 Xiulong Xu
Xiulong Xu Yurong Lu1
Yurong Lu1 Richard A. Prinz
Richard A. Prinz- 1College of Veterinary Medicine, Institute of Comparative Medicine, Yangzhou University, Yangzhou, China
- 2Department of Anatomy and Cell Biology, Rush University Medical Center, Chicago, IL, United States
- 3Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX, United States
- 4Department of Surgery, NorthShore University Health System, Evanston, IL, United States
Thyroid cancer is the most common malignancy of the endocrine system. The initiation of thyroid cancer is often triggered by a genetic mutation in the phosphortidylinositol-3 kinase (PI3K) or mitogen-activated protein kinase (MAPK) pathway, such as RAS and BRAF, or by the rearrangement of growth factor receptor tyrosine kinase genes such as RET/PTC. The sonic hedgehog (Shh) pathway is evolutionarily conserved and plays an important role in the embryonic development of normal tissues and organs. Gene mutations in the Shh pathway are involved in basal cell carcinomas (BCC). Activation of the Shh pathway due to overexpression of the genes encoding the components of this pathway stimulates the growth and spread of a wide range of cancer types. The Shh pathway also plays an important role in cancer stem cell (CSC) self-renewal. GDC-0449 and LDE-225, two inhibitors of this pathway, have been approved for treating BCC and are being tested as a single agent or in combination with other drugs for treating various other cancers. Here, we review the recent findings on activation of the Shh pathway in thyroid cancer and its role in maintaining thyroid CSC self-renewal. We also summarize the recent developments on crosstalk of the Shh pathway with the MAPK and PI3K oncogenic pathways, and its implications for combination therapy.
Introduction
Thyroid cancer is the fifth most common cancer in women in the USA. Approximately 64,000 patients were newly diagnosed in 2016 (1, 2). The incidence of thyroid cancer has risen sharply in the past two decades. Much of this increase is attributed to newer and more sensitive imaging equipment and to intensive surveillance (3). Types of thyroid cancer include well differentiated papillary thyroid carcinomas (PTCs), follicular thyroid carcinomas (FTCs), medullary thyroid carcinomas (MTCs), Hürthle cell carcinomas (HTCs), and poorly differentiated or anaplastic thyroid carcinomas (ATCs) (2). PTCs account for more than 80% of all thyroid cancers (2). Surgery, thyroid hormone therapy, and radioiodine can cure most well differentiated thyroid cancers (PTC and FTC) but are much less effective treating poorly differentiated thyroid cancers. In addition, approximately 15–20% of all thyroid cancer patients will develop recurrence in their lifetime. The 10-year survival rate for patients with recurrent disease is approximately 10% (2, 4). The undifferentiated anaplastic subtype of thyroid cancer is almost always fatal, with a mean survival of only 2–6 months. There were approximately 2,000 deaths from thyroid cancer in the USA in 2016 (3).
Genetic alterations in thyroid cancer are relatively well understood (Table 1). They include RET/PTC rearrangements and mutations in the RAS, RET, BRAF, PIK3CK, and TERT genes (2). Mutations of these genes lead to activation of two prominent signaling pathways, the mitogen-activated protein kinase (MAPK) and phosphortidylinositol-3 kinase (PI3K) pathways. Approximately 60% of PTCs have a BRAF V600E mutation. BRAF-mutated PTCs often present with the pathological features of the classical variant or tall cell subtype. Approximately 15% of PTCs have a gene rearrangement with a frequency of RET > NTRK > others (2, 5). Those with a RET or NTRK rearrangement are mainly classical PTCs (2). Approximately 13% of PTCs have a RAS mutation (NRAS > HRAS > KRAS) and have a follicular variant feature (2). PTCs with BRAF mutations tend to be associated with more aggressive clinicopathologic characteristics, such as increased local invasion and distal metastasis, advanced stage at diagnosis, decreased radioiodine uptake, and increased mortality (6). The BRAF gene is almost never mutated in FTC (2). Instead, activation mutations of the RAS and PIK3CA (the p110 catalytic subunit of the PI3K) genes or the inactivation mutations of the PTEN gene frequently occur in FTC. Recent studies have shown that approximately 10% of PTCs have a TERT gene mutation, whereas 40% of poorly differentiated thyroid cancers and 70% of ATCs have a TERT mutation (7–9). ATCs are thought to progress from some well-differentiated PTCs or FTCs (2). BRAF and RAS are mutated in 45 and 24% of ATCs, respectively. The majority of ATCs harbor mutations of the BRAF or RAS gene plus the TERT gene (2). Understanding these genetic alterations and the activation of these signaling pathways offers unique opportunities for targeted therapy of thyroid cancer. However, due to drug resistance and crosstalk between different signaling pathways, targeted therapy often achieves only moderate or limited success. Therefore, the prevailing consensus is that combination therapies are needed to simultaneously target multiple signaling pathways to overcome drug resistance.

Table 1. Major genetic alterations in thyroid follicular cell carcinomas.
The Sonic Hedgehog (Shh) Pathway
The Shh pathway is activated by three ligands [Shh, Indian hedgehog (Ihh), and Desert hedgehog (Dhh)] that bind to their shared Patched (Ptch) receptor. These ligands are synthesized as precursor proteins, which are then cleaved to produce an N-terminal signaling protein that allows dual lipid modifications (Figure 1) (10, 11). The first modification is the addition of a cholesterol moiety on the C-terminus of cleaved hedgehog (HH), which allows HH to be retained at the plasma membrane. The second modification is mediated by HH acyltransferase, which catalyzes the addition of a palmitoyl group to the cholesterol-modified HH (11–13). Lipidated HH tends to be retained in sterol-rich membrane microdomains (14–16). Dispatched (Disp), a large multi-pass transmembrane protein, cooperates with Scube2, a secreted glycoprotein, to release the HH ligands from the plasma membrane and shield it from the aqueous microenvironment (17, 18). In addition, the lipidated HH can form monomers or multimers through cholesterol linkages (19, 20). HH-interacting protein (Hhip1) and the glycophosphatidylinositol (GPI)-linked heparin sulfate proteoglycan, Glypican-3 (Gpc3), can sequester HH, thus preventing its binding to Ptch receptor and inhibiting its activity (21–23).
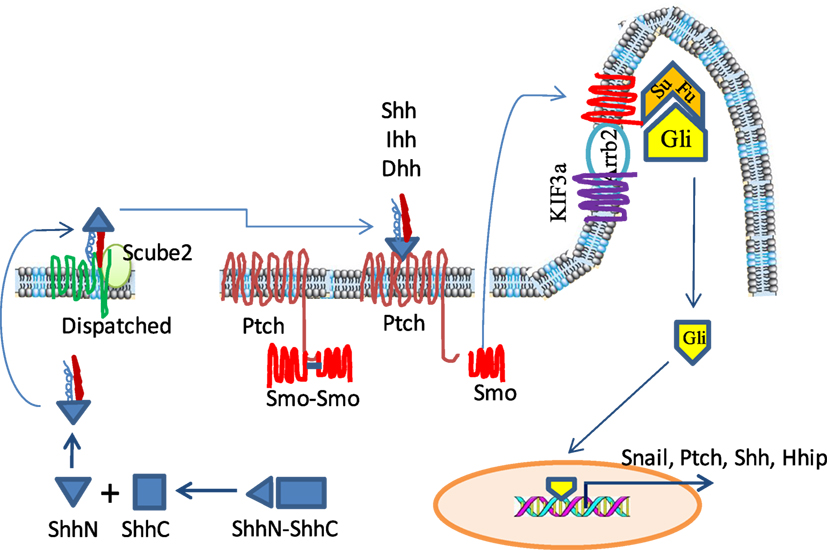
Figure 1. The sonic hedgehog (Shh) signaling pathway in a mammalian system. Hedgehog (HH) ligand proteins are processed in the cytosol by autoproteolytical cleavage to generate an N-terminal subunit, which is further modified by the addition of palmitoyl and cholesterol moieties. The lipidated Shh is stored in the lipid-rich microdomain on the cell surface but is released by cooperative action of Dispatched and Scube 2. In the absence of ligand binding, Patched (Ptch) restrains Smoothened (Smo) in the cytosol and keeps it as an inactive dimer. Glioma-associated oncogene (Gli) is located at the ciliary tip where it interacts with and is repressed by Suppressor of fused (SuFu). Upon HH binding, Ptch releases Smo and allows it to translocate into the cytoplasmic membrane of the ciliary tip where it cooperates with KIF3a and Arrb2 to disrupt the interaction of SuFu and Gli. Freed Gli is then translocated into the nucleus to activate the transcription of its target genes such as Snail, Shh, and Ptch.
Patched is a 12-pass transmembrane receptor in HH-responsive cells. In the absence of HH, Ptch constitutively represses Smoothened (Smo), a G-protein-coupled seven-pass transmembrane receptor, by preventing Smo translocation into the primary cilia (24–26). Therefore, in the absence of HH, Smo is inactive and is present in the cytoplasm as a dimer through the clustering of two amino acids in its C-terminus, arginine and asparagine (Figure 1). The Glioma-associated oncogene (Gli) proteins, including Gli1, Gli2, and Gli3, are a family of latent zinc-finger transcription factors. Gli forms complexes with the Suppressor of fused (Sufu). These complexes are located at the ciliary tip (Figure 1) (27, 28). Protein kinase A (PKA) and glycogen synthase kinase 3β (GSK3β) phosphorylate Gli2 and Gli3 but not Gli1 to create a binding site for the adaptor protein β-transducin repeat containing protein (β-TrCP) (29, 30). The Gli/β-TrCP complex is ubiquinated by the Cul1-based E3 ligase followed by partial proteasomal degradation. Truncated Gli2 and Gli3 translocate into the nucleus and usually function as transcriptional repressors through competing with Gli1 to bind the same DNA sequence (31). Since Gli1 cannot be processed in this way, it remains as a full-length transcriptional activator (32).
