 Michele Podetta1Marc Pusztaszeri2
Michele Podetta1Marc Pusztaszeri2 Christian Toso1Michel Procopiou3
Christian Toso1Michel Procopiou3 Frédéric Triponez4
Frédéric Triponez4 Samira Mercedes Sadowski4*
Samira Mercedes Sadowski4*
- 1Department of Visceral Surgery, University Hospitals of Geneva, Geneva, Switzerland
- 2Department of Clinical Pathology, University Hospitals of Geneva, Geneva, Switzerland
- 3Private Practice, Endocrinologist, Neuchâtel, Switzerland
- 4Department of Thoracic and Endocrine Surgery, University Hospitals of Geneva, Geneva, Switzerland
Adrenal oncocytoma (AO) is an extremely rare adrenocortical neoplasm and little is known about its malignant potential, secretory properties, and hereditary origin. We present the case of a benign AO with concomitant incidentally found papillary thyroid cancer (PTC) and review similar cases in the literature. Immunohistochemistry and next-generation sequencing (NGS) were performed. A 66-year-old women was incidentally found to have a large, androgen-secreting right adrenal mass. 18F-Fluorodeoxyglucose positron emission tomography showed intense uptake (SUVmax 88.7) of this mass and found a hypermetabolic right thyroid mass. Open adrenalectomy was performed for this highly suspicious adrenal mass. Histopathology revealed benign AO that was BRAFV600E negative, with low Ki-67, and no somatic mutation found on NGS. Thyroidectomy revealed invasive, BRAFV600E-positive PTC. At 6 months follow-up, androgen levels returned to normal, and no recurrence was seen on imaging. To our knowledge, this is the first report of an androgen-secreting AO with concomitant PTC. Possibly the simultaneous discovery of two independent neoplasms was observed. In conclusion, this case highlights that care should be given to exclude concomitant neoplasms. Long-term and regular imaging with biochemical follow-up is warranted, since the outcome and clinical behavior of AO remains uncertain.
Background
Oncocytic neoplasms occur in various organs such as the kidneys, thyroid, parathyroid, or salivary glands. An adrenal location, called adrenal oncocytoma (AO), is extremely rare with about 150 reported cases. They have been considered benign non-functional neoplasms; however, in about 20% of cases, they show malignant potential, and in 15% of cases, adrenocortical hormone secretion was detected (1). In most cases, these adrenal masses were detected incidentally at a large size and were thus suspicious for adrenocortical carcinoma (ACC) (1). Computed tomography (CT) scan or magnetic resonance imaging is weak in defining the nature of large adrenal masses, and the role of 18F-FDG positron emission tomography (PET) remains uncertain. Fine needle aspiration (FNA) biopsy does not allow for the diagnosis of AO, and since their biological behavior is uncertain, adrenalectomy remains the only reliable diagnostic and therapeutic option.
The incidence of thyroid cancer (TC) is about 3 cases per 100,000 population, and papillary thyroid cancer (PTC) represents 85% of TC, with a good prognosis (2). Mutations with activation of MAPK and PI3K-AKT signaling pathways are crucial in the pathogenesis of PTC. Common alterations are point mutations of BRAF (most commonly V600E) and RAS genes as well as RET/PTC and PAX8/PPARγ chromosomal rearrangements. The type of alteration in PTC seems to be linked to specific etiologic factors. Rearrangements have a strong association with exposure to ionizing radiation and possibly DNA fragility, whereas point mutations probably arise as a result of chemical mutagenesis (3).
No endocrine syndrome is known associating AO and PTC. Given the presentation of two rare endocrine tumors in this patient, a possible hereditary link was suggested, especially when initially suspecting an ACC. None of the known syndromes caused by hereditary predisposition specifically include PTC and ACC or adrenal adenoma. Multiple endocrine neoplasia (MEN) Type 1 (Menin gene) is known to present with multiples combinations, including ACC but not PTC (4) and MEN Type 2 syndromes (RET gene) include pheochromocytoma and medullary TC, but do not traditionally include PTC or ACC. ACC is known to appear in Li–Fraumeni syndrome with TP53 mutation. There are four reports that attribute the association of ACC and PTC to coincidence, but do not discount a potential genetic or hereditary link (5–8).
