated that the mice livers have altered cholesterol and bile homeostasis as SRSF2 stimulates expression of liver transcription factors Srebp1c, Cebpa, Ppara, Nr1i3 (CAR1), Nr1h4 (FXR), Mlxipl (CHREBBP), and Foxa2. Thus, SRSF2 has effects on liver RNA splicing that are not compensated by other SR proteins, unlike the role of SRSF1 that appears redundant.
Homozygous deletion of Hnrnpa1 causes perinatal lethality within 30 min of birth because of muscle developmental defects. Death was due to cardiac dysfunction with higher blood pressure and heart rate, but defects were also observed in smooth and skeletal muscle (58). No liver phenotype was reported. The polypyrimidine tract binding protein PTBP1 (HNRNPI) binds to intronic sequences upstream of the 3′ splice site and represses splicing of pre-mRNAs (59). The effect of PTBP1 on liver function in vivo has not been studied as the homozygous deletion of Ptbp1 is embryonic lethal at the implantation stage (60, 61). In HepG2 hepatoma cells, however, PTBP1 modulates splicing of multiple genes involved in cholesterol synthesis and uptake including LDLR, MVK, HMGCS1, and PSCK9. It also regulates splicing of the fatty acid desaturase genes 2 and 3 (FADS2 and 3) that are involved in fatty acid elongation and unsaturation (62, 63). Consequently, omega-3 and omega-6 poly-unsaturated fatty acids were reduced following Ptbp1 knockdown, but saturated and mono-unsaturated fatty acids were not altered. Interestingly, PTBP1 is upregulated during hepatitis B virus infection and reduces expression of the proapoptotic form of FAS, which may contribute to the survival of infected hepatocytes (64).
Changes in alternative splicing during the mesenchymal to epithelial differentiation have been attributed to the splicing factors ESRP1 and ESRP2 (65). Expression of Esrp2 was increased in the adult liver and ablation of Esrp2 led to impaired adult splicing patterns implicating this splicing factor in the fetal-to-adult transition in hepatocytes (39). The livers did not show changes in morphology, however, nor did they display signs of liver damage, or any alterations in lipid, cholesterol, or glucose metabolism.
Other splicing factors have also been implicated in hepatocyte differentiation. Mice with complete loss of Srsf1 or Srsf3 die during early embryogenesis, but mice carrying a hepatocyte-specific deletion of Srsf1 or Srsf3 are viable (66, 67). The hepatocyte-specific deletion of Srsf1 did not show an overt liver phenotype but loss of Srsf3 caused impaired hepatocyte maturation (57, 68). The impaired differentiation was consistent with mis-splicing of Hnf1a that is critical for liver development, leading to reductions in other liver-enriched transcription factors including HNF6α (Onecut1), HNF3α (Foxa3), and C/EBPα. Consequently, the livers continued to express fetal markers such as α-fetoprotein (Afp) and H19. The impaired differentiation was associated with disrupted hepatic architecture characterized by large irregular hepatocytes, with compressed sinusoidal spaces and bile canaliculi, and reduced binuclearity. Interestingly, expression of Esrp2 is significantly reduced in the Srsf3 knockout, which may partly explain the impaired differentiation phenotype. The entire phenotype cannot be explained by loss of Esrp2, however, as Esrp2 ablated mice do not show changes in liver morphology or histology. Loss of Srsf3 also causes alterations in glucose and lipid homeostasis characterized by reduced glycogen storage, fasting hypoglycemia, increased insulin sensitivity, and reduced cholesterol synthesis although the target genes are distinct from those altered in the Srsf2 knockout. Like the Srsf2 knockout, loss of Srsf3 causes endoplasmic reticulum stress, hepatocyte apoptosis and proliferation, and liver damage but did not cause the liver failure seen in the Srsf2 knockout.