With HH ligand binding, Ptch1 activates the G-protein-coupled receptor kinase-2 (Grk2) to phosphorylate the adjacent domain of the C-terminus of Smo, and converts it from the inactive to an open conformation by neutralizing the electrostatic interactions of Smo dimers (33, 34) (Figure 1). HH binding to the Ptch receptor can be strengthened by several coreceptors, including CAM-Related/Downregulated by Oncogenes (Cdon), Brother of Cdon (Boc), and growth arrest specific 1 (Gas1), which form a multimolecular complex with Ptch (35, 36). HH binding also leads to Ptch1 internalization and degradation by lysosomes. Active Smo interacts with β-Arrestin (Arrb2) (37, 38) and the intraflagellar microtubule motor protein Kif3α, and translocates within the ciliary membrane where it facilitates the release of transcriptionally active full-length Gli proteins (GliA) from Sufu, thus avoiding proteasomal proteolytic cleavage and processing (37, 39). Gli1 then translocates to the nucleus and transcriptionally activates HH target genes (Figure 1).
Crosstalk Between the MAPK and Shh Pathways
The MAPK pathway is activated by a variety of extracellular stimuli, such as growth factors, osmotic stress, UV irradiation, reactive oxygen species, cytokines, and integrins (40) There are three parallel MAPK pathways, the classical MAPK, JNK, and p38 kinase pathways (41, 42). The cascade of these three MAPK pathways involves the activation of multiple serine/threonine kinases in the order of MAP3K → MAP2K → MAPK (40) (Figure 2). The classical MAPK pathway is activated by the binding of growth factors or cytokines to their receptor tyrosine kinases, which activate Ras through two adaptor proteins, Grb2 and SOS (43). Ras activation leads to the activation of the MAPK cascade from RAF (B-Raf, C-Raf, Raf-1) → MEK1/2 → ERK1/1 (44).
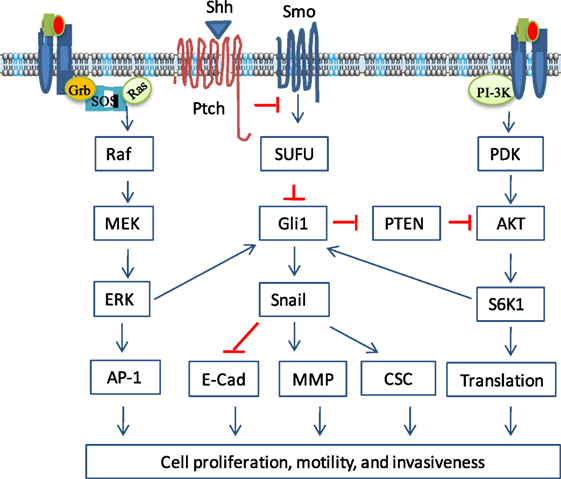
Figure 2. Non-canonical activation of the sonic hedgehog (Shh) pathway. Growth factor binding to their receptors activates two prominent oncogenic pathways, the phosphortidylinositol-3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways. In addition to the canonical activation, Gli1 can be activated by S6 kinase 1 (S6K1)-mediated phosphorylation at Serine 84, leading to nuclear translocation and induction of gene transcription. Gli1 can also be activated by ERK, probably through phosphorylation of its N-terminus by ERK2. Gli2 activity can be regulated by the MAPK pathway through increasing its stability. In addition, the MAPK pathway can activate the Shh pathway by inducing Shh expression through transcriptional upregulation. Gli1 can reciprocally activate the PI3K pathway indirectly by inducing Bmi1 expression, which represses PTEN expression. Crosstalk between the Shh and other oncogenic pathways regulates a variety of cellular functions, including cell proliferation, cell cycle progress, epithelial-to-mesenchymal transition, cell motility and invasiveness, and cancer stem cell (CSC) self-renewal.
Mutations of the genes in the MAPK pathway, such as RAS and BRAF, frequently take place in a wide variety of solid and hematological malignancies (45, 46). Several studies have shown that the Gli1 transcriptional activity can be enhanced by activation of the MAPK pathway (47). For example, Riobo et al. (48) reported that the expression of Gli target genes such as Gli1 itself and Ptch is enhanced in NIH3T3 cells transfected with constitutively active MEK mutants. The N-terminal region of Gli1, though not phosphorylated by MEK or its downstream ERK kinases, is required for sensing the MAPK pathway-mediated regulation (49). A later study showed that ERK2 may be responsible for the phosphorylation of a consensus site in the N-terminus of Gli1 (50). Ji et al. (51) reported that introduction of KRAS V12 into an immortalized human pancreatic epithelial cell line HPDE-c7 increases Gli1 expression levels and its transcriptional activity. Whereas inhibition of the MAPK pathway by the MEK1/2 inhibitor U0126 decreases Gli1 stability and suppresses the Gli1-mediated transcriptional activity in a KRAS-mutated pancreatic cancer cell line. KRAS cooperates with Gli1 to induce pancreatic cancer in a mouse model (52–54). The Shh pathway is activated in pancreatic cancers in mice transgenic for KRASG12D and p53R172H (52). Gli1 activation is required for tumor cell survival and KRAS-induced transformation in a second pancreatic mouse model (55). Inhibition of both Shh and MAPK pathways synergistically suppresses the proliferation of TE-1 gastric cancer cells (56). Inhibition of the MAPK pathway also leads to the inhibition of Gli1 transcriptional activity in an HT-29 colon cancer cell line (57, 58). Schnidar et al. (59) reported that the HH/GLI pathway cooperates with the epidermal growth factor receptor (EGFR) pathway to synergistically induce oncogenic transformation; and that pharmacologic inhibition of both EGFR and HH-Gli effectively reduces the growth of basal cell carcinoma (BCC) cell lines derived from mice with activated HH/GLI signaling. Similar to Gli1 regulation by K-Ras in pancreatic cancer, HRAS or NRAS mutation in melanoma stimulates Gli1 nuclear translocation by antagonizing the suppressive effect of SuFu through MEK1/2. Shh pathway inhibition by cyclopamine, a plant-derived teratogenic steroidal alkaloid that inhibits Smo (24–26), suppresses tumor growth in the tyrosinase-NRASQ61K:Ink4a−/− mouse model of melanoma (60, 61). Moreover, melanoma cell lines with a BRAF gene mutation are more sensitive to sonidegib than those without a BRAF mutation (62). Activation of the Shh pathway is also responsible for increased expression of PDGFRα in vemurafenib-resistant melanoma cell lines in vitro (63). PTCs have a high frequency of BRAF V600E mutation (6, 64, 65). Whether simultaneous inhibition of both Shh and MAPK pathways can synergistically inhibit thyroid tumor cell proliferation and tumor growth remains to be investigated.
Crosstalk Between the PI3K and Shh Pathways
The PI3K pathway plays important roles in tumor initiation, growth, and metastasis (66). It is activated by growth receptor tyrosine kinases, such as the insulin receptor, EGFR, and PDGFR (67) (Figure 3). These receptor tyrosine kinases phosphorylate the p85 subunit of the PI3K. Activated PI3K catalyzes the conversion of phosphoinositol (4,5) biphosphate (PIP2) to phosphoinositol (3,4,5) triphosphate (PIP3) (68). PIP3 interacts with the Plekstrin homology domain of AKT and recruits it to the cell membrane. Membrane-bound AKT changes its conformation and opens the C-terminal kinase domain for threonine 308 (T308) phosphorylation by phosphotidylinositol-dependent kinase (PDK). mTORC2 phosphorylates AKT at serine 473 (S473), the second site in the C-terminal hydrophobic motif, and fully activates AKT. However, the PI3K-mediated AKT activation can be antagonized by PTEN (phosphatase and tensin homolog deleted on chromosome 10), which dephosphorylates PIP3 to produce PIP2 (69). AKT is inactivated by protein phosphatase 2 A (PP2A), which dephosphorylates AKT at T308 (70), and by the Plekstrin homology domain leucine-rich repeat protein phosphatases (PHLPPs) 1 and 2, which dephosphorylate AKT at S473 (71). AKT phosphorylates tuberous sclerosis protein 2 (TSC2) and alleviates its repressive effect on RheB. RheB activates the mechanistic target of rapamycin (mTOR), a serine/threonine kinase involved in the formation of two complexes, mTORC1 and mTORC2 (72). mTORC1 consists of mTOR, mLST8, Raptor, Deptor, and PRAS40 (73) and phosphorylates the elF4E-binding protein (4F-BP) and p70 S6 kinase 1 (S6K1), a serine/threonine kinase that phosphorylates the ribosomal protein S6 (73). Both 4E-BP and S6 are involved in translation initiation and protein synthesis (Figure 2). mTORC2 consists of mTOR, Rictor, mLST8, Deptor, mSIN1, and Protor, and is responsible for AKT phosphorylation at S473.