We describe a case with two incidentally found endocrine tumors, an androgen-secreting AO mimicking ACC and a PTC.
Case Presentation
A 66-year-old women was admitted with a complaint of transient global amnesia and abdominal pain. She mentioned a 4-year history of fatigue with occasional headaches and epigastric burn-like pain persisting for 1 month. No previous episodes of pain, weight loss, or family history for endocrine malignancy were reported. Physical examination revealed painful upper right quadrant palpation and the absence of Cushing syndrome features or hirsutism. Abdominal ultrasound (US) revealed a hypoechoic mass, and CT scan revealed a 7.6 cm × 5.6 cm × 7.4 cm right adrenal mass, hyperdense (>50 Hounsfield units) with delayed wash out after contrast injection (Figures 1A,B). The left adrenal was normal. Further endocrine work-up revealed normal cortisol after 1 mg of dexamethasone (43 nmol/l), normal 24-hour urinary excretion (156 nmol/24 h), and normal free plasma metanephrines. Dehydroepiandrosterone sulfate (DHEAS) was increased after dexamethasone suppression at 21.7 μmol/l (range, 0.9–2.0), testosterone at 2.6 nmol/l (range, 0.5–1.9), and estradiol at 237 pmol/l (<103 postmenopausal). TSH and calcium levels were normal.
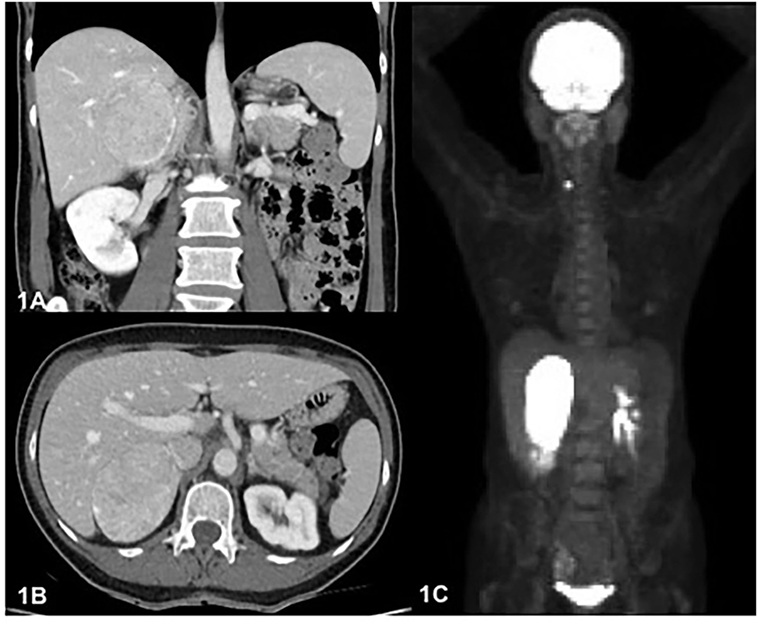
Figure 1. (A). Axial image on portal phase computed tomography (CT) scan showing right adrenal mass heterogeneously enhanced of 7.6 cm × 5.8 cm. (B). Cross-sectional image on CT scan showing right adrenal mass with imprint on the liver. (C). Maximum intensity projection image on FDG positron emission tomography showing the two hypermetabolic masses, right adrenal, and right thyroid lobe.
Because of suspected malignancy, a 18F-FDG PET was performed, revealing a highly metabolic 7.3 cm × 5.6 cm × 8.1 cm right adrenal mass (SUVmax 88.7) with a mass effect on the liver (Figures 1A–C and 2A). Hypodense areas within the mass suggested necrotic spots, however, without local infiltrative signs. In addition, a hypermetabolic right thyroid lesion of 1.2 cm was found (SUVmax 10.6; Figure 2B) without suspicious lymph nodes, and US-guided FNA revealed PTC.