Alternative Splicing and Fatty Liver
Overnutrition and obesity leads to non-alcoholic fatty liver disease (NAFLD) and its more severe form non-alcoholic steatohepatitis (NASH) (69, 70). These metabolic disturbances are becoming more common in the general population due to the current obesity epidemic (71–73). Both NAFLD and NASH are associated with the metabolic syndrome and insulin resistance, and are risk factors for type 2 diabetes, non-alcoholic liver cirrhosis, and for the development of hepatocellular carcinoma (HCC) (74, 75). So understanding the changes that occur in the fatty or NASH liver is important to elucidate mechanisms underlying the heightened risk for subsequent disease progression. Transcriptome profiling by microarray has been performed in humans with NAFLD (76, 77). While this allows gene expression changes to be monitored, most studies do not address changes in RNA alternative splicing (78). Toward the goal of understanding changes in splicing, Pihlajamaki et al. profiled gene expression in liver samples from insulin-resistant humans with obesity (79). The top-ranked pathway downregulated in obese liver samples related to RNA processing and splicing. A number of splicing factors were decreased including SRSF10, SRSF7, SF3A1, SRSF2, SFPQ, and HNRNPs A1, K, D, and H. The authors showed that knockdown of SRSF10 increased lipogenesis in vitro in HepG2 cells and that heterozygous loss of Srsf10 in mice increased plasma triglycerides due to increased secretion of VLDL and mis-splicing of the lipid storage protein LIPIN-1 (Lpin1). Homozygous deletion of Srsf10, however, causes embryonic lethality with liver degeneration (80). This was the first example of how a change in RNA splicing could cause a change in lipid metabolism in the obese liver. SRSF10 may also regulate the splicing of the scavenger receptor class B, member 1 gene (SCARB1) that encodes the SR-BI and SR-BII proteins that mediate reverse cholesterol transport (81). The loss of SRSF10 in obesity remains controversial, however, as it was not seen in another study (82).
Another large microarray study examined liver gene expression in 72 subjects with mild or advanced NAFLD, 10 normal liver, and 17 subjects with HBV-associated liver failure (77, 83, 84). Ninety-two splicing factor genes were altered in this dataset with 30 splicing factors being altered in either mild or advanced NAFLD. Many of these were also identified in the Pihlajamaki study. Another study has shown a decrease in SRSF4 in NASH (85). A systems biology weighted gene co-expression network analysis of 16 human NASH, 10 NAFLD, and 19 normal liver samples identified a highly significant module (p < 2 × 10−6) associated with RNA processing (86). These changes are not in all datasets, however, as a German study in 45 morbidly obese subjects with NAFLD or NASH did not show alterations in splicing factor expression (87, 88). Studies in mice have shown similar changes in the expression of splicing factors in diet-induced obesity and NASH models (79, 85, 88, 89). So NAFLD and NASH are associated with changes in RNA splicing factor expression in the liver, and this likely contributes to alterations in RNA splicing. Transcriptome profiling by RNA-seq could potentially provide a measure of RNA splicing although such an approach has not been published. It will be interesting to see whether these alterations in RNA splicing can contribute to the pathophysiology.
Alternative Splicing and HCC
Worldwide, more than 700,000 people are diagnosed and 600,000 people die each year of liver cancer. HCC is the most common primary liver cancer (70–85%) (90) and usually arises after years of liver disease and inflammation (91) either due to chronic hepatitis B or C virus (HBV/HCV) infection (92), or alcoholic and non-alcoholic cirrhosis. The relative importance of these HCC subtypes depends on geography. HCC in HBV/HCV endemic regions in Asia and Africa is 80–90% virus associated, compared to only 20–50% of HCC in the US (93–95). Approximately 15–25% of HBV-infected individuals will develop chronic liver disease including cirrhosis, liver cancer, or failure, and 5–20% of HCV-infected individuals develop cirrhosis. A large majority (80%) of patients with HCC have cirrhosis, so cirrhosis is a major risk factor, but only 8% of patients with cirrhosis will develop HCC (96). In addition to chronic alcoholism, cirrhosis can have viral or metabolic causes (97–99), and alcohol use by at-risk individuals substantially increases the risk of cirrhosis and HCC. From a metabolic standpoint, obesity, NAFLD, and NASH are all risk factors for cirrhosis (75).