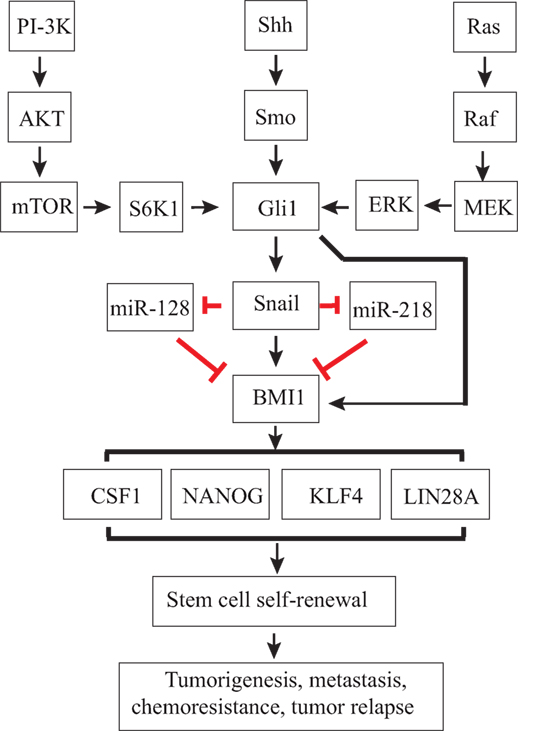
Figure 3. Regulation of thyroid cancer stem cell (CSC) self-renewal by the sonic hedgehog (Shh) pathway. Canonical or non-canonical Gli activation induces Snail expression. Gli1 and Snail may directly induce Bmi1 expression or indirectly induce Bmi1 expression through miRNAs, such as miR-128 or miR-218. Bmi1 is a master regulator that controls the expression of several stem cell-related genes, such as Sox2 and Nanog, and CSC self-renewal.
Several studies have shown that the PI3K and Shh pathways crosstalk with each other. Wang et al. (74) reported that S6K1 phosphorylates Gli1 at S84 and frees its sequestration from SuFu (Figure 2). S6K1 activation enhances Gli1 transcriptional activity and promotes its oncogenic function in esophageal cancer cell lines (74). Moreover, inhibition of both mTOR and Smo activities synergistically suppresses tumor growth (74). Upregulation of the PI3K pathway is in part responsible for drug resistance to sonidegib, a Shh pathway inhibitor used for treating medullablastoma (75). S6K1 activation induces Snail expression and epithelial-to-mesenchymal transition (EMT) in ovarian cancer cell lines (76). Gli1 activity is enhanced by AKT and by loss of tumor suppressors, such as p53 and PTEN (77). Gli1 is a key sensor that responds to both HH and an oncogenic load (77). Combined targeting of both the Shh and PI3K pathways achieves a synergistic therapeutic effect for a subgroup of chronic lymphocytic leukemia patients (78). The Shh and PI3K pathways synergistically promote the viability and growth of human PTEN-deficient glioblastomas (79). Co-inhibition of the PI3K and Shh pathways leads to mitotic catastrophy, tumor cell apoptosis, with a marked decrease of growth of PTEN-deficient glioblastomas in vitro and in vivo (79). The PI3K and Shh pathways also crosstalk in esophageal cancer (56). Cyclopamine inhibits EGF-stimulated AKT phosphorylation in TE-1 cells, an esophageal cell line; whereas Shh induces AKT phosphorylation, which is also partially inhibited by cyclopamine (56). Cyclopamine in combination with LY294002 synergistically inhibits the proliferation of three melanoma cell lines (WM-115, MeWo, and SK-Mel2) (61).
The PI3K pathway is highly activated in a variety of malignancies due to overexpression of growth factor receptor tyrosine kinases or due to mutations of the receptor tyrosine kinases (66, 80). Though PI3KCA and PTEN mutations only occur in ATCs and FTCs, the PI3K pathway is also highly activated in PTCs (68). Immunohistochemical staining and Western blot revealed AKT phosphorylation in >50% of thyroid cancers (68). Overexpression and hyperactivation of the growth factor receptor, RAS gene mutations, PIK3CA amplification, PTEN promoter hypermethylation, and RET/PTC rearrangements may all contribute to AKT activation (68). In addition, AKT nuclear localization is associated with thyroid cancer invasion and metastasis. AKT is detected in the nuclei of thyroid cancer cells, in particular in the region of tumor invasion (68).
Limited studies suggest that the PI3K and Shh pathways also crosstalk in thyroid cancer. AKT phosphorylation is decreased by inhibition of the Shh pathway with the Gli1 inhibitor GANT61 or with Shh/Gli1 knockdown, but it is increased in Gli1-overexpressing KAT-18 cells (81). Unexpectedly, cyclopamine inhibits Gli1 expression but has little effect on AKT phosphorylation in KAT-18 and SW1736 cells (81). It is possible that only Gli1 activation is responsible for AKT phosphorylation. A Smo inhibitor cannot, whereas a Gli1 inhibitor can inhibit AKT phosphorylation. Similarly, GANT61 inhibits AKT phosphorylation in embryonal and alveolar rhabdomyosarcomas (82). While these studies suggest that the Shh pathway regulates the activity of the PI3K pathway, whether the PI3K pathway also regulates the Shh pathway in thyroid cancer has not been investigated.
Shh Signaling in Thyroid Cancer
The Shh pathway plays an important role in tumorigenesis and is a valuable molecular target for cancer therapy (24, 25, 83). Activation of the Shh signaling pathway predisposes individuals to the development of the nevoid basal cell carcinoma syndrome (NBCCS), an autosomal-dominant disorder characterized by PTCH mutations (84–86). SMO and PTCH mutations are found in sporadic BCC and medulloblastomas in their early stage of tumor growth (86–89). Ligand-dependent activation of the Shh pathway occurs in early stage breast, prostate, digestive tract, and small cell lung cancers (90–94). The Shh pathway promotes tumorigenesis in part by stimulating cell proliferation via inducing the expression of the Cyclin D, N-Myc, Igf2, and Hes1 genes (24–26). Cyclopamine inhibits tumor cell proliferation and growth by inhibiting Smo activity (24–26).
Numerous studies suggest that the Shh pathway is involved in the growth and invasion of thyroid cancer in vitro and in vivo. We have reported that the Shh pathway is highly activated in approximately two-thirds of thyroid neoplasms and in thyroid cancer cell lines (95, 96). Shh, Ptch, Smo, and Gli1 are detected in approximately two-thirds of FTAs and PTCs and in the majority of ATC specimens (95). Greater than 77% of thyroid tumors remain simultaneously positive or negative for Shh, Ptch, Smo, and Gli1 (95). mRNAs and proteins of Shh, Ptch, Smo, and Gli1 were detected in three thyroid tumor cell lines (KAT-18, SW1736, and WRO82) (95). Inhibition of the Shh pathway by Shh or Gli1 knockdown or by Smo or Gli1 inhibitors significantly reduces cell proliferation (95). However, clinicopathological analysis shows no correlation between the activation of the Shh pathway and local invasion, distant metastasis, or tumor stage (95). By contrast, Bian et al. found that increased expression of the components of the Shh pathway is associated with increased thyroid tumor invasion and metastasis in a large number of PTC cases (97). The components of the Shh pathway are expressed in approximately 50% of anaplastic thyroid cancers and in two ATC cell lines, Hth 74 and C643 (98, 99). Both cell lines are very sensitive to cyclopamine, with the IC50 values between 1 and 4 µM. The components of the Shh pathway are also highly expressed in 7 MTC specimens; GDC-0449, a Smo inhibitor, inhibits the proliferation of TT cells, an MTC cell line (98, 99). These results collectively suggest that the Shh pathway is highly active in more than 50% of thyroid cancers to stimulate their cell proliferation.
Shh Signaling in Thyroid Cancer Stem Cells (CSCs)
Thyroid CSCs
Cancer is a very complex tissue (100). Cancer cell heterogeneity can be explained by two mutually non-exclusive stochastic and hierarchical models (100, 101). The former random model postulates that cancer development is initiated by the accumulation of genetic alterations in a single cancer cell, followed by a distinct gene mutation in a subpopulation of cells that are subsequently derived from this cell line (100, 102). The CSC model postulates that cancer comprises tumor cells in a hierarchy of cellular differentiation. The ability of CSCs to renew themselves confers CSC ability to resist chemotherapy, to metastasize, and to develop recurrent disease (103, 104). Thus, even if the initial response to chemotherapy or radiation is encouragingly robust, the patient may not be cured as long as CSC survives. Thus, these features of CSCs could explain well-observed but poorly understood phenomenon in cancer biology, including metastasis, recurrence, and therapeutic resistance (100).
Cancer stem cells are characterized by their remarkable capacity to develop tumors when implanted in immunodeficient mice and by their capacity to grow sphere-like aggregates in ultra-low attachment plates in the serum-free medium (100). Several markers have used to identify a unique group of stem cells in normal thyroid follicles and CSCs in thyroid cancers (101, 105–108). Stem cells identified in normal thyroid tissue in mice and humans are a side population in flow cytometry with Hoechst 33342 staining. These cells exhibit stem/progenitor cell-like characteristics, including the expression of stem cell markers, such as nucleostemin and Oct4, but do not express cell differentiation markers, such as thyroid peroxidase, thyroglobulin, and thyroid stimulating hormone receptor (109, 110). This side population identified in thyroid cancer cell lines has great capacity to form thyrospheres. It also expresses ATP-binding cassette sub-family G member 2 and exhibits increased clonality and invasive potential as well as drug resistance (111, 112). CD133 marks CSCs in several other tissues, but whether it could be used as a marker for thyroid CSCs remains controversial (101).