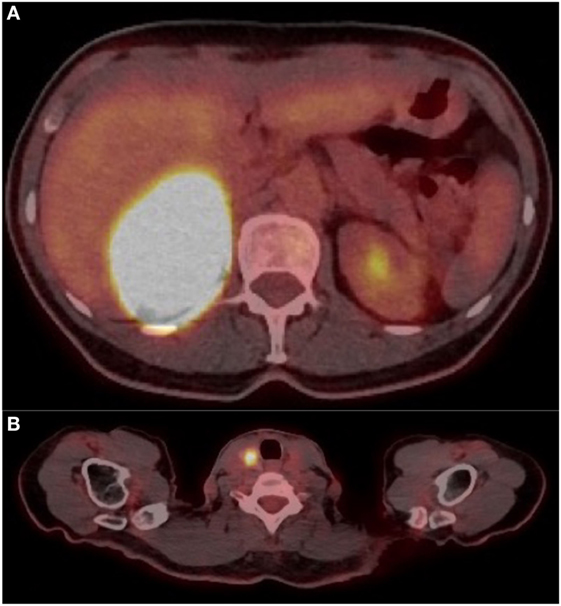
Figure 2. (A) Cross-sectional image on FDG positron emission tomography (PET) showing right adrenal mass at SUV max 88.7. (B) Cross-sectional image on FDG PET showing right thyroid mass at SUV max 10.6.
Based on the size, hypermetabolic activity, and androgen secretion, malignancy of the adrenal mass was suspected, and an open right adrenalectomy was performed. The adrenal mass was well encapsulated, without any intraoperative signs of local invasion. Synchronous total thyroidectomy and right level VI node dissection were performed. Isthmus and left thyroid lobe were found to be absent (thyroid hemiagenesis).
Histopathology of the adrenal showed a rounded, well-circumscribed, and encapsulated mass, measuring 8.5 cm, without necrosis (Figure 3A). The margins were negative on examination. Microscopically, the tumor was entirely composed of oncocytic cells (Figures 3B,C). The Lin–Weiss–Bisceglia criteria were absent, and the mass was classified a benign AO. Further evaluation using immunohistochemistry revealed negative BRAFV600E immunostaining and a low Ki-67 proliferation index (<2%) (Figure 3D). In addition, next-generation sequencing (NGS) was performed on the adrenal tissue, including known mutations in ACC [p53, RB1, and CTNNB1 (9)]; 2,800 (hotspot) mutations in a classic panel of 50 genes associated with different cancers (Ion Ampliseq Cancer Hotspot v2) were assessed using the Ion Proton™ system (see Table S1 in Supplementary Material for list of the 50 tested genes). NGS did not reveal any mutations for this adrenal mass. Histopathology of the thyroid gland revealed an invasive PTC of 1.2 cm, BRAFV600E positive, without lymphovascular invasion (Figures 4A–D), staged pT1b, N1a, R1 (3/4 positive central lymph nodes, R1: microscopically positive resection margin).