Alterations in RNA splicing in cancer have been known for over 30 years (11, 100, 101). Profiling the molecular alterations that occur in HCC has uncovered a number of targets with altered RNA splicing including the DNMT3b, AURKB, MDM2, TENSIN2, MAD1, KLF6, SVH, TP73, TP53, and FN1 genes (102). Many of these changes have been shown to have functional effects to promote proliferation, prevent apoptosis, and support transformation in cell culture experiments. RNA splicing is also important for HBV and HCV viral expression, and many viruses hijack the cellular splicing machinery to allow splicing of viral RNAs (103). More recent studies have utilized high-throughput technologies to survey the HCC transcriptome (104–107). A 2011 study sequenced the transcriptomes of 10 matched pairs of cancer and non-cancerous liver tissue from HBV-infected individuals (108). A total of 1,378 differentially expressed genes were identified in HCC, but more surprisingly 24,338 exons were differentially expressed, and the vast majority of differentially expressed genes also contained differentially expressed exons. A recent study utilized the RNA-seq data available through the TCGA database. Sequence data from 377 liver samples were reanalyzed to assess alterations in RNA splicing, uncovering ~45,000 alternative splicing events (109). These events were further filtered allowing the identification of 3,250 transcripts from 2,051 genes whose expression was altered in HBV-associated HCC, 1,380 transcripts from 907 genes that were altered in HCV-associated HCC, and 1,517 genes altered in non-viral HCC. Of these transcripts, 1,336 were shared by at least two groups. The authors also assessed splicing factor expression in these samples and found altered expression of 26 splicing factors, including ESRP2, SRSF2, CELF2, MBNL1, HNRNPA1, and HNRNPH, that were found altered in hepatocyte maturation study by Bhate et al. (39), that is consistent with oncofetal transformation. These studies are likely underestimates of the true dysregulated RNA splicing as most approaches rely on databases of known annotated RNA isoforms, so will exclude reads that do not correspond to known splicing events.
Conclusion and Future Perspectives
Although gross alterations in gene expression have been documented in nearly every disease state, recent data indicate that more subtle qualitative changes also occur, which may be just as important in disease pathogenesis. Recent high-throughput technologies are allowing a reassessment of these transcriptional changes with much higher resolution, providing a comprehensive documentation of individual transcript isoform identity and relative expression. These isoforms ultimately encode different proteins that could influence cellular function. Do these changes play a causal role in disease pathogenesis or are they simply a side effect of the disease? Traditionally, cancer was considered a disease of the genome and many of the changes in RNA splicing were thought to be a result of global alterations in gene expression in the cancer genome. Recent data, however, are indicating that subtle alterations in RNA splicing are observed in early disease, long before genomic alterations have occurred, and these alterations may play a role in predisposition to later disease. Data from mouse studies have suggested that altered splicing may cause cancer. Overexpression of the SR proteins SRSF1, SRSF3, and TRA2β (SRSF10) transforms fibroblasts and accelerates tumor growth in nude mice, and the proteins have been found to be elevated in certain cancers suggesting that they are proto-oncogenes. Interestingly, SRSF3 loss in hepatocytes also leads to liver cancer in mice (110), and SRSF3 is reduced in human HCC (111), so the properties of individual splicing factors may depend on cellular context. Aside from the SR proteins, other RNA-binding proteins that have been implicated as hnRNP proteins hnRNPA1, hnRNPA2, hnRNPH, and hnRNPI (PTB) are overexpressed in certain cancers (24, 100, 112–114), and knockdown of the proteins causes apoptosis in vitro. Overexpression of the zinc-finger protein MBNL2 in HCC correlates with smaller lower grade tumors and inhibits tumor growth and invasion in mice (115). Somatic mutations in splicing factor genes have also been found in cancers, the most frequently mutated being SF3B1, U2AF1, SRSF2, and ZRSR2 (116). Thus, dysregulation of RNA splicing may precede and predispose to carcinogenesis, and changes in splicing may be an early event in cancer initiation and warrant further investigation. Further studies testing individual transcript changes will be required to complete our understanding of the subtleties of gene expression that underlie early disease pathogenesis.
Author Contributions
NW conceived of and wrote the review.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
NW is funded by a VA Merit Review award (I01BX000130) and grants from the NIH (HD012303, CA155435, CA023100, CA196853, and DK033651).