Aldehyde dehydrogenase (ALDH) is a reliable marker of CSCs in several types of malignancies. ALDH-positive cells isolated by flow cytometry can be analyzed for their tumor-initiating capacity. Todaro et al. reported that thyroid CSCs constitute a unique population (1–3%) with highly invasive and metastatic behavior in different types of thyroid cancers (113). Poorly differentiated or undifferentiated thyroid cancers contain a higher percentage of ALDH-positive CSCs than benign adenomas and well differentiated thyroid cancers (113). ALDH+ CSCs can expand indefinitely in vitro as tumor spheres and retain their tumorigenic potential when implanted in immunocompromised mice. Several hundred CSCs injected into the thyroid of immunocompromised mice develop into thyroid cancer that recapitulate the behavior of the parental tumor, including the aggressive metastatic features of undifferentiated thyroid carcinomas (113). Consistent with these observations, Hardin et al. (114) recently characterized two ALDHhigh clones derived from an ATC cell line, THJ-16T, and found that these clones have a much higher capacity to form thyrospheres than their parental cells and are highly tumorigenic in immnocompromised mice. By contrast, Shimamura et al. (115) showed that inhibition of ALDH activity with a specific inhibitor or by siRNA knockdown reduces ALDH-positive cells but does not significantly lower the number and growth of thyrospheres of four thyroid cancer cell lines (FRO, ACT1, KTC3, and 8505C), suggesting that ALDH may be just a marker for thyroid CSCs and may not have a functional role in thyroid CSC self-renewal. Ma et al. (116) showed that SSEA-1 is also a specific marker for thyroid CSCs; and that SSEA-1-postive thyroid CSCs express high levels of stem cell-related genes, such as Nanog, Sox2, and Oct4, and are resistant to 5-fluorouracil cytotoxicity. These authors also showed that an injection of 10,000 SSEA-1-positive T238 cells, an ATC cell line, was needed to develop cancer into athymic mice (116).
Regulation of Thyroid CSC Self-Renewal by the Shh Pathway
The Shh pathway has been implicated in regulating the self-renewal of CSCs (117). In breast cancer, the Shh pathway plays a critical role in maintaining mammary stem cells (118, 119). The mRNA levels of the genes in the Shh pathway, including PTCH1, GLI1, and GLI2, are elevated in CD44+/CD24−/lowLin− breast CSCs (119). The Shh pathway is also required for the maintenance of self-renewal of embryonal rhabdomyosarcoma CSCs (120). Inhibition of the Shh pathway by GANT61 or by siRNA suppresses the formation of tumor spheres in vitro and the development of embryonal rhabdomyosarcoma in vivo (120). Activation of the Shh pathway in multiple myeloma cell lines NCI-H929 and KMS12 stimulates the self-renewal and expansion of CD138+CD19− CSCs; whereas inhibition of the Shh pathway by cyclopamine or by the Shh neutralizing antibody 5E1 decreases the clonal capacity of multiple myeloma cell lines and CD138− cells through the induction of plasma differentiation (121). Loss of Shh signaling by genetically disrupting SMO results in the inhibition of BCR-ABL expressing leukemic stem cells and prolongs their survival (122, 123). The Shh signaling pathway is also highly activated in glioblastoma CSCs, whereas cyclopamine or siRNA directed against the pathway components results in the loss of tumorigenic potential (124, 125). Thus, the Shh pathway dictates the fate of CSCs, including self-renewal and differentiation (117).
Studies by our group suggest that the Shh pathway is involved in regulating thyroid CSC self-renewal (126). Suppression of the Shh pathway by Shh or Gli1 knockdown in KAT-18 thyroid cancer cell line leads to decreased size and number of thyrospheres, whereas Gli1 overexpression leads to increased number and size of thyrospheres. Malaguarnera et al. (127) reported that insulin receptor and insulin-like growth factor receptor activation stimulates the formation of thyrospheres of normal and thyroid cancer cells and induces the expression of stemness-related genes. Chen et al. (128) reported that metformin inhibits the proliferation of thyroid carcinoma cells, suppresses the self-renewal of CSCs, and potentiates the therapeutic effect of chemotherapeutic agents. Mechanistic studies suggest that activation of AMPK by metformin and subsequent inhibition of mTOR is responsible for this inhibitory effect on cell proliferation and thyroid CSC self-renewal (128). It is highly likely that the stimulatory effect of insulin and the inhibitory effect of metformin on thyroid CSC self-renewal may hinge on the activation of the Shh pathway through its crosstalk with the PI3K and MAPK pathways.
A hallmark of CSCs is their ability to resist chemo- and radiation therapy (129). Overexpression of Gli1 in KAT-18 cells significantly increases the number of surviving colonies after irradiation, compared to vector-transfected control cells. CSCs are highly invasive and metastatic due to the expression of several molecules involved in tumor cell motility and invasion. For example, CSCs express high levels of CXCR4, a chemokine receptor for CXCL12/SDF1 ligand which facilitates bone metastasis (130). CSCs that survive chemo- and radiation therapy play a critical role in tumor recurrence and metastasis (129). Todaro et al. showed that c-Met and AKT are highly activated and required for stimulating thyroid CSC invasion and metastasis (113). Williamson et al. (81) showed that suppression of the Shh pathway by miRNA targeting either Shh or Gli1 in a KAT-18 anaplastic cancer cell line decreases motility and invasiveness in Matrigel. By contrast, Gli1 overexpression in KAT-18 cells increases motility and invasive potential, compared to the cells transfected with the empty expression vector (81). These observations suggest that activation of the Shh pathway stimulates the motility and invasiveness of thyroid CSCs.
Mechanisms by Which the Shh Pathway Regulates CSC Self-Renewal
The mechanisms by which the Shh pathway maintains the self-renewal of stem cells are not fully understood. Numerous studies suggest that the Shh pathway promotes CSC self-renewal by inducing the expression of the stemness-related genes. Activation of the Shh pathway promotes breast CSC self-renewal by inducing the expression of the polycomb gene Bmi1, a master regulator of CSCs (131). Cyclopamine reduces the capacity of neurosphere formation and decreases the expression of Nanog, Sox2, and Oct4 in glioblastoma cell lines (132). Inactivation of the Shh pathway by Huaier extract reduces the number of CD44+/CD24− cells and decreases the levels of stem cell markers (OCT4, NESTIN, and NANOG) (133). Inhibition of the Shh pathway by cyclopamine leads to the downregulation of NANOG mRNA in a HCT-116 colon cancer cell line and decreases tumor spheres (134). Epigallocatechin-3-gallate (EGCG), an active compound in green tea that inhibits the expression of the components of the Shh pathway (Smo, Ptch, Gli1, and Gli2) and Gli transcriptional activity, inhibits the self-renewal capacity of pancreatic CSCs by inhibiting the expression of pluripotency maintaining transcription factors (Nanog, c-Myc, and Oct4) (135). Mechanistic studies suggest that Sox2 is transcriptionally upregulated by Gli1 binding to a cis-element in the SOX2 promoter (136).
Snail is a transcriptional factor whose expression is induced by Gli1 (137, 138). Snail is best known for its role in inducing EMT (139), but emerging evidence suggests that Snail plays an important role in regulating CSC self-renewal (139). Snail expression in immortalized mammary epithelial cells leads to the enrichment of CD44hiCD24lo breast CSCs (140). Snail expression in a human squamous cell carcinoma cell line induces EMT and CSC-like properties (141). Increased Snail expression in ComBit transgenic mice leads to the spontaneous development of thyroid cancer and increases the thyroid cancer incidence rate after irradiation (142). Heiden et al. (126) reported that activation of the Shh pathway leads to increased Snail expression in thyroid cancer cell lines, and that suppression of Snail expression by siRNA decreases the number of ALDHHigh thyroid CSCs in SW1736 and KAT-18 cells, two anaplastic thyroid cancer cell lines. These findings suggest that Snail plays a critical role in the Shh pathway-mediated maintenance of CSC self-renewal in ATC cell lines. Consistently, Ma et al. (116) reported that Snail expression is significantly higher in SSEA-1-positive thyroid CSCs than in SSEA-1-negative non-CSCs. Intriguingly, Yasui et al. (143) reported that Snail overexpression in an ACT-I thyroid tumor cell line increases the number of thyrospheres but decreases the number of ALDHHigh cells. Baquero et al. (144) reported that BRAF V600E mutation leads to increased Snail expression and decreased E-cadherin expression in thyroid cancer cell lines. Though the underlying molecular mechanisms are not clear, it is highly likely that non-canonical Gli1 activation through the MAPK pathway may be responsible for BRAF mutation-induced Snail expression (Figure 3).
Bmi1 is a member of the polycomb gene group that regulates gene expression by chromatin modification (145). Bmi1 inhibits PTEN expression in nasopharyngeal cancers, subsequently activating AKT (146). Bmi1 stimulates the proliferation of hepatocellular carcinomas by suppressing INK4A/ARF gene expression (147). Bmi1 stimulates CSC self-renewal in hepatocellular carcinomas (147, 148), pancreatic cancer (149, 150), and head and neck squamous cell carcinomas (151, 152). Artemisinin, an antimalarial drug, inhibits tumor cell proliferation of head and neck squamous cell carcinomas by inhibiting Bmi1 expression (153). A recent study showed that Bmi1 induces an invasive signature that promotes metastasis and chemoresistance in melanoma (154).