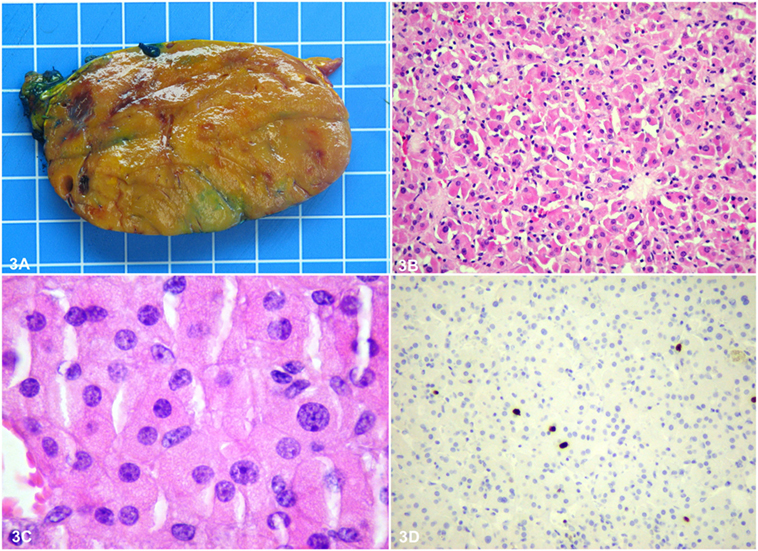
Figure 3. Macroscopically, the adrenal tumor was a rounded, well-circumscribed orange brown homogeneous mass, measuring 8.5 cm × 6 cm × 4.5 cm (A). The size of the grid in the background is 1 cm × 1 cm. Microscopically, it was well circumscribed and partially encapsulated and consisted entirely of bland looking oncocytic cells, with granular eosinophilic cytoplasm and round nuclei with prominent nucleoli, arranged in a solid pattern (B,C). Parenchymatous or venous invasion, high mitotic rate, and atypical mitoses were not observed. Therefore, according to the Lin–Weiss–Bisceglia criteria, it was diagnosed as a benign oncocytic adrenocortical adenoma. Further evaluation using immunohistochemistry revealed negative BRAF V600E immunostaining and a low Ki-67 proliferation index (<2%) (D).
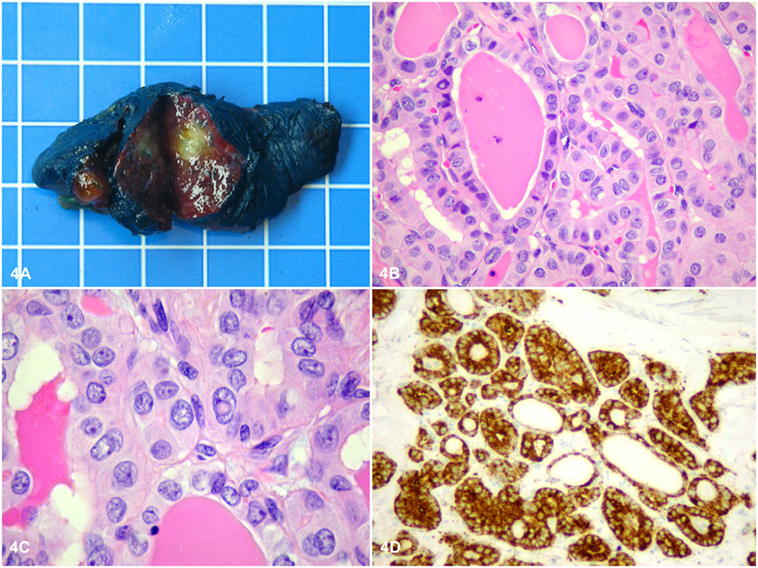
Figure 4. Macroscopically, the thyroid tumor was a rounded, well-circumscribed white intrathyroidal nodule measuring 1.2 cm × 1 cm × 0.8 cm (A). The size of the grid in the background is 1 cm × 1 cm. Microscopically, the tumor consisted of cuboidal cells arranged mostly in follicles with a few papillae, showing classical nuclear features of papillary thyroid cancer (PTC), including chromatin pallor, numerous nuclear grooves, and several nuclear pseudoinclusions (B,C). Further evaluation using immunohistochemistry revealed positive BRAF V600E immunostaining in the tumor cells (D), consistent with the classical type of PTC (BRAF-like PTC).
Postoperative recovery was uneventful, and patient was started on thyroid hormone replacement therapy. According to ATA guidelines (2) and local invasiveness, the patient underwent I131 radioactive iodine therapy. Postoperative work-up at 2 months showed normalization of DHEAS to 1.7 μmol/l and morning cortisol at 464 nmol/l, as well as testosterone level at 0.8 nmol/l. Corrected calcium was normal at 2.32 mmol/l and thyroglobulin at <0.10 μg/l. For the thyroid lesion, the patient will be followed up by measuring stimulated thyroglobulin after radioactive iodine therapy, and aiming for a suppressed TSH, as well as by performing a neck ultrasonography and thyroglobulin at 6-month intervals. For the adrenal lesion, at 6-month follow-up, the androgen levels stayed normal (1.7 μmol/l), and no recurrence was seen on CT scan imaging. Regular imaging and biochemical follow-up at 12 and 18 months for a minimum of 5 years are planned.