References
1. Ezkurdia I, Juan D, Rodriguez JM, Frankish A, Diekhans M, Harrow J, et al. Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum Mol Genet (2014) 23:5866–78. doi:10.1093/hmg/ddu309
2. C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science (1998) 282:2012–8. doi:10.1126/science.282.5396.2012
3. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science (2000) 287:2185–95. doi:10.1126/science.287.5461.2185
4. Fritz-Laylin LK, Prochnik SE, Ginger ML, Dacks JB, Carpenter ML, Field MC, et al. The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell (2010) 140:631–42. doi:10.1016/j.cell.2010.01.032
6. Roy B, Haupt LM, Griffiths LR. Review: alternative splicing (AS) of genes as an approach for generating protein complexity. Curr Genomics (2013) 14:182–94. doi:10.2174/1389202911314030004
7. Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. Function of alternative splicing. Gene (2013) 514:1–30. doi:10.1016/j.gene.2012.07.083
8. Light S, Elofsson A. The impact of splicing on protein domain architecture. Curr Opin Struct Biol (2013) 23:451–8. doi:10.1016/j.sbi.2013.02.013
9. Tavares R, de Miranda Scherer N, Pauletti BA, Araujo E, Folador EL, Espindola G, et al. SpliceProt: a protein sequence repository of predicted human splice variants. Proteomics (2014) 14:181–5. doi:10.1002/pmic.201300078
10. Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink JJ, Lopez G, et al. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res (2013) 41:D110–7. doi:10.1093/nar/gks1058
11. Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet (2007) 8:749–61. doi:10.1038/nrg2164
12. Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett (2005) 579:1900–3. doi:10.1016/j.febslet.2005.02.047
13. Philips AV, Cooper TA. RNA processing and human disease. Cell Mol Life Sci (2000) 57:235–49. doi:10.1007/PL00000687
14. Lee Y, Rio DC. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem (2015) 84:291–323. doi:10.1146/annurev-biochem-060614-034316
15. Hertel KJ. Combinatorial control of exon recognition. J Biol Chem (2008) 283:1211–5. doi:10.1074/jbc.R700035200
16. Berget SM. Exon recognition in vertebrate splicing. J Biol Chem (1995) 270:2411–4. doi:10.1074/jbc.270.6.2411
17. Chabot B. Directing alternative splicing: cast and scenarios. Trends Genet (1996) 12:472–8. doi:10.1016/0168-9525(96)10037-8
18. Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet (2002) 18:186–93. doi:10.1016/S0168-9525(01)02626-9
19. Yeo G, Hoon S, Venkatesh B, Burge CB. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A (2004) 101:15700–5. doi:10.1073/pnas.0404901101
20. Ibrahim el C, Schaal TD, Hertel KJ, Reed R, Maniatis T. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc Natl Acad Sci U S A (2005) 102:5002–7. doi:10.1073/pnas.0500543102
21. Stark JM, Cooper TA, Roth MB. The relative strengths of SR protein-mediated associations of alternative and constitutive exons can influence alternative splicing. J Biol Chem (1999) 274:29838–42. doi:10.1074/jbc.274.42.29838
22. Lam BJ, Hertel KJ. A general role for splicing enhancers in exon definition. RNA (2002) 8:1233–41. doi:10.1017/S1355838202028030
23. Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma (2013) 122:191–207. doi:10.1007/s00412-013-0407-z
24. Han SP, Tang YH, Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J (2010) 430:379–92. doi:10.1042/BJ20100396
25. Barreau C, Paillard L, Mereau A, Osborne HB. Mammalian CELF/Bruno-like RNA-binding proteins: molecular characteristics and biological functions. Biochimie (2006) 88:515–25. doi:10.1016/j.biochi.2005.10.011
26. Font J, Mackay JP. Beyond DNA: zinc finger domains as RNA-binding modules. Methods Mol Biol (2010) 649:479–91. doi:10.1007/978-1-60761-753-2_29
27. Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem (2005) 94:5–24. doi:10.1002/jcb.20204
28. Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1’s multifunctional regulatory roles. RNA (2010) 16:1449–62. doi:10.1261/rna.2254110
29. Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol (2009) 10:741–54. doi:10.1038/nrm2777
30. Han J, Ding JH, Byeon CW, Kim JH, Hertel KJ, Jeong S, et al. SR proteins induce alternative exon skipping through their activities on the flanking constitutive exons. Mol Cell Biol (2011) 31:793–802. doi:10.1128/MCB.01117-10
31. Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, et al. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat Struct Mol Biol (2011) 18:1435–40. doi:10.1038/nsmb.2143
32. Nellore A, Jaffe AE, Fortin JP, Alquicira-Hernandez J, Collado-Torres L, Wang S, et al. Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the sequence read archive. Genome Biol (2016) 17:266. doi:10.1186/s13059-016-1118-6
33. Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol (2004) 5:R74. doi:10.1186/gb-2004-5-10-r74
34. Swain A, Inoue T, Tan KS, Nakanishi Y, Sugiyama D. Intrinsic and extrinsic regulation of mammalian hematopoiesis in the fetal liver. Histol Histopathol (2014) 29:1077–82. doi:10.14670/HH-29.1077
35. Enomoto H, Yoshida K, Kishima Y, Okuda Y, Nakamura H. Participation of hepatoma-derived growth factor in the regulation of fetal hepatocyte proliferation. J Gastroenterol (2002) 37(Suppl 14):158–61. doi:10.1007/BF03326437
36. Gruppuso PA, Bienieki TC, Faris RA. The relationship between differentiation and proliferation in late gestation fetal rat hepatocytes. Pediatr Res (1999) 46:14–9. doi:10.1203/00006450-199907000-00003
37. Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell (2010) 18:175–89. doi:10.1016/j.devcel.2010.01.011
38. Lemaigre FP. Mechanisms of liver development: concepts for understanding liver disorders and design of novel therapies. Gastroenterology (2009) 137:62–79. doi:10.1053/j.gastro.2009.03.035
39. Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun (2015) 6:8768. doi:10.1038/ncomms9768
40. Peng L, Yoo B, Gunewardena SS, Lu H, Klaassen CD, Zhong XB. RNA sequencing reveals dynamic changes of mRNA abundance of cytochromes P450 and their alternative transcripts during mouse liver development. Drug Metab Dispos (2012) 40:1198–209. doi:10.1124/dmd.112.045088
41. Gunewardena SS, Yoo B, Peng L, Lu H, Zhong X, Klaassen CD, et al. Deciphering the developmental dynamics of the mouse liver transcriptome. PLoS One (2015) 10:e0141220. doi:10.1371/journal.pone.0141220
42. Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans (2009) 37:1281–6. doi:10.1042/BST0371281
43. Cho DH, Tapscott SJ. Myotonic dystrophy: emerging mechanisms for DM1 and DM2. Biochim Biophys Acta (2007) 1772:195–204. doi:10.1016/j.bbadis.2006.05.013
44. Salvatori S, Furlan S, Fanin M, Picard A, Pastorello E, Romeo V, et al. Comparative transcriptional and biochemical studies in muscle of myotonic dystrophies (DM1 and DM2). Neurol Sci (2009) 30:185–92. doi:10.1007/s10072-009-0048-4
45. Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science (2003) 302:1978–80. doi:10.1126/science.1088583
46. Hao M, Akrami K, Wei K, De Diego C, Che N, Ku JH, et al. Muscleblind-like 2 (Mbnl2)-deficient mice as a model for myotonic dystrophy. Dev Dyn (2008) 237:403–10. doi:10.1002/dvdy.21428
47. Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet (2006) 15:2087–97. doi:10.1093/hmg/ddl132
48. Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron (2012) 75:437–50. doi:10.1016/j.neuron.2012.05.029
49. Cheng AW, Shi J, Wong P, Luo KL, Trepman P, Wang ET, et al. Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood (2014) 124:598–610. doi:10.1182/blood-2013-12-542209
50. Kress C, Gautier-Courteille C, Osborne HB, Babinet C, Paillard L. Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol Cell Biol (2007) 27:1146–57. doi:10.1128/MCB.01009-06