Bmi1 is an important target gene of the Shh pathway. Activation of the Shh pathway induces Bmi1 expression in medulloblastoma and breast cancer (131, 155). Gli2 overexpression in mammosphere-initiating cells results in the production of ductal hyperplasia (131). Modulation of Bmi1 expression in mammosphere-initiating cells alters mammary development in a humanized non-obese diabetic-severe combined immunodeficient mouse model (131). Using chromatin immunoprecipitation assay, Wang et al. reported that Gli1 directly binds the promoter of Bmi1 in medulloblastoma (155). Gopinath et al. (156) reported that cathespin and uPAR proteinase induce Sox2 and Bmi1 expression and promotes the self-renewal of glioma CSCs by Gli1. Liu et al. (131) showed that activation of the Shh pathway induces Bmi1 expression in breast cancer through Gli1. Taken together, these observations suggest that the Shh pathway regulates the expression of Bmi1 by Gli1 directly or indirectly by multiple pathways (Figure 3).
Anaplastic thyroid carcinomas contain a higher percentage of CSCs than other types of thyroid cancer (113). Several CSC-related genes, SOX2, SOX4, NANOG, c-MYC, and ABCG, are highly expressed in ATC (157). Some of these molecules, in particular, Sox2, Nanog, CD133, and ABC G2, are expressed in much higher levels in thyrospheres from fresh thyroid tumors than in monolayers (127) and are expressed at higher levels in ALDH+ than ALDH− thyroid cancer cell lines (114). Ferretti et al. (154) recently reported that Bmi1 confers resistance to a B-Raf inhibitor by activation of the non-canonical Wnt pathway in melanoma and that a Bmi1-induced gene signature predicts metastasis and the clinical outcome of melanoma patients. Our ongoing studies suggest that Bmi1, Sox2, and Nanog are highly expressed in thyroid cancer and can be regulated by the Shh pathway. Thus, the Shh pathway may regulate thyroid CSC self-renewal by inducing the expression of stem cell-related genes through Bmi1.
Implications and Perspectives
The Shh pathway is activated due to gene mutations or overexpression of the components of the Shh pathway, or due to cross-activation by other signaling pathways in a wide range of malignancy (33, 158). The Shh pathway is also important in the maintenance of CSC self-renewal (16). Since CSCs play essential roles in tumor initiation, drug resistance, recurrence, and metastasis, the inhibitors of the Shh pathway have been explored for treating various cancers. GDC-0449 (Vismodegib/Erivedge) and LDE-225 (Erismodegib/Sonidegib/Odomzo), two Smo inhibitors, were approved in 2012 and 2015, respectively, by the Federal Drug Administration for treating metastatic or locally invasive BCC (159). These two drugs, along with a dozen other inhibitors targeting the Shh pathway, are in various stages of clinical trials as mono- or combination therapy for a variety of malignancies (117, 158, 159). These clinical trials are promising but have achieved only modest responses (159). Better understanding of the underlying cell biology is needed to improve the efficacy of the inhibitors of the Shh pathway. In particular, most inhibitors in the Shh pathway target Smo. Non-canonical Gli1 activation by the PI3K and MAPK pathways may bypass Smo inhibition and may be responsible for the poor performance of Smo inhibitors in clinical trials (160, 161).
Although several studies suggest that the Shh pathway is highly activated in thyroid neoplasms and plays an important role in thyroid tumor cell proliferation and CSC self-renewal, many critical questions remain unanswered. First, the role of the Shh pathway in thyroid CSC self-renewal and tumor initiation has not been investigated in vivo. It remains unclear if blockade of the Shh pathway will eliminate CSCs and prevent thyroid tumor metastasis. Second, whether the Shh pathway is involved in thyroid tumor initiation or facilitates tumor development initiated by oncogenes, such as mutant BRAF, RAS, or RET, needs to be verified in a transgenic mouse model. Third, the crosstalk between the Shh and PI3K/MAPK pathways needs to be more thoroughly investigated. Better understanding of the interaction between the Shh and other oncogenic pathways will help design novel combination therapies for poorly differentiated or anaplastic thyroid cancers. Fourth, exactly how the Shh pathway regulates the self-renewal of CSCs in the thyroid and other organs remains vague and needs to be studied in detail. Finally, Smo and Gli1 inhibitors have not been tested in clinically relevant models such as patient-derived orthotopic xenografts. Better understanding of how the Shh signaling pathway regulates CSC self-renewal may offer unique opportunities for thyroid cancer therapy.
Author Contributions
XX conducted literature review and wrote the manuscript. YLu contributed to the conception and design of the work in the manuscript. YLi contributed to review, discussion, and conception of the work. RP critically read, revised, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by a Natural Science Foundation of China (81672643) and the Priority Academic Program Development of Jiangsu Higher Education Institutions to XX, and by NIH R01 (CA204926) to YLi.
References
1. Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer (2006) 6:292–306. doi:10.1038/nrc1836
2. Fagin JA, Wells SA Jr. Biologic and clinical perspectives on thyroid cancer. N Engl J Med (2016) 375:1054–67. doi:10.1056/NEJMra1501993
3. Cabanillas ME, McFadden DG, Durante C. Thyroid cancer. Lancet (2016) 388:2783–95. doi:10.1016/S0140-6736(16)30172-6
4. Cabanillas ME, Patel A, Danysh BP, Dadu R, Kopetz S, Falchook G. Braf inhibitors: experience in thyroid cancer and general review of toxicity. Horm Cancer (2015) 6:21–36. doi:10.1007/s12672-014-0207-9
5. Prescott JD, Zeiger MA. The ret oncogene in papillary thyroid carcinoma. Cancer (2015) 121:2137–46. doi:10.1002/cncr.29044
6. Xing M. Braf mutation and thyroid cancer recurrence. J Clin Oncol (2015) 33:2482–3. doi:10.1200/JCO.2015.61.4016
7. Shen X, Liu R, Xing M. A six-genotype genetic prognostic model for papillary thyroid cancer. Endocr Relat Cancer (2017) 24:41–52. doi:10.1530/ERC-16-0402
8. Liu R, Xing M. Tert promoter mutations in thyroid cancer. Endocr Relat Cancer (2016) 23:R143–55. doi:10.1530/ERC-15-0533
9. Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality risk stratification by combining braf v600e and tert promoter mutations in papillary thyroid cancer: genetic duet of braf and tert promoter mutations in thyroid cancer mortality. JAMA Oncol (2017) 3(2):202–8. doi:10.1001/jamaoncol.2016.3288
10. Perler FB. Protein splicing of inteins and hedgehog autoproteolysis: structure, function, and evolution. Cell (1998) 92:1–4. doi:10.1016/S0092-8674(00)80892-2
11. Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, et al. Identification of a palmitic acid-modified form of human sonic hedgehog. J Biol Chem (1998) 273:14037–45. doi:10.1074/jbc.273.22.14037
12. Chamoun Z, Mann RK, Nellen D, von Kessler DP, Bellotto M, Beachy PA, et al. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science (2001) 293:2080–4. doi:10.1126/science.1064437
13. Chen X, Tukachinsky H, Huang CH, Jao C, Chu YR, Tang HY, et al. Processing and turnover of the hedgehog protein in the endoplasmic reticulum. J Cell Biol (2011) 192:825–38. doi:10.1083/jcb.201008090
14. Taylor FR, Wen D, Garber EA, Carmillo AN, Baker DP, Arduini RM, et al. Enhanced potency of human sonic hedgehog by hydrophobic modification. Biochemistry (2001) 40:4359–71. doi:10.1021/bi002487u
15. Callejo A, Torroja C, Quijada L, Guerrero I. Hedgehog lipid modifications are required for hedgehog stabilization in the extracellular matrix. Development (2006) 133:471–83. doi:10.1242/dev.02217
16. Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers (Basel) (2015) 7:1554–85. doi:10.3390/cancers7030851
17. Tukachinsky H, Kuzmickas RP, Jao CY, Liu J, Salic A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep (2012) 2:308–20. doi:10.1016/j.celrep.2012.07.010
18. Amanai K, Jiang J. Distinct roles of central missing and dispatched in sending the hedgehog signal. Development (2001) 128:5119–27.