Discussion
We describe a female patient with a highly suspicious adrenal mass that reveals to be a benign lesion based on the pathological criteria, without somatic mutations, and who presents with a simultaneous endocrine malignancy of the thyroid gland.
Adrenal oncocytoma is a rare neoplasm arising from the adrenal cortex. Malignant oncocytoma and ACC are large, heterogeneous, with internal necrosis and associated with slow wash out, such as found in this case’s preoperative imaging. The concern for metastatic disease led us to perform a 18F-FDG, which revealed a highly metabolic adrenal mass and the possibility of another synchronous primitive tumor or metastasis (Figures 1C, 2B); ultimately it led to the detection of concomitant PTC. Adrenal metastases from PTC have been described (10, 11). In reported cases, metastases were smaller than this case’s mass, and the adrenal tumors were detected by 18F-FDG, but were not as metabolically active (10). Little is known about the 18F-FDG behavior of AO. Some cases of high metabolic uptake have been reported, with uptake explained by intense activity of glucose metabolism or the high number of mitochondria known to be present in oncocytic cells (12, 13); and high uptake has been described in other oncocytic lesions such as in lung carcinoids (14).
The elements such size, hypermetabolic activity, and androgen secretion led us to suspect a malignant primary adrenal mass or a metastatic tumor, possibly invading the liver, and, therefore, open adrenalectomy was preferred for radical tumor resection (15). The safety of laparoscopic techniques for large AO remains a matter of debate.
Adrenal oncocytoma are usually non-functional, and only about 10–20% of cases present with adrenocortical hormone production. This case presented oversecretion of androgen pathway hormones with elevated plasma levels of DHEAS, testosterone, and estradiol. As has been shown in published reports, androgen-secreting lesions increase the likelihood of adrenal malignancy, a finding considered to be suspicious by the authors (16, 17). In literature, very few cases of virilizing AO have been reported in female children or women; onset symptomatology reported was pseudoprecocious puberty, hirsutism, or deepening of the voice (18–20). Another clinical presentation of functional AO may be Cushing’s syndrome (21) or pheochromocytoma-like syndrome (22, 23). In few cases, a co-secretion of cortisol with testosterone and aldosterone was reported (24, 25).
Histopathology characteristics of this case meet the typical features of AO, assessed by the Lin–Weiss–Bisceglia criteria. If one or more of the major criteria (a mitotic rate of more than 5 mitoses per 50 high power fields, any atypical mitosis or venous invasion) is detected, the tumor is considered malignant. Large-sized tumors (>10 cm and/or >200 g), necrosis, capsular, and sinusoidal invasion are minor criteria to define an oncocytic tumor as borderline/uncertain malignant potential (1). Our patient presented a benign oncocytic tumor, in which situation, the prognosis seems excellent, and no adjuvant treatment was recommended after radical surgery. Considering the benign pattern of AO, we do not believe the outcome of PTC to be worse for this patient. Regular imaging follow-up at 12 and 18 months for a minimum of 5 years is planned as well as biochemical follow-up for this adrenal lesion of uncertain clinical behavior.
To our knowledge, no hereditary syndrome is known comprising AO and PTC. The AO was BRAFV600E negative, and no somatic mutation was found, while the PTC was BRAFV600E positive. However, we did not perform germline mutation analysis. In literature, two cases of functional adrenal adenoma (5, 6) and two cases of ACC presenting simultaneously with PTC have been reported (7, 8). Wanta et al. (8) performed germline mutation analysis and found wild-type p53 and no mutation in the Menin gene, thus concluding the absence of a hereditary cancer syndrome. Only one case of borderline malignant non-functional AO with simultaneous PTC has been reported (26). However, no mutation analyses had been performed.