51. Wang GL, Salisbury E, Shi X, Timchenko L, Medrano EE, Timchenko NA. HDAC1 promotes liver proliferation in young mice via interactions with C/EBPbeta. J Biol Chem (2008) 283:26179–87. doi:10.1074/jbc.M803545200
52. Jones K, Timchenko L, Timchenko NA. The role of CUGBP1 in age-dependent changes of liver functions. Ageing Res Rev (2012) 11:442–9. doi:10.1016/j.arr.2012.02.007
53. Wu X, Wu X, Ma Y, Shao F, Tan Y, Tan T, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun (2016) 7:13498. doi:10.1038/ncomms13498
54. Cibois M, Boulanger G, Audic Y, Paillard L, Gautier-Courteille C. Inactivation of the Celf1 gene that encodes an RNA-binding protein delays the first wave of spermatogenesis in mice. PLoS One (2012) 7:e46337. doi:10.1371/journal.pone.0046337
55. Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet (2010) 19:3614–22. doi:10.1093/hmg/ddq277
56. Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, et al. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. EMBO J (2004) 23:885–96. doi:10.1038/sj.emboj.7600054
57. Cheng Y, Luo C, Wu W, Xie Z, Fu X, Feng Y. Liver-specific deletion of SRSF2 caused acute liver failure and early death in mice. Mol Cell Biol (2016) 36:1628–38. doi:10.1128/MCB.01071-15
58. Liu TY, Chen YC, Jong YJ, Tsai HJ, Lee CC, Chang YS, et al. Muscle developmental defects in heterogeneous nuclear ribonucleoprotein A1 knockout mice. Open Biol (2017) 7:160303. doi:10.1098/rsob.160303
59. Kafasla P, Mickleburgh I, Llorian M, Coelho M, Gooding C, Cherny D, et al. Defining the roles and interactions of PTB. Biochem Soc Trans (2012) 40:815–20. doi:10.1042/BST20120044
60. Shibayama M, Ohno S, Osaka T, Sakamoto R, Tokunaga A, Nakatake Y, et al. Polypyrimidine tract-binding protein is essential for early mouse development and embryonic stem cell proliferation. FEBS J (2009) 276:6658–68. doi:10.1111/j.1742-4658.2009.07380.x
61. Suckale J, Wendling O, Masjkur J, Jager M, Munster C, Anastassiadis K, et al. PTBP1 is required for embryonic development before gastrulation. PLoS One (2011) 6:e16992. doi:10.1371/journal.pone.0016992
62. Reardon HT, Park WJ, Zhang J, Lawrence P, Kothapalli KS, Brenna JT. The polypyrimidine tract binding protein regulates desaturase alternative splicing and PUFA composition. J Lipid Res (2011) 52:2279–86. doi:10.1194/jlr.M019653
63. Medina MW, Krauss RM. Alternative splicing in the regulation of cholesterol homeostasis. Curr Opin Lipidol (2013) 24:147–52. doi:10.1097/MOL.0b013e32835cf284
64. Liu W, Lin YT, Yan XL, Ding YL, Wu YL, Chen WN, et al. Hepatitis B virus core protein inhibits Fas-mediated apoptosis of hepatoma cells via regulation of mFas/FasL and sFas expression. FASEB J (2015) 29:1113–23. doi:10.1096/fj.14-263822
65. Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J (2010) 29:3286–300. doi:10.1038/emboj.2010.195
66. Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell (2005) 120:59–72. doi:10.1016/j.cell.2004.11.036
67. Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol (1999) 9:899–902. doi:10.1016/S0960-9822(99)80394-7
68. Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun (2013) 4:1336. doi:10.1038/ncomms2342
69. Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol (2003) 98:2042–7. doi:10.1111/j.1572-0241.2003.07659.x
70. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology (2006) 43:S99–112. doi:10.1002/hep.20973
71. Munteanu MA, Nagy GA, Mircea PA. Current management of NAFLD. Clujul Med (2016) 89:19–23. doi:10.15386/cjmed-539
72. Sherif ZA, Saeed A, Ghavimi S, Nouraie SM, Laiyemo AO, Brim H, et al. Global epidemiology of nonalcoholic fatty liver disease and perspectives on US minority populations. Dig Dis Sci (2016) 61:1214–25. doi:10.1007/s10620-016-4143-0
73. Ahmed M. Non-alcoholic fatty liver disease in 2015. World J Hepatol (2015) 7:1450–9. doi:10.4254/wjh.v7.i11.1450