19. Gallet A, Rodriguez R, Ruel L, Therond PP. Cholesterol modification of hedgehog is required for trafficking and movement, revealing an asymmetric cellular response to hedgehog. Dev Cell (2003) 4:191–204. doi:10.1016/S1534-5807(03)00031-5
20. Goetz JA, Singh S, Suber LM, Kull FJ, Robbins DJ. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J Biol Chem (2006) 281:4087–93. doi:10.1074/jbc.M511427200
21. Capurro MI, Shi W, Filmus J. Lrp1 mediates hedgehog-induced endocytosis of the gpc3-hedgehog complex. J Cell Sci (2012) 125:3380–9. doi:10.1242/jcs.098889
22. Capurro MI, Xu P, Shi W, Li F, Jia A, Filmus J. Glypican-3 inhibits hedgehog signaling during development by competing with patched for hedgehog binding. Dev Cell (2008) 14:700–11. doi:10.1016/j.devcel.2008.03.006
23. Chang SC, Mulloy B, Magee AI, Couchman JR. Two distinct sites in sonic hedgehog combine for heparan sulfate interactions and cell signaling functions. J Biol Chem (2011) 286:44391–402. doi:10.1074/jbc.M111.285361
24. Hooper JE, Scott MP. Communicating with hedgehogs. Nat Rev Mol Cell Biol (2005) 6:306–17. doi:10.1038/nrm1622
25. Taipale J, Beachy PA. The hedgehog and wnt signalling pathways in cancer. Nature (2001) 411:349–54. doi:10.1038/35077219
26. van den Brink GR. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev (2007) 87:1343–75. doi:10.1152/physrev.00054.2006
27. Barnfield PC, Zhang X, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of gli1 and gli2 activator function by suppressor of fused through multiple mechanisms. Differentiation (2005) 73:397–405. doi:10.1111/j.1432-0436.2005.00042.x
28. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet (2010) 11:331–44. doi:10.1038/nrg2774
29. Niewiadomski P, Kong JH, Ahrends R, Ma Y, Humke EW, Khan S, et al. Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep (2014) 6:168–81. doi:10.1016/j.celrep.2013.12.003
30. Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, et al. Phosphorylation by double-time/ckiepsilon and ckialpha targets cubitus interruptus for slimb/beta-trcp-mediated proteolytic processing. Dev Cell (2005) 9:819–30. doi:10.1016/j.devcel.2005.10.006
31. Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, et al. Multiple ser/thr-rich degrons mediate the degradation of ci/gli by the cul3-hib/spop e3 ubiquitin ligase. Proc Natl Acad Sci U S A (2009) 106:21191–6. doi:10.1073/pnas.0912008106
32. Ruiz i Altaba A, Palma V, Dahmane N. Hedgehog-gli signalling and the growth of the brain. Nat Rev Neurosci (2002) 3:24–33. doi:10.1038/nrn704
33. Briscoe J, Therond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol (2013) 14:416–29. doi:10.1038/nrm3598
34. Chen Y, Sasai N, Ma G, Yue T, Jia J, Briscoe J, et al. Sonic hedgehog dependent phosphorylation by ck1alpha and grk2 is required for ciliary accumulation and activation of smoothened. PLoS Biol (2011) 9:e1001083. doi:10.1371/journal.pbio.1001083
35. Allen BL, Song JY, Izzi L, Althaus IW, Kang JS, Charron F, et al. Overlapping roles and collective requirement for the coreceptors gas1, cdo, and boc in shh pathway function. Dev Cell (2011) 20:775–87. doi:10.1016/j.devcel.2011.04.018
36. Izzi L, Levesque M, Morin S, Laniel D, Wilkes BC, Mille F, et al. Boc and gas1 each form distinct shh receptor complexes with ptch1 and are required for shh-mediated cell proliferation. Dev Cell (2011) 20:788–801. doi:10.1016/j.devcel.2011.04.017
37. Kim J, Kato M, Beachy PA. Gli2 trafficking links hedgehog-dependent activation of smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A (2009) 106:21666–71. doi:10.1073/pnas.0912180106
38. Chen W, Ren XR, Nelson CD, Barak LS, Chen JK, Beachy PA, et al. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and grk2. Science (2004) 306:2257–60. doi:10.1126/science.1104135
39. Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of gli transcription factors. Development (2005) 132:3103–11. doi:10.1242/dev.01894
40. Yang SH, Sharrocks AD, Whitmarsh AJ. Map kinase signalling cascades and transcriptional regulation. Gene (2013) 513:1–13. doi:10.1016/j.gene.2012.10.033
41. Caunt CJ, Sale MJ, Smith PD, Cook SJ. Mek1 and mek2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer (2015) 15:577–92. doi:10.1038/nrc4000
42. Burotto M, Chiou VL, Lee JM, Kohn EC. The mapk pathway across different malignancies: a new perspective. Cancer (2014) 120:3446–56. doi:10.1002/cncr.28864
43. Farooq A, Zhou MM. Structure and regulation of mapk phosphatases. Cell Signal (2004) 16:769–79. doi:10.1016/j.cellsig.2003.12.008
44. Malumbres M, Barbacid M. Ras oncogenes: the first 30 years. Nat Rev Cancer (2003) 3:459–65. doi:10.1038/nrc1097
45. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339:1546–58. doi:10.1126/science.1235122
46. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the raf-erk signaling pathway by oncogenic mutations of b-raf. Cell (2004) 116:855–67. doi:10.1016/S0092-8674(04)00215-6
47. Rovida E, Stecca B. Mitogen-activated protein kinases and hedgehog-gli signaling in cancer: a crosstalk providing therapeutic opportunities? Semin Cancer Biol (2015) 35:154–67. doi:10.1016/j.semcancer.2015.08.003
48. Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr. Phosphoinositide 3-kinase and akt are essential for sonic hedgehog signaling. Proc Natl Acad Sci U S A (2006) 103:4505–10. doi:10.1073/pnas.0504337103
49. Riobo NA, Haines GM, Emerson CP Jr. Protein kinase c-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control gli activation in hedgehog signaling. Cancer Res (2006) 66:839–45. doi:10.1158/0008-5472.CAN-05-2539
50. Whisenant TC, Ho DT, Benz RW, Rogers JS, Kaake RM, Gordon EA, et al. Computational prediction and experimental verification of new map kinase docking sites and substrates including gli transcription factors. PLoS Comput Biol (2010) 6:e1000908. doi:10.1371/journal.pcbi.1000908
51. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic kras activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem (2007) 282:14048–55. doi:10.1074/jbc.M611089200
52. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53r172h and krasg12d cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell (2005) 7:469–83. doi:10.1016/j.ccr.2005.04.023
53. Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/ras interactions regulate early stages of pancreatic cancer. Genes Dev (2006) 20:3161–73. doi:10.1101/gad.1470806
54. Lau J, Kawahira H, Hebrok M. Hedgehog signaling in pancreas development and disease. Cell Mol Life Sci (2006) 63:642–52. doi:10.1007/s00018-005-5357-z
55. Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, et al. GLI1 is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev (2009) 23:24–36. doi:10.1101/gad.1753809
56. Wei L, Xu Z. Cross-signaling among phosphinositide-3 kinase, mitogen-activated protein kinase and sonic hedgehog pathways exists in esophageal cancer. Int J Cancer (2011) 129:275–84. doi:10.1002/ijc.25673
57. Mazumdar T, Devecchio J, Agyeman A, Shi T, Houghton JA. Blocking hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res (2011) 71:5904–14. doi:10.1158/0008-5472.CAN-10-4173
58. Mazumdar T, DeVecchio J, Agyeman A, Shi T, Houghton JA. The GLI genes as the molecular switch in disrupting hedgehog signaling in colon cancer. Oncotarget (2011) 2:638–45. doi:10.18632/oncotarget.310
59. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, et al. Epidermal growth factor receptor signaling synergizes with hedgehog/gli in oncogenic transformation via activation of the mek/erk/Jun pathway. Cancer Res (2009) 69:1284–92. doi:10.1158/0008-5472.CAN-08-2331
60. Stecca B, Ruiz IAA. Context-dependent regulation of the gli code in cancer by hedgehog and non-hedgehog signals. J Mol Cell Biol (2010) 2:84–95. doi:10.1093/jmcb/mjp052
61. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require hedgehog-gli signaling regulated by interactions between gli1 and the ras-mek/akt pathways. Proc Natl Acad Sci U S A (2007) 104:5895–900. doi:10.1073/pnas.0700776104
62. O’Reilly KE, de Miera EV, Segura MF, Friedman E, Poliseno L, Han SW, et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel) (2013) 6:1429–50. doi:10.3390/ph6111429
63. Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D, et al. Pdgfralpha up-regulation mediated by sonic hedgehog pathway activation leads to braf inhibitor resistance in melanoma cells with braf mutation. Oncotarget (2014) 5:1926–41. doi:10.18632/oncotarget.1878
64. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer (2013) 13:184–99. doi:10.1038/nrc3431
65. Xing M. Braf mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev (2007) 28:742–62. doi:10.1210/er.2007-0007
66. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase akt pathway in human cancer. Nat Rev Cancer (2002) 2:489–501. doi:10.1038/nrc839
67. Wullschleger S, Loewith R, Hall MN. Tor signaling in growth and metabolism. Cell (2006) 124:471–84. doi:10.1016/j.cell.2006.01.016
68. Robbins HL, Hague A. The pi3k/akt pathway in tumors of endocrine tissues. Front Endocrinol (2015) 6:188. doi:10.3389/fendo.2015.00188
69. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. Pten, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275:1943–7. doi:10.1126/science.275.5308.1943
70. Newton AC, Trotman LC. Turning off akt: Phlpp as a drug target. Annu Rev Pharmacol Toxicol (2014) 54:537–58. doi:10.1146/annurev-pharmtox-011112-140338
71. Nitsche C, Edderkaoui M, Moore RM, Eibl G, Kasahara N, Treger J, et al. The phosphatase phlpp1 regulates akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology (2012) 142(377–387):e371–5. doi:10.1053/j.gastro.2011.10.026
72. Bhaskar PT, Hay N. The two torcs and akt. Dev Cell (2007) 12:487–502. doi:10.1016/j.devcel.2007.03.020
73. Schmelzle T, Hall MN. Tor, a central controller of cell growth. Cell (2000) 103:253–62. doi:10.1016/S0092-8674(00)00117-3
74. Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The crosstalk of mtor/s6k1 and hedgehog pathways. Cancer Cell (2012) 21:374–87. doi:10.1016/j.ccr.2011.12.028
75. Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the pi3k pathway in medulloblastoma. Sci Transl Med (2010) 2:51ra70. doi:10.1126/scitranslmed.3001599
76. Pon YL, Zhou HY, Cheung AN, Ngan HY, Wong AS. P70 s6 kinase promotes epithelial to mesenchymal transition through snail induction in ovarian cancer cells. Cancer Res (2008) 68:6524–32. doi:10.1158/0008-5472.CAN-07-6302
77. Ruiz i Altaba A. Hedgehog signaling and the gli code in stem cells, cancer, and metastases. Sci Signal (2011) 4:t9. doi:10.1126/scisignal.2002540
78. Kern D, Regl G, Hofbauer SW, Altenhofer P, Achatz G, Dlugosz A, et al. Hedgehog/gli and pi3k signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene (2015) 34:5341–51. doi:10.1038/onc.2014.450
79. Filbin MG, Dabral SK, Pazyra-Murphy MF, Ramkissoon S, Kung AL, Pak E, et al. Coordinate activation of shh and pi3k signaling in pten-deficient glioblastoma: new therapeutic opportunities. Nat Med (2013) 19:1518–23. doi:10.1038/nm.3328
80. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the pik3ca gene in human cancers. Science (2004) 304:554. doi:10.1126/science.1096502
81. Williamson AJ, Doscas ME, Ye J, Heiden KB, Xing M, Li Y, et al. The sonic hedgehog signaling pathway stimulates anaplastic thyroid cancer cell motility and invasiveness by activating akt and c-met. Oncotarget (2016) 7:10472–85. doi:10.18632/oncotarget.7228
82. Srivastava RK, Kaylani SZ, Edrees N, Li C, Talwelkar SS, Xu J, et al. Gli inhibitor gant-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting shh/akt-mtor axis. Oncotarget (2014) 5:12151–65. doi:10.18632/oncotarget.2569
83. Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, et al. Selective modulation of hedgehog/gli target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol (2006) 26:6283–98. doi:10.1128/MCB.02317-05
84. Unden AB, Zaphiropoulos PG, Bruce K, Toftgard R, Stahle-Backdahl M. Human patched (ptch) mRNA is overexpressed consistently in tumor cells of both familial and sporadic basal cell carcinoma. Cancer Res (1997) 57:2336–40.
85. Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, et al. Mutations of the patched gene in several types of sporadic extracutaneous tumors. Cancer Res (1997) 57:2369–72.
86. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature (1998) 391:90–2. doi:10.1038/34201
87. Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain ptch mutations. Cancer Res (1997) 57:842–5.
88. Zurawel RH, Allen C, Wechsler-Reya R, Scott MP, Raffel C. Evidence that haploinsufficiency of ptch leads to medulloblastoma in mice. Genes Chromosomes Cancer (2000) 28:77–81. doi:10.1002/(SICI)1098-2264(200005)28:1<77::AID-GCC9>3.0.CO;2-Y
89. Zurawel RH, Allen C, Chiappa S, Cato W, Biegel J, Cogen P, et al. Analysis of ptch/smo/shh pathway genes in medulloblastoma. Genes Chromosomes Cancer (2000) 27:44–51. doi:10.1002/(SICI)1098-2264(200001)27:1<44::AID-GCC6>3.0.CO;2-V
90. Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res (2004) 64:6071–4. doi:10.1158/0008-5472.CAN-04-0416
91. Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, et al. Inhibition of prostate cancer proliferation by interference with sonic hedgehog-gli1 signaling. Proc Natl Acad Sci U S A (2004) 101:12561–6. doi:10.1073/pnas.0404956101
92. Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature (2004) 431:707–12. doi:10.1038/nature02962
93. Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature (2003) 425:846–51. doi:10.1038/nature01972
94. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature (2003) 425:851–6. doi:10.1038/nature02009
95. Xu X, Ding H, Rao G, Arora S, Saclarides CP, Esparaz J, et al. Activation of the sonic hedgehog pathway in thyroid neoplasms and its potential role in tumor cell proliferation. Endocr Relat Cancer (2012) 19:167–79. doi:10.1530/ERC-11-0305
96. Nelson KK, Gattuso P, Xu X, Prinz RA. Expression of the sonic hedgehog pathway molecules in synchronous follicular adenoma and papillary carcinoma of the thyroid gland in predicting malignancy. Surgery (2010) 148:654–60; discussion 660. doi:10.1016/j.surg.2010.07.030
97. Bian XH, Sun H, Xue H, Zhang G, Zhang CH, Liu XL, et al. Expression and clinical significance of shh/gli-1 in papillary thyroid carcinoma. Tumour Biol (2014) 35(10):10523–8. doi:10.1007/s13277-014-2365-3
98. Hinterseher U, Wunderlich A, Roth S, Ramaswamy A, Bartsch DK, Hauptmann S, et al. Expression of hedgehog signalling pathway in anaplastic thyroid cancer. Endocrine (2014) 45:439–47. doi:10.1007/s12020-013-0015-y
99. Bohinc B, Michelotti G, Diehl AM. Hedgehog signaling in human medullary thyroid carcinoma: a novel signaling pathway. Thyroid (2013) 23:1119–26. doi:10.1089/thy.2012.0474
101. Zane M, Scavo E, Catalano V, Bonanno M, Todaro M, De Maria R, et al. Normal vs cancer thyroid stem cells: the road to transformation. Oncogene (2016) 35:805–15. doi:10.1038/onc.2015.138
102. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414:105–11. doi:10.1038/35102167
103. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (2011) 331:1559–64. doi:10.1126/science.1203543
104. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med (2011) 17:313–9. doi:10.1038/nm.2304
105. Nagayama Y, Shimamura M, Mitsutake N. Cancer stem cells in the thyroid. Front Endocrinol (2016) 7:20. doi:10.3389/fendo.2016.00020
106. Guo Z, Hardin H, Lloyd RV. Cancer stem-like cells and thyroid cancer. Endocr Relat Cancer (2014) 21:T285–300. doi:10.1530/ERC-14-0002
107. Davies TF, Latif R, Minsky NC, Ma R. Clinical review: the emerging cell biology of thyroid stem cells. J Clin Endocrinol Metab (2011) 96:2692–702. doi:10.1210/jc.2011-1047
108. Klonisch T, Hoang-Vu C, Hombach-Klonisch S. Thyroid stem cells and cancer. Thyroid (2009) 19:1303–15. doi:10.1089/thy.2009.1604
109. Hoshi N, Kusakabe T, Taylor BJ, Kimura S. Side population cells in the mouse thyroid exhibit stem/progenitor cell-like characteristics. Endocrinology (2007) 148:4251–8. doi:10.1210/en.2006-0490
110. Lan L, Cui D, Nowka K, Derwahl M. Stem cells derived from goiters in adults form spheres in response to intense growth stimulation and require thyrotropin for differentiation into thyrocytes. J Clin Endocrinol Metab (2007) 92:3681–8. doi:10.1210/jc.2007-0281
111. Zheng X, Cui D, Xu S, Brabant G, Derwahl M. Doxorubicin fails to eradicate cancer stem cells derived from anaplastic thyroid carcinoma cells: characterization of resistant cells. Int J Oncol (2010) 37:307–15. doi:10.3892/ijo_00000679
112. Mitsutake N, Iwao A, Nagai K, Namba H, Ohtsuru A, Saenko V, et al. Characterization of side population in thyroid cancer cell lines: cancer stem-like cells are enriched partly but not exclusively. Endocrinology (2007) 148:1797–803. doi:10.1210/en.2006-1553
113. Todaro M, Iovino F, Eterno V, Cammareri P, Gambara G, Espina V, et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res (2010) 70:8874–85. doi:10.1158/0008-5472.CAN-10-1994
114. Hardin H, Yu XM, Harrison AD, Larrain C, Zhang R, Chen J, et al. Generation of novel thyroid cancer stem-like cell clones: effects of resveratrol and valproic acid. Am J Pathol (2016) 186:1662–73. doi:10.1016/j.ajpath.2016.02.003
115. Shimamura M, Kurashige T, Mitsutake N, Nagayama Y. Aldehyde dehydrogenase activity plays no functional role in stem cell-like properties in anaplastic thyroid cancer cell lines. Endocrine (2017) 55:934–43. doi:10.1007/s12020-016-1224-y
116. Ma R, Minsky N, Morshed SA, Davies TF. Stemness in human thyroid cancers and derived cell lines: the role of asymmetrically dividing cancer stem cells resistant to chemotherapy. J Clin Endocrinol Metab (2014) 99:E400–9. doi:10.1210/jc.2013-3545
117. Merchant AA, Matsui W. Targeting hedgehog – a cancer stem cell pathway. Clin Cancer Res (2010) 16:3130–40. doi:10.1158/1078-0432.CCR-09-2846
118. Woodward WA, Chen MS, Behbod F, Rosen JM. On mammary stem cells. J Cell Sci (2005) 118:3585–94. doi:10.1242/jcs.02532
119. Kasper M, Jaks V, Fiaschi M, Toftgard R. Hedgehog signalling in breast cancer. Carcinogenesis (2009) 30:903–11. doi:10.1093/carcin/bgp048
120. Satheesha S, Manzella G, Bovay A, Casanova EA, Bode PK, Belle R, et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene (2016) 35:2020–30. doi:10.1038/onc.2015.267
121. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A (2007) 104:4048–53. doi:10.1073/pnas.0611682104
122. Naka K, Hoshii T, Hirao A. Novel therapeutic approach to eradicate tyrosine kinase inhibitor resistant chronic myeloid leukemia stem cells. Cancer Sci (2010) 101:1577–81. doi:10.1111/j.1349-7006.2010.01584.x
123. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature (2009) 458:776–9. doi:10.1038/nature07737
124. Nicolis SK. Cancer stem cells and “stemness” genes in neuro-oncology. Neurobiol Dis (2007) 25:217–29. doi:10.1016/j.nbd.2006.08.022
125. Xu Q, Yuan X, Liu G, Black KL, Yu JS. Hedgehog signaling regulates brain tumor-initiating cell proliferation and portends shorter survival for patients with pten-coexpressing glioblastomas. Stem Cells (2008) 26:3018–26. doi:10.1634/stemcells.2008-0459
126. Heiden KB, Williamson AJ, Doscas ME, Ye J, Wang Y, Liu D, et al. The sonic hedgehog signaling pathway maintains thyroid cancer stem cell self-renewal by inducing snail expression. J Clin Endocrinol Metab (2014) 99(11):E2178–87. doi:10.1210/jc.2014-1844
127. Malaguarnera R, Frasca F, Garozzo A, Giani F, Pandini G, Vella V, et al. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab (2011) 96:766–74. doi:10.1210/jc.2010-1255
128. Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. J Clin Endocrinol Metab (2012) 97:E510–20. doi:10.1210/jc.2011-1754
129. Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol (2012) 198:281–93. doi:10.1083/jcb.201202014
130. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell (2007) 1:313–23. doi:10.1016/j.stem.2007.06.002
131. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res (2006) 66:6063–71. doi:10.1158/0008-5472.CAN-06-0054
132. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. Hedgehog-gli1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17:165–72. doi:10.1016/j.cub.2006.11.033
133. Wang X, Zhang N, Huo Q, Sun M, Dong L, Zhang Y, et al. Huaier aqueous extract inhibits stem-like characteristics of mcf7 breast cancer cells via inactivation of hedgehog pathway. Tumour Biol (2014) 35:10805–13. doi:10.1007/s13277-014-2390-2
134. Batsaikhan BE, Yoshikawa K, Kurita N, Iwata T, Takasu C, Kashihara H, et al. Cyclopamine decreased the expression of sonic hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res (2014) 34:6339–44.
135. Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer (2012) 131:30–40. doi:10.1002/ijc.26323
136. Santini R, Pietrobono S, Pandolfi S, Montagnani V, D’Amico M, Penachioni JY, et al. Sox2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene (2014) 33:4697–708. doi:10.1038/onc.2014.71
137. Li X, Deng W, Lobo-Ruppert SM, Ruppert JM. Gli1 acts through snail and e-cadherin to promote nuclear signaling by beta-catenin. Oncogene (2007) 26:4489–98. doi:10.1038/sj.onc.1210241
138. Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, et al. Snail induction is an early response to gli1 that determines the efficiency of epithelial transformation. Oncogene (2006) 25:609–21. doi:10.1038/sj.onc.1209077
139. Baulida J, Garcia de Herreros A. Snail1-driven plasticity of epithelial and mesenchymal cells sustains cancer malignancy. Biochim Biophys Acta (2015) 1856:55–61. doi:10.1016/j.bbcan.2015.05.005
140. Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein kinase c alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell (2013) 24:347–64. doi:10.1016/j.ccr.2013.08.005
141. Zhu LF, Hu Y, Yang CC, Xu XH, Ning TY, Wang ZL, et al. Snail overexpression induces an epithelial to mesenchymal transition and cancer stem cell-like properties in scc9 cells. Lab Invest (2012) 92:744–52. doi:10.1038/labinvest.2012.8
142. Hardy RG, Vicente-Duenas C, Gonzalez-Herrero I, Anderson C, Flores T, Hughes S, et al. Snail family transcription factors are implicated in thyroid carcinogenesis. Am J Pathol (2007) 171:1037–46. doi:10.2353/ajpath.2007.061211
143. Yasui K, Shimamura M, Mitsutake N, Nagayama Y. Snail induces epithelial-to-mesenchymal transition and cancer stem cell-like properties in aldehyde dehydroghenase-negative thyroid cancer cells. Thyroid (2013) 23:989–96. doi:10.1089/thy.2012.0319
144. Baquero P, Sanchez-Hernandez I, Jimenez-Mora E, Orgaz JL, Jimenez B, Chiloeches A. (v600e)braf promotes invasiveness of thyroid cancer cells by decreasing e-cadherin expression through a snail-dependent mechanism. Cancer Lett (2013) 335:232–41. doi:10.1016/j.canlet.2013.02.033
145. Siddique HR, Saleem M. Role of bmi1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells (2012) 30:372–8. doi:10.1002/stem.1035
146. Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, et al. The polycomb group protein bmi-1 represses the tumor suppressor pten and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest (2009) 119:3626–36. doi:10.1172/JCI39374
147. Chiba T, Seki A, Aoki R, Ichikawa H, Negishi M, Miyagi S, et al. Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both ink4a/arf-dependent and -independent manners in mice. Hepatology (2010) 52:1111–23. doi:10.1002/hep.23793
148. Chiba T, Miyagi S, Saraya A, Aoki R, Seki A, Morita Y, et al. The polycomb gene product bmi1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res (2008) 68:7742–9. doi:10.1158/0008-5472.CAN-07-5882
149. Proctor E, Waghray M, Lee CJ, Heidt DG, Yalamanchili M, Li C, et al. Bmi1 enhances tumorigenicity and cancer stem cell function in pancreatic adenocarcinoma. PLoS One (2013) 8:e55820. doi:10.1371/journal.pone.0055820
150. Yu CC, Lo WL, Chen YW, Huang PI, Hsu HS, Tseng LM, et al. Bmi-1 regulates snail expression and promotes metastasis ability in head and neck squamous cancer-derived aldh1 positive cells. J Oncol (2011) 2011:1–16. doi:10.1155/2011/609259
151. Chou CH, Yang NK, Liu TY, Tai SK, Hsu DS, Chen YW, et al. Chromosome instability modulated by bmi1-aurka signaling drives progression in head and neck cancer. Cancer Res (2013) 73:953–66. doi:10.1158/0008-5472.CAN-12-2397
152. Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, et al. Bmi1 is essential in twist1-induced epithelial-mesenchymal transition. Nat Cell Biol (2010) 12:982–92. doi:10.1038/ncb2099
153. Wu J, Hu D, Yang G, Zhou J, Yang C, Gao Y, et al. Down-regulation of bmi-1 cooperates with artemisinin on growth inhibition of nasopharyngeal carcinoma cells. J Cell Biochem (2011) 112:1938–48. doi:10.1002/jcb.23114
154. Ferretti R, Bhutkar A, McNamara MC, Lees JA. Bmi1 induces an invasive signature in melanoma that promotes metastasis and chemoresistance. Genes Dev (2016) 30:18–33. doi:10.1101/gad.267757.115
155. Wang X, Venugopal C, Manoranjan B, McFarlane N, O’Farrell E, Nolte S, et al. Sonic hedgehog regulates bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene (2011) 31:187–99. doi:10.1038/onc.2011.232
156. Gopinath S, Malla R, Alapati K, Gorantla B, Gujrati M, Dinh DH, et al. Cathepsin b and upar regulate self-renewal of glioma-initiating cells through gli-regulated sox2 and bmi1 expression. Carcinogenesis (2013) 34:550–9. doi:10.1093/carcin/bgs375
157. Carina V, Zito G, Pizzolanti G, Richiusa P, Criscimanna A, Rodolico V, et al. Multiple pluripotent stem cell markers in human anaplastic thyroid cancer: the putative upstream role of sox2. Thyroid (2013) 23:829–37. doi:10.1089/thy.2012.0372
158. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat Med (2013) 19:1410–22. doi:10.1038/nm.3389
159. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and gli inhibitors. Cancers (Basel) (2016) 8:1–23. doi:10.3390/cancers8020022
160. Pandolfi S, Stecca B. Cooperative integration between hedgehog-gli signalling and other oncogenic pathways: implications for cancer therapy. Expert Rev Mol Med (2015) 17:e5. doi:10.1017/erm.2015.3
Keywords: thyroid neoplasms, sonic hedgehog, cancer stem cells, phosphortidylinositol-3 kinase, MAP kinase signaling system, BRAF
Citation: Xu X, Lu Y, Li Y and Prinz RA (2017) Sonic Hedgehog Signaling in Thyroid Cancer. Front. Endocrinol. 8:284. doi: 10.3389/fendo.2017.00284
Received: 14 July 2017; Accepted: 10 October 2017;
Published: 30 October 2017
Edited by:
Angela Hague, University of Bristol, United KingdomReviewed by:
Marialuisa Appetecchia, Istituti Fisioterapici Ospitalieri (IRCCS), ItalyMichaela Luconi, University of Florence, Italy
Copyright: © 2017 Xu, Lu, Li and Prinz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiulong Xu, xxl@yzu.edu.cn, xxu@rush.edu