Concluding Remarks
In conclusion, we present the first case of an androgen-secreting AO with concomitant PTC. We probably observed the simultaneous incidental discovery of two independent endocrine neoplasms. Regular and long-term imaging with biochemical follow-up is recommended, since the outcome and clinical behavior of AO remain uncertain.
Ethics Statement
The patient provided written informed consent for research participation and for the publication of indirectly identifiable data.
Author Contributions
Wrote the first draft of the manuscript: MPod. Contributed to the writing of the manuscript: SS. Made contributions to the acquisition of the clinical data: MPod, MPu, CT, MPro, FT, and SS. Performed pathological analyses: MPu. Agreed with manuscript results and conclusions: MPod, MPu, CT, MPro, FT, and SS. Made critical revisions and approved final version: MPu, FT, and SS. All the authors revised and approved the final manuscript and agreed to be accountable for the content of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JL and handling editor declared their shared affiliation.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fendo.2017.00384/full#supplementary-material.
References
1. Bisceglia M, Ludovico O, Di Mattia A, Ben-Dor D, Sandbank J, Pasquinelli G, et al. Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol (2004) 12(3):231–43. doi:10.1177/106689690401200304
2. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association Guidelines Task Force on thyroid nodules and differentiated thyroid cancer. Thyroid (2016) 26(1):1–133. doi:10.1089/thy.2015.0020
3. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol (2011) 7(10):569–80. doi:10.1038/nrendo.2011.142
4. Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, et al. Multiple endocrine neoplasia type 1 (MEN1): not only inherited endocrine tumors. Genet Med (2009) 11(12):825–35. doi:10.1097/GIM.0b013e3181be5c97
5. Casula G, Angioy F, Sirigu P, Sirigu F. Carcinoma of the thyroid gland, adenoma of the adrenal cortex and peptic ulcer: an unreported association. Tumori (1976) 62(6):665–72.
6. Fukushima A, Okada Y, Tanikawa T, Kawahara C, Misawa H, Kanda K, et al. Virilizing adrenocortical adenoma with Cushing’s syndrome, thyroid papillary carcinoma and hypergastrinemia in a middle-aged woman. Endocr J (2003) 50(2):179–87. doi:10.1507/endocrj.50.179
7. Noordzij MJ, de Heide LJ, Links TP, Jager PL, Wolfenbuttel BH. [Four patients with incidentalomas of the thyroid discovered on 18-fluoro-deoxyglucose positron-emission tomography (FDG-PET)]. Ned Tijdschr Geneeskd (2007) 151(42):2337–41.
8. Wanta SM, Basina M, Chang SD, Chang DT, Ford JM, Greco R, et al. A rare case of an aldosterone secreting metastatic adrenocortical carcinoma and papillary thyroid carcinoma in a 31-year-old male. Rare Tumors (2011) 3(4):e45. doi:10.4081/rt.2011.e45
9. Nicolson NG, Man J, Carling T. Advances in understanding the molecular underpinnings of adrenocortical tumors. Curr Opin Oncol (2017) 30(1):16–22. doi:10.1097/CCO.0000000000000415
10. Wagenaar N, Oosterhuis JW, Rozendaal L, Comans E, Simsek S. Adrenal metastasis from a primary papillary thyroid carcinoma. Intern Med (2008) 47(24):2165–8. doi:10.2169/internalmedicine.47.1582
11. Batawil N. Papillary thyroid cancer with bilateral adrenal metastases. Thyroid (2013) 23(12):1651–4. doi:10.1089/thy.2013.0065
12. Kim DJ, Chung JJ, Ryu YH, Hong SW, Yu JS, Kim JH. Adrenocortical oncocytoma displaying intense activity on 18F-FDG-PET: a case report and a literature review. Ann Nucl Med (2008) 22(9):821–4. doi:10.1007/s12149-008-0199-z
13. Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: why choose inefficient mitochondria? Biochim Biophys Acta (2011) 1807(6):633–42. doi:10.1016/j.bbabio.2010.08.006
14. Kadowaki T, Yano S, Araki K, Tokushima T, Morioka N. A case of pulmonary typical carcinoid with an extensive oncocytic component showing intense uptake of FDG. Thorax (2011) 66(4):361–2. doi:10.1136/thx.2010.135525
15. Gaujoux S, Mihai R; Joint Working Group of ESES and ENSAT. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma. Br J Surg (2017) 104(4):358–76. doi:10.1002/bjs.10414
16. Moreno S, Montoya G, Armstrong J, Leteurtre E, Aubert S, Vantyghem MC, et al. Profile and outcome of pure androgen-secreting adrenal tumors in women: experience of 21 cases. Surgery (2004) 136(6):1192–8. doi:10.1016/j.surg.2004.06.046
17. Messer CK, Kirschenbaum A, New MI, Unger P, Gabrilove JL, Levine AC. Concomitant secretion of glucocorticoid, androgens, and mineralocorticoid by an adrenocortical carcinoma: case report and review of literature. Endocr Pract (2007) 13(4):408–12. doi:10.4158/EP.13.4.408
18. Gumy-Pause F, Bongiovanni M, Wildhaber B, Jenkins JJ, Chardot C, Ozsahin H. Adrenocortical oncocytoma in a child. Pediatr Blood Cancer (2008) 50(3):718–21. doi:10.1002/pbc.21090
19. Tahar GT, Nejib KN, Sadok SS, Rachid LM. Adrenocortical oncocytoma: a case report and review of literature. J Pediatr Surg (2008) 43(5):E1–3. doi:10.1016/j.jpedsurg.2007.12.067
20. Sahin SB, Yucel AF, Bedir R, Ogullar S, Ayaz T, Algun E. Testosterone- and cortisol-secreting adrenocortical oncocytoma: an unusual cause of hirsutism. Case Rep Endocrinol (2014) 2014:206890. doi:10.1155/2014/206890
21. Lee SS, Baek KH, Lee YS, Lee JM, Kang MI, Cha BY, et al. Subclinical Cushing’s syndrome associated with an adrenocortical oncocytoma. J Endocrinol Invest (2008) 31(7):675–9. doi:10.1007/BF03345624
22. Goel T, Thomas J, Garg S, Rao AC, Reddy S. Adrenal oncocytoma masquerading as a functional tumor. Indian J Urol (2007) 23(1):77–8. doi:10.4103/0970-1591.30275
23. Kiriakopoulos A, Papaioannou D, Linos D. Adrenal cortical oncocytoma mimicking pheochromocytoma. Hormones (Athens) (2011) 10(1):76–9. doi:10.14310/horm.2002.1296
24. Ali AE, Raphael SJ. Functional oncocytic adrenocortical carcinoma. Endocr Pathol (2007) 18(3):187–9. doi:10.1007/s12022-007-9000-4
25. Logasundaram R, Parkinson C, Donaldson P, Coode PE. Co-secretion of testosterone and cortisol by a functional adrenocortical oncocytoma. Histopathology (2007) 51(3):418–20. doi:10.1111/j.1365-2559.2007.02780.x
Keywords: adrenal oncocytoma, papillary thyroid cancer, hereditary cancer syndrome, functional adrenal neoplasm, concomitant neoplasia
Citation: Podetta M, Pusztaszeri M, Toso C, Procopiou M, Triponez F and Sadowski SM (2018) Oncocytic Adrenocortical Neoplasm with Concomitant Papillary Thyroid Cancer. Front. Endocrinol. 8:384. doi: 10.3389/fendo.2017.00384
Received: 30 August 2017; Accepted: 28 December 2017;
Published: 22 January 2018
Edited by:
Leon Bach, Monash University, AustraliaReviewed by:
Matthias Kroiss, University of Würzburg, GermanySalvatore Ulisse, Sapienza Università di Roma, Italy
James Cheng-yen Lee, Monash University, Australia
Copyright: © 2018 Podetta, Pusztaszeri, Toso, Procopiou, Triponez and Sadowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samira Mercedes Sadowski, samira.sadowski@hcuge.ch