74. Torres DM, Harrison SA. Nonalcoholic steatohepatitis and noncirrhotic hepatocellular carcinoma: fertile soil. Semin Liver Dis (2012) 32:30–8. doi:10.1055/s-0032-1306424
75. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med (2002) 346:1221–31. doi:10.1056/NEJMra011775
76. Lake AD, Novak P, Fisher CD, Jackson JP, Hardwick RN, Billheimer DD, et al. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos (2011) 39:1954–60. doi:10.1124/dmd.111.040592
77. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology (2014) 59:471–82. doi:10.1002/hep.26661
78. Shackel NA, Seth D, Haber PS, Gorrell MD, McCaughan GW. The hepatic transcriptome in human liver disease. Comp Hepatol (2006) 5:6. doi:10.1186/1476-5926-5-6
79. Pihlajamaki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab (2011) 14:208–18. doi:10.1016/j.cmet.2011.06.007
80. Feng Y, Valley MT, Lazar J, Yang AL, Bronson RT, Firestein S, et al. SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Dev Cell (2009) 16:528–38. doi:10.1016/j.devcel.2009.02.009
81. Zhang X, Moor AN, Merkler KA, Liu Q, McLean MP. Regulation of alternative splicing of liver scavenger receptor class B gene by estrogen and the involved regulatory splicing factors. Endocrinology (2007) 148:5295–304. doi:10.1210/en.2007-0376
82. Brosch M, von Schonfels W, Ahrens M, Nothnagel M, Krawczak M, Laudes M, et al. SFRS10 – a splicing factor gene reduced in human obesity? Cell Metab (2012) 15:265–6. doi:10.1016/j.cmet.2012.02.002
83. Zhu R, Baker SS, Moylan CA, Abdelmalek MF, Guy CD, Zamboni F, et al. Systematic transcriptome analysis reveals elevated expression of alcohol-metabolizing genes in NAFLD livers. J Pathol (2016) 238:531–42. doi:10.1002/path.4650
84. Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology (2013) 145:1076–87. doi:10.1053/j.gastro.2013.07.047
85. Lopez-Vicario C, Gonzalez-Periz A, Rius B, Moran-Salvador E, Garcia-Alonso V, Lozano JJ, et al. Molecular interplay between Delta5/Delta6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut (2014) 63:344–55. doi:10.1136/gutjnl-2012-303179
86. Ye H, Liu W. Transcriptional networks implicated in human nonalcoholic fatty liver disease. Mol Genet Genomics (2015) 290:1793–804. doi:10.1007/s00438-015-1037-3
87. Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab (2013) 18:296–302. doi:10.1016/j.cmet.2013.07.004
88. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology (2016) 151:513–25. doi:10.1053/j.gastro.2016.05.051
89. Smalling RL, Delker DA, Zhang Y, Nieto N, McGuiness MS, Liu S, et al. Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am J Physiol Gastrointest Liver Physiol (2013) 305:G364–74. doi:10.1152/ajpgi.00077.2013
90. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin (2011) 61:69–90. doi:10.3322/caac.20107
91. El-Serag HB. Hepatocellular carcinoma. N Engl J Med (2011) 365:1118–27. doi:10.1056/NEJMra1001683
92. Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol (2006) 45:529–38. doi:10.1016/j.jhep.2006.05.013
93. Di Bisceglie AM. Hepatitis B and hepatocellular carcinoma. Hepatology (2009) 49:S56–60. doi:10.1002/hep.22962
94. Nguyen VT, Law MG, Dore GJ. Hepatitis B-related hepatocellular carcinoma: epidemiological characteristics and disease burden. J Viral Hepat (2009) 16:453–63. doi:10.1111/j.1365-2893.2009.01117.x
95. Harnois DM. Hepatitis C virus infection and the rising incidence of hepatocellular carcinoma. Mayo Clin Proc (2012) 87:7–8. doi:10.1016/j.mayocp.2011.11.004
96. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology (2004) 127:S35–50. doi:10.1053/j.gastro.2004.09.014
97. Lin CW, Lin CC, Mo LR, Chang CY, Perng DS, Hsu CC, et al. Heavy alcohol consumption increases the incidence of hepatocellular carcinoma in hepatitis B virus-related cirrhosis. J Hepatol (2013) 58:730–5. doi:10.1016/j.jhep.2012.11.045
98. Campbell PT, Newton CC, Patel AV, Jacobs EJ, Gapstur SM. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care (2012) 35:1835–44. doi:10.2337/dc12-0002
99. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med (2003) 348:1625–38. doi:10.1056/NEJMoa021423
100. Kaida D, Schneider-Poetsch T, Yoshida M. Splicing in oncogenesis and tumor suppression. Cancer Sci (2012) 103:1611–6. doi:10.1111/j.1349-7006.2012.02356.x
101. Venables JP. Aberrant and alternative splicing in cancer. Cancer Res (2004) 64:7647–54. doi:10.1158/0008-5472.CAN-04-1910
102. Berasain C, Goni S, Castillo J, Latasa MU, Prieto J, Avila MA. Impairment of pre-mRNA splicing in liver disease: mechanisms and consequences. World J Gastroenterol (2010) 16:3091–102. doi:10.3748/wjg.v16.i25.3091
103. Karakama Y, Sakamoto N, Itsui Y, Nakagawa M, Tasaka-Fujita M, Nishimura-Sakurai Y, et al. Inhibition of hepatitis C virus replication by a specific inhibitor of serine-arginine-rich protein kinase. Antimicrob Agents Chemother (2010) 54:3179–86. doi:10.1128/AAC.00113-10
104. Lin KT, Shann YJ, Chau GY, Hsu CN, Huang CY. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene (2014) 33:4786–94. doi:10.1038/onc.2013.424
105. Burchard J, Zhang C, Liu AM, Poon RT, Lee NP, Wong KF, et al. microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol (2010) 6:402. doi:10.1038/msb.2010.58
106. Feng H, Qin Z, Zhang X. Opportunities and methods for studying alternative splicing in cancer with RNA-Seq. Cancer Lett (2013) 340:179–91. doi:10.1016/j.canlet.2012.11.010
107. Shiraishi Y, Fujimoto A, Furuta M, Tanaka H, Chiba K, Boroevich KA, et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS One (2014) 9:e114263. doi:10.1371/journal.pone.0114263
108. Huang Q, Lin B, Liu H, Ma X, Mo F, Yu W, et al. RNA-Seq analyses generate comprehensive transcriptomic landscape and reveal complex transcript patterns in hepatocellular carcinoma. PLoS One (2011) 6:e26168. doi:10.1371/journal.pone.0026168
109. Tremblay MP, Armero VE, Allaire A, Boudreault S, Martenon-Brodeur C, Durand M, et al. Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma. BMC Genomics (2016) 17:683. doi:10.1186/s12864-016-3029-z
110. Sen S, Langiewicz M, Jumaa H, Webster NJ. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology (2015) 61:171–83. doi:10.1002/hep.27380
111. Elizalde M, Urtasun R, Azkona M, Latasa MU, Goni S, Garcia-Irigoyen O, et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J Clin Invest (2014) 124:2909–20. doi:10.1172/JCI74382
112. Shkreta L, Bell B, Revil T, Venables JP, Prinos P, Elela SA, et al. Cancer-associated perturbations in alternative pre-messenger RNA splicing. Cancer Treat Res (2013) 158:41–94. doi:10.1007/978-3-642-31659-3_3
113. Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene (2014) 33:5311–8. doi:10.1038/onc.2013.533
114. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev (2010) 24:2343–64. doi:10.1101/gad.1973010
115. Lee YH, Jhuang YL, Chen YL, Jeng YM, Yuan RH. Paradoxical overexpression of MBNL2 in hepatocellular carcinoma inhibits tumor growth and invasion. Oncotarget (2016) 7:65589–601. doi:10.18632/oncotarget.11577
Keywords: non-alcoholic fatty liver disease, RNA splicing, hepatocellular carcinoma, splicing factors, microarrays
Citation: Webster NJG (2017) Alternative RNA Splicing in the Pathogenesis of Liver Disease. Front. Endocrinol. 8:133. doi: 10.3389/fendo.2017.00133
Received: 31 January 2017; Accepted: 30 May 2017;
Published: 21 June 2017
Edited by:
Gaetano Santulli, Columbia University, United StatesReviewed by:
Ulla Hansen, Boston University, United StatesJean Mazella, Centre national de la recherche scientifique (CNRS), France
Anne-Francoise Burnol, Institut national de la santé et de la recherche médicale (INSERM), France
Manuel D. Gahete, Instituto Maimónides de Investigación Biomédica de Córdoba, Spain
Copyright: © 2017 Webster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas J. G. Webster, nwebster@ucsd.edu